
#23 | Protein-polymer conjugates as a new type of artificial enzymes |
|
| Presenting author: | Changzhu WU of UNIVERSITY OF SOUTHERN DENMARK |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial enzyme / Artificial polyenzyme / polymer-protein conjugate / Asymmetric reaction |
| Purpose: | Artificial enzymes, usually created by incorporating one or two abiological catalytic species into protein hosts, are becoming promising tools for biocatalysis. However, they are still far behind natural enzymes due to their limited active sites with low activity and selectivity. Here, I present a new approach to preparing artificial enzymes by combining proteins with catalytically active polymers, generating so-called “artificial polyenzymes” (ArPoly), see Figure 1. Different from traditional artificial enzymes, ArPoly can be easily tailored for their structure, composition, catalysis loading, and active sites. This tunability allows ArPoly to carry out the catalysis with not only high reactivity but also new-to-nature selectivity. Our first proof-of-concept study is to combine protein scaffolds with proline polymers using the atom transfer radical polymerization (ATRP) method to graft polymer from proteins.[1] Surprisingly, the resultant ArPoly is highly water-soluble, allowing for the asymmetric aldol reaction in pure water, which is the first report for water-soluble proline catalysts. Our study further suggests that this new-to-nature reactivity is due to the synergetic effects between proline catalysts and protein scaffolds. Taking advantage of high controllability, the polymer structure on ArPoly can be tailored. In this context, polystyrene and polyproline are copolymerized from the protein surface with their composition finely adjusted.[2] Since the hydrophobic microenvironment promotes proline-catalyzed aldol reactions, the tailored ArPoly containing polystyrene contributes to remarkable catalytic efficiency and selectivity (i.e., 94% conversion and 98% ee), which is a significant improvement compared to the prototype ArPoly. In addition to catalytic polymers, active ligands are polymerized to protein, thereafter, coordinated with metal ions, generating metal-containing artificial polyenzymes. To demonstrate this possibility, ArPoly is conjugated with proline polymers that are in situ coordinated with Cu (II) to form a metal complex during the ARTP preparation.[3] The resulting ArPoly is then used as a clickase for highly efficient click reactions. Importantly, the artificial clickase is biocompatible, causing no cytotoxicity cells, thus becoming promising catalysts for bioorthogonal chemistry. Moreover, we further expand our toolbox by polymerizing chiral ligands from proteins and then coordinating with Ru metal.[4] We demonstrate that the metal-containing ArPoly allows for asymmetric hydrogenation with almost 100% yield and 93% ee. More interestingly, the metal-containing polymer can be grafted from transaminase, which enables the enzyme to transform amine to chiral alcohol, creating the new-to-nature reactivity that the parental enzyme doesn’t have. To summarize, my group is dedicated to developing artificial polyenzymes (ArPoly) for catalysis by combining active polymers with proteins/enzymes. We have showcased four different types of ArPoly, in which polymer active units, structures, and compositions are tailored to enable not only high activity and selectivity but also new-to-nature reactions. |
| References: | [1] N. Zhang, Z. Sun, C. Wu, ACS Catal. 2022, 12, 4777-4783. [2] N. Zhang, C. Wu, ACS Synth. Biol. 2022, In press. [3] N. Zhang, P. Bessel, C. Wu, Bioconjugate Chem. 2022, 33, 1892-1899. [4] S. Wang, C. Wu, In preparation 2023. |
| Figures: | 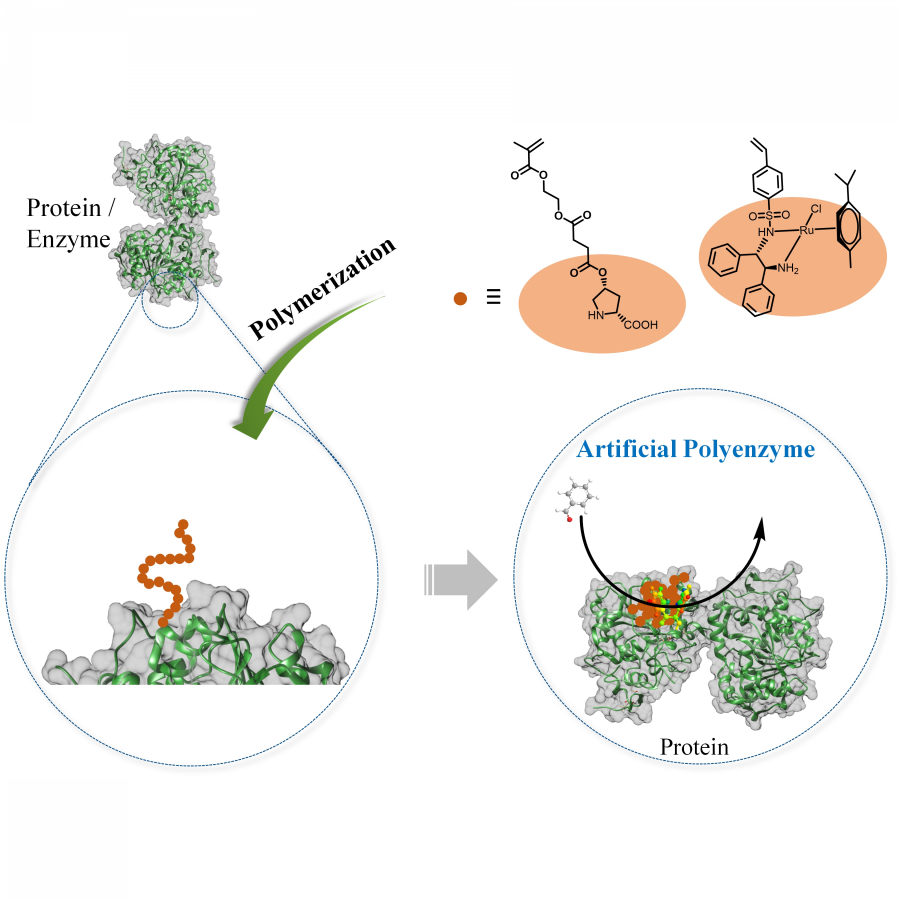 Figure 1 Artificial polyenzymes (ArPoly) combining proteins and polymers |
#39 | Pyrrolnitrin congeners: Selective access via biocatalytic halogenation |
|
| Presenting author: | Thomas CLASSEN of FORSCHUNGSZENTRUM JÜLICH GMBH |
| Other authors: | Jan GEBAUER of RWTH AACHEN Jörg PIETRUSZKA of HEINRICH HEINE UNIVERSITY DUESSELDORF |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Halogenation / Antifungal Agent / flavin-dependent halogenase / |
| Purpose: | Pyrrolnitrin is an antifungal agent that requires two halogenating enzymes in its biosynthesis.[1,2] Halogenating enzymes have evolved several times in nature,[3] yet their biocatalytic use remains limited. Obvious arguments are that biocatalytic use may be more sustainable, which needs to be proven, but undisputed is the argument that toxic elemental halogens are avoided here. In this study, however, we were able to show above all that halogenating enzymes may well have advantages, particularly in regioselectivity. The flavin-dependent halogenase PrnC from Pseudomonas protegens Pf-5 was successfully heterologously expressed in Escherichia coli and isolated. The enzyme requires an electron transport protein namely a flavin reductase for function. The use of the E. coli homolog SsuE[4] failed because of solubility issues during hmologoues expression, thus, the natural flavoprotein PrnF from the same biosynthetic cluster proved to be advantageous for biocatalytic conversions. Overall, an in vitro system consisting of the halogenase, the flavoreductase, and a glucose dehydrogenase for cofactor recycling was optimized by a design-of-experiment in such a way that it could be used for the synthesis of non-natural congeners. It is worth noting that these halogenations could not be introduced by chemical methods, so that this approach provides access to such pyrrolnitrin analogs for the first time. |
| References: | ex : [1] j. gribble, g., chemosphere. 2003, 52, 289-297. ex : [2] j. gribble, g., j. chem. edu. 2004, 81, 1441. ex : [3] j. fejzagić, a., molecules. 2019, 24, 4008. ex : [4] j. gao b., biochem. biophys. bes. commun. 2005, 331, 1137-1145. |
| Figures: | 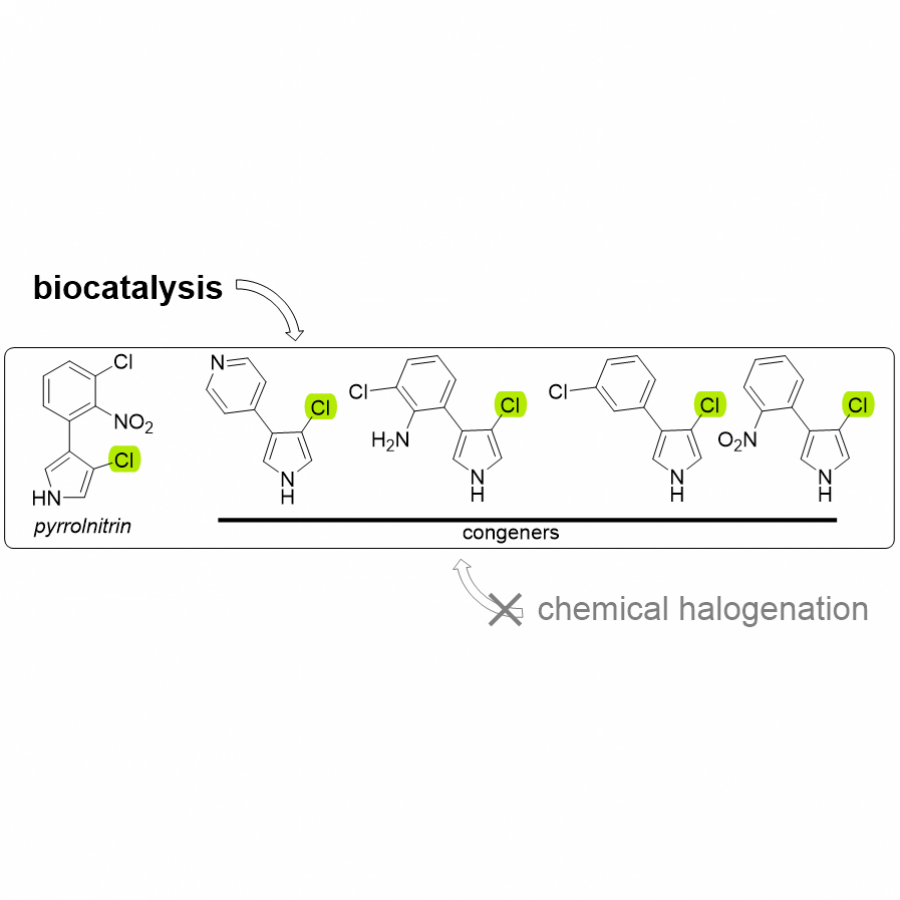 Pyrrolnitrin and synthesized congeners Marked in green is the halogen that has been introduced by PrnC. |
#61 | Enzymatic Crosslinking of Yeast Derived Proteins by Laccase and Effects on Emulsification Properties |
|
| Presenting author: | Kelly LIGHT of MCGILL UNIVERSITY |
| Corresponding author: | Salwa KARBOUNE of MCGILL UNIVERSITY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Laccase / Phenolic mediator / Yeast protein / Emulsion |
| Purpose: | Laccase has shown great potential in the cross-linking or conjugation of protein, contributing to the modulation of the protein-containing biopolymer matrix. Yeast derived cytoplasmic proteins and yeast cell wall mannoproteins have been investigated for their promising emulsification potential. In this work, laccase from two fungal sources is being used to cross link yeast derived proteins from saccharomyces cerevisiae in order to improve their characteristics and broaden their applications. Commercial laccase from Trametes versicolor and laccase produced in house from Coriolus hirsutus were combined with yeast cytoplasmic protein to achieve varying extents of cross linking based on reaction time course. Ferulic acid was included as a phenolic mediator to enhance the extent of cross linking. Reaction kinetics were monitored by fluorescence spectroscopy and the resulting curves were fitted with Michaelis-Menten and Hill models. Emulsification properties of native and modified proteins were evaluated turbidimetrically. Particle size and zeta potential were evaluated by DLS and solubility was evaluated by precipitation with PEG-8000. Reaction with laccase from T. versicolor resulted in greater extent of crosslinking at 3h than laccase from C. hirsutus, and addition of ferulic acid enhanced the crosslinking reaction. The end-product profile with or without ferulic acid mediator was dependent on the reaction time and the type of biocatalysts. Reactions run with laccase from T. versicolor and ferulic acid up to 24h showed significant increase in the molecular weight fraction above 150 kDa. Modeled reaction kinetics fitted very similarly with both applied models, with slightly better fit for the Hill model. Protein modified with ferulic acid showed improved emulsion activity index at pH 8 and decreased emulsion particle size at pH 4. Yeast derived proteins can be successfully crosslinked using laccase from T. versicolor with the addition of a phenolic mediator and extent of cross linking can be controlled by varying the reaction conditions. These modified proteins show promising improvements in emulsification potential and may be used as green biopolymer based emulsifiers in food and pharmaceutical applications. The understanding of the relationship between the extent of cross-linking by laccase and the functional properties will provide the capability to generate enhanced protein-containing biopolymer matrix. |
| References: | [1] light, k., karboune, s., crit. rev. food sci. nutr. 2021, 62, 8199-8229. [2] li, m., karboune, s., light, k., & kermasha, s., innov. food sci. emerg. technol. 2021, 71, 102723. [3] vélez-erazo, e. m., saturno, r. p., marson, g. v., & hubinger, m. d., food res. int. 2021, 140, 109853. |
| Figures: | 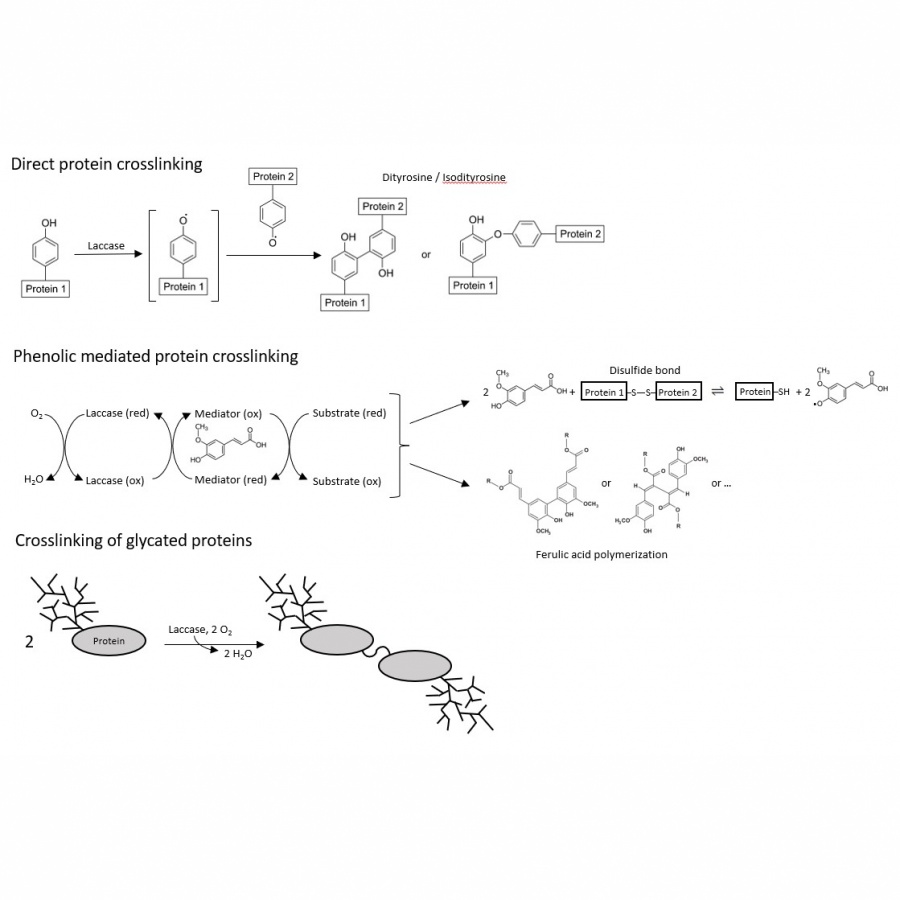 Laccase mediated cross linking of yeast derived proteins |
#64 | A Fully- Integrated Technology and Process Platform for Production of Functional Ingredients |
|
| Presenting author: | TRISH CHOUDHARY of WILLOW BIOSCIENCES |
| Other authors: | CHRISTOPHER SAVILE of WILLOW BIOSCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Strain Engineering / Upstream/Downstream Process Optimization / Functional Ingredients / Vertically-Integrated Technology Platform |
| Purpose: | Willow Biosciences is a synthetic biology company that produces pure, consistent, and sustainable functional ingredients using precision-fermentation technology. Our FutureGrown® yeast-based platform is vertically integrated with state-of-the-art biological, computational, and automation technologies to produce cost-effective compounds for consumer care, pharmaceutical, and food and beverage applications. The platform was initially utilized to manufacture the first functional ingredient, cannabigerol (CBG), a non-intoxicating cannabinoid found in the Cannabis plant genus. The natural pathway begins with the activation of hexanoic acid (HA) by hexanoyl-CoA synthetase and is proceeded by a polyketide synthase (PKS), an olivetolic acid cyclase (OAC), and completed by a prenyltransferase to produce cannabigerolic acid (CBGA). A proof-of-concept strain was created by integrating the pathway into Saccharomyces cerevisiae capable of producing mg per liter titers of cannabigerolic acid (CBGA) from hexanoic acid (HA). This parent strain was then engineered using multiple strategies at the enzyme, pathway, and genome levels resulting in multi-gram per liter strains. Bottlenecks in the pathway were identified and select enzymes were subjected to site saturation mutagenesis and further recombination of beneficial mutations to improve individual enzyme performance. Flux through the pathway was optimized by modulating gene expression through promoter optimization and balancing copy number. Genome-wide approaches focussed on increasing endogenous substrate availability and improving host robustness. The multiplexed HTP approach utilized a combination of computational tools, automated robotics, and high-speed mass spectrometry enabling rapid and efficient screening of thousands of strains per week. As part of the development cycle, improved strains were validated using benchtop fermenters, allowing for precise control of upstream parameters (growth and production) essential for the successful scale-up of engineered strains. In parallel, downstream chemical processing of the resulting biomass was optimized to achieve >98% purity of CBG, enabling rapid tech transfer and commercial-scale production with a large-scale manufacturing partner. Willow Biosciences has continued to utilize its FutureGrown® platform to expand its product portfolio, both internally and externally through partnerships and produce them at economically viable titers. |
#75 | REGIOSELECTIVE GLUCOSYLATION OF (+)-CATECHIN USING A NEW VARIANT OF THE SUCROSE PHOSPHORYLASE OF BIFIDOBACTERIUM ADOLESCENTIS |
|
| Presenting author: | Marie DEMONCEAUX of NANTES UNIVERSITÉ |
| Other authors: | Marine GOUX of NANTES UNIVERSITÉ Claude SOLLEUX of NANTES UNIVERSITÉ Émilie LORMEAU of NANTES UNIVERSITÉ Frédéric CADET of UNIVERSITÉ PARIS CITÉ Johann HENDRICKX of NANTES UNIVERSITÉ Bernard OFFMANN of NANTES UNIVERSITÉ Corinne MIRAL of NANTES UNIVERSITÉ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flavonoids / Regioselectivity / Sucrose phosphorylase / Modelling |
| Purpose: | Catechins belong to the flavan-3-ols class, a sub-family of flavonoids which have a role in preventing chronic inflammation responsible to neurodegenerative, cardiovascular or viral diseases.[1] (+)-Catechin (3,3’,4’,5,7-pentahydroxyflavan) is distributed in a wide variety of plants and has been reported to have a stronger antioxidant activity compared to other flavonoids thanks to its catechol moiety (C3’-OH and C4’-OH). It can scavenge free radicals and inhibit production of reactive oxygen species by transfer of a single electron to electron-accepting radicals.[2] Nevertheless, its low aqueous solubility prevents (+)-catechin from being well-absorbed by the human body. During the last steps of their biosynthesis, flavonoids are functionalized and, in most cases, they are beta-glucosylated to increase their biodisponibility in living organisms. While these glucosylated forms of (+)-catechin present interest, their extraction from plants as well as their chemical synthesis is not viable as they both lead to low yields of purified products. Thus, enzymatic synthesis is a solution as it is known to be efficient, specific and eco-friendly. Controlling the regioselectivity of the enzymatic glucosylation of flavonoids remains nevertheless a methodological challenge for scientists. Sucrose phosphorylases (SPs) are enzymes from the Glycoside Hydrolase family GH13 subfamily 18 according to CAZY database (EC 2.4.1.7). They catalyze in vivo reversible phosphorolysis of sucrose into alpha-D-glucose-1-phosphate and D-fructose via a glucosyl-enzyme intermediate. Their natural substrate, sucrose, is a cheap and efficient glucose donor. It has been shown that SP from Bifidobacterium adolescentis (BaSP) could synthesize alpha-glucosylated phenolic compounds using other acceptors than phosphate.[3] The mutation of native BaSP Gln345 by a phenylalanyl residue (mutant Q345F) increased the size of the catalytic site entry, allowing the glucosylation of flavonoids. Three alpha-glucosylated products of (+)-catechin were synthesized, but with no control on the reaction regioselectivity.[4] In order to control the regioselectivity of the transglucosylation reaction of (+)-catechin, we engineered a new mutant of Q345F containing a second mutation and obtained the variant P134D/Q345F. Using this new mutant as catalyst, we observed a modification of the products proportions favoring the formation of (+)-catechin-3’-O-alpha-D-glucoside compare to (+)-catechin-5-O-alpha-D-glucoside. Indeed, we showed, using standard molecular modelling and docking protocols, that P134D mutation impacted the local geometry of the acceptor site leading to the formation of the majority product by destabilizing one possible orientation of (+)-catechin acceptor. This original combination of theorical and experimental complementary approaches can be used to predict preferred orientations of flavonoids in acceptor sites, hence providing a rational basis for building variants towards a better control of regioselective glucosylation using SPs. |
| References: | [1] S. M. Ahmadi, R. Farhoosh, A. Sharif and M. Rezaie, Journal of Food Science, 2020, 85, 298-305 [2] H. Çelik and M. Kosar, Chemico-Biological Interactions, 2012, 197, 103-109 [3] M. Kraus, J. Görl, M. Timm and J. Seibel, Chemical Communications, 2016, 52, 4625-4627 [4] M. Kraus, C. Grimm and J. Seibel, ChemBioChem, 2016, 17, 33-36 |
| Figures: | 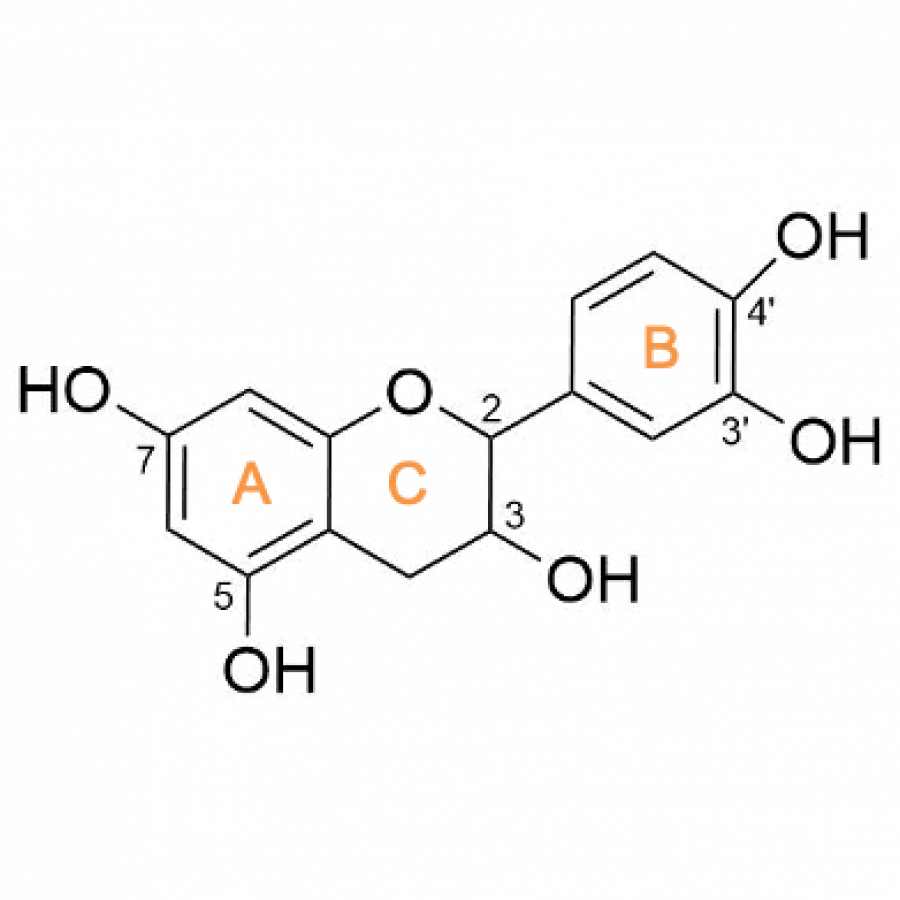 Molecular structure of (+)-catechin (+)-catechin is composed of three rings designated by letters A, B and C. Ring B is the one with the catechol moiety. |
#79 | The terpene mini-path: a new artificial terpene biosynthetic pathway |
|
| Presenting author: | Gilles IACAZIO of AIX-MARSEILLE UNIVERSITY |
| Other authors: | Julie COUILLAUD of CHALMERS UNIVERSITY OF TECHNOLOGY Létitia LEYDET of AIX-MARSEILLE UNIVERSITY Agnès AMOURIC of AIX-MARSEILLE UNIVERSITY Elise COURVOISIER-DEZORD of AIX-MARSEILLE UNIVERSITY Carole AVESQUE of AIX-MARSEILLE UNIVERSITY Mireille ATTOLINI of AIX-MARSEILLE UNIVERSITY Katia DUQUESNE of AIX-MARSEILLE UNIVERSITY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Terpene mini-path / Terpene production / Enzymatic cascade / Kinases |
| Purpose: | Terpenes are the largest class of natural products with more than 100,000 different structures described to date. Due to the diversity of their biological and physicochemical properties, terpenes are the preferred targets of many industries. However, due to their rarity and/or their low concentration in living organisms, access to these molecules is problematic in relation to the protection of living species and the environment. On the other hand, due to their structural complexity, the chemical synthesis of these molecules is hardly possible industrially. Thus, the microbiological production of terpenes using the natural biosynthetic pathways of these compounds (mevalonate and/or methyl erythritol phosphate pathways) starting from glucose, has been developed in recent years. One limitation of this approach is that glucose serves both as a carbon source for terpene production and as a carbon and energy source for the microorganism, making optimization of terpene production difficult. Several groups, including ours, have independently developed what we have called the terpene mini-path, which allows the decoupling of terpene biosynthesis and central metabolism in vivo, but also makes it possible to consider with more simplicity the enzymatic synthesis of terpenes in vitro. We have developed a two-enzymes cascade (2 kinases) to generate from their corresponding alcohols the two universal precursors of terpenes, dimethylallyl diphosphate (DMAPP) and isopentenyl diphosphate (IPP). We were able to apply this enzymatic cascade to the in vivo and in vitro synthesis of a cytotoxic prenyl derivative of brevianamide F, tryprostatin B, to the in vitro synthesis of farnesyl diphosphate (FPP), the precursor of all sesquiterpenes and triterpenes, and to the in vivo synthesis of delta-cadinol, a sesquiterpene of interest. On the other hand, the structure of the terpene mini-path also allows a simplified access to non-natural terpenes. We were thus able to synthesize the cyclobutyl derivatives of both FPP and tryprostatin B. These results, as well as those of other groups, allow to envisage the industrial implementation of a new and very general way of producing terpenes, either in vitro or in vivo. |
| References: | [1] J. Couillaud et al. ACS Omega 2019, 4, 4, 7838-7849 [2] J. Couillaud et al. Genes 2021, 12(12), 1974 [3] J. Couillaud et al. ChemBioChem 2022, 23, e2021006 [4] J. Couillaud et al. ChemBioChem 2022, 23, e2022005 |
| Figures: | 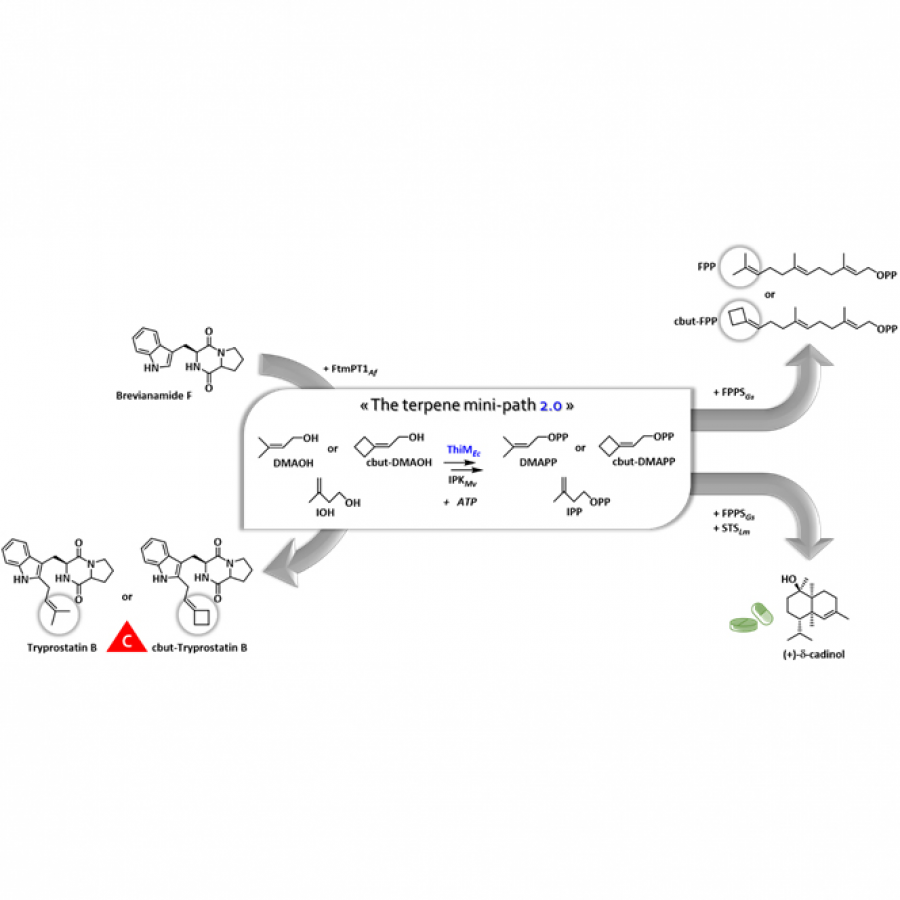 The terpene mini-path The Terpene mini-path, a very short enzymatic cascade to access the universal precursors of all terpenes. |
#81 | An intimate look at immobilized enzymes toward the design of robust and efficient heterogeneous biocatalysts |
|
| Presenting author: | Fernando LÓPEZ GALLEGO of COOPERATIVE BIOMATERIALES INSTITUTE CIC BIOMAGUNE |
| Other authors: | Eleftheria DIAMANTI of CIC BIOMAGUNE Daniel ÁNDRES-SANZ of CIC BIOMAGUNE |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme immobilization / ketoreductases / transamianses / enzyme kinetics |
| Purpose: | In applied biocatalysis, protein immobilization is a key technology to enhance biotransformations. In the last decade, single- and multi-enzyme systems together with their cofactors have been co-immobilized on solid materials with the aim of boosting the throughput of enzymatic cascades.[1] In one of the most striking advances for the design and assembly of multienzyme cascades reported in recent years, Merck and Codexis in a joint effort have brought together 5 enzymes, co-immobilizing two of them, for the synthesis of the antiviral Islatravir.[2] The heterogenization of enzymes and their corresponding cofactors to carry out chemical reactions without the exogenous supply of the latter gives rise to a new generation of self-sufficient heterogeneous biocatalysts that facilitate the separation and recycling of all the elements that form the biocatalysts. However, the immobilization process in biocatalysis is still too empirical and enzyme behavior (kinetics and stability) inside solid materials remains as a black box that we barely understand. In this work, we advance the characterization of the spatiotemporal behavior of heterogeneous biocatalysts to better understand the stability and kinetic properties of enzymes when confined within the solid surface of porous materials. We have exploited image processing derived from time-lapse fluorescence microscopy experiments to determine the intraparticle kinetics of self-sufficient heterogeneous biocatalysts composed of different enzymes (oxidoreductases, hydrolases and transaminases) and their corresponding cofactors (NAD(P)H, PLP or FAD).[3] The resulting self-sufficient heterogeneous biocatalysts were analyzed under static and in operando conditions to investigate the thermodynamics of cofactor binding, enzyme density and apparent Michaelis-Menten (MM) kinetics of the enzyme, both at single particle and intraparticle level. Furthermore, we revealed that unexpected migration of immobilized enzymes across the solid surface occurs when the enzymes are reversibly bound, and that this process takes place both during storage of the biocatalysts and during their operational performance. Our studies also revealed that enzyme concentration and spatial organization are the main sources of functional variability in these self-sufficient systems and have a significant impact on their catalytic efficiency. This knowledge, which could only be revealed by single particle microscopic studies, led us to optimize highly efficient heterogeneous biocatalysts in which both enzyme(s) and cofactor(s) are reused either discontinuously (batch reactors)[1] or continuously (flow reactors),[4] and can reach total turnover numbers as high as 105 and 104, respectively, exhibiting industrially relevant productivities (50 g L-1 h-1). |
| References: | [1] A. H. Orrego, D. Andrés-Sanz, S. Velasco-Lozano, M. Sanchez-Costa, J. Berenguer, J. M. Guisan, J. Rocha-Martin, F. López-Gallego, Catal. Sci. Tech. 2021, 11, 3217-3230. [2] M. A. Huffman, A. Fryszkowska, O. Alvizo, M. Borra-Garske, K. R. Campos, K. A. Canada, P. N. Devine, D. Duan, J. H. Forstater, S. T. Grosser, H. M. Halsey, G. J. Hughes, J. Jo, L. A. Joyce, J. N. Kolev, J. Liang, K. M. Maloney, B. F. Mann, N. M. Marshall, M. McLaughlin, J. C. Moore, G. S. Murphy, C. C. Nawrat, J. Nazor, S. Novick, N. R. Patel, A. Rodriguez-Granillo, S. A. Robaire, E. C. Sherer, M. D. Truppo, A. M. Whittaker, D. Verma, L. Xiao, Y. Xu, H. Yang, Science 2019, 366, 1255-1259. [3] E. Diamanti, J. Santiago-Arcos, D. Grajales-Hernández, N. Czarnievicz, N. Comino, I. Llarena, D. Di Silvio, A. L. Cortajarena, F. López-Gallego, ACS Catal. 2021, 11, 15051-15067. [4] A. I. Benítez-Mateos, M. L. Contente, S. Velasco-Lozano, F. Paradisi, F. López-Gallego, ACS Sustain. Chem. Eng. 2018, 6, 13151-13159. |
| Figures: | 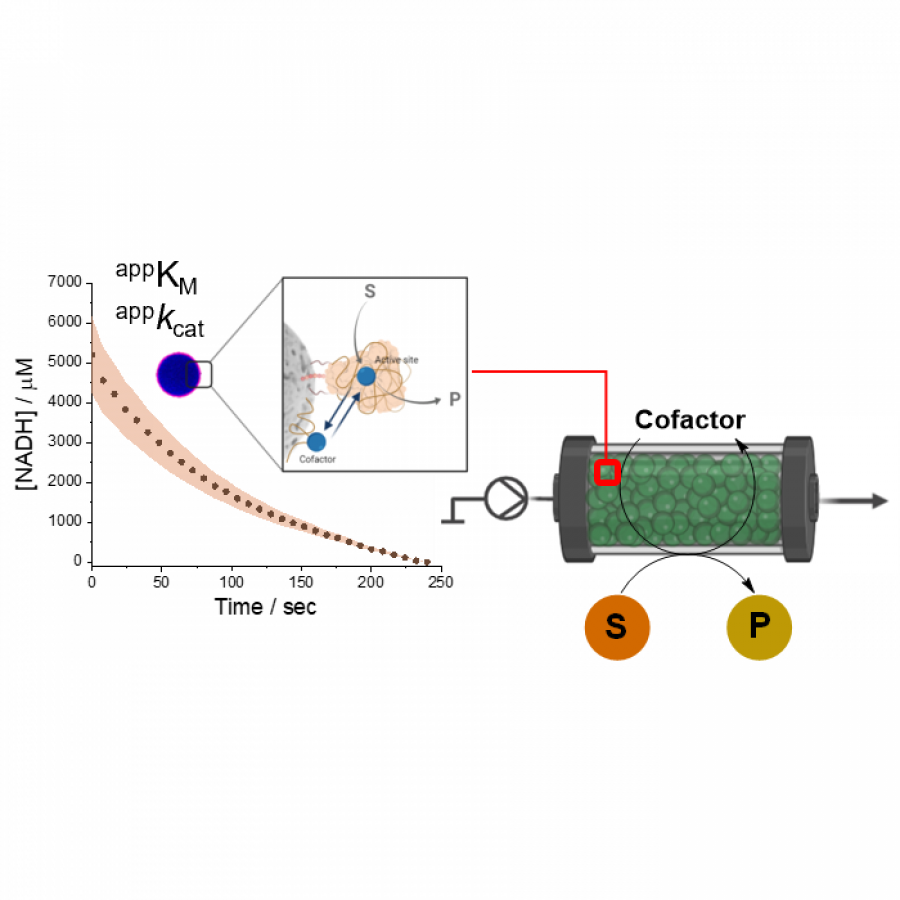 Intraparticle kinetics of self-sufficient heterogeneous biocatalysts to be applied in flow-reactors The heterogeneous biocatalyst packed into a plug-flow column can be analyzed in operando through fluorescence microscopy to determine the enzyme kinetics |
#83 | Carbonyl Reduction by Carboxylic Acid Reductases: An unsurprising Surprise? |
|
| Presenting author: | Margit WINKLER of ACIB GMBH AND GRAZ UNIVERSITY OF TECHNOLOGY |
| Other authors: | Bastian DANIEL of UNIVERSITY OF GRAZ Chiam HASHEM of GRAZ UNIVERSITY OF TECHNOLOGY Ludmila MARTÍNKOVÁ of CZECH ACADEMY OF SCIENCES Tea PAVKOV-KELLER of UNIVERSITY GRAZ Hannah G. BREUER of TUGRAZ, I. F. MOLEKULARE BIOTECHNOL |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Carboxylic Acid Reductase (CAR) / aldehyde / alcohol / enzymatic synthesis |
| Purpose: | Carboxylic acid reductase enzymes (CARs) are well known for the reduction of a wide range of carboxylic acids to the respective aldehydes. The current understanding is that CARs release aldehydes, because they intrinsically circumvent the kinetic preference of aldehyde versus carboxylic acid reduction, owing to their reaction mechanism. CARs are comprised of three domains: an adenylation domain, a phosphopantetheinyl-binding domain and a reductase-domain and acid reduction is a multi-step cascade starting with carboxylate activation, which is followed by the formation of an enzyme-tethered thioester and finally, this thioester is reduced. One of our long-term goals is to gain deep insight into the structure-function-relationship of CARs and we subjected various single-domains, di-domains and full-length CARs to crystallization trials. We solved the structure of the R-domain of a fungal CAR from Neurospora crassa (NcCAR, (PDB-code 8AEP). Its resemblance to short chain dehydrogenases (SDRs) triggered the question, why this R-domain releases a carbonyl compound while SDRs reduce carbonyl compounds to alcohols although both active sites were highly similar. Others postulated an on/off mechanism triggered by a particular amino acid that differed between SDRs and a crystallized R-domain of a bacterial CAR [1]. This particular amino acid, however, is not present in the NcCAR R-domain or any other R-domains of fungal CARs. We therefore sought to proof that the NcCAR R-domain was not acting as carbonyl reductase by incubation of a few carbonyl compounds in the presence of highly pure R-domain and NADPH. The unsurprising surprise was that the R-domain is in fact able to reduce carbonyls, including aldehydes, which are typically considered to be the final product of carboxylic acid reduction by CAR. We discovered that the respective full length NcCAR was able to reduce aldehydes [2]. Herein, we shed light on the structural elements, substrate requirements and an extended substrate scope of CARs (Figure 1). Acknowledgements: This research was supported by the Austrian Science Fund (FWF): CATALOX [doc.funds46], V-00415 and I-04607. The work was also supported by COMET center: acib: Next Generation Bioproduction is funded by BMK, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria and Vienna Business Agency in the framework of COMET - Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency FFG. |
| References: | [1] D. Gahloth, M. S. Dunstan, D. Quaglia, E. Klumbys, M. P. Lockhart-Cairns, A. M. Hill, S. R. Derrington, N. S. Scrutton, N. J. Turner, D. Leys, Nat. Chem. Biol. 2017, 13, 975-981. [2] B. Daniel, C. Hashem, M. Leithold, T. Sagmeister, A. Tripp, H. Stolterfoht-Stock, J. Messenlehner, R. Keegan, C. K. Winkler, J. G. Ling, S. H. H. Younes, G. Oberdorfer, F. D. A. Bakar, K. Gruber, T. Pavkov-Keller, M. Winkler, ACS Catal. 2022, 12, 15668-15674. |
| Figures: | 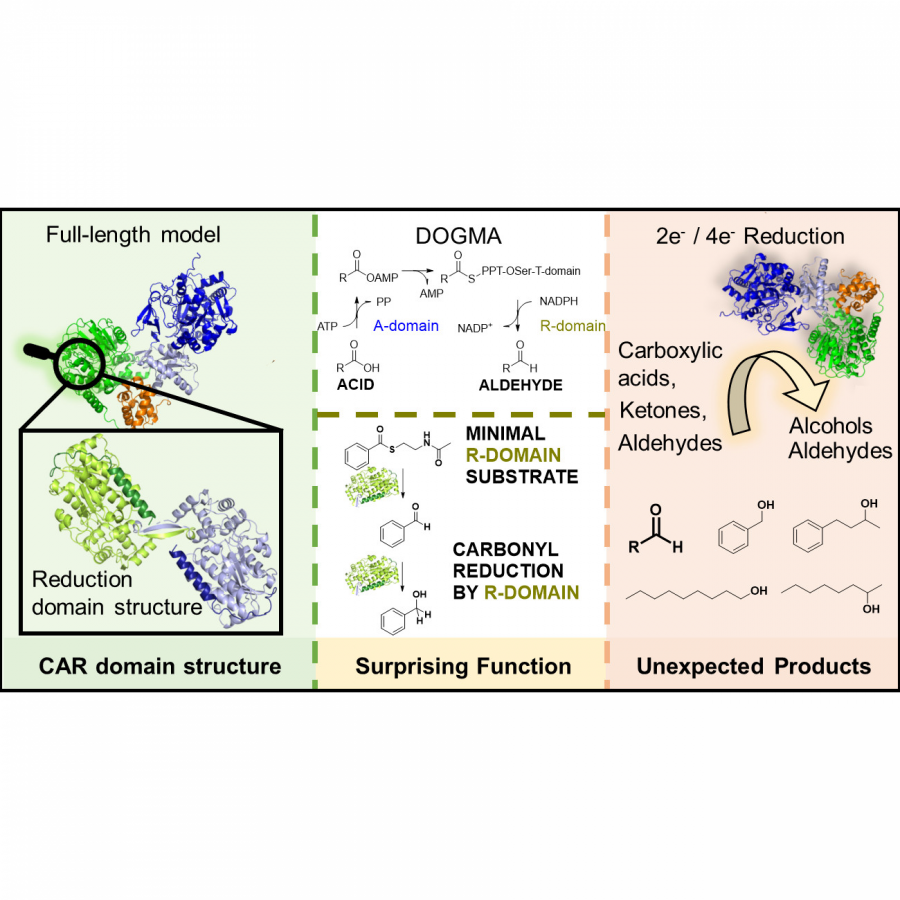 Figure 1 Structure and function of a CAR R-domain |
#84 | Structure Elucidation and Characterization of Patulin Synthase, Insights into the Formation of a Fungal Mycotoxin |
|
| Presenting author: | Gwen TJALLINKS of UNIVERSITY OF GRONINGEN |
| Other authors: | Alessandro BOVERIO of UNIVERSITY OF GRONINGEN Ivana MARIC of UNIVERSITY OF GRONINGEN Henriette ROZEBOOM of UNIVERSITY OF GRONINGEN Mark ARENTSHORST of LEIDEN UNIVERSITY Jaap VISSER of LEIDEN UNIVERSITY Arthur RAM of LEIDEN UNIVERSITY Andrea MATTEVI of UNIVERSITY OF PAVIA Marco FRAAIJE of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flavoprotein / Patulin / Crystallography / Biocatalysis |
| Purpose: | Elucidating the three-dimensional structure of biological macromolecules yields a wealth of information facilitating developments in biotechnology such as modern drug design.[1] Patulin synthase is an enzyme involved in the biosynthesis of the secondary metabolite patulin, a mycotoxin often present in apples and apple-derived products.[2,3] Penicillium expansum, also termed as apple blue mold, is the major contributor to the presence of patulin on apples and has resulted in significant postharvest losses.[4] Hence, understanding and characterizing the functioning of patulin synthase involved in the key step of patulin production is of utmost importance. In this study, the P. expansum patE gene was expressed in Aspergillus niger, purified via a C-terminally added His-Tag to show that patulin synthase is active exclusively on aromatic alcohols including 5-hydroxymethyl furfural (HMF) and the natural substrate ascladiol. By elucidating its crystal structure (Figure 1), details on its catalytic mechanism were revealed. Several aspects of the active site architecture are reminiscent of that of fungal aryl alcohol oxidases (Figure 2). Overall, this study provides detailed insights into the functioning of patulin synthase and has shown that it can also be used for selective oxidation of a large number of aromatic compounds. This can be of help in strategies to lower the risk on formation of this mycotoxic, for example by developing patulin synthase inhibitors, while it may also develop as a useful biocatalyst for synthetic chemistry, e.g. for conversion of HMF. |
| References: | [1] H. Zheng, J. Hou, M.D. Zimmerman, A. Wlodawer, W. Minor, Expert Opin Drug Discov, 2014, 9, 125-137. [2] B. Li, Y. Cheng, Y. Zong, Y. Shang, Z. Zhang, X. Xu, X. Wang, M. Long, S. Tian, Environ Microbiol, 2019, 21, 1124-1139. [3] O. Puel, P. Galtier, I.P. Oswald, Toxins (Basel), 2010, 2, 613-631. [4] H. Morales, S. Marín, A.J. Ramos, V. Sanchis, Food Control, 2010, 21, 953-962. [5] J. Carro, P. Amengual-Rigo, F. Sancho, M. Medina, V. Guallar, P. Ferreira, A.T. Martínez, Sci Rep, 2018, 8, 1-12. |
| Figures: | 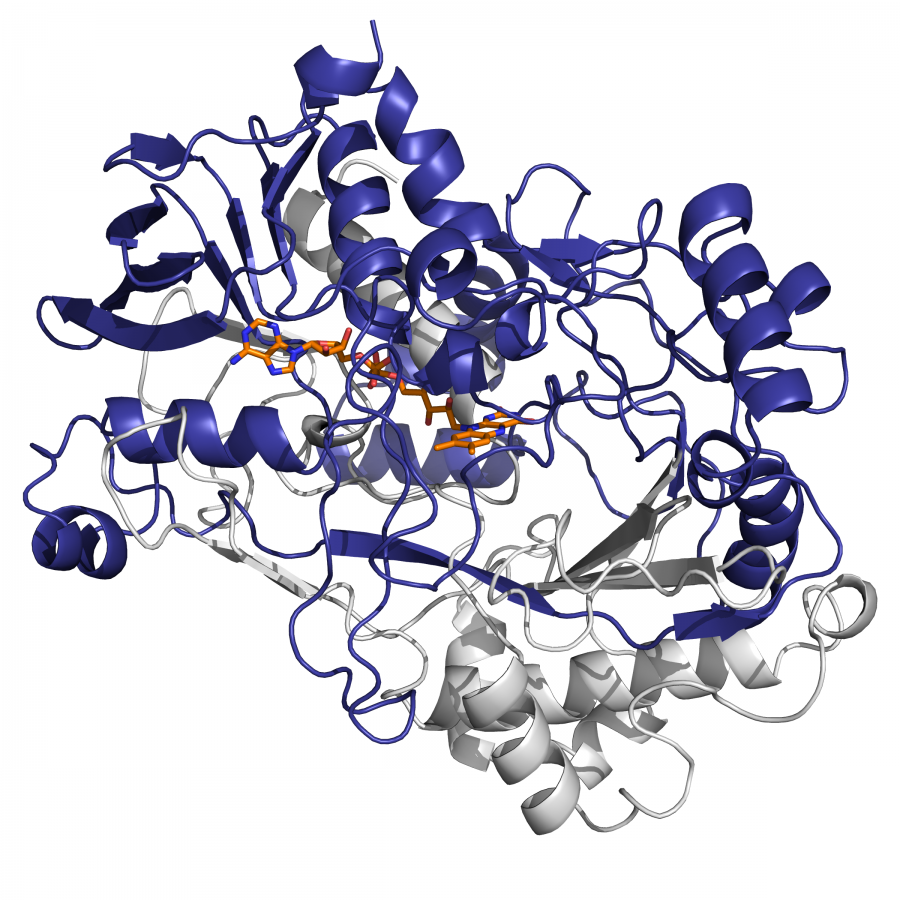 Monomeric structure of patE. In deep blue the flavin-domain, in gray the substrate-domain and in orange the FAD cofactor is represented. - 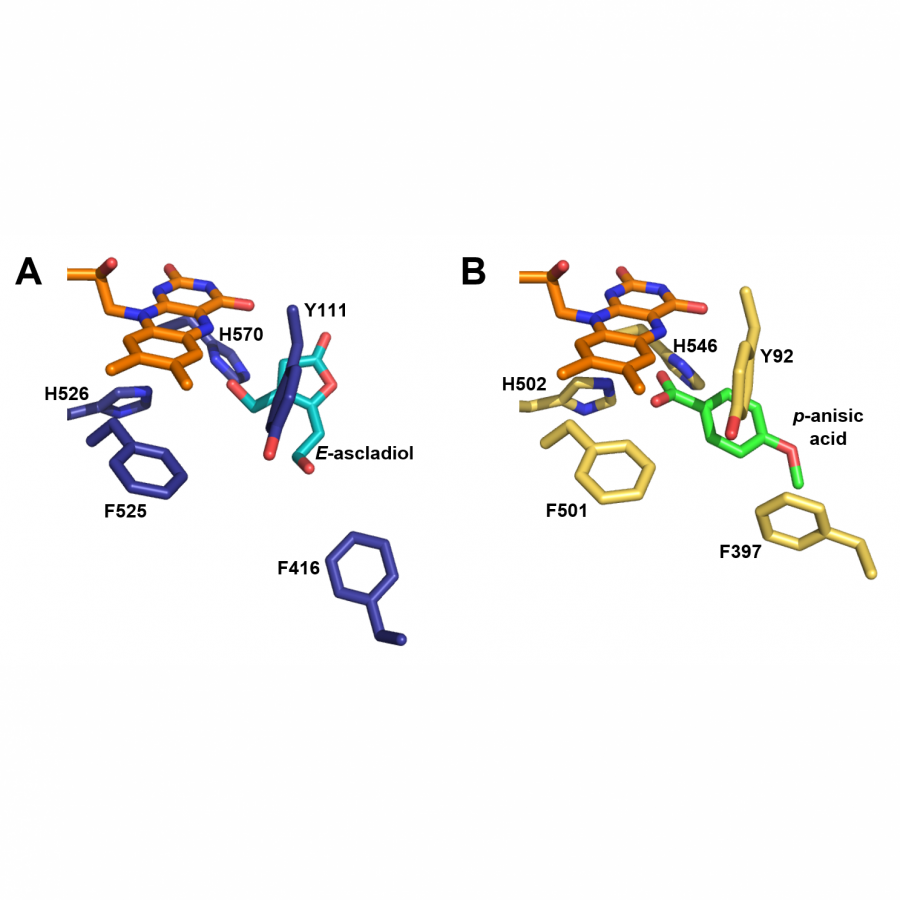 A: docked binding pose of (E)-ascladiol (blue sticks) in the active site of patE (deep blue). B: binding pose of p-anisyl (green sticks) in the active site of aryl-alcohol oxidase from P. eryngii (yellow, PDB ID: 5OC1) [5]. - |
#88 | Cross-linked cellulase aggregates for the efficient hydrolysis of lignocellulosic residue biomass and organic fraction of municipal waste |
|
| Presenting author: | Io ANTONOPOULOU of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Other authors: | KYRIAKI ELEFTHERIA VERGITSI of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY ULRIKA ROVA of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY PAUL CHRISTAKOPOULOS of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cross Linked Enzyme Aggregates / Cellulases / Cellulose hydrolysis / Advanced biofuels |
| Purpose: | By 2030, the EU aims to increase the share of renewable energy in transport to at least 14%, including a minimum share of 3.5% of advanced biofuels (1). It is anticipated that advanced biofuels, mainly 2nd generation biofuels, will boost EU economy by creating 1.2 million jobs and a revenue of 400 billion euros by 2030. As such, feedstock costs need to remain competitive, and sustainability criteria need to be met. Enzymes are one of the biggest cost in producing fermentable sugars, accounting up to 20% of the cost for traditional 2nd generation bioethanol production (2). Enzyme immobilization could reduce significantly the use of cellulase to achieve a total cost reduction of 10% (3). When applying immobilized enzymes within heterogeneous processes where the substrate is initially insoluble, the efficient enzyme separation and recovery is crucial for enzyme reuse. Enzyme immobilization offers many advantages over enzymes in solution, including economic convenience, higher stability, and the possibility to be easily removed from the reaction mixture leading to pure product isolation. Aim of this work is the preparation, characterization and application of Cross Linked Enzyme Aggregates (CLEAs) based on commercial multi-enzymatic bends with cellulase activity, Cellic® Ctec2 and Cellic® CTec3 HS (Novozymes A/S, Bagsværd, Denmark). CLEA preparation includes two distinct steps: a precipitation step, where the enzyme is aggregated with the addition of a saturated salt solution or organic solvent and a crosslinking step, where the aggregates are stabilized by a crosslinker e.g. glutaraldehyde. Each step was optimized investigating the effect of different parameters on the recovery of cellulase activity (filter paper activity, endoglucanase activity, exoglucanase activity and β-glucosidase activity). The parameters investigated in the precipitation step were the type of precipitant, concentration of precipitant and time of precipitation, while in the cross-linking step the parameters investigated were the glutaraldehyde concentration, enzyme load, time and temperature for crosslinking. Moreover, the magnetic CLEA (m-CLEAs) formation using magnetic nanoparticles (4) was investigated. The prepared CLEAs and m-CLEAs were characterized in terms of optimal temperature performance, temperature stability and were compared to the free enzyme. Then, they were applied in the hydrolysis of two renewable lignocellulosic feedstocks, a cellulose fraction obtained from organosolv pretreated beechwood and an organic fraction of municipal waste. The substrate concentration, enzyme load and hydrolysis time were optimized. The formed CLEA and m-CLEAs showed good performance when compared to the free enzyme and their reusability potential was investigated for >5 cycles. This work is supported by the Horizon 2020 funded project FLEXI-GREEN FUELS (Flexible and resilient integrated biofuel processes for competitive production of green renewable jet and shipping fuels, Grant agreement ID 101007130). |
| References: | 1. European commission. Renewable Energy Directive (EU) 2018/2001. 2. Johnson E (2016) Biofuels. Bioprod. Bioref 10:164-174. 3. Datta S et al. (2013) 3 Biotech. 3(1): 1-9. 4. Jia J et al. (2017) Molecules 22, 269. |
#89 | Synthesis of high-value natural benzaldehydes by Basidiomycete-mediated reduction of the corresponding benzoic acids |
|
| Presenting author: | Filip BORATYŃSKI of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Stefano MARZORATI of CONSIGLIO NAZIONALE DELLE RICERCHE, ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE Stefano SERRA of CONSIGLIO NAZIONALE DELLE RICERCHE (C.N.R.), ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE Tomasz STRZAŁA of DEPARTMENT OF GENETICS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Ewa SZCZEPAŃSKA of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | natural flavours / Basidiomycetes / biotransformations / CAR activity |
| Purpose: | Substituted benzaldehydes of phenylpropanoid origin are the natural products of utmost industrial importance [1,2]. These compounds have been widely employed for food flavouring. Their extraction from the natural sources or chemical synthesis can be considered as the fundamental processes in the flavours and fragrances field. Benzaldehyde (1), anisaldehyde (2), vanillin (3) and piperonal (5) (Figure 1) are considered as the most important aromatic compounds in this aldehydes class and their production has steadily increased over the years [3]. The consumer’s preference for ‘natural or organic’ aromas has increased the request for flavours possessing the ‘natural’ status. The resulting shortage of aromatic aldehydes of extractive origin, such as vanillin (3), veratraldehyde (4) and piperonal (5), can be offset by developing a new biotechnological synthesis method. Here, we report a study on the microbiological reduction of five natural benzoic acid derivatives, namely p-anisic, vanillic, veratric, piperonylic, and eudesmic acids, to produce the corresponding fragrant aldehydes. We found that different Basidiomycota strains, including white-rot, brown-rot saprophytic, and ectomycorrhizal species can efficiently perform this transformation, with good chemical selectivity and tolerance to the toxicity of substrates and products. Besides confirming the carboxylic acid reductase activity of the already exploited fungi Pycnoporus cinnabarinus, we discovered that other species such as Pleurotus eryngii, Pleurotus sapidus, Laetiporus sulphurens as well as the saprophytic fungi Lepista nuda are valuable microorganisms for the synthesis of anisaldehyde (2), vanillin (3), veratraldehyde (4), piperonal (5) and 3,4,5-trimethoxybenzaldehyde (6) from the corresponding acids. For these flavours, a preparative biocatalytic process was proposed. Starting from benzoic acids of natural origin, the obtained aldehydes can be commercialized as high-value natural flavours, in compliance with the European and USA regulation of food flavouring substances. The research was conducted under the LIDER XII program financed by the National Centre for Research and Development (NCRD) grant agreement no. LIDER/44/0228/L-12/20/NCBR/2021. |
| References: | [1] T. Vogt, Molecular Plant, 2010, 3 (1). [2] H. Surburg, J. Panten, John Wiley & Sons, Weinheim, 2016. [3] G.A. Burdock, Fenaroli’s Handbook of Flavor Ingredients, 6th ed., 2010. |
| Figures: | 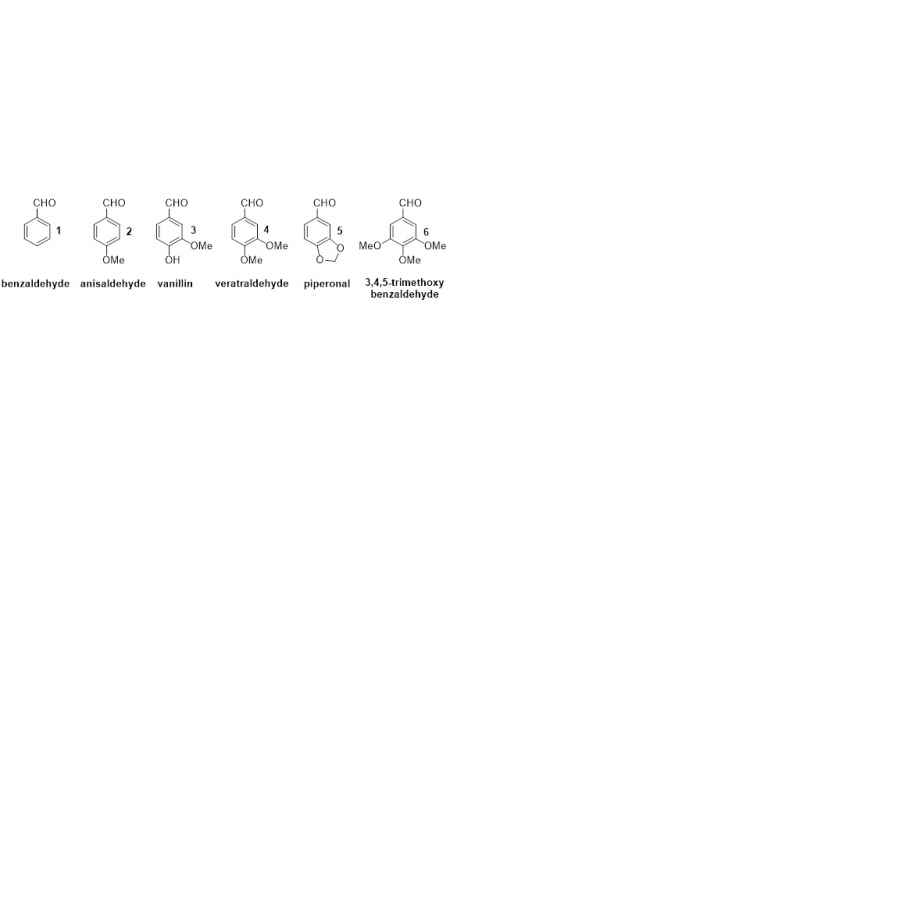 Some relevant natural benzaldehydes of interest in F&F field. - |
#100 | Stereodivergent synthesis 2-hydroxy-3-substituted-4-butyrolactones by tandem of aldolases and oxidoreductases. |
|
| Presenting author: | CARLOS J MORENO of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA (IQAC-CSIC) |
| Other authors: | Karel HERNADEZ of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY Samantha GITTINGS of PROZOMIX LTD. Michael BOLTE of INSTITUT FÜR ANORGANISCHE CHEMIE Jesús JOGLAR of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY Jordi BUJONS of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY Teodor PARELLA of SERVEI DE RESSONÀNCIA MAGNÈTICA NUCLEAR. UNIVERSITAT AUTÒNOMA DE BARCELONA Pere CLAPES of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | 2-Oxoacid aldolase / Ketoreductases / Aldol addition / 2-Hydroxy-4-butyrolactones |
| Purpose: | Stereodivergent synthesis 2-hydroxy-3-substituted-4-butyrolactones by tandem of aldolases and oxidoreductases. Homochiral 2-hydroxy-4-butyrolactone derivatives are important structural motifs frequently found in naturally occurring biologically active products, synthetic drugs, biodegradable polymers as well as building blocks and chiral auxiliaries in asymmetric organic synthesis (1). Among the methods described in the literature, the stereoselective reduction of 4-hydroxy-2-oxoacid derivatives to produce 2-hydroxy-4-butyrolactone derivatives remains unexplored. In this communication we report a tandem biocatalytic stereodivergent route for the preparation of these compounds using two stereocomplementary aldolases and oxidoreductases, using simple and achiral starting materials. The strategy comprises (Scheme 1) (i) aldol addition reaction of 2-oxoacids to formaldehyde using both 3-methyl-2-oxobutanoate hydroxymethyltransferase and 2-keto-3-deoxy-L-rhamnonate aldolase and variants thereof, and (ii) subsequent 2-oxogroup reduction of the aldol adduct by both ketopantoate reductase and a delta-1-piperidine-2-carboxylate/delta-1-pyrroline-2-carboxylate reductase with uncovered promiscuous ketoreductase activity. Finally, the formation of the corresponding lactone takes place during the work up and purification steps. Yields, enantiomeric excesses and diasteromeric ratios for a total of 23 structurally diverse 2-hydroxy-3-substituted-4-butyrolactones will be discussed. Moreover, one-pot one-step cascade reactions with the aldolases and reductases operating in tandem will be presented. |
| References: | [1] (a) Hu, Z.-Q.; Li, X.; Liu, L.-X., et al., J. Org. Chem. 2021, 86, 17453-17461; (b) Gröger, H., Adv. Synth. Catal. 2001, 343, 547-558; (c) Heidlindemann, M.; Hammel, M.; Scheffler, U., et al., J. Org. Chem. 2015, 80, 3387-3396; (d) Bourgeois, F.; Medlock, J. A.; Bonrath, W., et al., Org. Lett. 2020, 22, 110-115; (e) Yamaguchi, S.; Matsuo, T.; Motokura, K., et al., Chem. Asian J. 2016, 11, 1731-1737; (f) Evans, D. A.; Wu, J.; Masse, C. E., et al., Org. Lett. 2002, 4, 3379-3382. |
| Figures: | 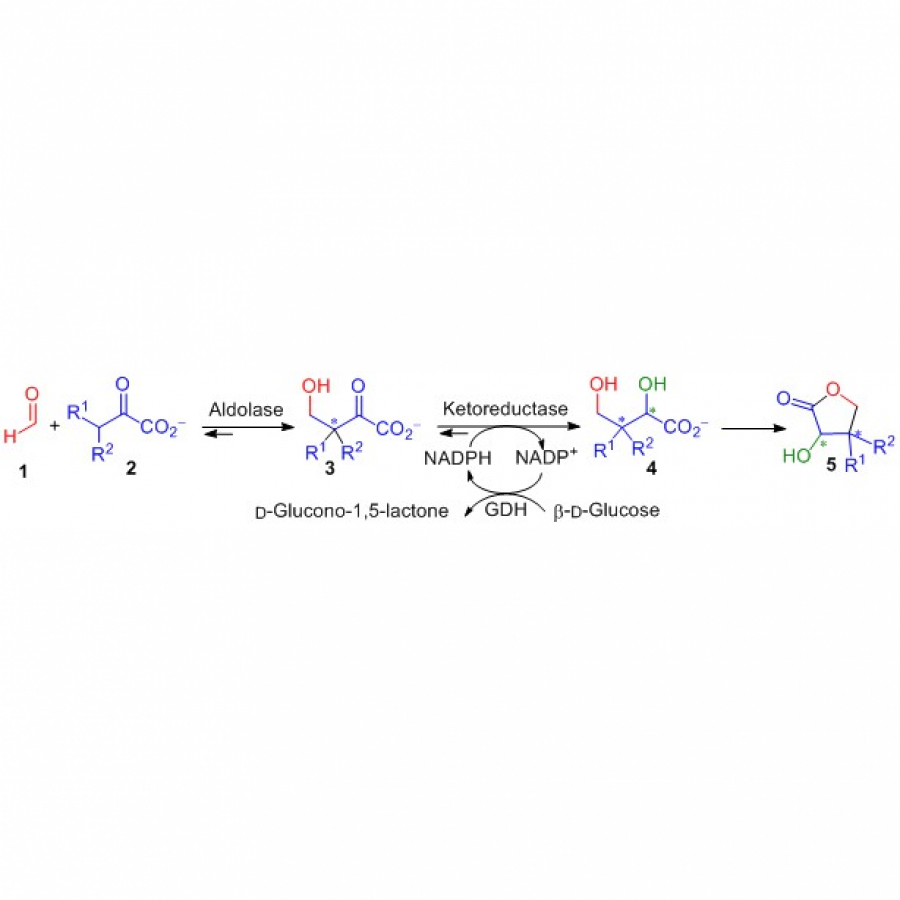 Scheme 1. Biocatalytic synthesis of 3-substituted-2-hydroxy-4-butyrolactones 5 by tandem aldolase-ketoreductase biocatalytic reactions starting from formaldehyde 1 and 2-oxoacids 2. |
#113 | POLYCYPS: BIOCATALYTIC TOOLS FOR THE OPTIMISATION OF DRUG LEADS |
|
| Presenting author: | Vincent POON of HYPHA DISCOVERY LIMITED |
| Other authors: | Aksana KHAN of HYPHA DISCOVERY LIMITED Emily HOPKINS of HYPHA DISCOVERY LIMITED Tetsuo KOKUBUN of HYPHA DISCOVERY LIMITED Kinga NYTKO of HYPHA DISCOVERY LIMITED Richard PHIPPS of HYPHA DISCOVERY LIMITED Frank SCHEFFLER of HYPHA DISCOVERY LIMITED Julia SHANU-WILSON of HYPHA DISCOVERY LIMITED Liam EVANS of HYPHA DISCOVERY LIMITED Jonathan STEELE of HYPHA DISCOVERY LIMITED Stephen WRIGLEY of HYPHA DISCOVERY LIMITIED |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cytochrome P450 / Biocatalyst / Drug metabolism / Late-stage functionalisation |
| Purpose: | PolyCYPs® enzymes are bacterial Class I and Class III cytochrome P450s (P450s) successfully mined from actinomycete bacteria in Hypha’s biotransformation panel. Eighteen of the best performing P450s as well as human aldehyde oxidase and flavin-containing monooxygenase 3 were developed into an easy-to-use lyophilised cell-free extract kit. PolyCYPs enzymes are available in screening and scale-up format which enables the production of low milligram amounts of oxidised metabolites for purification and identification. Multi-gram quantities of metabolites and oxidised derivatives can be generated using a Streptomyces lividans whole cell biotransformations expressing the specific PolyCYP enzyme. The poster features the application of PolyCYP enzymes to produce multiple oxidised metabolites and derivatives of drug compounds in parallel and provides opportunities to: 1. Boost DMPK properties, particularly metabolic stability 2. Empirically discover new polar interactions in binding sites, improving potency and selectivity 3. Establish if metabolites are active before deprioritising a scaffold due to metabolic instability 4. Utilise hydroxylated derivatives/metabolites as a handle for late-stage functionalisation e.g. fluorination 5. Rapidly expand polar SAR and broaden IP coverage, including the exemplification of active metabolites. |
| References: | J. Boström, D. Brown, R. Young, and G. M. Keserü, Nat. Rev. Drug. Discov., 2018 17, 709-727 J. Shanu-Wilson, L. Evans, S. Wrigley, J. Steele, J. Atherton, and J. Boer, ACS Med. Chem. Lett., 2020, 11, 2087-2107 |
| Figures: | 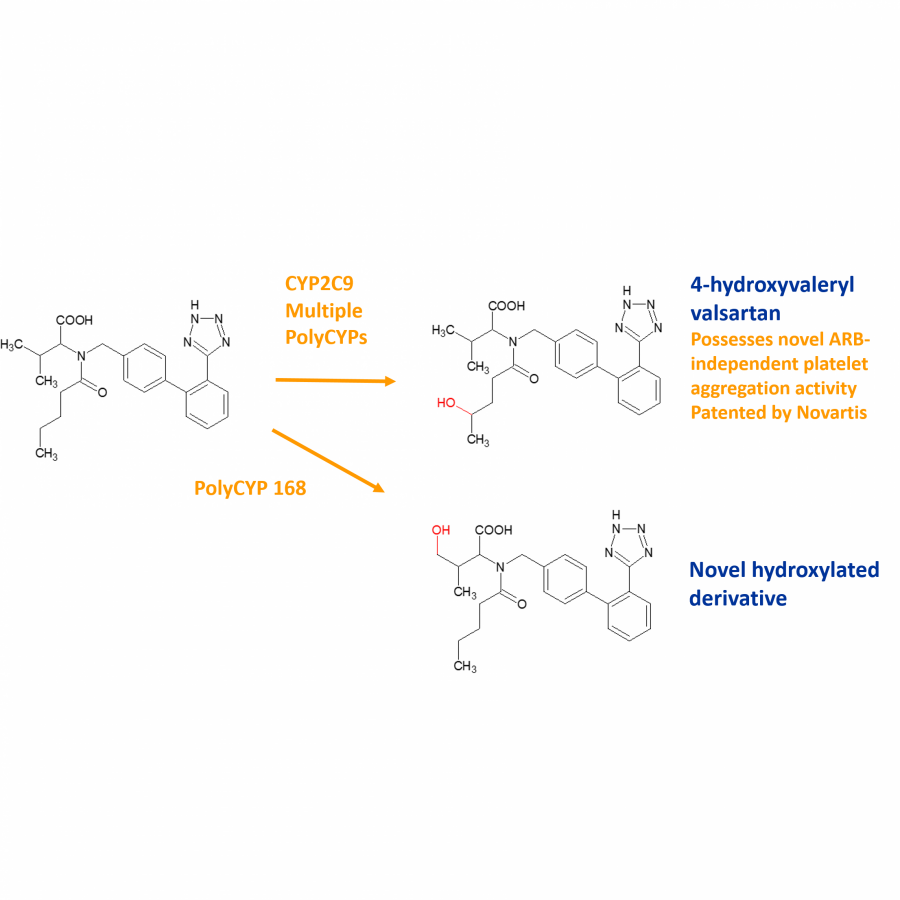 Valsartan case study PolyCYPs used to quickly generate metabolites and oxidised derivatives with enhanced properties that could widen IP coverage.
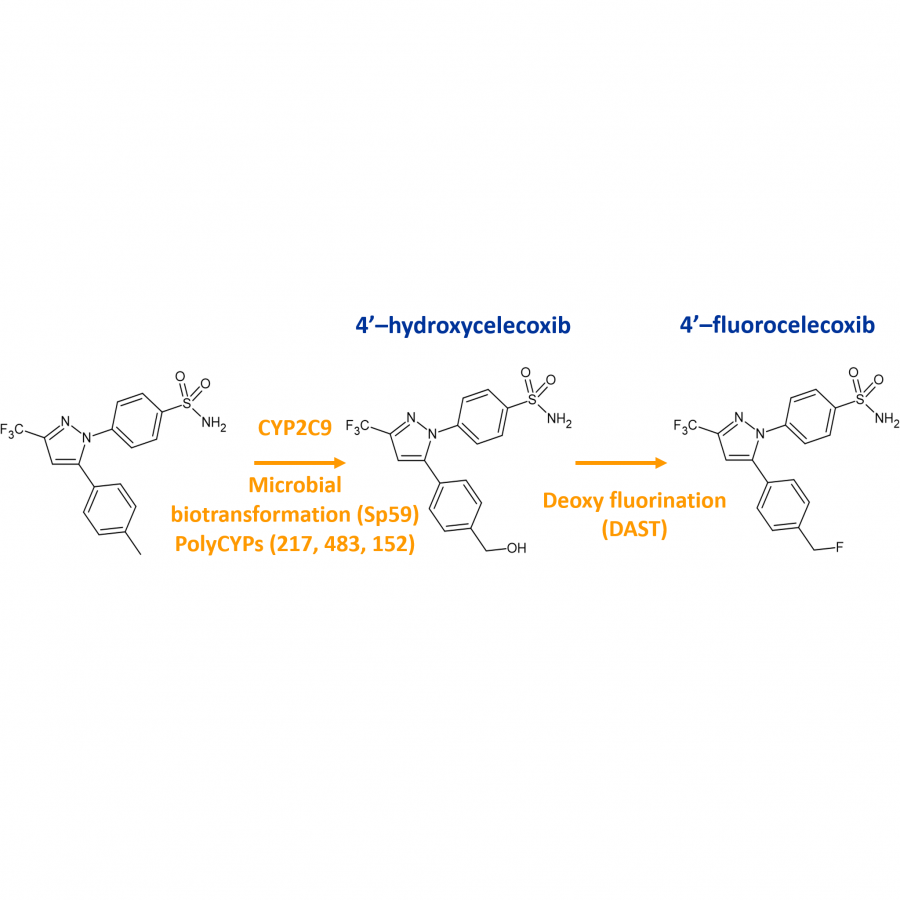 Celecoxib case study PolyCYPs providing handles for late-stage hydroxylation-fluorination strategy |
#120 | Styrene Oxide Isomerase-Catalyzed Meinwald Rearrangement and its Applications in Enantioselective Cascade Biotransformations |
|
| Presenting author: | Willy W. L. SEE of NATIONAL UNIVERSITY OF SINGAPORE |
| Other authors: | Zhi LI of NATIONAL UNIVERSITY OF SINGAPORE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Styrene oxide isomerase / Biocatalytic cascade reactions / Enantioselective synthesis / Dynamic kinetic resolution |
| Purpose: | The Meinwald rearrangement of epoxides is a synthetically useful reaction to produce aldehydes and ketones, but typically requires harsh conditions and toxic reagents, while also lacking regioselectivity and stereo-control. Styrene oxide isomerase (SOI) catalyzes the Meinwald rearrangement of aryl epoxides to carbonyl compounds with high selectivity under mild conditions, offering an effective biocatalytic alternative to chemical Meinwald rearrangement. Herein, we describe our recent applications of SOI-catalyzed epoxide isomerization as a key step in enantioselective cascade biotransformations. We report the discovery of a new type of SOI-catalyzed Meinwald rearrangement, which involves the isomerization of internal epoxides via a 1,2-methyl shift without 1,2-H shift to produce the corresponding aldehydes or cyclic ketone. SOI-catalyzed isomerization was determined to proceed via a 1,2-trans-shift in a stereospecific manner, thereby retaining the substrate’s enantio-configuration. The synthetic utility of this unique enzymatic Meinwald rearrangement involving a stereospecific 1,2-methyl shift was demonstrated by its incorporation into enantioselective cascades, to convert trans-methyl styrenes into (R)-configured 2-arylpropanols, 2-arylpropionic acids, or 2-arylpropyl amines with high enantioselectivity and yield. In addition, new types of one-pot enzymatic cascades involving SOI-catalyzed Meinwald rearrangement and dynamic kinetic resolution were developed to convert readily available racemic epoxides into valuable chiral products. SOI-catalyzed isomerization of racemic trans-methyl epoxides (via 1,2-methyl shift) or alpha-methyl epoxides (via 1,2-H shift) generated 2-arylpropanal in situ, which was followed by spontaneous racemization and enantioselective alcohol dehydrogenase-catalyzed oxidation or transaminase-catalyzed amination, producing a wide range of pharmaceutically relevant (S)-2-arylpropionic acids, (R)- and (S)-2-arylpropyl amines with high enantioselectivity and yield. The cascade reactions were performed with isolated enzymes or whole-cell biocatalysts, and the use of SOI to generate the aldehyde intermediate in situ effectively minimized side reactions related to aldehyde instability. Finally, chemoenzymatic cascades involving SOI-catalyzed Meinwald rearrangement were also developed. The combination of enantioselective whole-cell cascade biotransformation and metal-catalyzed coupling reactions successfully produced several examples of drug-related molecules. |
| References: | [1] R. Xin+, W. W. L. See+, H. Yun, X. Li, Z. Li. Angew. Chem. Int. Ed. 2022, 61, e202204889. [2] W. W. L. See, X. Li, Z. Li, Adv. Synth. Catal. 2023, 365, 68-77. [3] W. W. L. See, Z. Li, Chem. Eur. J. 2023, e202300102. |
| Figures: | 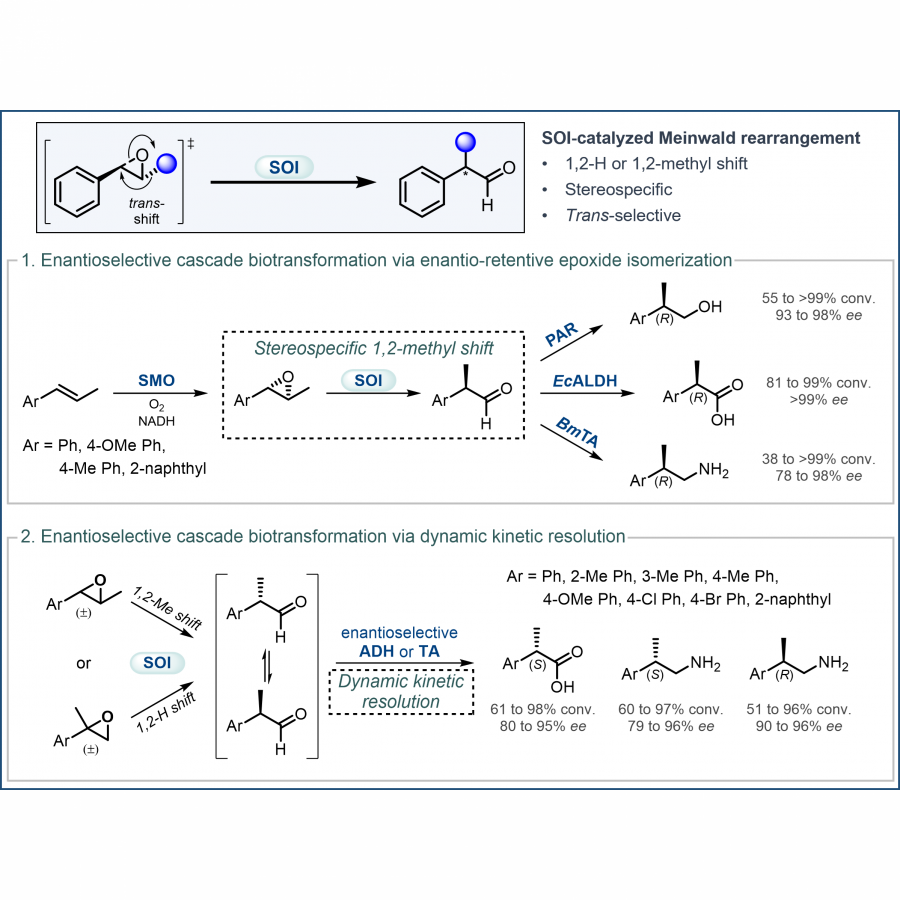 SOI-catalyzed Meinwald rearrangement and its applications in enantioselective cascade biotransformations Part 1. Discovery and application of SOI-catalyzed stereospecific epoxide isomerization via 1,2-methyl shift
Part 2. Development of enantioselective cascades involving SOI-catalyzed epoxide isomerization and dynamic kinetic resolution |
#123 | Optimization of a multi-step enzymatic synthesis of lauryl(3-hydroxypropyl)-succinate, precursor for the synthesis of extended carbohydrate-based surfactants |
|
| Presenting author: | ALEXIS SPALLETTA of UNIV. ARTOIS, UNILASALLE, ULR7519 - UNITÉ TRANSFORMATIONS & AGRO-RESSOURCES |
| Other authors: | NICOLAS JOLY of UNIV. ARTOIS, UNILASALLE, ULR7519 - UNITÉ TRANSFORMATIONS & AGRO-RESSOURCES PATRICK MARTIN of UNIV. ARTOIS, UNILASALLE, ULR7519 - UNITÉ TRANSFORMATIONS & AGRO-RESSOURCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzymatic cascade / multi-step / surfactant / spacer |
| Purpose: | The actual worldwide need for a more sustainable and renewable consumption leads conventional chemistry to undergo profound changes to meet nowadays and future challenges, especially processes decarbonization. Biocatalyzed processes can fit with this purpose of more sustainable industrial processes [1], as in surfactant-producing industry. As example of “green” surfactants, non-ionic biobased fatty acid carbohydrate esters are used in multiple fields, such as cosmetics, food industry, and pharmacology and biocontrol [2]. These multi-applications have created a worldwide market for such bioproducts and consequently expectations for their production. As a reminder, a surfactant is generally composed of a polar part, a carbohydrate for example, a lipophilic moiety with in-between, but rarely, a spacer/linker conferring an additional property to the surfactant, such as improved amphiphilic properties and a lower CMC (critical micellar concentration) value (Figure 1) [3]. Beyond the surfactant aspects, add a spacer would allow to integrate molecular patterns to classical surfactants and thus create a molecule-application relationship, with for example Pathogen-Associated Molecular Patterns (PAMP). The present work, as a part of biocatalyzed synthesis of carbohydrate-based surfactants, deals with the selective conception of a spacer-lipophilic synthon in two steps. In a first phase, the purpose is to condense a fatty alcohol, dodecan-1-ol, onto succinic acid using a lipase as catalyst. Then, propan-1,3-diol is added to the reaction medium to react with the products obtained in the first step to obtain lauryl(3-hydroxypropyl)succinate (Figure 1). The reference synthesis is performed in 2-methylbutan-2-ol, at 56°C, under stirring (240 rpm) and using 1% (w/v) supported Aspergillus Niger lipase. The first step provides mono-laurylsuccinate and di-laurylsuccinate, with molar yields of 49% and 25%, respectively. A kinetic study shows that a maximum yield is obtained after only 2h reacting. Then, the addition of propan-1,3-diol allows to obtain the lauryl(3-hydroxypropyl)succinate. The decrease of monolaurylsuccinate ratio shows its conversion to the product, lauryl(3-hydroxypropyl)succinate, and thus the interest to favor its synthesis at the expense of dilaurylsuccinate (Figure 2). An optimization based-on reagent ratios variation, solvent and co-solvent ratios, reaction media water content, as well as enzymatic cocktails has been performed and reaction kinetics were controlled using liquid and gas chromatography. To summarize few results, different ratios were tested, with diethylsuccinate/dodecan-1-ol molar ratio ranging from 3/1 to 1/3. To maximize the formation of mono-laurylsuccinate and minimize di-laurylsuccinate, the ratio that gave the best yield is 1/2. A temperature effect study highlighted an optimal temperature of 56°C for supported Aspergillus Niger lipase and supported CalB (Novozym 435). Various solvents were tested. For example, 2-methyltetrahydrofuran-3-one, a biobased solvent, reduced the amount of di-laurylsuccinate produced but the yield of mono-laurylsuccinate decreased from 49% to 30%. The addition of water to the reaction medium also limits the formation to di-laurylsuccinate (Figure 2). For instance, with CalB, the addition of 0.5% (V/V) water increased the mono-laurylsuccinate/di-laurylsuccinate ratio from 2 to 4. We have therefore mapped various parameters and so we can propose a multi-step one-pot synthesis of lauryl(3-hydroxypropyl)-succinate in 4h, without treatment. We are now working on the grafting lauryl(3-hydroxypropyl)-succinate onto carbohydrate using a β glucosidase, to implement an enzymatic cascade. Other parameters, such as the effect of bioprinting on monodispersity, are currently evaluated. At least, the impact of these optimizations on the recycling and reuse of enzymes will also be studied. |
| References: | [1] H.; Santi, C. Flow Biocatalysis: A Challenging Alternative for the Synthesis of APIs and Natural Compounds. International Journal of Molecular Sciences 2021, 22, 990, doi:10.3390/ijms22030990. [2] Sachdev, D.P.; Cameotra, S.S. Biosurfactants in Agriculture. Appl Microbiol Biotechnol 2013, 97, 1005-1016, doi:10.1007/s00253-012-4641-8. [3] Chaveriat, L.; Gosselin, I.; Machut, C.; Martin, P. Synthesis, Surface Tension Properties and Antibacterial Activities of Amphiphilic D-Galactopyranose Derivatives. Eur J Med Chem 2013, 62, 177-186, doi:10.1016/j.ejmech.2012.12.032. |
| Figures: | 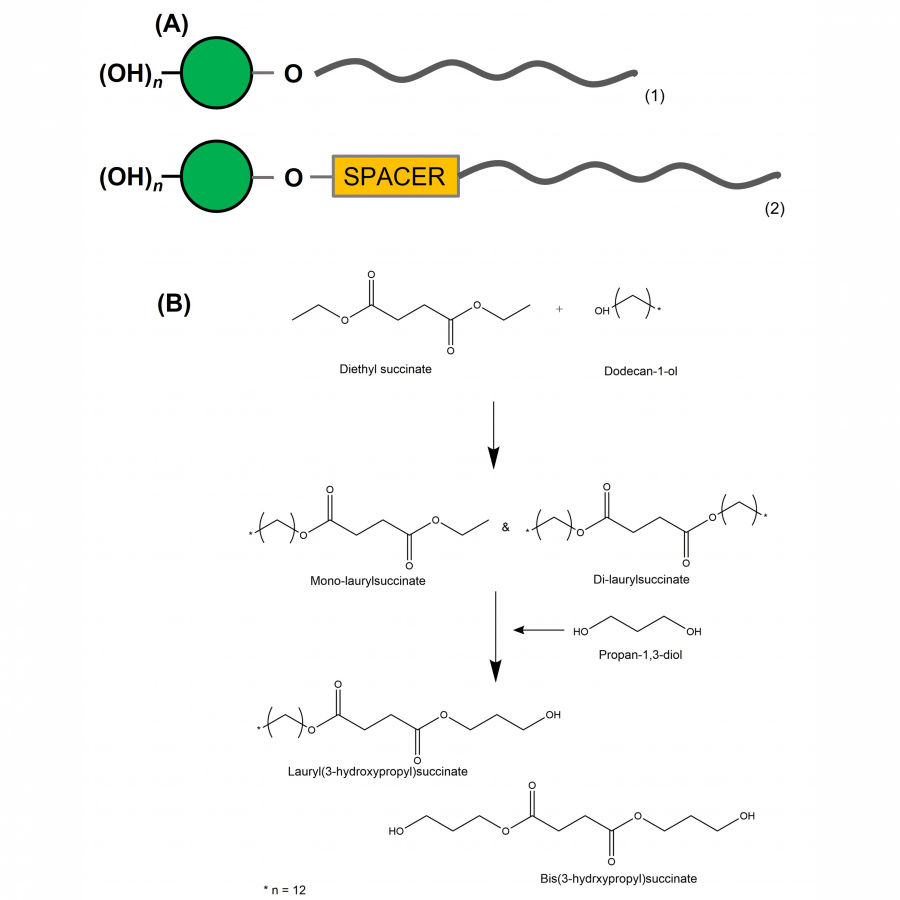 Synthesis pathway (A) Classical structure of a surfactant (1), surfactant with spacer (2), with polar part in green, and lipophilic part in grey. (B) Synthesis pathway for lipase-catalyzed synthesis of lauryl(3-hydroxypropyl) succinate. 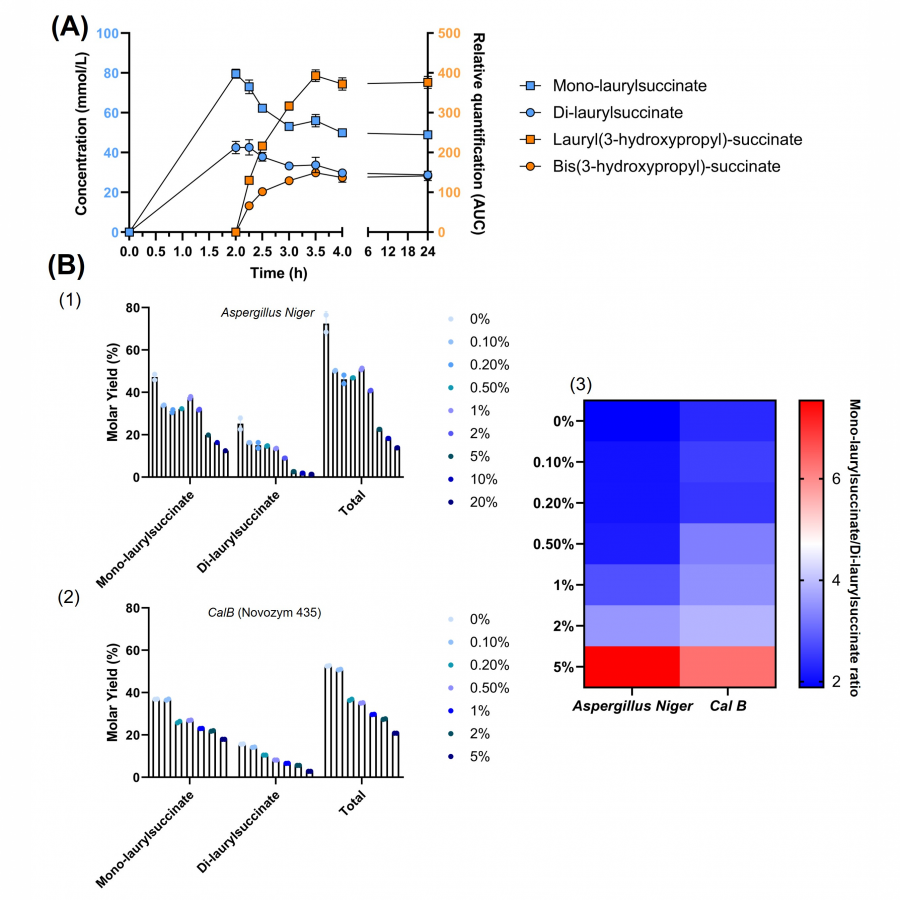 Kinetics study of formation and conversion of mono-laurylsuccinate and di-laurylsuccinate (A) Kinetics study of formation and conversion of mono-laurylsuccinate and di-laurylsuccinate in mmol/L to lauryl(3-hydroxypropyl) in AUC.
(B) Evolution of yields obtained for the synthesis of mono-laurylsuccinate and di-laurylsuccinate according to varia |
#131 | A cell-free platform for the synthesis of the active form of Vitamin B6 from xylose |
|
| Presenting author: | Renan Yuji MIYAMOTO of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY |
| Corresponding author: | Leticia ZANPHORLIN of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY |
| Other authors: | Vivian Pascal WILLERS of TECHNICAL UNIVERSITY OF MUNICH Ricardo Rodrigues DE MELO of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY Gabriela Felix PERSINOTI of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY Volker SIEBER of TECHNICAL UNIVERSITY OF MUNICH |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cascade / Synthetic biochemistry / Xylose / Vitamin |
| Purpose: | Vitamin B6 is a fundamental nutrient involved in more bodily processes than any other vitamin. Although vitamin B6 is essential for all forms of life, humans cannot synthesize it, so this micronutrient must be obtained from foods or isolated supplements. Currently, technological routes through chemical or fermentative processes can produce vitamin B6. Chemical synthesis employs expensive and/or hazardous resources while biological pathways still suffer to produce high titers due to the toxic features of its intermediates, and because vitamin B6 itself can interact with a wide range of metabolic reactions. Cutting-edge biotechnology relies on a genetically modified bacteria from the Rhizobium genus to produce vitamin B6. This process takes seven days and uses glucose as the building block in a complex media. Alternatively, the synthetic biochemistry approach can handle metabolic engineering cases that face highly complex challenges, but usually requires a formidable amount of enzymes. Here we propose a bio-based strategy via a synthetic biochemistry approach to produce the active form of vitamin B6 that uses xylose and ammonium sulfate as substrates and requires only six enzymes. Thermodynamical analysis demonstrated the cascade’s feasibility, giving us the initial conditions to test in vitro. Because the obtained yield was only 15.6 % (0.4 mM), all the enzymes of the cascade were characterized. Two possible limiting steps were identified regarding the low thermal stability of the phosphoketolase and the low activity of the PLP synthase. The rational prospection of novel targets was performed using the sequence similarity network (SSN) tool and genome mining, crossing those data with thermophilic organisms available in our laboratory strain collection. This way, the final phosphoketolase and synthase presented melting temperatures 35 and 13 °C higher, and their activities improved by 21 and 2-fold, respectively. The optimized system was able to produce 74 % higher vitamin B6 titers. Several approaches were made to optimize even more the cascade, including three ATP recycling systems, relieving the side-reaction of the phosphoketolase, and providing an amidotransferase partner for the synthase. However, none of those strategies was effective in the further improvement of the cascade. Although there is still room for improvement to reach higher titers, our system to produce vitamin B6 from xylose presented higher productivity than the state-of-art bio-based technology that uses glucose as a building block (15.9 vs 7.7 mg L-1 h-1), and higher yields (27.2 vs 2.2 %). |
| References: | Rosenberg J, Ischebeck T, Commichau FM. Biotechnol. Adv., 2017 Acevedo-Rocha CG et al, Curr. Opin. Biotechnol., 2019 Guédez G et al, Structure, 2012. |
| Figures: | 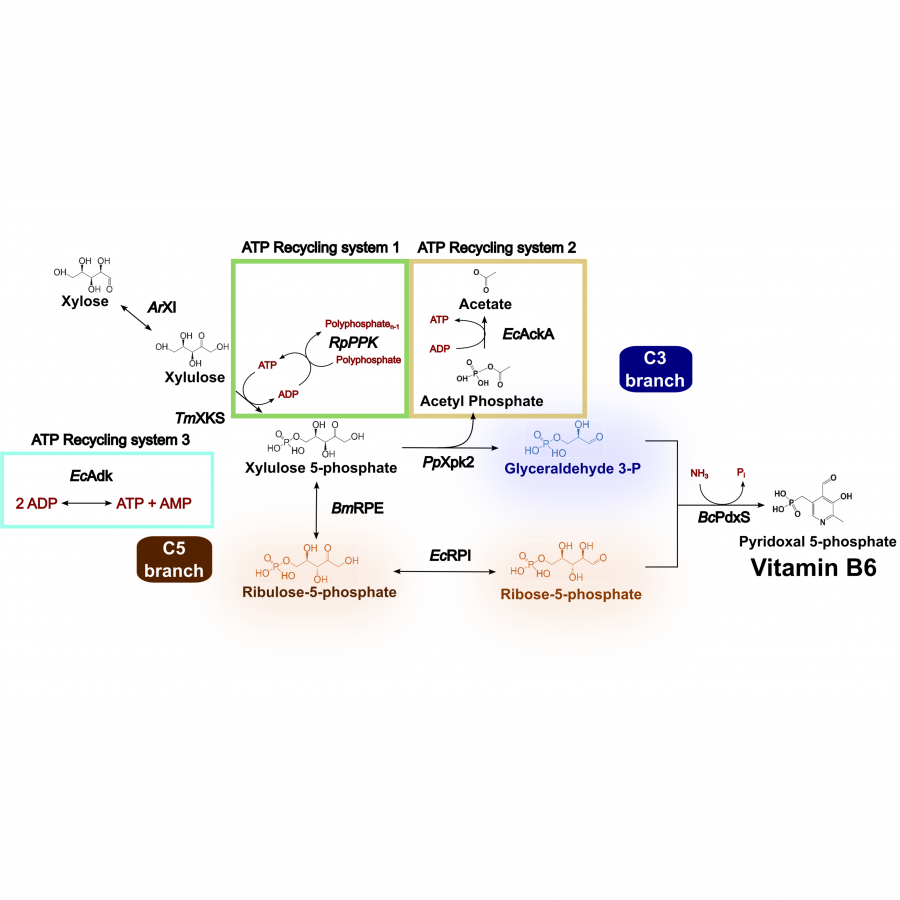 Enzymatic cascade Description of the cascade starting from xylose |
#135 | Exploring sequence space to improve transglycosylation efficiency in CGTase |
|
| Presenting author: | Gareth SURMAN of UNIVERSITY OF MANCHESTER |
| Other authors: | Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Glycoscience / Ancestral Sequence Reconstruction / CGTase |
| Purpose: | Cyclodextrin glycosyltransferases (CGTases) are multifunctional enzymes, performing four related reactions that each involve stereoselective α-glucosyl transfer on to an alcohol acceptor [1]. It has been shown to catalyse the glycosylation of a variety of acceptors [2-4]. However, this multifunctionality creates a challenge when trying to create polyglycosylated products biocatalytically, as the products are hydrolysed down to those containing fewer glucose units. We have developed a high throughput assay system in order to assess the degree of glycosylation, and applied this to study both directed evolution and metagenomics of CGTase. ____ [1] B. A. Van Der Veen et al, Eur. J. Biochem., 2000, 267, 658–665 [2] W. M. J. Kloosterman et al, Macromol. Biosci., 2014, 14, 1268–1279 [3] X. Tao et al, Crit. Rev. Biotechnol., 2019, 39, 249–257 [4] D. Svensson et al, Biotechnol. Bioeng., 2009, 104, 854–861 |
| References: | [1] B. A. Van Der Veen et al, Eur. J. Biochem., 2000, 267, 658-665 [2] W. M. J. Kloosterman et al, Macromol. Biosci., 2014, 14, 1268-1279 [3] X. Tao et al, Crit. Rev. Biotechnol., 2019, 39, 249-257 [4] D. Svensson et al, Biotechnol. Bioeng., 2009, 104, 854-861 |
#136 | Rational design of cyclohexanone dehydrogenase for enhanced α, β-desaturation and substrate specificity |
|
| Presenting author: | Gary BLACK of NORTHUMBRIA UNIVERSITY |
| Other authors: | Warispreet SINGH of NORTHUMBRIA UNIVERSITY Nicola BROWN of NORTHUMBRIA UNIVERSITY Hannah MCCUE of UNIVERSITY OF LIVERPOOL Sophie MARRIOTT of UNIVERSITY OF LINCOLN Justin PERRY of NORTHUMBRIA UNIVERSITY Richard WILSON of UNIVERSITY OF YORK Johan TURKENBURG of UNIVERSITY OF YORK Kshatresh DUBEYE of SHIV NADAR UNIVERSITY Stephen PRIOR of UNIVERSITY OF LINCOLN Andrew CARNELL of UNIVERSITY OF LIVERPOOL Edward TAYLOR of UNIVERSITY OF LINCOLN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cyclic ketones / molecular dynamics / rational engineering / X-ray crystallography |
| Purpose: | The selective α, β-desaturation of cyclic carbonyl compounds, which are found in the core of many steroid and bioactive molecules, using green chemistry is highly desirable. To achieve this task, we have for the first time described and solved the de novo structure of a member of a new enzyme class, cyclohexanone dehydrogenases (Figure 1). The breadth of substrate specificity was investigated by assaying the cyclohexanone dehydrogenase against several cyclic ketones, lactones and lactams. To investigate substrate binding, a catalytic mutant was generated and used to obtain a crystallographic complex with the natural substrate, cyclohexanone. This revealed substrate-active site interactions, as well as the proximity of the cofactor, flavin adenine dinucleotide, and enabled us to propose a mechanistic function to key amino acids. We then used molecular dynamic simulations to guide design to add functionality to the cyclohexanone dehydrogenase enzyme. The resulting mutant had overall improved enzyme activity and substrate scope, i.e., accepting the bulkier carbonyl compound, lactone dihydrocoumarin. Structural analysis of the mutant revealed a broader, more open active site, which helped explain the modified substrate specificity. This work paves the way for future bespoke regioselective α, β-desaturation in the synthesis of important bioactive molecules via rational enzyme engineering. |
| Figures: | 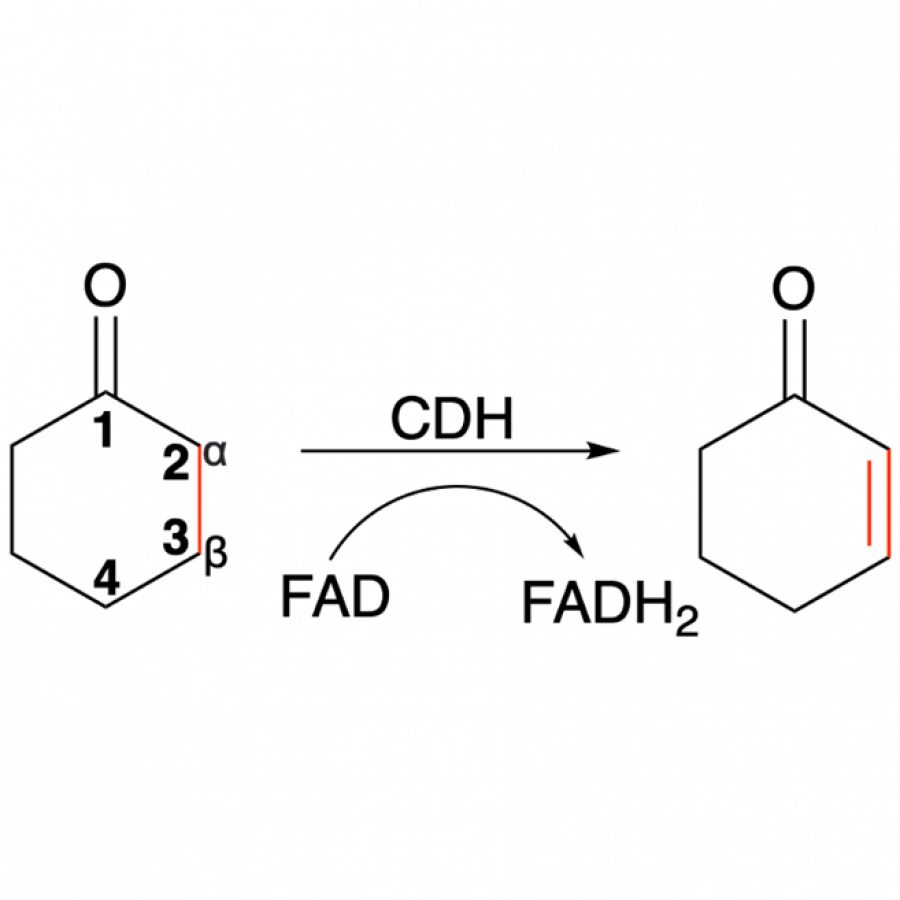 Figure 1 The α, β-desaturation of cyclohexanone to 2-cyclohexen-1-one catalysed by cyclohexanone dehydrogenase (CDH) in the presence of flavin adenine dinucleotide (FAD), which is reduced to FADH2. |
#140 | NOVEL METHODS AND PROCESSES TO SUPPLY AND REGENERATE BIOCHEMICAL COFACTORS |
|
| Presenting author: | Monika MÜLLER of INNOSYN B.V. |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Industral Biocatalysis / Cofactor dependent enzymes / Regeneration strategies / |
| Purpose: | Biocatalysis is one of the core technologies at InnoSyn to develop chemo-enzymatic processes for the production of small molecules which are applied in pharmaceutical and chemical industries [1-3]. Hydrolases like lipases, esterases and proteases are not that popular in academic research anymore, but represent the work horses of industrially applied biocatalysis since decades because of their unmet stability and efficiency [4]. Alcohol dehydrogenases in the reductive mode come close to the efficiencies of hydrolases but require NAD(P)H cofactor regeneration. Recently we focused on more challenging co-factor and co-substrate supply and regeneration strategies for cost efficient industrial biocatalysis such as the in situ regeneration of NAD(P)+ for oxidative ADH reactions at pilot plant scale and other co-factors and co-substrates. Regio-, chemo- and enantioselective alcohol oxidations with pure O2 as terminal oxidant were scaled up to pilot plant production achieving industrially relevant product concentrations of 40 - 200 g L-1 and high volumetric productivities of 1.5 - 14 g L-1 h-1, respectively [5-6]. Finally, proof-of-principle of scalable and efficient ATP regeneration with stable and cheap phosphoenolpyruvate formulations, 2-ketoglutarate production from L-glutamate at up to 1 mol L-1 for 2-KG/Fe(II) dependent oxygenase reactions and UDP-glucose supply and recycling from sucrose for the multi-gram scale production high-value glucosylated natural products were obtained [7] and will be presented. |
| References: | [1] H. E. Schoemaker, D. Mink, M. G. Wubbolts, Science 2003, 299, 1694. [2] M. Schürmann, M. Wolberg, S. Panke, H. Kierkels, in Green Chemistry in the Pharmaceutical Industry (eds P. J. Dunn, A. S. Wells and M. T. Williams) 2010, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. [3]M. Schürmann, in Industrial Enzyme Applications (eds A. Vogel and O. May) 2019, Wiley‐VCH Verlag GmbH & Co. KGaA, Weinheim, Germany [4] M. Schürmann and I. Kaluzna, Specialty Chemicals Magazine 2022, 42(1), 28-30 [6] S. Bartsch, J. Brummund, S. Köpke, H. Straatman, A. Vogel, M. Schürmann, Biotechnol. J. 2020, 15, 2000171 [7] M. Biermann, M. Schürmann, T. Schmitges, A. Vogel, J. Brummund, Org. Process Res. Dev. 2022, 26 (7), 2021-2029 [8] M. Schürmann and M. Müller, Chimica Oggi - Chemistry Today 2021, 39(6), 12-16 |
#145 | A chemo-enzymatic CO2 capture process by ionic liquid technology |
|
| Presenting author: | Pedro LOZANO of UNIERSIDAD DE MURCIA (SPAIN) |
| Other authors: | ROCIO VILLA of UNIVERSIDAD DE MURCIA SUSANA NIETO of UNIVERSIDAD DE MURCIA FRANCISCO J RUIZ of UNIVERSIDAD DE MURCIA FRANCISCO VELASCO of UNIVERSIDAD DE MURCIA JAIRTON DUPONT of UNIVERSIDAD DE MURCIA ANTONIO DONAIRE of UNIVERSIDAD DE MURCIA RAUL PORCAR of UNIVERSIDAD JAUME I EDUARDO GARCIA-VERDUGO of UNIVERSIDAD JAUME I |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lipase / ionic liquids / co2 capture / chemo-enzymatic process |
| Purpose: | The reduction of the CO2 concentration in the atmosphere is one of the main global challenges of this century , that requires the development of technological solutions for CO2 capture, and its conversion into added-value products. In the same context of Sustainable Development, the substitution of recalcitrant plastics (e.g. polyurethanes produces a serious ecological threats, etc.) for bio-based and biodegradable polymeric materials fulfilling circular economy criteria (e.g. water-based non-isocyanate polyurethane, etc) is another global challenge.[1] Glycerol is a renewable chemical from biomass of great interest because of its large availability as a by-product of the biodiesel industry, which can be used as a raw material in the synthesis of numerous products (e.g. polymers, solvents, fuel additives, etc.). Among its derivatives, glycerol carbonate (GC), resulting from the net incorporation of a carbon dioxide molecule to glycerol, is a versatile molecule, which can also be modified by introducing other reactive moieties (e.g. acrylate groups) to produce new functionalized monomers for polymer chemistry.[2] A sustainable chemo-enzymatic process for producing both glycerol carbonate acrylate (GCA) and glycerol carbonate methacrylate (GCMA), as useful monomers for preparation of biodegradable plastic materials, has been carried out taking advantage of the ionic liquids (ILs) technologies.[3] The process consisted in two consecutive catalytic steps, which can be carried out by either sequential or one-pot experimental approaches. The glycidyl (meth)acrylate was firstly synthesized by enzymatic transesterification from (meth)acrylate vinyl ester with glycidol in Sponge Like Ionic Liquids (SLILs) as the reaction medium (100% yield after 6 h at 60 ºC). SLILs not only provided a suitable reaction medium but also allowed the simple isolation of the resulting glycidyl esters as an IL-free pure fraction through a cooling / centrifugation straightforward protocol. The second step consisted in the GCA, or GCMA, synthesis as the outcome of the cycloaddition of CO2 to the obtained glycidyl acrylate or glycidyl methacrylate, respectively, catalysed by a covalently attached 1-decyl-2-methylimidazolium moiety (Supported Ionic Liquids-Like Phase, SILLP) in a solvent-free system and under mild conditions (60 ºC, 1-10 bar), leading to up to 100 % yield after 6 h. The components of the reaction system (biocatalyst/SLIL/SILLP) can be fully recovered and reused for at least 6 cycles with unchanged catalytic performance (see Fig. 1).[4] This technology has also been successfully used for the synthesis of other cyclic glycerol carbonate derivatives. The combination of both ILs and (bio)catalyst technologies can pave a new sustainable way to contribute to the reduction of the CO2 concentration in the atmosphere through its capture and incorporation into added-value products of interest.. Acknowledgements This work was partially supported by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021- 124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN –European Union Next Generation EU-PRTR (TED2021-129626B-C21/C22) and SENECA (21884/PI/22) for financial support. J. D. is fellow of the “Maria Zambrano program” at the University of Murcia (Spain |
| References: | [1] Zanatta, N. M. Simon and J. Dupont. The nature of carbon dioxide in bare ionic liquids. ChemSusChem. 2020, 13, 3101-3109. [2] R. Villa, E. Alvarez, R. Porcar, E. Garcia-Verdugo, S. V. Luis, P. Lozano. Ionic liquids as an enabling tool to integrate reaction and separation processes. Green Chem. 2019, 21, 6527-6544. [3] P. Migowski, P. Lozano and J Dupont. Imidazolium based ionic liquid-phase green catalytic reactions. Green Chem. 2023, DOI: 10.1039/d2gc04749g. [4] R. Villa, R. Porcar, S. Nieto, A. Donaire, E. Garcia-Verdugo, P Lozano. Sustainable chemo-enzymatic synthesis of glycerol carbonate (meth)acrylate from glycidol and CO2 enabled by ILs technologies. Green Chem. 2021, 23, 4191-4200 |
| Figures: | 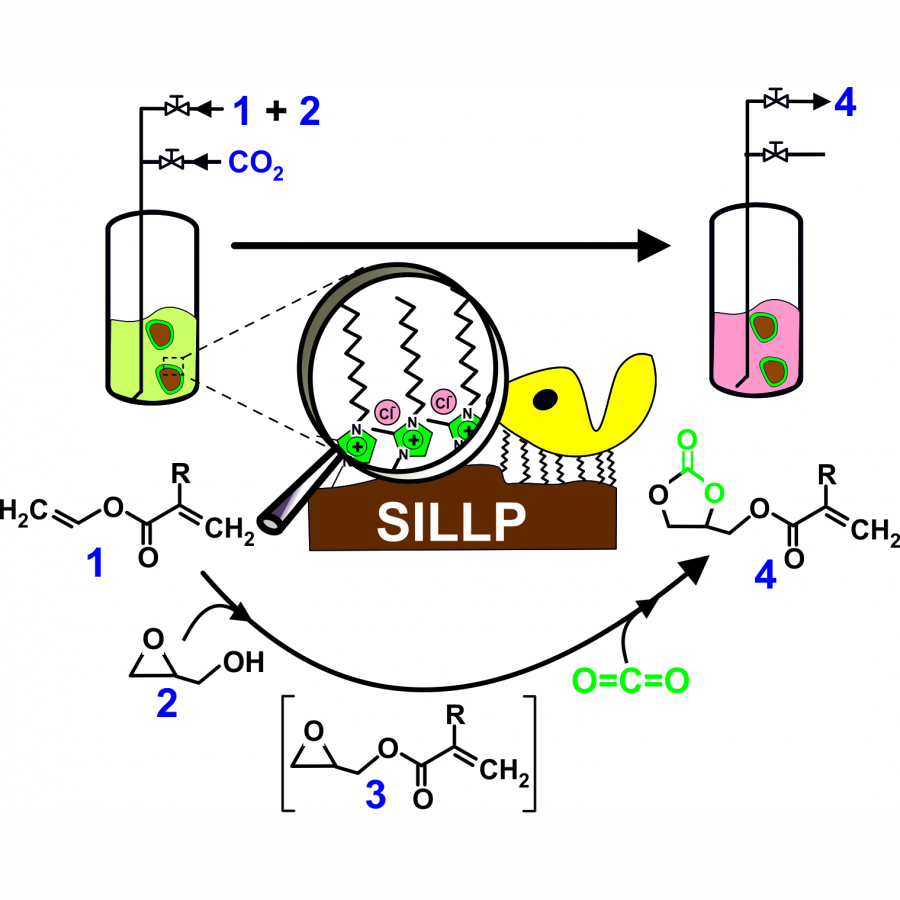 Figure 1 Schema of the one-pot chemo-enzymatic synthesis of GCA under solvent-free conditions, catalyzed through two consecutive reactions carried out by immobilized CALB onto 1-alkyl-3-methylimidazolium-based SILLPs, as a dual catalyst |
#150 | Anti-Markovnikov oxidation of unactivated, aliphatic alkenes by directed enzyme evolution |
|
| Presenting author: | Cindy KLAUS of BIELEFELD UNIVERSITY |
| Corresponding author: | Stephan HAMMER of BIELEFELD UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme engineering / Biocatalysis / Directed evolution / Cytochrome P450 |
| Purpose: | The direct aerobic oxidation of alkenes to carbonyls is an important, yet, in part very challenging transformation in synthesis.[1] Our research group has recently started to engineer cytochrome P450 enzymes for direct alkene to carbonyl oxidations. This includes enzymes that perform anti Markovnikov alkene oxidation of styrenes to generate the corresponding aldehydes,[2] as well as regioselective oxidation of internal aryl alkenes to the corresponding ketones.[3] The engineered enzymes fully exploit a catalytic cycle that has largely eluded small molecule catalysis. This is achieved by conformational control over a key radical intermediate to prevent the dynamically favored alkene epoxidation.[4,5] Here we expand this chemistry towards unactivated aliphatic alkenes through directed enzyme evolution.[6] The initially low activity and selectivity for the aliphatic 1-alkene was improved over 14 rounds of directed evolution, introducing 21 beneficial mutations. The final catalyst was characterized by exploring the substrate scope and its applicability in synthesis as well as determining kinetic parameters and solving the crystal structure. |
| References: | [1] J.J. Dong, W.R. Browne, B.L. Feringa, Angew. Chem. Int. Ed. 2015, 54, 734. [2] S.C. Hammer, G. Kubik, E. Watkins, S. Huang, H. Minges, F.H. Arnold, Science 2017, 358, 215. [3] S. Gergel, J. Soler, A. Klein, K.H. Schülke, B. Hauer, M. Garcia-Borràs, S.C. Hammer, ChemRxiv 2022, DOI: 10.26434/chemrxiv-2022-dp94p [4] J. Soler, S. Gergel, C. Klaus, S.C. Hammer, M. Garcia-Borràs, J. Am. Chem. Soc. 2022, 144, 15954. [5] C. Klaus, S.C. Hammer, Trends Chem. 2022, 4, 363. [6] C. Klaus, G. Kubik, Y. Gumulya, S.C. Hammer, unpublished data. |
| Figures: | 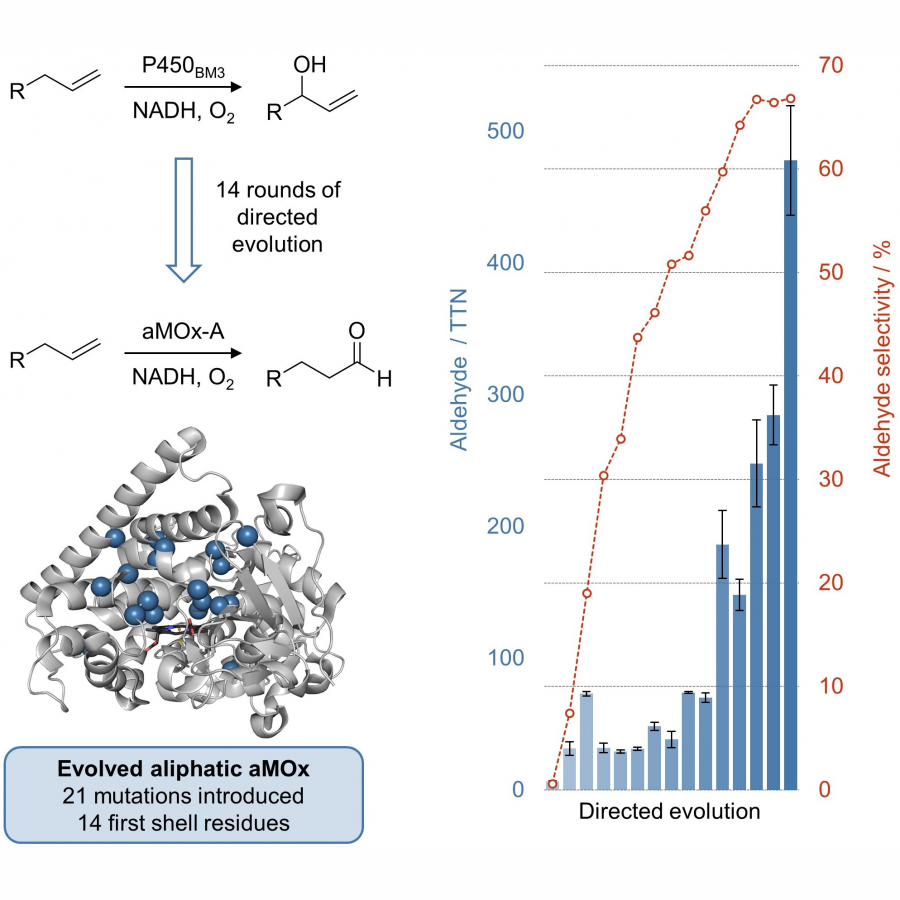 Directed evolution of an anti-Markovnikov oxidase for unactivated, aliphatic alkenes. Starting from P450BM3 which converts aliphatic 1-alkenes into the corresponding allylic alcohol an aldehyde selective anti-Markovnikov oxygenase (aMOx-A) was obtained in 14 rounds of directed evolution. |
#151 | Artificial [Mn]-hydrogenase for Asymmetric Transfer Hydrogenation |
|
| Presenting author: | Weijin WANG of EPFL |
| Corresponding author: | XILE HU of EPFL |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Artificial hydrogenase / Manganese catalysis / |
| Purpose: | Natural hydrogenases exclusively choose Ni and Fe in [NiFe]-, [FeFe]- and [Fe]-hydrogenases for (asymmetric) hydrogenation reactions. However, these enzymes are generally with limited substrate scope. In contrast, artificial metalloenzymes (ArMs) can incorporate abiotic metal complexes into a protein scaffold, leading to a wider substrate diversity. Although synthetic Mn complexes have been successfully incorporated into natural [Fe]-hydrogenase (Fig. 1A), achieving enantioselective hydrogenation reactions has remained a challenge. Herein, we report the development of an enantioselective [Mn]-hydrogenase based on biotin-streptavidin technology (Fig. 1B). Through chemogenetic optimzation of the reaction conditions, we achieved up to 98% enantiomeric excess (ee) and > 99% yield. Additionally, this artificial metalloenzyme displayed excellent functional group tolerance and broad substrate scope, catalyzing asymmetric transfer hydrogenation of ketones with high yield and enantioselectivity (Fig. 2). By combining QM/MM calculation and X-ray crystallography, out findings suggest that the S112Y-K121M mutations, along with the chemical structure of the Mn cofactor, play critical roles in enhancing the reactivity and enantioselectivity of the enzyme. These results highlight the potential of manganese as a metal cofactor in the development of artificial metalloenzymes, and demonstrate the power of chemogenetic optimization for enzyme engineering. |
| Figures: | 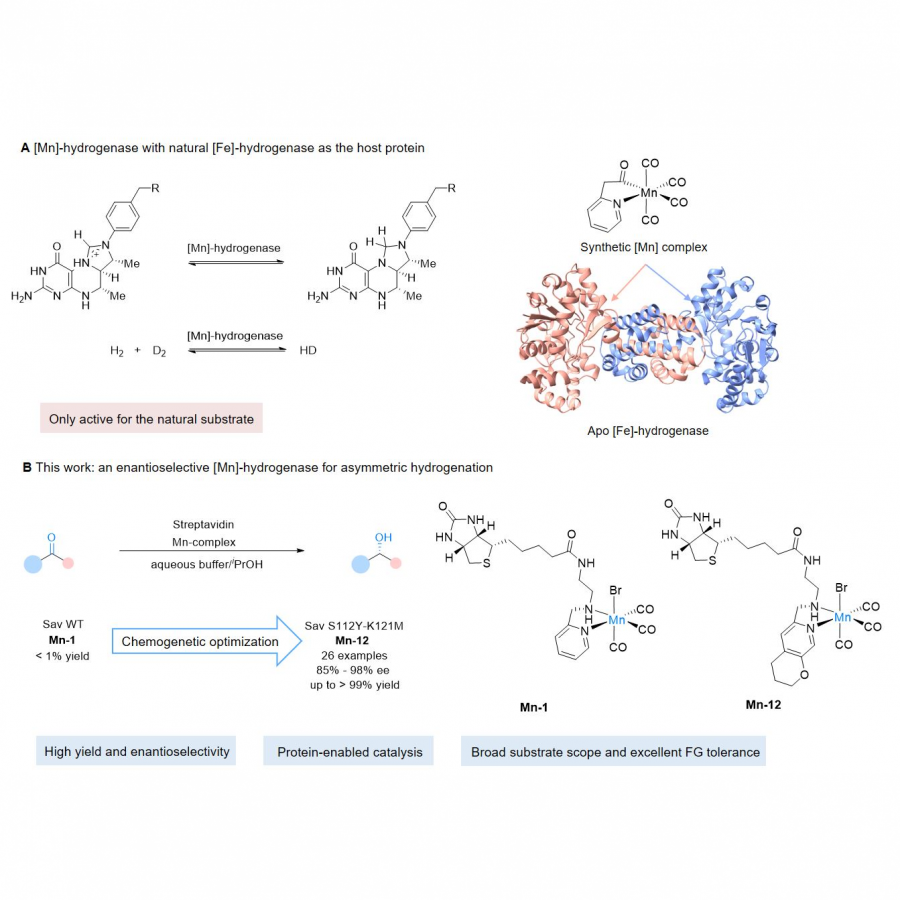 Development of artificial [Mn]-hydrogenase A, previous work on artificial [Mn]-hydrogenase with natural [Fe]-hydrogenase as the host protein; B, this work: an enantioselective [Mn]-hydrogenase for asymmetric hydrogenation
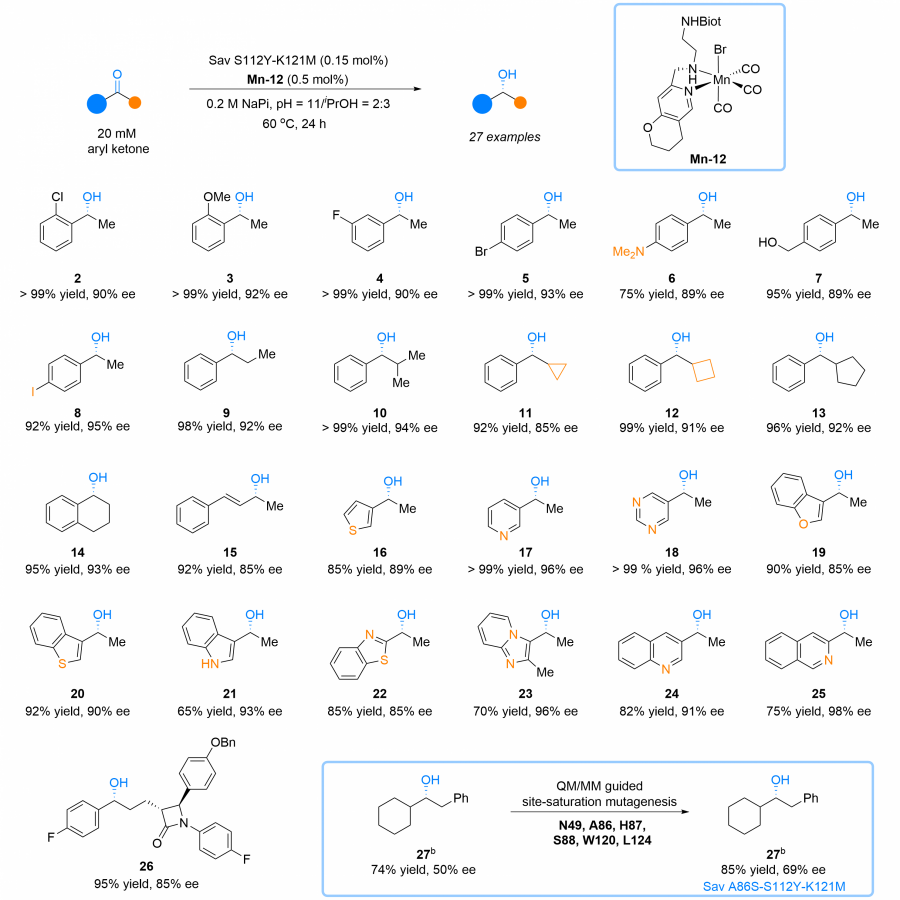 Substrate scope of [Mn]-hydrogenase This artificial metalloenzyme displayed excellent functional group tolerance and broad substrate scope, catalyzing asymmetric transfer hydrogenation of ketones with high yield and enantioselectivity |
#153 | Dimethylallyl tryptophan synthase RePT from Rasamsonia emersonii catalyzes prenylation on tryptophan, tyrosine, and plant phenolics |
|
| Presenting author: | Pimvisuth CHUNKRUA of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY |
| Other authors: | Kai LESCHONSKI of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY Alejandro GRAN-SCHEUCH of MOLECULAR ENZYMOLOGY GROUP, UNIVERSITY OF GRONINGEN Gijs VREEKE of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY Jean-Paul VINCKEN of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY Marco FRAAIJE of MOLECULAR ENZYMOLOGY GROUP, UNIVERSITY OF GRONINGEN Willem VAN BERKEL of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY Wouter DE BRUIJN of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY Mirjam KABEL of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | DMATS / prenylation / plant phenolics / stilbenes |
| Purpose: | Dimethylallyl tryptophan synthases (DMATSs) are aromatic prenyltransferases (PTs) that catalyze the transfer of a prenyl (isoprenoid) moiety from a donor to an aromatic acceptor. Aside from their natural role in prenylating tryptophan derivatives in fungal indole alkaloid biosynthesis, DMATSs also act on structurally diverse aromatic substrates [1]. This capability makes DMATSs a potential biotechnological tool to produce biologically active compounds in a wide range of applications, such as antimicrobial plant phenolics [2]. Our study explored the substrate scope and product profile of a recombinant RePT, a novel DMATS from Rasamsonia emersonii, a thermophilic fungus (Figure 1). RePT was successfully produced via a His6-SUMO-RePT construct, showing a molecular weight of 66.4 kDa with 99.1% purity. Among a variety of (plant) aromatic substrates, RePT showed the highest substrate conversion for L-tryptophan and L-tyrosine (>90%), both yielding two mono-prenylated products. Eight phenolics from diverse phenolic subclasses were accepted with a noticeable conversion (>10%) with three stilbenes showing the highest conversion: oxyresveratrol (55%), pinostilbene (39%), and resveratrol (37%). Besides stilbenes, also (+)-catechin, (-)-epicatechin, coumestrol, (±)-equol, and phloretin showed 11-25% conversion. The position of prenylation by RePT on L-tryptophan was determined using 1H, 13C, and 2D-NMR spectroscopy to be either C7-prenylation (80% relative abundance), or reverse N1-prenylation (20%). For plant phenolics, the position of prenylation as annotated using MS2 fragmentation patterns, showed mainly mono-O-prenylation. Moreover, RePT was tolerant to organic solvents and to some extent to a higher temperature, yielding higher than 90% L-tryptophan conversion in the presence of 20% (v/v) methanol or DMSO, or at 50°C. Our findings indicate that RePT may be a promising biocatalyst with potential applications in generating valuable bioactive prenylated aromatics for food, cosmetic, and pharmaceutical industries. |
| References: | [1] Chen, R. et al., Nat. Chem. Biol., 2017, 13, 226-234. [2] J. C. de Bruijn, W. et al., Trends Biotechnol., 2020, 38, 917-934. |
| Figures: | 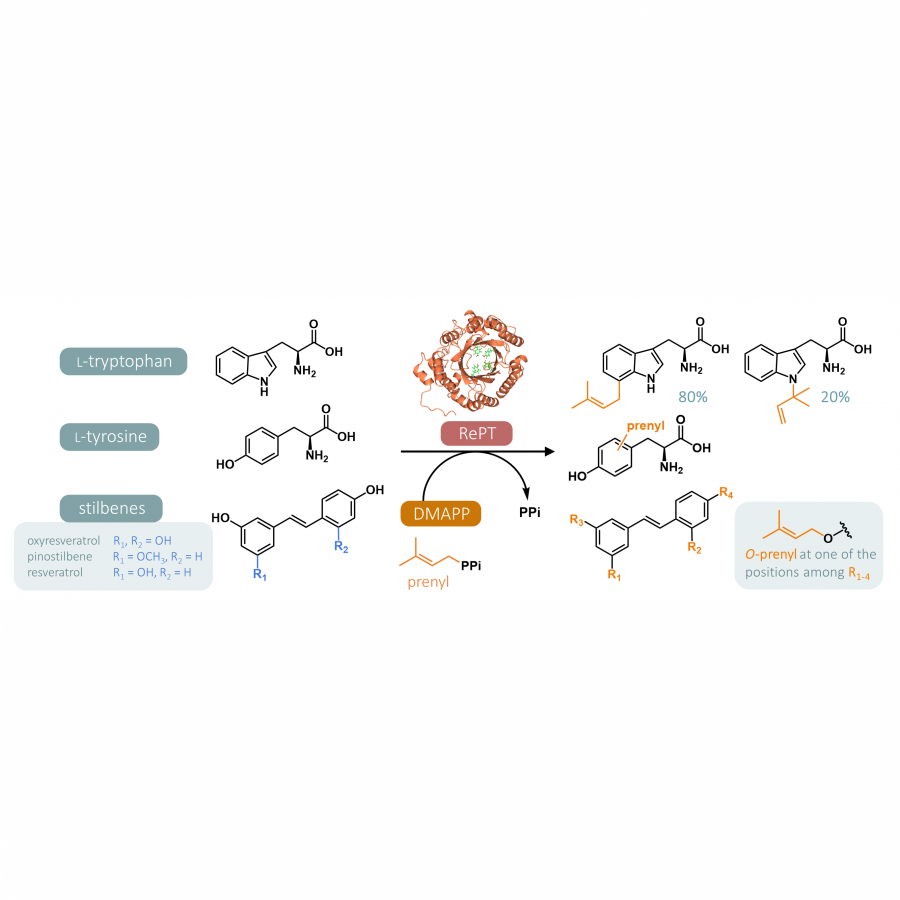 Figure 1. Prenylation of aromatic compounds by RePT in the presence of dimethylallyl pyrophosphate (DMAPP) as a prenyl donor. The three-dimensional structure of RePT is displayed with the strictly conserved four tyrosine residues among DMATSs (represented as green |
#162 | Bright and Flashy Figures of the Inclusive Kind |
|
| Presenting author: | Felix KASPAR of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | data / inclusive / color vision deficiency / |
| Purpose: | Biocatalysis has its roots in chemistry and microbiology; visual sciences. In biocatalysis, we use schemes to illustrate chemical transformations, protein structures to depict complex 3-dimensional architectures and graphs to convey complex relationships and datasets. In doing so, we often use color to highlight certain parts of a figure, guide the reader’s eye or transmit information. Colors are often easier to perceive and differentiate than shapes and can be particularly helpful to enhance recognition of recurring elements or intuitively link to concepts. Colors are incredibly useful. However, improper color choices can lead to a misrepresentation or inaccessibility of the underlying information. This can either bias the reader’s perception of the displayed data and/or make the figure inaccessible to readers with color vision deficiencies. This contribution discusses how a mindful and informed use of color in scientific illustrations – in particular through scientific color maps – yields more inclusive, accessible and accurate papers, grant proposals and talks. This one is not about enzymes and chemical transformations. It is about how to visualize them for everyone. |
| References: | [1] Kaspar, F., Crameri, F., Angew. Chem. Int. Ed. 2022, e202114910 |
| Figures: |  Figure 1. Improper color choices misrepresent the underlying data. |
#164 | GTHNL CATALYZES OXIDATIVE C=C BOND CLEAVAGE |
|
| Presenting author: | Ulf HANEFELD of TECHNISCHE UNIVERSITEIT DELFT |
| Other authors: | Peter-Leon HAGEDOORN of TECHNISCHE UNIVERSITEIT DELFT Jose COLOMA of TECHNISCHE UNIVERSITEIT DELFT |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Hydroxynitrle lyase / oxidative cleavage / promiscuity / |
| Purpose: | GtHNL is a hydroxynitrile lyase that was successfully utilized for the synthesis of cyanohydrins (Scheme 1B) and nitro aldol products, in batch and in flow [1,2,3]. GtHNL has moderate similarity (35%) with Tm1459 and the same catalytic metal ion, Mn(II) (Scheme 1A). Enzymes display high selectivity for one type of reaction. Therefore, lyases are not expected to catalyze oxidative C=C bond cleavage reactions as Tm1459 does (Scheme 1C) [4]. However, examining the structure of GtHNL there is no obvious reason why it should not catalyze the oxidative C=C bond cleavage, as described for Tm1459. Equally a hydroxynitrile lyase activity for Tm1459 cannot be ruled out. GtHNL was employed under condition earlier successful for Tm1459 [4]. tert-butyl hydroperoxide (TBHP) was used as oxidizing reagent and GtHNL satisfactorily catalyzed the oxidative cleavage of a number of styrene derivatives. Enzyme and reaction engineering led to improved selectivities and yields. On the other hand Tm1459 displayed only modest hydroxynitrile lyase activity without selectivity. These results question our concepts of enzyme selectivity and at the same time open new routes to novel enzyme activities. ____ |
| References: | [1] I. Hajnal, et al, FEBS J., 2013, 280, 5815-5828. [2] J. Coloma, et al, Catal. Sci. Technol., 2020, 10, 3613-3621. [3] J. Coloma, et al, Catalysts 2022, 12, 161. [4] I. Hajnal, et al, Adv. Synth. Catal. 2015, 357, 3309 -3316. |
| Figures: | 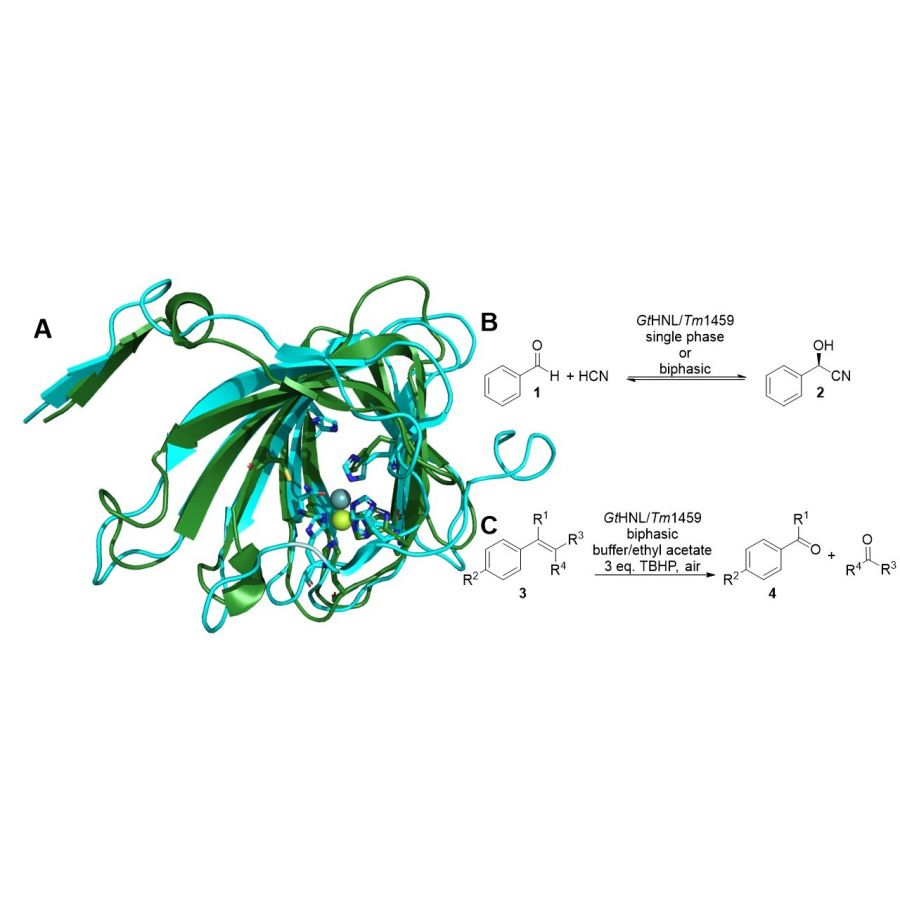 Scheme 1 A) Superimposition of GtHNL (PDB code: 4BIF) in blue with Mn(II) in grey coordinated by four histidines and glutamine and Tm1459 (PDB code: 1VJ2) in green with Mn(II) in lime coordinated by four histidines. B) Cyanohydrin synthesis C) Oxidative cleavage. |
#171 | Production of selected chiral diols via synthetic enzymes cascade in unconventional media |
|
| Presenting author: | Maria ALLAHHAM of FORSCHUNGSZENTRUM JÜLICH/ IBG-1 |
| Other authors: | Jan-Dirk SPÖRING of ENZYMASTER DEUTSCHLAND GMBH Chiara AUFDERHEIDE of FORSCHUNGSZENTRUM JÜLICH/ IBG-1 Torsten SEHL of FORSCHUNGSZENTRUM JÜLICH/ IBG-1 Lisa SEIBT of FORSCHUNGSZENTRUM JÜLICH/ IBG-1 Marco BOCLA of ENZYMASTER DEUTSCHLAND GMBH Neha VERMA of ENZYMASTER DEUTSCHLAND GMBH Thomas DAUSMANN of ENZYMASTER DEUTSCHLAND GMBH Jan WIESENTHAL of INSTITUTE OF TECHNICAL AND MACROMOLECULAR CHEMISTRY Niklas GAELINGS of INSTITUTE OF TECHNICAL AND MACROMOLECULAR CHEMISTRY Jürgen KLANKERMAYER of INSTITUTE OF TECHNICAL AND MACROMOLECULAR CHEMISTRY Dörte ROTHER of FORSCHUNGSZENTRUM JÜLICH/ IBG-1 |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzymatic cascade / vicinal chiral diols / carboligation / oxidoreduction |
| Purpose: | Stereopure vicinal diols are important building blocks used for fine chemicals and pharmaceutical compounds.1 2,3-Butanediol is a well-known chiral diol, which is used in practical applications such as plasticizers and antifreeze agents.2 These diols can also serve as precursors for bio-fuels starting from bio-resources. For this application, product concentration is more important than stereoselectivity.3 Due to the high selectivity of enzymes and their high reactivity under mild conditions, enzymes are potent catalysts for green chiral diol synthesis.4 In our enzymatic cascade we start with a ThDP dependent lyase forming a C C bond between two aldehydes molecules resulting in a 2-hydroxy ketone. By using an oxidoreductase in the second step, the carbonyl group is reduced and a diol with two chiral centers is formed. In a recent study, enzymatic and chemical steps are combined to form diols and cyclic acetals in an environment-friendly manner.3 Biotransformation in a green organic solvent was done to provide a reaction environment suitable for the enzymatic and the chemical step. The integration of H2 and CO2 into the reaction renders the process even more ecological. Here, we show a method of synthesizing diols with mainly excellent stereoselectivity by using enzymes. In addition, intensification of the process including the use of unconventional media led to an increase in concentrations and conversion from 84 mM and 84% to 900 mM and 90%, respectively. Results Stereoselective formation of all diols as precursors for fine chemicals The need of stereoselective synthesis has increased in the last decades as more than 50% of used drugs are chiral.5 Due to the high stereoselectivity of modularly combined enzymes, we were able to successfully gain all 12 out of 12 possible stereoisomers of 4 diols with different chain length with good to high concentrations and good to excellent isomeric contents (ic) (Table 1). For example, full conversion was achieved by producing almost 100 mM of (5S,6S)-decanediol with an ic value > 99 %. Pure stereoisomers of all diols as precursors for bio-fuels Due to the highly negative impact of CO2 produced from transportations on the climate, alternatives to fossil fuels help cutting CO2 emissions. Diols are seen as suitable precursors for bio-hybrid fuels, as starting material for the diols can be aldehydes produced via microbial cell factories from second generation feedstocks.6 Besides their chiral building block potential, the diols shown in table 1 can be further transformed to dioxolane via chemocatalysis integrating CO2. These dioxolanes show high potential due to optimal properties as blends for bio-fuels.3 When used as blend, yields are more important than selectivities. For this aim, only the ligation step was done enzymatically, the further reduction and cyclisation will be performed using (unselective) ruthenium catalysis. By intensifying the enzymatic carboligation step, an increase of the product concentration from 84 mM up to 900 mM was achieved. This was mainly achieved using enzyme catalysis in a micro-aqueous reaction system. micro-aqueous reaction system (MARS) Aqueous buffers are the first choice when it comes to reaction media with enzymes, as most of them are evolved in an aqueous environment. However, many interesting substrates are hydrophobic and this results in low substrate loads and therewith product concentration, when a second phase should be avoided. Applying biocatalysis in an organic solvent, like cyclopentyl methyl ether (CPME), allows to overcome this problem. Therefore, enzymes are formulated as lyophilized whole cells. Protecting the entrapped enzymes in the remaining cell envelope to a certain extent enables high yields under these reaction conditions. Moreover, the MARS is a convenient environment to combine enzymatic and chemical steps for dioxolane synthesis in one reaction environment and additionally facilitates product purification.7 |
| References: | [1] Zhang, J. et al. Characterization of four diol dehydrogenases for enantioselective synthesis of chiral vicinal diols. Chin J Chem Eng (2022) 47, 145-154. [2] Celińska, E. & Grajek, W. Biotechnological production of 2,3-butanediol—Current state and prospects. Biotechnol Adv (2009) 27, 715-725. [3] Spöring, J. et al. Effective Production of Selected Dioxolanes by Sequential Bio‐ and Chemocatalysis Enabled by Adapted Solvent Switching. ChemSusChem. (2022) 16. [4] Enzyme Biocatalysis. (Springer Netherlands, 2008). [5] Nguyen, L. A., He, H. & Pham-Huy, C. Chiral drugs: an overview. Int J Biomed Sci (2006) 2, 85-100 . [6] Mengers, H. G., Westarp, W. G. von, Brücker, D., Jupke, A. & Blank, L. M. Yeast-based production and in situ purification of acetaldehyde. Bioprocess Biosyst Eng (2022) 45, 761-769. [7] van Schie, M. M. C. H., Spöring, J.-D., Bocola, M., Domínguez de María, P. & Rother, D. Applied biocatalysis beyond just buffers - from aqueous to unconventional media. Options and guidelines. Green Chemistry (2021) 23, 3191-3206. |
| Figures: | 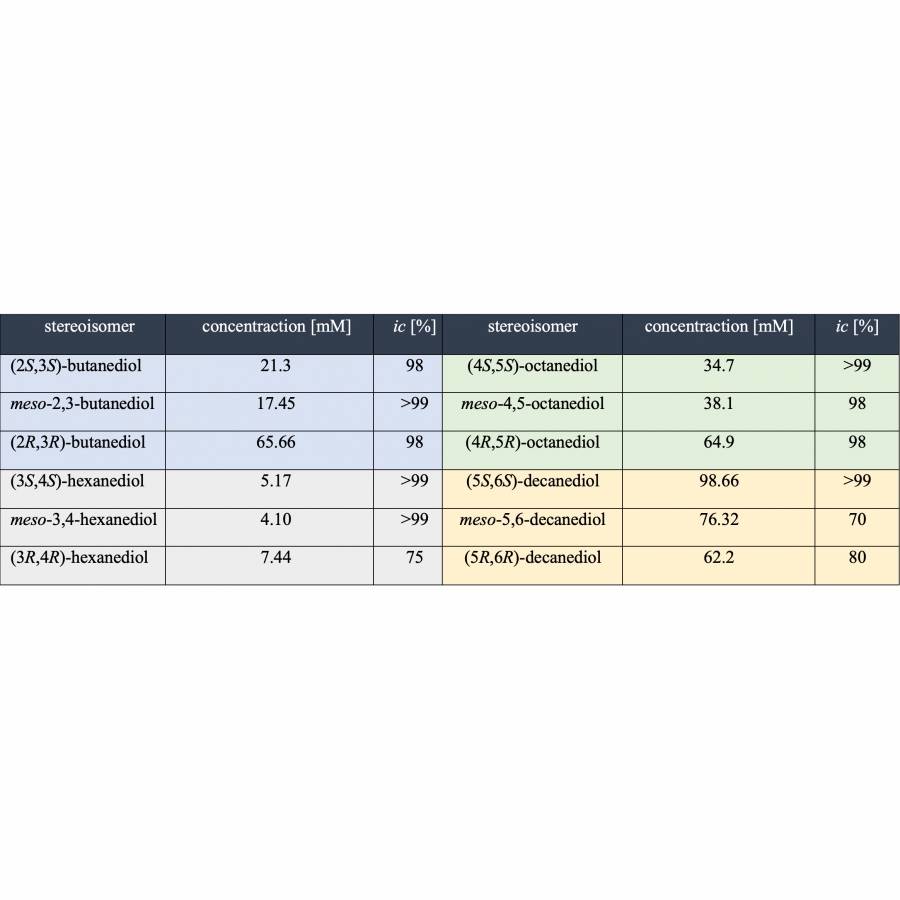 Table 1: Concentrations and ic values of gained stereoisomers. ic= isomeric content (target isomer/sum of all isomers [%]). Starting concentration of substrate: 200 mM 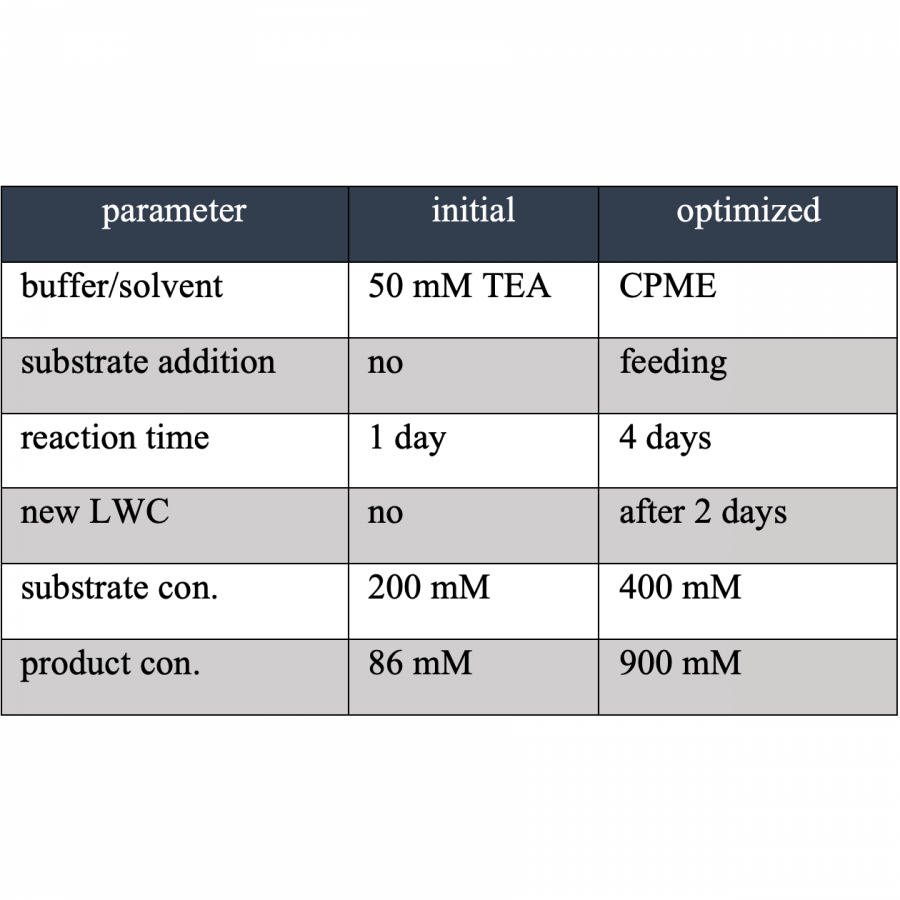 Table 2: Optimized parameters of the carboligation step. LWC: lyophilized whole cells. CPME: cyclopentyl methyl ether. TEA: triethanolamine |
#173 | Enzyme engineering of alkane monooxygenase for improved activity in a key reaction step of the synthesis of the alpha-methylene lactone Tulipalin A |
|
| Presenting author: | Robert KOURIST of GRAZ UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | bio-based polymer / membrane-bound enzyme / alkane monooxygenase / rational protein desigh |
| Purpose: | The synthesis of renewable and sustainable polymeric materials as replacement of petroleum-based raw materials has been receiving increasing attention. The alpha-methylene lactone Tulipalin A has two polymerizable functional moieties and is a potential substitute of (meth)acrylates in vinyl-addition polymerization and (co)monomer for lactone ring-opening polymerization. While Tulipalin A can be isolated from the flowers of tulips and alstroemerias, its biosynthesis remains unknown. We propose a synthesis from isoprenyl acetate, which itself can be produced via the microbial hemiterpenoid metabolism. Selective hydroxylation of isoprenyl acetate in C4-position and subsequent oxidation of the intermediate hydroxy group gives rise to 4-acetoxy-2-methylene butyric acid, whose hydrolysis and cyclization then leads to Tulipalin A. We identified bacterial alkane monooxygenases that catalyze the hydroxylation without, albeit with lower activity than the terminal hydroxylation of linear alkanes. Undesired epoxidation of the exo-methylene group was not observed. In order to increase the activity of the membrane-bound dioxygenase, we used de novo structure prediction to generate a structural model. Site-directed mutagenesis of the active-site cavity inspired by molecular docking allowed a substantial increase of the activity of the monooxygenase. We envision that the engineered enzyme variants will find application in a future whole-cell process, unlocking the supply of Tulipalin A as future bio-based monomer. |
| Figures: | 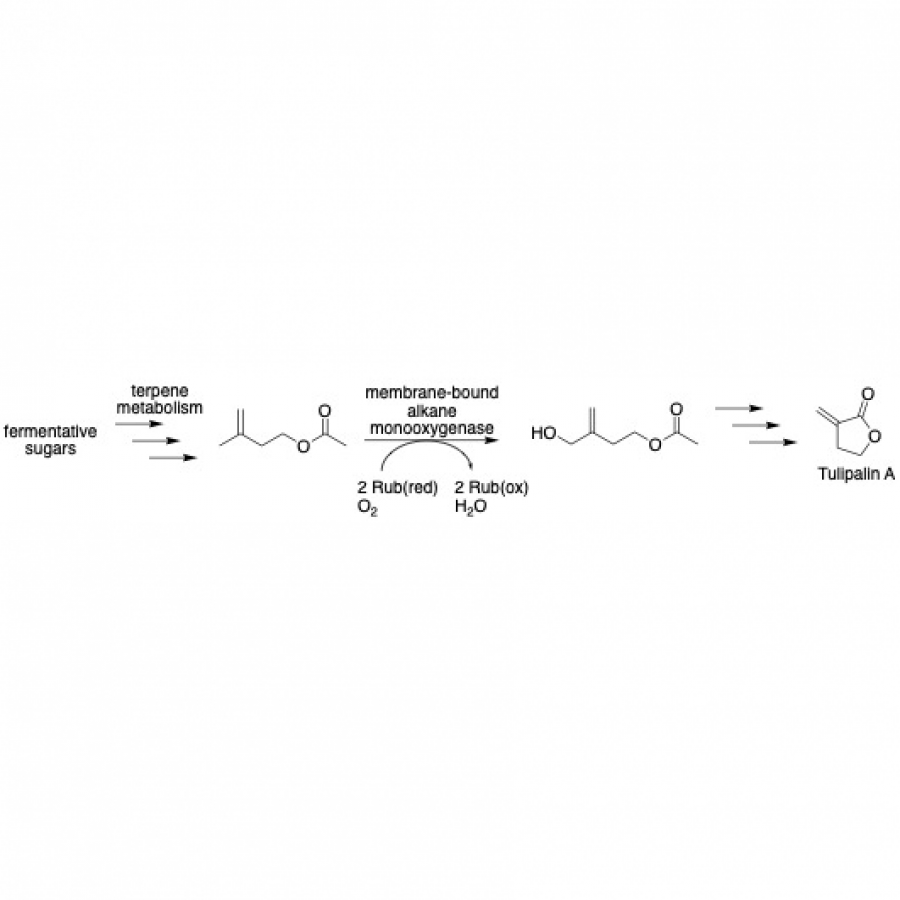 Figure 1 Proposed pathway for the synthesis of Tulipalin A |
#194 | Multienzyme coimmobilization on heterofunctional supports |
|
| Presenting author: | Susana VELASCO-LOZANO of UNIVERSIDAD DE ZARAGOZA - ISQCH |
| Other authors: | Javier SANTIAGO-ARCOS of CICBIOMAGUNE Fernando LOPEZ-GALLEGO of CICBIOMAGUNE |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | multienzyme systems / coimmobilization / cascade biotransformations / enzyme stabilization |
| Purpose: | Multienzyme cascade biotransformations in one pot are gaining momentum since they have demonstrated enhanced catalytic performance than traditional step-by-step transformations requiring sequential pots. Although their evident advantages, the coimmobilization of several enzymes requiring different anchoring chemistries and stability conditions is still challenging. In this work, we exploited a heterofunctional carrier activated with three different chemical functionalities in order to immobilize a wide variety of different enzymes under mild conditions [1]. This support is based on agarose microbeads activated with aldehyde, amino and cobalt moieties thus allowing a fast and irreversible immobilization of enzymes (Figure 1) and in the major cases accompanied by thermal stabilization (up to 21-fold higher than the soluble one). We demonstrated the effectiveness of the described support by coimmobilizing a multienzyme system composed by an alcohol dehydrogenase, a NADH oxidase and a catalase. The confined enzymatic system demonstrates higher performance than the soluble enzyme counterparts achieving a total turnover number (TTN) of 1x10^5 during five batch cycles under operation conditions. Finally, we expand the versatility of the described exploited heterogeneous chemistry to other frequently used immobilization carriers as cellulose microbeads and commercial methacrylate. We envision this solid material as a platform for coimmobilizing multienzyme systems with enhanced properties to catalyze stepwise biotransformations. |
| References: | Santiago-Arcos, J.; Velasco-Lozano, S.; López-Gallego, F. Multienzyme Coimmobilization on Triheterofunctional Supports. Biomacromolecules 2023, 24 (2), 929-942. |
| Figures: | 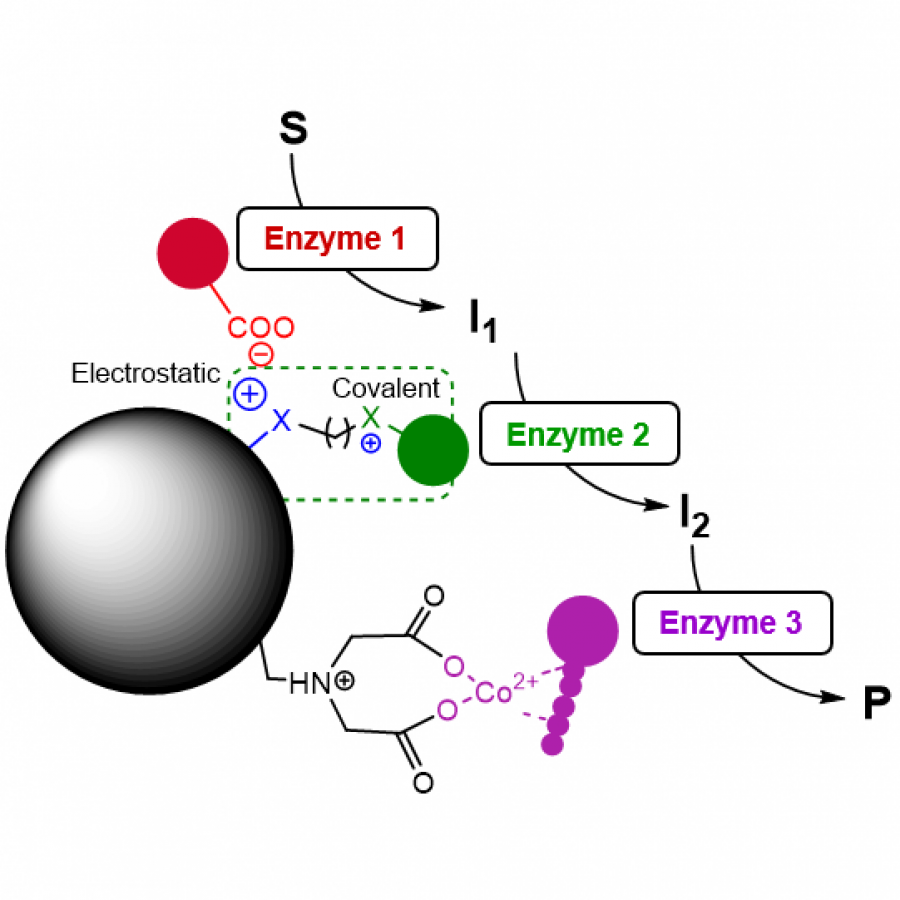 Figure 1. Triheterofunctional support to coimmobilize several enzymes through three different immobilization chemistries |
#195 | Engineering Amine Dehydrogenases (AmDHs) for the Synthesis of Vicinal Amino Alchols |
|
| Presenting author: | Ruth BRADSHAW ALLEN of MANCHESTER INSTITUTE OF BIOTECHNOLOGY - UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | ENGINEERING AMINE DEHYDROGENASES (AMDHS) FOR THE SYNTHESIS OF VICINAL AMINO ALCOHOLS Ruth T. Bradshaw Allen, William R Birmingham, and Nicholas J. Turner Manchester Institute of Biotechnology, University of Manchester, Manchester, United Kingdom Enantiopure vicinal amino alcohols are prevalent in a plethora of bioactive molecules and are thus a major target in biocatalytic synthesis. Amine dehydrogenases (AmDHs) are a growing class of biocatalysts capable of synthesising chiral amines via the reductive amination of prochiral ketones using ammonia as an inexpensive amino donor. AmDHs have previously been shown to exhibit excellent reductive amination activity towards a diverse collection of linear ketones and a-hydroxyketones. Herein, an amine dehydrogenase developed from a leucine amino acid dehydrogenase (L-AADH) is shown to catalyse the synthesis of cyclic vicinal amino alcohols from the corresponding a-hydroxy ketones with good stereoselectivity. Additionally, as biocatalytic cascades offer a sustainable synthetic route to vicinal amino alcohols from readily available starting materials, a multienzyme cascade featuring this AmDH has also been designed for the synthesis of 2-aminocyclohexanol with high atom efficiency. |
| References: | [1] Chen, F. F.; Zheng, G. W.; Liu, L.; Li, H.; Chen, Q.; Li, F. L.; Li, C. X.; Xu, J. H. ACS Catal. 2018, 8 (3), 2622-2628. [2] Chen, F. F.; Cosgrove, S. C.; Birmingham, W. R.; Mangas-Sanchez, J.; Citoler, J.; Thompson, M. P.; Zheng, G. W.; Xu, J. H.; Turner, N. J. ACS Catal. 2019, 9 (12), 11813-11818. |
#196 | Development of an enzymatic process for alkyl hydroxycinnamates synthesis |
|
| Presenting author: | Horiya Nassiba HAM of URD ABI AGROPARISTECH |
| Other authors: | Nabila IMATOUKENE of URD ABI AGROPARISTECH Michel LOPEZ of URD ABI AGROPARISTECH Louis MOUTERDE of URD ABI AGROPARISTECH |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alkyl hydroxycinnamates (AHCs) / p-hydroxycinnamic acid (HCA) / CoA-ligase (4CL) / Acyl transferase (ACT) |
| Purpose: | Alkyl hydroxycinnamates (AHCs) are hydroxycinnamic acid (HCA) esters aliphatic derivatives [1] found in plants associated with suberin and cutin. [2] The presence of the aliphatic moiety on AHCs modulates their hydrophilic/lipophilic balance and improves their integration in oil-based formulations and their biological activities compared to their phenolic acid parents.[3] These features open a broad panel of applications for these molecules as antioxidants, antimicrobials, and UV protectants among others.[1] Lipases are usually used to graft aliphatic moieties on phenolic acids. However, it has been reported that lipase’s activity is inhibited in the case of p-hydroxycinnamic acids due to the simultaneous presence of the p-hydroxyl and the double bound near the carboxylic function conjugated to the aromatic cycle on the phenolic acid.[4] Our work seeks to develop an enzymatic process for the synthesis of AHCs by miming their biosynthetic pathway in plants. We expressed two recombinant enzymes in a 5 L fermenter using transformed E. coli competent cells. The first one is a CoA-ligase (4CL) implicated in the activation of p-HCAs, and the second one is an acyltransferase (ACT) responsible for the transfer of the acyl moiety of the activated phenolic acid on an aliphatic moiety (acyl acceptor) for AHCs synthesis (Figure 1). We tested the enzymatic activity of the purified 4CL and ACT on different HCAs and acyl acceptors, respectively. HPLC and LC-MS results showed the successful activation of HCAs and AHCs synthesis. We also optimized the ATP concentration and the reaction time for HCAs activation using design of experiment method to minimize them as much as possible to make the process economically viable. We are currently working on ACT kinetic parameters determination and we aim to scale up the reaction and purify the final product. |
| References: | [1] D. A., Grajales-Hernández; M. A., Armendáriz-Ruiz; F. L., Gallego, and J. C., Mateos-Díaz, Applied Microbiology and Biotechnology, 2021, 105(10), 3901-3917. [2] F., Domergue and D. K. Kosma, Plants, 6(3), 25-41. [3] Y. G., Shi; Y.J., Zhu; S. Y., Shao; R. R., Zhang; Y., Wu; C. M., Zhu; X. R., Liang, and W. Q., Cai, Journal of Agricultural and Food Chemistry, 2018, 66 (45), 12088-12101. [4] P., Villeneuve, Biotechnology Advances,2007, 25(6), 515-536. |
| Figures: |  Reactions implicated in the enzymatic synthesis of AHCs in plants 4CL: 4-coumarate CoA ligase; ACT: Acyl transferase |
#197 | Glucose production from cellulose by immobilizing cellulase enzyme on chitosan beads |
|
| Presenting author: | Byung-Ki NA of CHUNGBUK NATIONAL UNIVERSITY |
| Other authors: | Beom Soo KIM of CHUNGBUK NATIONAL UNIVERSITY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cellulase / Immobilization / Chitosan beads / Cellurose |
| Purpose: | Recently, interest in an efficient biomass pretreatment and saccharification process is increasing for the production of biofuels and biochemicals from renewable non-edible biomass resources. In this study, we tried to produce glucose from cellulose by immobilizing the cellulase enzyme on chitosan beads, which are reported to have high pH and temperature stability. Polyvinyl alcohol modified with maleic anhydride was coated on chitosan beads to form an immobilized support and then cellulase enzyme was immobilized thereon. The immobilized amount of the enzyme according to the concentration of the cellulase enzyme solution and the specific activity of the immobilized enzyme at each concentration were investigated. In addition, conversion from cellulose to glucose was performed under conditions that showed the maximum cellulase specific activity. The domain immobilization method can be completed by a capture method that physically confines the domain and a binding method that adheres to the surface of a support or covalent bond. In this study, since the time division must be paid back when the capture method is implemented, the method of Dincer and Telefonce was used to limit the acceleration in order to contact the Wise mark and immobilize it when applied to the chitosan bead surface to overcome the extreme. As the concentration of the cellulase solution increased, the amount of cellulase immobilized on the chitosan beads increased linearly. The production of glucose increased to 7.2 g/L from 1% carboxymethyl cellulose (CMC) substrate when immobilized in 20% cellulase solution. The maximum specific activity value of the enzyme was 0.37 unit/mg protein when immobilized in 8% cellulase solution. The optimum reaction composition at pH 7 and 37 °C was 0.5 g beads/L from 1% CMC substrate, and conversion to glucose was completed in about 20 minutes. |
| References: | 1. Dincer, A., Telefoncu, A., 2007, J. Mol. Catal. B-Enzym. 45: 10-14. 2. Limayem, A., Ricke, S. C., 2012, Prog. Energ. Combust. 38: 449-467. |
#199 | Boronic-Acid Catalysis in an Artificial Enzyme |
|
| Presenting author: | Reuben LEVESON-GOWER of UNIVERSITY OF GRONINGEN |
| Other authors: | Lars LONGWITZ of UNIVERSITY OF GRONINGEN Gerard ROELFES of UNIVERSITY OF GRONINGEN |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial Enzyme / Unnatural Amino Acid / Directed Evolution / Crystallography |
| Purpose: | Due to their unparalleled activities, selectivities and benign operating conditions, enzymes will play an important role in the transition towards greener chemical manufacture[1]. However, their reaction scope still lags behind that of chemical catalysts, a problem that is addressed through the creation of artificial enzymes: protein scaffolds equipped with abiological catalytic moieties[2]. Here we disclose the design and evolution of an artificial enzyme exhibiting boronic-acid catalysis using a low-valent boron species as catalytic moiety (Figure 1)[3]. We mutated a non-enzymatic protein with a promiscuous hydrophobic pore to incorporate a genetically encoded boronic-acid containing unnatural amino acid[4]. This new side chain catalyses oxime-formation between alpha-hydroxy ketones and hydroxylamine. The protein scaffold boosts activity, as well as providing selectivity in the transformation, allowing a kinetic resolution to be performed. By screening cell-free lysates from site-saturation libraries in a plate-reader based assay, we performed several rounds of directed evolution and identified a triple mutant with greatly improved activity and E value over 100. Through 11B NMR spectroscopy, we could characterise the hybridisation state of the catalytic residue and its binding to several ligands. These adducts could also be observed with mass spectrometry. X-ray crystallography revealed a large structural change in the protein induced by the unnatural catalytic residue. This study paves the way for realisation of many new biocatalytic activities exploiting boronic acid catalytic residues. |
| References: | [1] R.A. Sheldon, J.M., Woodley Chem. Rev., 2018, 118, 801-838. [2] I. Drienovská, G. Roelfes, Nat. Catal., 2020, 3, 193-202. [3] D.G. Hall, Chem. Soc. Rev., 2019, 48, 3475-3496. [4] E. Brustad, M.L. Bushey, J.W. Kee, D. Groff, W. Liu, P.G. Schultz, Angew. Chem. Int. Ed., 2008, 47, 8220-8223. |
| Figures: |  Figure 1 Design and crystal structure of a boronic-acid based artificial enzyme with genetically encoded boronic-acid residue catalysing oxime formation of alpha-hydroxy ketones and its subsequent directed evolution to optimise the protein environment surrounding |
#200 | Study of two new DHA aldolases from acidophilic organisms |
|
| Presenting author: | Léo PAULAT of INSTITUT DE CHIMIE DE CLERMONT-FERRAND |
| Other authors: | Cédric GASTALDI of UNIVERSITÉ CATHOLIQUE DE LOUVAIN Marielle LEMAIRE of INSTITUT DE CHIMIE DE CLERMONT-FERRAND Virgil HELAINE of INSTITUT DE CHIMIE DE CLERMONT-FERRAND Lionel NAUTON of INSTITUT DE CHIMIE DE CLERMONT-FERRAND Jean-Louis PETIT of GENOSCOPE – UMR 8030 Véronique DE BERARDINIS of GENOSCOPE – UMR 8030 Christine GUERARD-HELAINE of INSTITUT DE CHIMIE DE CLERMONT-FERRAND |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / C-C bond formation / Biodiversity / extremophilic organisms |
| Purpose: | Biocatalysis is a tool to access new products of interest in a greener and more eco-friendly way. It allows us to respond to many current issues related to the 12 principles of green chemistry. In this context, Aldolases are C-C bond forming enzymes of particular interest for synthetic applications. Indeed, the aldol reaction allows to generate up to 2 asymmetric centers, providing chiral adducts. Depending on the aldolases used, the stereochemistry of these asymmetric centers can be controlled [1]. Fructose-6-phosphate aldolase (FSA) belonging to class I aldolases, was discovered in E. coli by Shürmann and Sprenger[2] in the 2000s. It was demonstrated as the first aldolase able to use hydroxyacetone[3], as nucleophile substrate, and furthermore particularly robust, efficient and versatile towards other nucleophiles such as dihydroxyacetone, hydroxybutanone[4] and glycolaldehyde[5]. These discoveries were the basis for mutagenesis[1] work or for the search from biodiversity for new aldolases presenting for example different stereochemistries. In the framework of our collaboration with the Génoscope (Evry), two FSA from acidophilic organisms are studied, one from, Acidobacteria Bacterium (A0A399XV01) and one from Acidiplasma Aeolicum (A0A0Q0RVA3). These catalysts have revealed atypical properties. Molecular modeling and their kinetic constants determination helped their characterization. These results will be presented here, as well as some synthetic applications. |
| References: | [1] Hélaine, V.; Gastaldi, C.; Lemaire, M.; Clapés, P.; Guérard-Hélaine, C. ACS Catal. 2022, 12 (1), 733-761. https://doi.org/10.1021/acscatal.1c04273. [2] Schürmann, M.; Sprenger, G. A. Journal of Biological Chemistry 2001, 276 (14), 11055-11061. https://doi.org/10.1074/jbc.M008061200. [3] Schürmann, M.; Schürmann, M.; Sprenger, G. A. Journal of Molecular Catalysis B: Enzymatic 2002, 19-20, 247-252. https://doi.org/10.1016/S1381-1177(02)00174-1. [4] Sugiyama, M.; Hong, Z.; Liang, P.-H.; Dean, S. M.; Whalen, L. J.; Greenberg, W. A.; Wong, C.-H. J. Am. Chem. Soc. 2007, 129 (47), 14811-14817. https://doi.org/10.1021/ja073911i. [5] Garrabou, X.; Castillo, J. A.; Guérard-Hélaine, C.; Parella, T.; Joglar, J.; Lemaire, M.; Clapés, P. Angew. Chem. Int. Ed. 2009, 48 (30), 5521-5525. https://doi.org/10.1002/anie.200902065 |
| Figures: |  Aldolisation reaction catalysed by aldolase Aldolisation reaction catalysed by aldolase  FSA : Decameric structure FSA : Decameric structure |
#203 | Green synthesis of a dibasic ester using nitrilase and lipase. |
|
| Presenting author: | Hannah BAKER of NORTHUMBRIA UNIVERSITY |
| Corresponding author: | Hannah BAKER of NORTHUMBRIA UNIVERSITY |
| Other authors: | Graeme TURNBULL of NORTHUMBRIA UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lipase / nitrilase / ester / flow |
| Purpose: | During the synthesis of adiponitrile, for nylon production, 2-methylglutaronitrile (2-MGN) is created as a by-product. The conversion of 2-MGN to 2-methylglutaric acid (2-MGA) through the use of a nitrilase has been shown by previous work at Northumbria University.[1] More usefully, the acid may be converted to a diester for use as a solvent in paint, however the current biocatalytic route is incomplete with several amide by-products produced (Scheme 1). Converting 2-MGA to the corresponding diester can be completed through a Fischer esterification. However, chemical esterification when 4-APA is still present will produce the highly corrosive ammonium bisulfite, which corrodes commercial steel vessels. In this work we are screening lipase enzymes for the synthesis of diesters of 2-MGA as this will avoid the production of corrosive by-products and will lead to a fully biocatalytic synthesis. |
| References: | [1] Chemoxy International Ltd, Process for Converting Nitriles, United Kingdom Pat., 2554708, 2016. |
| Figures: | 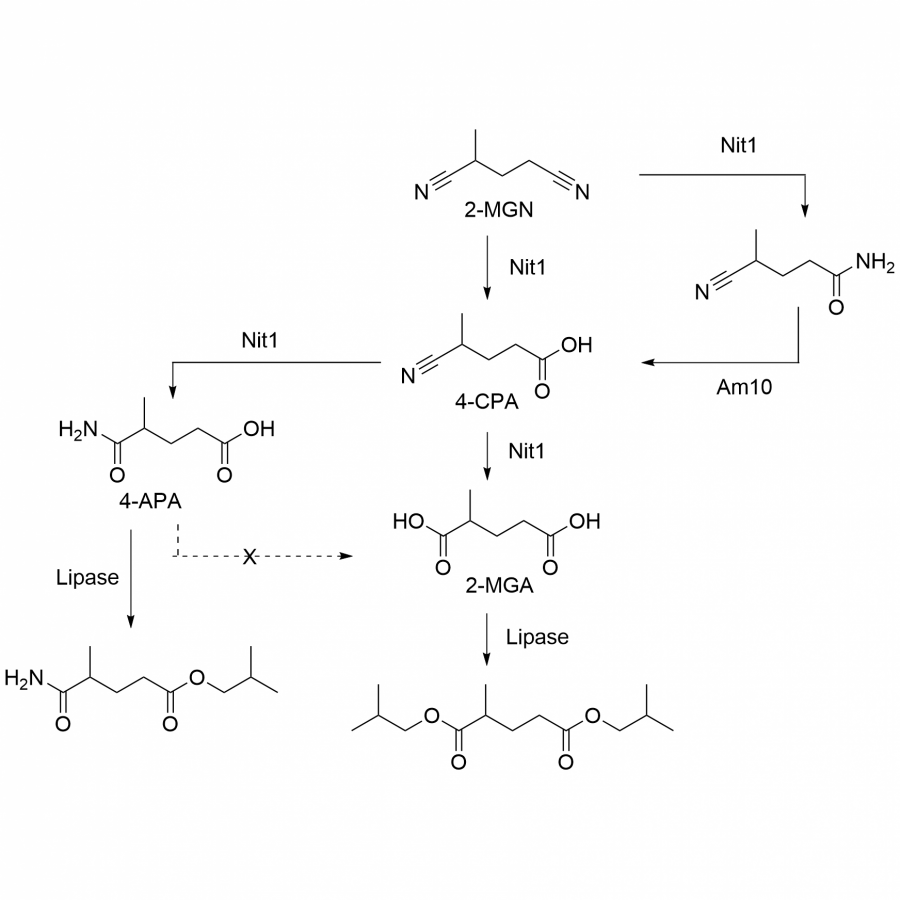 Scheme 1 The biocatalytic route from 2-MGN to diisobuyl-2-methylglutarate. |
#213 | From lignocellulosic biomass to value-added chemicals: A one-pot, whole-cell biocatalysis approach for vanillin production using lignin oil |
|
| Presenting author: | Ivana MARIC of UNIVERSITY OF GRONINGEN |
| Corresponding author: | Marco FRAAIJE of UNIVERSITY OF GRONINGEN |
| Other authors: | Yiming GUO of UNIVERSITY OF GRONINGEN Maximilian FÜRST of UNIVERSITY OF GRONINGEN Korneel VAN AELST of KU LEUVEN Sander VAN DEN BOSCH of KU LEUVEN Mario DE SIMONE of ITQB-NOVA UNIVERSIDADE NOVA DE LISBOA Lígia MARTINS of ITQB-NOVA UNIVERSIDADE NOVA DE LISBOA Bert SELS of KU LEUVEN |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Whole-cell catalysis / Lignin valorization / Enzymatic cascade / Vanillin production |
| Purpose: | Vanillin is among the most 'tasteful' and universally appreciated aromatic chemicals worldwide. It is extensively used as a flavour for food and beverages and a fragrance ingredient in perfumes and cosmetics. However, almost total global vanillin demand is satisfied by petroleum-derived guiacol. [1] The process is hazardous to the environment and unsustainable with the growing depletion of fossil fuels. Therefore, developing environmentally friendly, efficient, and sustainable routes to biobased vanillin is essential. Here, we report on vanillin production from 4-n-propyl guaiacol, one of the main components in lignin oil obtained through reductive catalytic fractionation (RCF) [2,3] by employing recombinant Escherichia coli cells. Conversion is based on the expression of two engineered oxidative enzymes: a 4-n-propyl guaiacol oxidase that can convert 4-n-propyl guaiacol to isoeugenol [4] and an isoeugenol oxygenase that transforms isoeugenol into the desired product, vanillin [5] (Figure 1). To identify the suitable conditions of the whole-cell cascade, we fine-tuned different reaction conditions, such as temperature, buffer systems, pH values and timing of biocatalyst addition, using 4-n-propyl guiacol as a model compound. Finally, a high vanillin yield (64%) was obtained (Figure 2), employing optimized conditions on a complex starting material, e.g. RCF lignin oil. This high-performance strategy was readily scaled up to produce vanillin in more than 10% yield based on lignin in spruce, without any by-products. The whole-cell bioconversion process shows good tolerance even at high loadings of starting material and in the presence of compounds that can potentially poison or compete for the catalyst, showcasing the robustness and applicability of the employed biocatalysts. Furthermore, we show that cells in a growth medium could be used as ready-to-use biocatalysts, making this one-pot lignin oil conversion into vanillin a rather facile process. This work paves the way for the efficient production of high-titer vanillin using depolymerized lignin as the feedstock. |
| References: | [1] M. Fache, B. Boutevin, S. Caillol, ACS Sustain. Chem. Eng. 2016, 4, 35-46. [2] Z. Sun, B. Fridrich, A. de Santi, S. Elangovan, K. Barta, Chem. Rev. 2018, 118, 614-678. [3] S. Van den Bosch, W. Schutyser, R. Vanholme, T. Driessen, S.-F. Koelewijn, T. Renders, B. De Meester, W. J. J. Huijgen, W. Dehaen, C. M. Courtin, B. Lagrain, W. Boerjanbc, B. F. Sels, Energy Environ. Sci. 2015, 8, 1748-1763. [4] Y. Guo, L. Alvigini, M. Trajkovic, L. Alonso-Cotchico, E. Monza, S. Savino 1, I. Marić, A. Mattevi ,M. W. Fraaije, Nat. Commun. 2022, 13, 7195. [5] M. De Simone, L. Alvigini, L. Alonso-Cotchico, V. Brissos, J. Caroli, M. Fátima Lucas, E. Monza, E. Pinho Melo, A. Mattevi, L. O. Martins, Biochemistry, 2022, 62, 419-428. |
| Figures: | 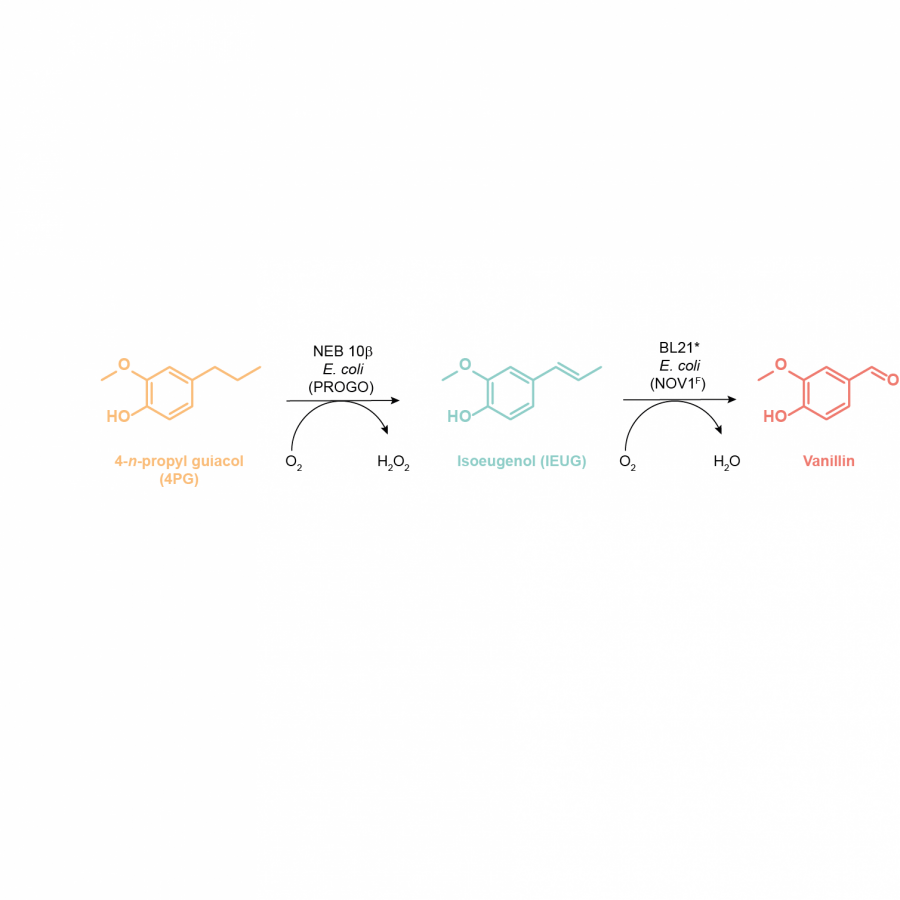 The whole-cell cascade for vanillin production from 4-n-propyl guaiacol The first step of the one-pot cascade, e.g. the conversion of 4-n-propyl guaiacol to isoeugenol is catalyzed by 4-n-propyl guaiacol oxidase expressing cells, followed by the transformation of isoeugenol to vanillin by isoeugenol oxygenase expressing cells 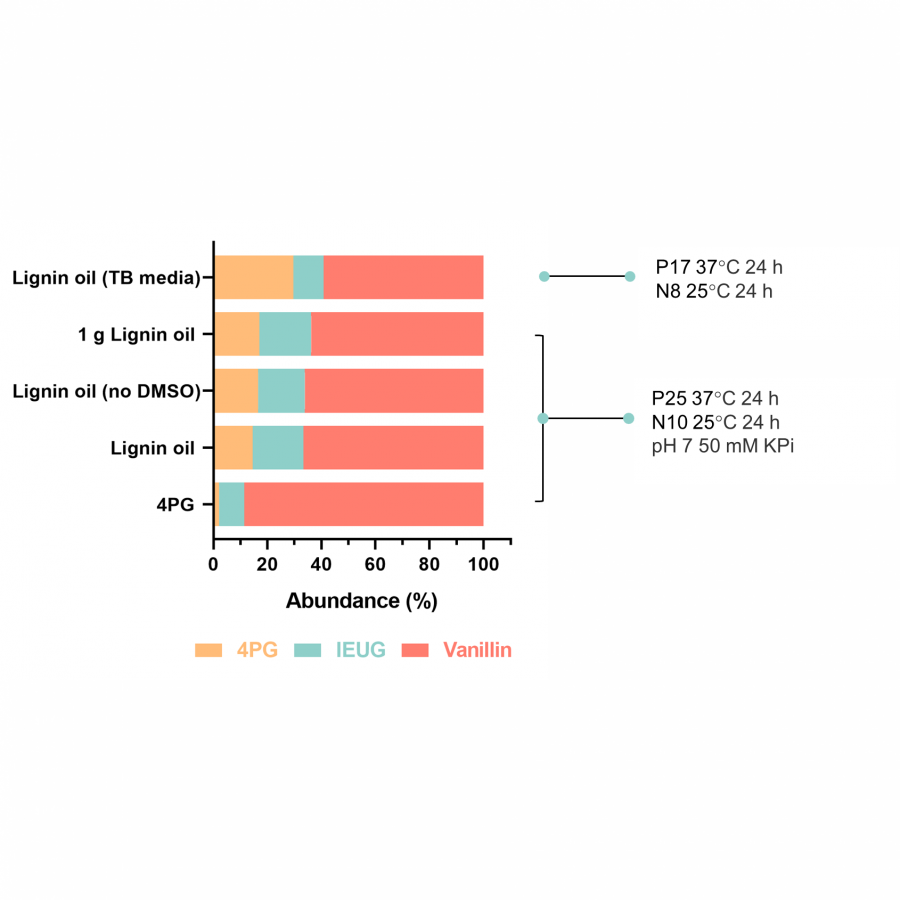 One-pot conversions of commercial 4-n-propyl guaiacol and lignin-derived oils by PROGO and NOV1-S283F expressing cells Experimental conditions (from bottom to top) for conversions of : pure 4-n-propyl guaiacol; lignin oil in the presence of DMSO; lignin oil in the absence of DMSO, 1g of lignin oil, and lignin oil in presence of cells in growth media. |
#222 | Degradation of PET microplastic to monomers in human serum by designer enzymes |
|
| Presenting author: | Ximena LOPEZ LORENZO of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Per-Olof SYREN of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Other authors: | David HUETING of KTH ROYAL INSTITUTE OF TECHNOLOGY Eliott BOSSHARD of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme engineering / Sustainability / Biocatalysis / Chemical biology |
| Purpose: | 400 Mt of plastic years is generated per year [1,2] with a large amount of this plastic entering to bodies of water. Aa a consequence, around 3 million tons of plastic debris enters our oceans from rivers which humans are exposed daily to hundreds of microplastics (plastic particles with a size < 5 mm) upon inhalation and digestion. Due to its small size, this microplastics can translocate from the gut to body fluids and all the way to the organs [3,4]. Recently, studies have shown that plastic particles of ≥700 nm in diameter are present in human whole blood [5]. Accordingly, the way synthetic polymers are discarded could pose a health risk to humans. Moreover, because of the prolonged exposure to these microplastic particles are found in human blood and other bodily fluids. As of now, there is a shortage of data regarding the hazards that could come due this exposure. Despite a lack of toxicity studies regarding microplastics, harmful effects for humans seem plausible and cannot be excluded. Since the possible health risks associated with microplastics can’t be determined due to lack of exposure data, there are no conclusions to which extent these microplastics represent a risk [4]. However, the European Environment Agency issued a report called Late Lessons from Early Warnings, highlighting how inaction regarding environmental issues due to a lack of risk data can pose a danger to society [6]. For this reason and based on the recent study by Leslie et. al. showing occurrence of microplastics in blood , in this study first steps towards a possible therapeutic measure to depolymerize microplastics in human serum was explored. In this study, we investigated the use of an enzyme-based treatment of serum that could constitute a promising avenue to clear synthetic polymers and their responding oligomers by degradation into monomers. Still, the activity of plastic degrading enzymes in serum remains unknown. Herein, we report how engineered PETases can depolymerize microplastic-like particles of the commodity polymer polyethylene terephthalate (PET) into its non-toxic monomer terephthalic acid (TPA) in human serum at 37°C. As WT PETase is active at ambient temperature with a melting point close to 40°C [7,8], this enzyme was chosen as a model system to investigate enzymatic activity under blood-like conditions. Therefore, we sought to test the activity of PETases in human serum, still representing a relatively simple model of blood without complex clotting factors and blood cells. Our results demonstrate that PETases are highly active in serum, especially over the initial timepoints. Moreover, these initial results are exciting yet further research is needed regarding the interaction of this bacterial enzyme with the blood components to try and avoid the enzyme to be neutralised when introduced to the bloodstream. By developing an efficient method to depolymerize microplastics in vitro, our work takes a step closer to find a solution to the problem that microplastics in the bloodstream may pose in the future. |
| References: | [1] Jonsson, C. et al. Biocatalysis in the Recycling Landscape for Synthetic Polymers and Plastics towards Circular Textiles. ChemSusChem 14, 4028-4040 (2021). [2] Geyer, R., Jambeck, J. R. & Law, K. L. Production, use, and fate of all plastics ever made. Sci. Adv. 3, e1700782-e1700782 (2017). [3] Yong, C. Q., Valiyaveettil, S. & Tang, B. L. Toxicity of Microplastics and Nanoplastics in Mammalian Systems. Int. J. Environ. Res. Public Health 17, 1509 (2020). [4] Vethaak, A. D. & Legler, J. Microplastics and human health. Science 371, 672-674 (2021). [5] Leslie, H. A. et al. Discovery and quantification of plastic particle pollution in human blood. Environ. Int. 163, 107199 (2022). [6] Leslie, H. A. & Depledge, M. H. Where is the evidence that human exposure to microplastics is safe? Environ. Int. 142, 105807 (2020). [7] Guo, B. et al. Conformational Selection in Biocatalytic Plastic Degradation by PETase. ACS Catal. 12, 3397-3409 (2022). [8] Guo et al. Fast depolymerization of PET bottle mediated by microwave pre-treatment and an engineered PETase. (2023). Under Revision |
| Figures: | 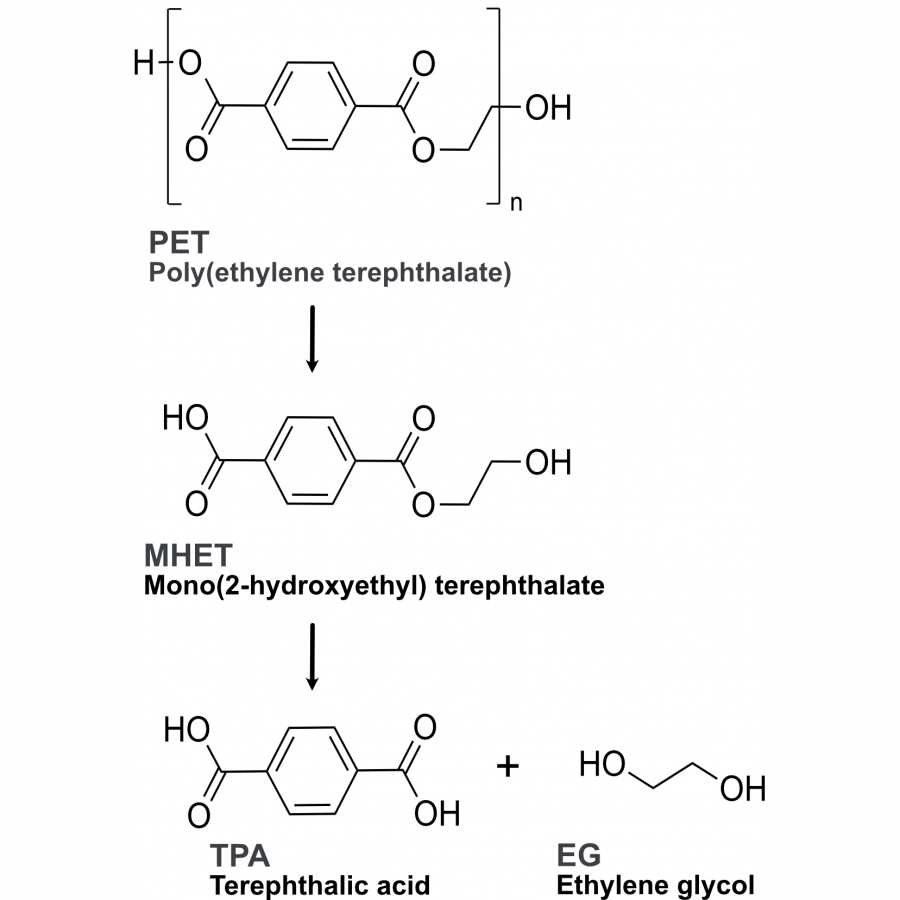 Degradation of PET Depolymerization steps of PET into its monomers terephthalic acid (TPA) and ethylene glycol (EG) by PETase. For simplicity, only the intermediate MHET is shown. 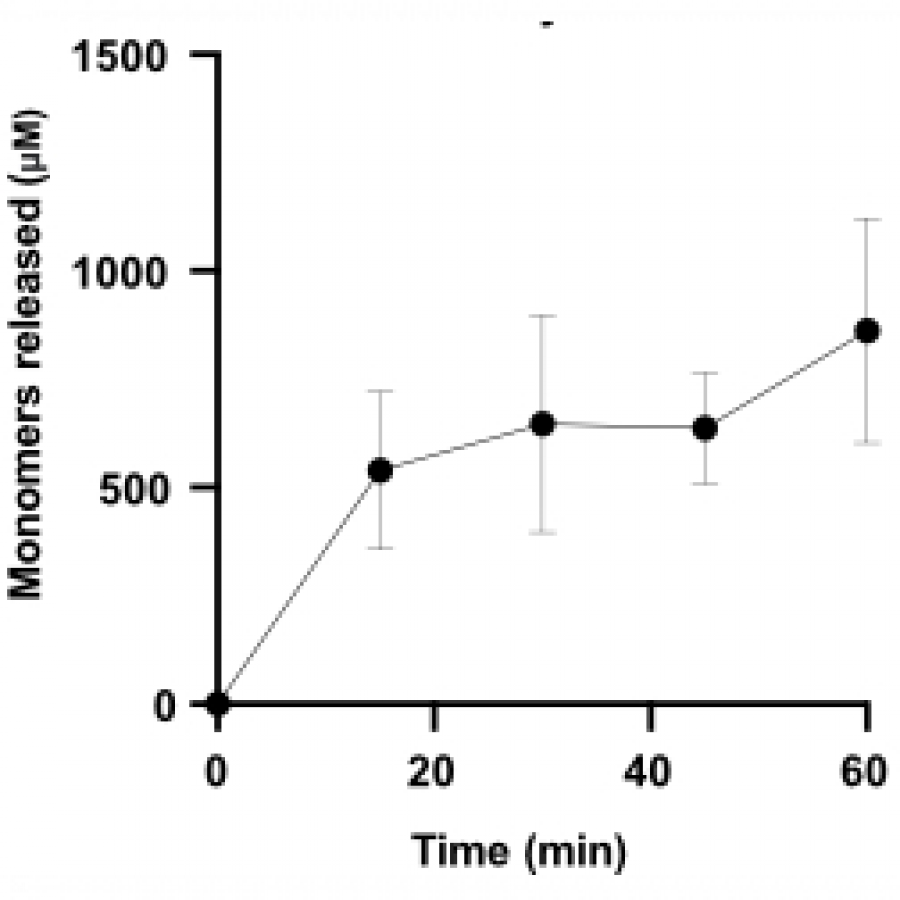 PETase activity in human serum S238A activity in serum. For each reaction 1.5 ug mL-1 of enzyme was added to 1 mL of human serum with a substrate concentration of 2 mg mL-1. The samples were incubated at 37C. All reactions were stopped by heat inactivation (at 95C). Samples were take |
#229 | UPOs for the oxidative valorization of HMF |
|
| Presenting author: | Alexander SWOBODA of ACIB C/O UNIVERSITY OF GRAZ |
| Other authors: | Zerina DUHOVIC of ACIB C/O UNIVERSITY OF GRAZ Silvie ZWÖLFER of UNIVERSITY OF GRAZ Moritz BÜRGLER of BISY GMBH Katharina EBNER of BISY GMBH Anton GLIEDER of BISY GMBH Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Unspecific Peroxygenases (UPOs) / 5-Hydroxymethylfurfural (HMF) / 2,5-Furandicarboxylic acid (FDCA) / Enzymatic Cascades |
| Purpose: | 2,5-Furandicarboxylic acid (FDCA) is a versatile platform chemical that has its main application in the polymer industry.[1] The enormous interest in this building block is largely due to the fact that it can be accessed via renewable starting materials. One of the most prominent strategies for the synthesis of FDCA is the catalytic oxidation of biomass derived 5-hydroxymethylfurfural (HMF).[2] The inclusion of FDCA in the list of the top 12 biobased chemicals by the U.S. Department of Energy (DOE) back in 2004[3] triggered a wave of research which resulted in a remarkable toolbox of catalytic oxidation methods towards FDCA. One of the most prominent strategies for the synthesis of FDCA is the catalytic oxidation of biomass derived HMF.[2] The main advantages of biocatalysts in this regard are their selectivity and benign reaction conditions, which are a prerequisite for a sustainable process. Yet, the challenge of enzymatic processes is to bring product titers to competitive levels. In this respect, unspecific peroxygenases (UPOs) have shown great potential as oxidation catalysts.[4] Despite the ever increasing interest in UPOs, up until now only AaeUPO has been used in cascades with oxidases for the oxidation of HMF.[5] We screened 23 different UPOs and report that HspUPO[6] and UPOx8 are the first UPOs which are capable of performing three consecutive oxidation steps from HMF to FDCA on their own (Figure 1). We also observed that the chemoselectivity in the first oxidation step can be enhanced by the use of cosolvents, although accompanied by lower conversions. The combination of HspUPO with HMFO[7] and variants thereof[8] in a self-sufficient cascade leads to increased efficiency of the system. Our results to date underscore the potential of UPOs for the synthesis of FDCA and thereby contribute to the development of sustainable and efficient methods for the production of value-added chemicals from biomass-derived feedstocks. |
| References: | [1] A. F. Sousa, C. Vilela, A. C. Fonseca, M. Matos, C. S. R. Freire, G.-J. M. Gruter, J. F. J. Coelho, A. J. D. Silvestre, Polym. Chem. 2015, 6, 5961. [2] H. Cong, H. Yuan, Z. Tao, H. Bao, Z. Zhang, Y. Jiang, Di Huang, H. Liu, T. Wang, Catalysts 2021, 11, 1113. [3] T. Werpy, G. Petersen, Top Value Added Chemicals from Biomass: Volume I -- Results of Screening for Potential Candidates from Sugars and Synthesis Gas, 2004. [4] F. Tonin, F. Tieves, S. Willot, A. van Troost, R. van Oosten, S. Breestraat, S. van Pelt, M. Alcalde, F. Hollmann, Org. Process Res. Dev. 2021, 25, 1414. [5] a) J. Carro, E. Fernández-Fueyo, C. Fernández-Alonso, J. Cañada, R. Ullrich, M. Hofrichter, M. Alcalde, P. Ferreira, A. T. Martínez, Biotechnology for biofuels 2018, 11, 86; b) A. Karich, S. B. Kleeberg, R. Ullrich, M. Hofrichter, Microorganisms 2018, 6, 5. [6] L. Rotilio, A. Swoboda, K. Ebner, C. Rinnofner, A. Glieder, W. Kroutil, A. Mattevi, ACS Catal. 2021, 11511. [7] W. P. Dijkman, D. E. Groothuis, M. W. Fraaije, Angew. Chem. Int. Ed. 2014, 126, 6633. [8] M. Pickl, A. Swoboda, E. Romero, C. K. Winkler, C. Binda, A. Mattevi, K. Faber, M. W. Fraaije, Angew. Chem. Int. Ed. 2018, 57, 2864. |
| Figures: | 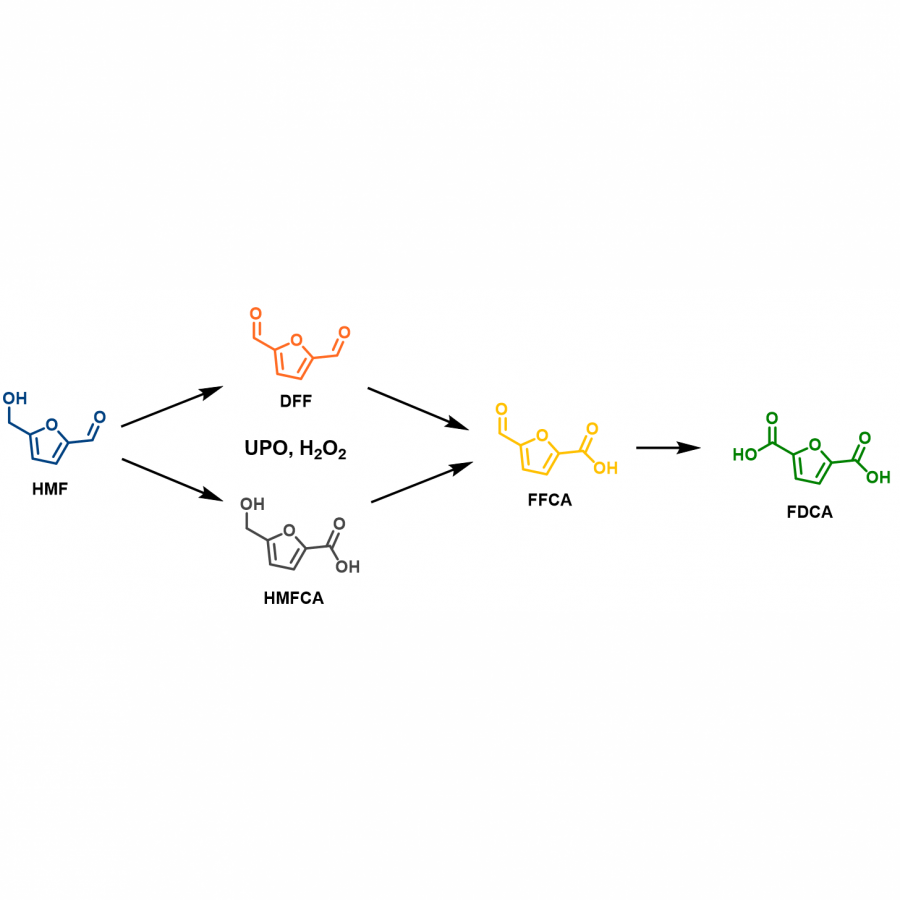 Oxidation of HMF to FDCA by UPO. HMF = 5-Hydroxymethylfurfural, DFF = 2,5-Diformylfuran, HMFCA = 5-Hydroxymethyl-2-furancarboxylic acid, FFCA = 5-Formyl-2-furancarboxylic acid, FDCA = 2,5-Furandicarboxylic acid |
#232 | A single biocatalyst for an alcohol oxidation – conjugated addition cascade reaction |
|
| Presenting author: | Mathias PICKL of UNIVERSITY OF GRAZ |
| Corresponding author: | Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Other authors: | Andrea RAAB of UNIVERSITY OF GRAZ Jasper S. MÖHLER of ETH ZÜRICH Helma B. WENNEMERS of ETH ZÜRICH |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cascade / Alcohol Oxidation / Conjugated Addition / |
| Purpose: | Aldehydes offer the reactivity as starting material to access many compound classes. Biocatalysis has a strong record in carbonyl modifying redox chemistry and a plethora of potent catalysts is available to produce e.g. aldehydes from primary alcohols via oxidation. However, utilizing aldehydes in carbon-carbon bond formations, biocatalysis is outperformed by the reaction portfolio of organocatalysis. The enzymatic equivalents for reactions such as Michael additions, Mannich reactions or Knoevenagel condensations are inconceivably underexplored.[1] Remarkably, many organocatalytic reactions such as the Michael addition are mediated by proteinogenic amino acids or peptides.[2] Hence, we envisioned that a biocatalyst can be designed that has the ability to oxidize a primary alcohol to the corresponding aldehyde and in parallel enables enamine catalysis allowing the -carbon of the aldehyde to act as a nucleophile in a Michael addition. Several alcohol dehydrogenases and alcohol oxidases were screened for their ability to mediate the redox reaction.[3] To enable the carboligation, a peptide sequence known to mediate Michael additions was N-terminally attached, or alternatively, it was tested whether amino acid side chains such as the -amino groups of lysine can catalyze a Michael addition (Figure 1). It turned out that several designed candidates mediate the desired conjugated addition. In case an appropriate co-factor recycling system is chosen, a one-pot, one-step cascade system is feasible starting from aliphatic alcohol and yielding g-nitro aldehydes. |
| References: | [1] G. Xu, G. J. Poelarends, Angew. Chem. Int. Ed.2022, 61, e202203613. [2] M. Wiesner, J. D. Revell, H. Wennemers, Angew. Chem. Int. Ed. 2008, 47, 1871-1874. [3] J. Dong, E. Fernández-Fueyo, F. Hollmann, C. Paul, M. Pasic, S. Schmidt, Y. Wang, S. Younes, W. Zhang, Angew. Chem. Int. Ed. 2018, 57, 9238-9261. |
| Figures: | 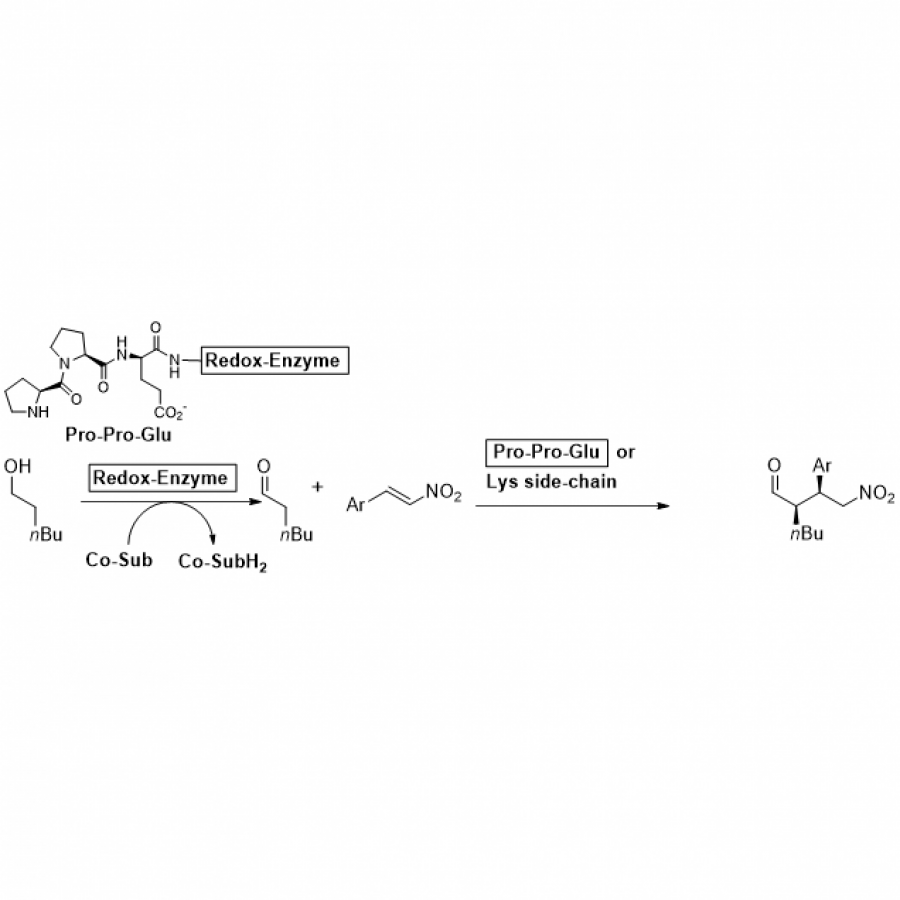 Scheme 1 A redox enzyme with an N-terminal tripeptide catalyst performing the oxidation of n-hexanol to n-hexanal and a subsequent conjugated addition to a -nitroaldehyde |
#237 | Cascade Processes Merging Chemical and Enzyme Catalysis |
|
| Presenting author: | Juan MANGAS SANCHEZ of UNIVERSITY OF OVIEDO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cascade / organocatalysis / oxidoreductases / one-pot |
| Purpose: | Organocatalysis, i.e., the use of small molecules to catalyse chemical transformations, has proven to be an excellent tool in asymmetric synthesis, allowing a wide variety of reactions under mild conditions. [1] Despite the relatively late development when compared with other modalities of catalysis, the field has greatly expanded in the last 15 years and has proved to be especially useful to make asymmetric C-C bonds. Even though both technologies present moderate similarities concerning reaction conditions as well as the mode of action, and display a broad synthetic complementarity, examples in the literature on cascade processes are still scarce compared to those of metal and enzyme catalysis. [2] These processes are an attractive strategy to rapidly build molecular complexity and circumvent the need to isolate reaction intermediates, allowing higher efficiencies into synthetic routes and simpler experimental setups.[3] We are particularly interested in using these synthetic strategies to make chiral compounds starting from simple starting materials and in this oral communication, we will provide the audience with an overview of our latest findings on the preparation of chiral 1,4 [4] and 1,2-nitro alcohols as well as chiral 1,2-hydroxy phosphonates via the one-pot sequential combination of oxidoreductases and different organocatalysts. |
| References: | [1] S. Ardevines, E. Marqués-López, R. P. Herrera. Catalysts 2022, 12, 101. [2] C. Ascaso-Alegre, J. Mangas-Sánchez. Eur. J. Org. Chem. 2022, e202200093. [3] S. González-Granda, L. Escot, I. Lavandera, V. Gotor-Fernández. Angew. Chem.Int. Ed. 2023, 62, e202217713. [4] C. Ascaso-Alegre, R. P. Herrera, J. Mangas-Sánchez. Angew. Chem. Int. Ed. 2022, 61, e202209159. |
| Figures: | 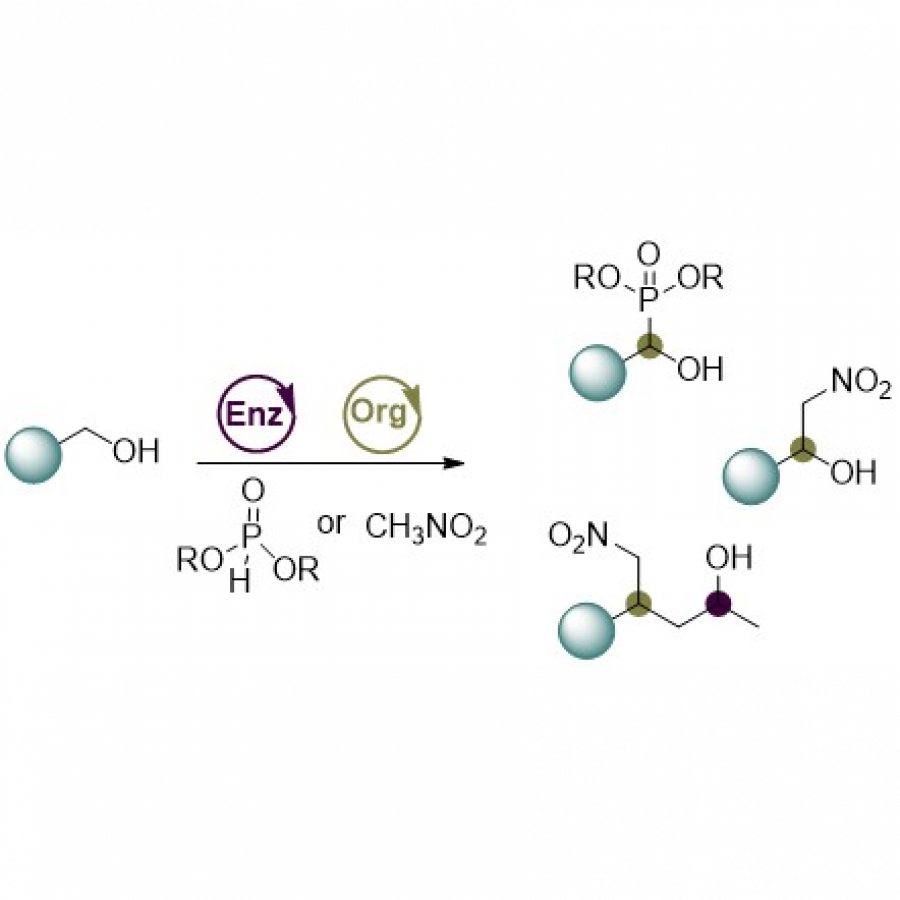 Scheme 1 Cascade processes involving enzyme and organocatalysis to access chiral compounds |
#240 | Natural and synthetic transaminase fusions and their use in biocatalysis |
|
| Presenting author: | Luba PROUT of UCL |
| Corresponding author: | John WARD of UCL |
| Other authors: | Helen HAILES of UCL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transaminase / fusion / engineering / Pseudomonas |
| Purpose: | Multi-enzyme cascades provide the means for generating complex amine compounds from abundant or cheap simple starting materials and are becoming the standard for many biotransformation processes. However, in nature, enzymes have evolved to function in unity with other enzymes in specific environments, meaning that their isolation and use in artificial settings may reduce their efficiency. To get around this, nature-based synthetic biology solutions are being developed and implemented. Natural multifunctional enzymes represent a part of multi-step biosynthetic pathways that ensure a one-way flux of reactants. In vivo, they confer selective advantages via increased reaction rates and chemical stabilities or the prevention of toxicities arising from reactive intermediates. Here we report the identification and characterisation of a natural transaminase fusion, PP_2782, from Pseudomonas putida KT2440, as well as three of its thermophilic homologs from Thermaerobacter marianensis, Thermaerobacter subterraneus, and Thermincola ferriacetica. The information on these systems was then used to design and engineer novel synthetic enzyme fusions comprising (1) Chromobacterium violaceum DSM 30191 ω-transaminase, and (2) Equus caballus alcohol dehydrogenase, (3) Geobacillus stearothermophilus 22 alcohol dehydrogenase, and (4) Thalictrum flavum subsp. glaucum (S)-norcoclaurine synthase enzymes, to enable efficient amine synthesis. The analysis of both the natural and synthetic fusion activity is presented. |
| References: | [1] Kim, J., et al. (2006). Journal of Microbiology and Biotechnology: 16, 450-456. [2] Chung, E. J., et al. (2008). Appl. Environ. Microbiol.: 74, 723-730. [3] Davis, E., et al. (2017). MicrobiologyOpen: 6, 1-9 . [4] Lozano, G. L., et al. (2019). Appl. Environ. Microbiol.: 85 (10). [5] Couturier, M., et al. (2021). RSC Chem. Biol.: 2, 551-555. [6] Richardson, S. M., et al. (2022). ACS Catal.: 12, 12701-12710. [7] Schlapschy, M., et al. (2013). Protein Engineering, Design & Selection: 26, 489-501. [8] Lerchner, A., et al. (2016). Protein Engineering, Design & Selection: 29, 1-6. |
| Figures: |  P. putida KT2440 natural transaminase fusion as a template for novel synthetic enzyme fusion engineering. |
#244 | Homospermidine synthase facilitated N-cross coupling and applications in N-heterocycle synthesis. |
|
| Presenting author: | Joe SHARRATT of UNIVERSITY OF MANCHESTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / heterocycle / poly-amine / enzyme cascades |
| Purpose: | Polyamines (PAs) are a diverse and ubiquitous class of molecules with essential cellular functions including cell proliferation, apoptosis, and DNA stabilization. Additionally they are a utilitarian intermediates for the biosynthesis of pyrrolizidine and indolizidine alkaloids, a potent class of therapeutics with antiviral and anticancer properties. Despite the abundance and diversity of polyamines in nature, bulk chemical syntheses of such molecules remains a challenge due to selectivity issues associated with traditional chemical synthesis. Homospermidine synthase (HSS) is a key enzyme in the biosynthesis of PAs, with HSS from Blastochloris viridis (BvHSS) able to catalyse the synthesis of homospermidine from two molecules of putrescine in a hydrogen borrowing type mechanism that can be described as an N-cross coupling reaction. In this work, we present the use of BvHSS as a biocatalyst to produce homospermidine, and structurally related PAs, as well as several non-natural PA analogues via BvHSS catalysed N-cross coupling between diamine donors and non-biogenic amine acceptors including amino alcohols, amino acids, and alkylamines. Subsequently, we combined BvHSS with diamine oxidases, transaminases, and choline oxidase in order to obtain a number of N-heterocycles. |
| References: | Krossa, S., Faust, A., Ober, D. et al. . Sci Rep 6, 19501 (2016). Ramsden, J., Turner, N J,. et al.. J. Am. Chem. Soc. 141, 3, 1201-1206 (2019) |
#245 | Lipase-catalyzed chemoselective acylation of sesquiterpene lactones: towards new ester derivatives |
|
| Presenting author: | Juan RODRIGUEZ MOSHEIM of UMR TRANSFRONTALIÈRE BIOECOAGRO N1158, UNIV. LILLE, INRAE, UNIV. LIÈGE, UPJV, JUNIA, UNIV. ARTOIS, UNIV. LITTORAL CÔTE D’OPALE, INSTITUT CHARLES VIOLLETTE |
| Other authors: | Francesca RUGGIERI of UNIVERSITY OF LILLE, INSERM, INSTITUT PASTEUR DE LILLE Egon HEUSON of UNIV. LILLE, CNRS, CENTRALE LILLE, UNIV. ARTOIS, UMR 8181—UCCS—UNITÉ DE CATALYSE ET CHIMIE DU SOLIDE Renato FROIDEVAUX of UMR TRANSFRONTALIÈRE BIOECOAGRO N1158, UNIV. LILLE, INRAE, UNIV. LIÈGE, UPJV, JUNIA, UNIV. ARTOIS, UNIV. LITTORAL CÔTE D’OPALE, INSTITUT CHARLES VIOLLETTE Jean-Louis HILBERT of UMR TRANSFRONTALIÈRE BIOECOAGRO N1158, UNIV. LILLE, INRAE, UNIV. LIÈGE, UPJV, JUNIA, UNIV. ARTOIS, UNIV. LITTORAL CÔTE D’OPALE, INSTITUT CHARLES VIOLLETTE Philippe HANCE of UMR TRANSFRONTALIÈRE BIOECOAGRO N1158, UNIV. LILLE, INRAE, UNIV. LIÈGE, UPJV, JUNIA, UNIV. ARTOIS, UNIV. LITTORAL CÔTE D’OPALE, INSTITUT CHARLES VIOLLETTE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lipase acylation / terpenoids / chicory / biocatalytic functionalization |
| Purpose: | Terpenes are the most abundant and diverse family of natural compounds with over 64000 structures identified to date. Plants rich in terpenes have been used for medicinal purposes by various human cultures around the world, and many terpenes and terpenoids are now recognized for their biological activities, such as antimicrobial, anti-inflammatory, and antitumor properties. The chicory plant has a long medicinal history, as it was used as a herbal remedy by ancient civilizations for the treatment of various pathologies. Its root is rich in secondary metabolites with biological activities, mainly polyphenolic compounds and sesquiterpene lactones (STL). The functionalization of these compounds is a promising approach for pharmaceutical applications, as well as in the agri-food sector. Out of the thousands of terpenoids compounds that have been characterized, it is notable that a primary allyl alcohol moiety is highly frequent among the terpene family, serving as a promising starting point for the addition of side chains or various linkers. Our global strategy was then to use the anchor points to substitute them with various chains and chemical functions, converting the STL into versatile building blocks bearing different physicochemical properties. Our approach is based on the use of enzymes to maximize the selectivity of the reactions, as terpenes often exhibit more than one accessible hydroxyl group on their structure. As an example, we present here one of our most promising achievements, based on the use of an immobilized lipase to introduce aliphatic esters onto STL, with the primary goal of modulating their lipophilicity. This precise reaction was selected in the perspective to later evaluate their antibiotic activity, as some terpenes already shown to exhibit interesting actions onto bacterial membranes. For this study, the rather classical Novozyme 435 (immobilized CAL-B), well known for its successful application in ester synthesis, was selected among a lipase panel based on preliminary tests on several monoterpenes, but also for its high availability, stability and ease of use, for further industrial development. Following our first screening, the enzymatic reaction conditions were first optimized (1mL of a mixture of MTBE and ACN (3:1) at 37 °C with 5 angstrom molecular sieves) using (S)-perillyl alcohol, a cheap cyclic monoterpenoid carrying an allyl alcohol moiety. Afterward, we applied our synthesis to the transformation of the very expensive main four STL found in chicory root (lactucin, dihydrolactucin, lactucopicrin and dihydrolactucopicrin), which were never functionalized enzymatically by any means until then. We started with the simplest ester derivative, a methyl ester, for which we compared the reactivity of two common types of acetyl donors, acetic acid and vinyl acetate. While 76 % yield could be achieved with the first in our conditions, a complete transformation could be observed with the latter, as we could expect from its higher reactivity. In both cases, acyl donors were introduced in 10:1 equivalent to maximize the consumption of the expensive STL. Then, we continued with dihydrolactucin, varying the length of the aliphatic chain in order to study both the selectivity of the lipase, but also to target a range of different properties for the synthesized products. Chloroacetate (92 % yield), propanoate (100 % yield), hexanoate (74 % yield) and octanoate (69 % yield) ester derivatives could be obtained after 48h hours of reaction. In addition to these high yields, very high selectivity was observed with no substitution on the secondary alcohol of our 4 STL, and the naturally present ester groups of lactucopicrin and dihydrolactucopicrin were not hydrolyzed during the reaction. In conclusion, we report here the first example of enzymatic synthesis methodology to functionalize STLs from chicory with very high selectivity and a range of aliphatic esters. |
| References: | [1] Matsumi, R.; Atomi, H.; Driessen, A. J. M.; van der Oost, Research in Microbiology 2011, 162 (1), 39-52. [2] Gershenzon, J.; Dudareva, N., Nat Chem Biol 2007, 3 (7), 408-414. [3] Salakhutdinov, N. F.; Volcho, K. P.; Yarovaya, O. I., Pure and Applied Chemistry 2017, 89 (8), 1105-1117. [4] Yang, W.; Chen, X.; Li, Y.; Guo, S.; Wang, Z.; Yu, X., Natural Product Communications 2020, 15 (3), 1934578X20903555. [5] Zielinska-Blajet, M.; Feder-Kubis, J., IJMS 2020, 21 (19), 7078. [6] Chaturvedi, D., In Discovery and Development of Therapeutics from Natural Products Against Neglected Tropical Diseases; Elsevier, 2019; pp 49-85. [7] Al-Snafi, A., IOSR Journal of Pharmacy 2016, 6, 41-56. [8] Wang, Q. P, African Journal of Biotechnology 2011, 10 (11), 1966-1977. [9] Baert, J. R. A.; Van Bockstaele, E. J., Industrial Crops and Products 1992, 1 (2-4), 229-234. [10] Gupta, A. K.; Mamta; Bhatia, I. S., Phytochemistry 1985. [11] Ortiz, C.; Ferreira, M. L.; Barbosa, O.; dos Santos, J. C. S.; Rodrigues, R. C.; Berenguer-Murcia, Á.; Briand, L. E.; Fernandez-Lafuente, R., Catal. Sci. Technol. 2019, 9 (10), 2380-2420. |
| Figures: |  Representation of the selective transesterification of DHLc Selective transesterification of DHLc, a sesquiterpene lactone from chicory, using Novozyme 435. 100% conversion was obtained within 24h  Yield of different alkyl esters of dihydrolactucin for each acyl donor Conversion (%) after 48h at 37°C, 35rpm in 1mL MBTE-ACN (3:1) with 10mM of DHLc, 100mM of vinyl ester, 20mg of N435 and 2 molecular sieves 5 angstrom. Error margin = +/- 5% |
#246 | Selective methylations and alkylations using methyltransferases |
|
| Presenting author: | Matthew SALINGER of UNIVERSITY COLLEGE LONDON |
| Other authors: | Helen HAILES of UNIVERSITY COLLEGE LONDON Thomas MOODY of ALMAC SCIENCES John WARD of UNIVERSITY COLLEGE LONDON Jack JEFFRIES of UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Methyltransferases / Bioalkylations / Green Chemistry / Directed Evolution |
| Purpose: | Research into the utilisation of methyltransferase enzymes to perform selective methylations and alkylations in chemical synthesis is rapidly gaining momentum, in part due to the stringent substrate stereoselectivities and regioselectivities these biological catalysts offer [1]. In addition, these methyltransferases operate under mild conditions and avoid the use of toxic chemical reagents, such as methyl iodide and dimethyl sulfate [2,3,4], that would otherwise be used in equivalent traditional synthetic methods [5]. Therefore, there is a significant green chemistry component to this field of research. A range of S-adenosylmethionine (SAM)-dependent methyltransferases are being explored to assess their ability to selectively methylate an array of valuable small molecule substrates of pharmaceutical relevance, given the impressive enhancements in bioactivity of drugs commonly observed post-methylation, known as the "magic methyl effect" [5]. Furthermore, an enlargement of the scope of non-methyl alkyl groups that these enzymes can transfer to substrates is being investigated, to probe the further expansion of chemical space in a selective manner. This is being carried out by the generation of SAM analogues that have their methyl group substituted for various alkyl moieties. Importantly, alkylations are performed in the context of a multi-enzyme in vitro SAM generation cascade that is necessary to ensure a continuous and immediate supply of SAM and its analogues [6], which are relatively unstable molecules and expensive to procure. This cascade also ensures the efficient removal of the methyltransferase inhibitor S-adenosylhomocysteine (SAH) generated by SAM mediated alkylations [7,8]. Furthermore, the properties of the methyltransferases and the accessory enzymes in the SAM generation cascades associated with them are being enhanced by directed evolution to improve activities, substrate selectivities and tolerance towards SAM analogues. |
| References: | [1] F. Subrizi, Y. Wang, B. Thair, D. Méndez-Sánchez, R. Roddan, M. Cárdenas-Fernández, J. Siegrist, M. Richter, J. N. Andexer, J. M. Ward and H. C. Hailes, Angew. Chem. Int. Ed., 2021, 60, 18673-18679. [2] A. Sidana, A. Singh, N. Sawal, B. S. Chavan and R. Gupta, Indian J. Psychiatry, 2020, 62, 97-99. [3] M. Guo and S. Gao, J. Environ. Qual., 2009, 38, 513-519. [4] J. C. R. Rippey and M. I. Stallwood, Emerg. Med. J., 2005, 22, 878-879. [5] H. Schönherr and T. Cernak, Angew. Chem. Int. Ed., 2013, 52, 12256-12267. [6] J. Siegrist, S. Aschwanden, S. Mordhorst, L. Thöny-Meyer, M. Richter and J. N. Andexer, ChemBioChem, 2015, 16, 2576-2579. [7] R. Kumar, R. Srivastava, R. Kumar Singh, A. Surolia and D. N. Rao, Bioorg. Med. Chem., 2008, 16(5), 2276-2285. [8] H. Chen, B. Zhou, M. Brecher, N. Banavali, S.A. Jones, Z. Li, J. Zhang, D. Nag, L.D. Kramer, A.K. Ghosh and H. Li, PLoS One, 2013, 8(10):e76900. |
| Figures: |  Multi-enzyme Cascades The scheme for the MAT-MT-MTAN bioalkylation cascade. MAT = Methionine adenosyltransferase, MT = Methyltransferase, MTAN = Methylthioadenosine nucleosidase, SAH = S-adenosylhomocysteine. 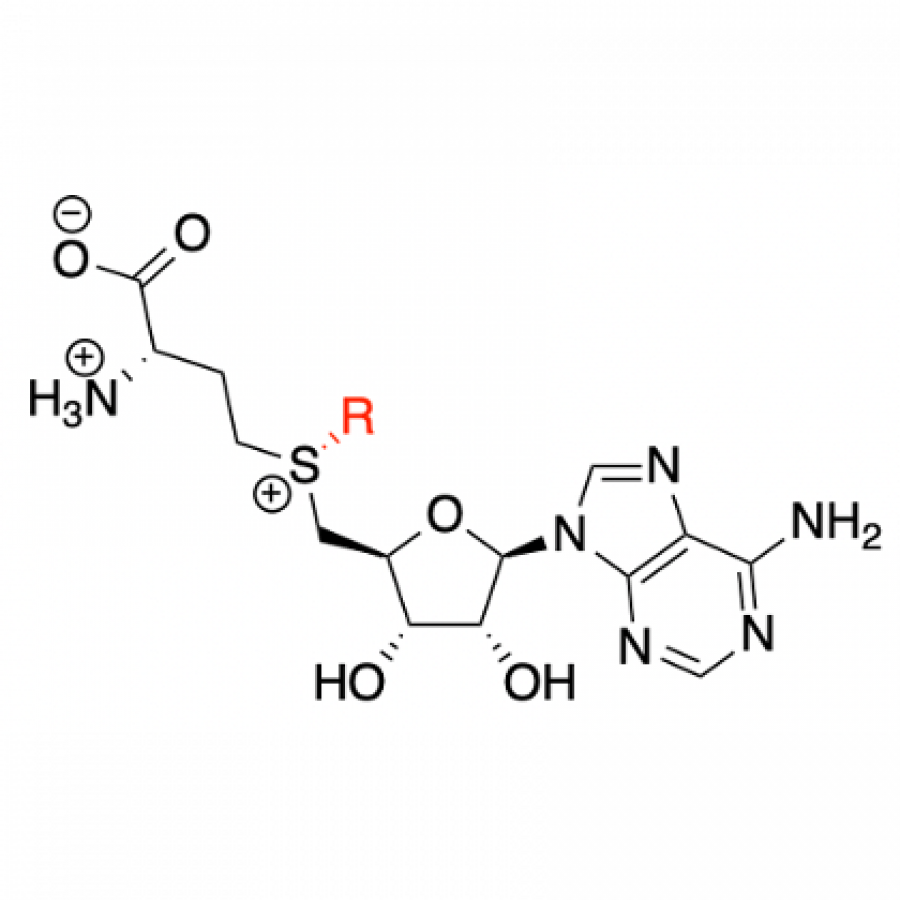 SAM Analogues The SAM analogue molecular structure. Methyltransferases catalyse the transfer of the R-group (highlighted red) to various substrates, with R representing various alkyl groups. |
#247 | Discovery and characterization of a novel Baeyer-Villiger monooxygenase using sequence similarity network analysis |
|
| Presenting author: | Thaleia SAKOLEVA of UNIVERSITY OF GREIFSWALD |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Baeyer-Villiger monooxygenase / Aliphatic ketones / Biocatalysis / |
| Purpose: | Baeyer-Villiger monooxygenases (BVMOs) are flavin binding and NAD(P)H-dependent enzymes performing oxygen insertion reactions leading to valuable products.[1] As has been previously reported in several studies, BVMOs are usually unstable during application, preventing their wider usage in biocatalysis.[2,3] Here, we discovered a novel NADPH-dependent BVMO which originates from Halopolyspora algeriensis and was discovered using sequence similarity networks (SSNs). The enzyme is stable over days and yields the normal ester product. The investigation of the substrate scope revieled acceptance of a broad range of aliphatic ketones. The enzyme was biochemically characterized to identify the optimum reaction conditions. The highest conversion (86%) was found in in vitro reactions with 2-dodecanone. This new BVMO could be applied in enzymatic cascades, to utilize aliphatic alkanes as a bioprocess feedstock. |
| References: | [1]F. Leipold, R. Wardenga, U. T. Bornscheuer, Appl. Microbiol. Biotechnol. 2012, 94, 705-717. [2]Z. Lin, Z. Xu, Y. Li, Z. Wang, T. Chen, X. Zhao, Microb. Cell Fact. 2014, 13, 104. [3]S. Milker, L. C. P. Goncalves, M. J. Fink, F. Rudroff, Front. Microbiol. 2017, 8, 2201. |
| Figures: |  BVMO discovery and characterization. Process of the sequence retrieving, heterologous expression and characterization of the BVMO originated from Halopolyspora algeriensis. |
#249 | Bioconversion of oleic acid into 7,10-dihydroxy-8(E)-octadecenoic acid |
|
| Presenting author: | Beom Soo KIM of CHUNGBUK NATIONAL UNIVERSITY |
| Other authors: | Ji Wan JEONG of CHUNGBUK NATIONAL UNIVERSITY Mamata SINGHVI of CHUNGBUK NATIONAL UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | 7,10-dihydroxy-8(E)-octadecenoic acid / Pseudomonas aeruginosa / extracellular enzyme / fed-batch process |
| Purpose: | 7,10-Dihydroxy-8(E)-octadecenoic acid (DOD) was produced using an extracellular enzyme present in the supernatant of Pseudomonas aeruginosa [1]. DOD concentration and productivity were improved by using extracellular enzyme obtained from P. aeruginosa culture medium compared to the conventional whole cell culture method. Among the carbon sources used during cell preculture, glycerol showed higher DOD production than glucose. In the batch process, the highest concentration of DOD (8.82 g/L) was achieved using supernatants derived from a 12-hour pre-culture of P. aeruginosa after 72 hours of bioconversion process. To further improve DOD production, a fed-batch process was used with the addition of surfactants and concentrated enzymes. The fed-batch process using a 4-fold concentrated enzyme solution containing Tween 80 yielded the highest DOD concentration (27.5 g/L at 72 hours) with an 8.28-fold increase in DOD production compared to using whole cells. This research is expected to accelerate the development of DOD production using inexpensive substrates such as crude glycerol on a large scale. |
| References: | [1] J.W. Jeong, M. Singhvi, B.S. Kim, Biotechnology and Bioprocess Engineering 2022, 27, 415-422 |
#251 | One-Pot Efficient Synthesis of (R)-Mandelic Acids and Other Valuable Chiral Chemicals from Racemic Epoxides via Cascade Biotransformations |
|
| Presenting author: | Jieran YI of NATIONAL UNIVERSITY OF SINGAPORE |
| Corresponding author: | Zhi LI of NATIONAL UNIVERSITY OF SINGAPORE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Sustainable catalysis / Enzymatic cascade / Enantioselective synthesis / Racemic epoxides |
| Purpose: | Enzymatic cascades bear great potential for offering economical and sustainable synthesis of valuable enantiopure chemicals from cheap starting substrates, due to their advantages of mild reaction conditions, and less toxic waste generation. Enantiopure (R)-mandelic acid is a highly valuable precursor in the synthesis of many pharmaceutically active molecules such as antibiotics.. Its chemical synthesis usually requires expensive and toxic reagents while giving unsatisfactory enantiomeric excess (e.e.) and generating problematic by-products. A few existing biocatalytic routes to this compound are reported, including cascade reactions from styrene – they are greener, but also suffer low efficiencies in many cases. Here we report a novel and efficient artificial cascade consisting of enantioconvergent epoxide hydrolysis, enantioselective diol oxidation, and aldehyde oxidation to produce enantiopure (R)-mandelic acids in high yields and high ee from easily available racemic epoxides. Using coupled E. coli (StEH) and E. coli (EcALDH-Aldo(M)) strains, (R)-mandelic acid was produced in up to 175mM, 88%-92% yields, and >99% ee from racemic styrene oxide. This represents the highest (R)-mandelic acid titre produced from simple chemicals via green synthesis. The cascade reaction was also successfully applied to convert other racemic epoxides to the corresponding (R)-4-fluoromandelic acid, (R)-4-chloromandelic acid, and (R)-4-bromomandelic acid in >99% ee, demonstrating the first enzymatic syntheses of these valuable molecules. With high yields and high product concentrations, the new cascade developed in this work offers a greener and practical synthetic route to useful and valuable enantiopure hydroxy acids, being potentially applicable for industrial production. Racemic styrene oxides could also be converted to other valuable chiral chemicals such as amino acids and amino alcohols, by cascading different enzymes after the enantioconvergent StEH. However, the yields of such cascades are still limited by one or more enzymes involved. Directed evolution of these yield-limiting enzymes using our newly developed ultrahigh-throughput microfluidics-based screening platforms are currently in progress. |
| References: | [1] Yi, J., Wang, Z., & Li, Z.*, ACS Catal. 2022, 12, 13697-13702 [2] Wang, Z., Li, X., & Li, Z.*, ChemCatChem 2022, 14, e202200418 |
| Figures: |  One-Pot Efficient Synthesis of (R)-Mandelic Acids from Racemic Epoxides vis Cascade Biotransformations StEH - epoxide hydrolase from Solanum tuberosum
Aldo(M) - V133M/G236V/V250I/G399K mutant of alditol oxidase from Streptomyces coelicolor
EcALDH - phenylacetaldehyde dehydrogenase from Escherichia coli |
#258 | Enzymatic synthesis, purification and immobilization of coenzymes of interest: the example of NADP(H). |
|
| Presenting author: | Celestin BOURGERY of URD ABI - AGROPARISTECH |
| Other authors: | David MENDOZA of BIORESOURCE PROCESSING RESEARCH INSTITUTE OF AUSTRALIA (BIOPRIA) Gil GARNIER of BIORESOURCE PROCESSING RESEARCH INSTITUTE OF AUSTRALIA (BIOPRIA) Louis MOUTERDE of URD ABI - AGROPARISTECH Florent ALLAIS of URD ABI - AGROPARISTECH |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / NADP(H) / Membrane-based purification / Cofactor immobilization |
| Purpose: | Biocatalysis is significantly developing these last decades due to its great potential to strengthen the chemical and pharmaceutical industries through the use of enzymes, which can (1) lead to sustainable and energy-efficient production methods, and (2) enable the production of high-value fine chemicals and pharmaceutical compounds that would otherwise be inaccessible. Although some enzymes do not require any additional chemicals for their catalytic activity other than their amino acid residues, many require a complementary component called cofactor. Therefore, to study or take benefit of all naturally occurring enzymes, it is necessary to have an affordable access to these cofactors. Although the use of inorganic ions seems straightforward, it is not so obvious when it comes to coenzymes. Indeed, their price can be prohibitive: 1600 €/g, 30 000 €/g or 370 €/g for Coenzyme A (CoA), its disulfide (CoAS2) and Nicotinamide Adenine Dinucleotide Phosphate (NADP(H)), respectively. Oxidoreductases, which represent one of the largest classes of enzymes (25% of all known enzymes), are of real interest. Indeed, thanks to their enantioselectivity and intrinsic specificity, they are able to promote bio oxido/reduction reactions that are vital for the global pharmaceutical and chemical market. However, these enzymatic oxido/reduction depend on a coenzyme: the nicotinamide adenine dinucleotide NAD(H) or its phosphorylated form NADP(H). Due to theirs high costs, the use of the NAD(P)+ /NAD(P)H-dependent enzymes is not viable on an industrial scale without an efficient and adapted regeneration system. Inspired by previous works on another coenzyme of interest, the coenzyme A1, a new pathway towards NADP(H) involving an in vitro enzymatic cascade has been designed to greatly decrease the price of this coenzyme and allow its use at a stochiometric level. Moreover, membrane-based purification to access high purity NADP+ was developed2. Finally, in collaboration with Monash University, we developed an efficient click-chemistry-based synthetic strategy3 to graft Adenosine, Adenosine mono- and triphosphate onto cellulose nanocrystals, that offers a novel sustainable approach for cofactor immobilization. The latter opens the way for the development of fast-growing fields such as flow chemistry applied to biocatalysis. |
| References: | [1] Mouterde, L. M. M.; Stewart, J. D. Org. Proc. Res. Dev. 2016, 20 (5), 954-959. [2] Bourgery et al. Combining the power of biocatalysis and membrane-based purification to access NADP+. ACS Sustainable Chem. Eng. 2023, accepted [3 ]Joram Mendoza et al. 2020. « Grafting Nature‐Inspired and Bio‐Based Phenolic Esters onto Cellulose Nanocrystals Gives Biomaterials with Photostable Anti‐UV Properties ». ChemSusChem 13 (24): 6552‑61. https://doi.org/10.1002/cssc.202002017. |
| Figures: |  Enzymatic synthesis, purification and immobilization of coenzymes of interest: the example of NADP(H). |
#265 | Toward scalable biocatalytic conversion of 5-hydroxymethylfurfural by galactose oxidase using coordinated reaction and enzyme engineering[1] |
|
| Presenting author: | William BIRMINGHAM of UNIVERSITY OF MANCHESTER |
| Other authors: | Asbjørn TOFTGAARD PEDERSEN of TECHNICAL UNIVERSITY OF DENMARK Mafalda DIAS GOMES of TECHNICAL UNIVERSITY OF DENMARK Mathias BØJE MADSEN of TECHNICAL UNIVERSITY OF DENMARK Michael BREUER of BASF SE John WOODLEY of TECHNICAL UNIVERSITY OF DENMARK Nicholas TURNER of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Galactose Oxidase / 5-Hydroxymethylfurfural / |
| Purpose: | 5-Hydroxymethylfurfural (HMF) has emerged as a crucial bio-based chemical building block in the drive towards developing materials from renewable resources, due to its direct preparation from sugars and its readily diversifiable scaffold. A key obstacle in transitioning to bio-based plastic production lies in meeting the necessary industrial production efficiency, particularly in the cost-effective conversion of HMF to valuable intermediates. Toward addressing the challenge of developing scalable technology for oxidizing crude HMF to more valuable chemicals, here we report coordinated reaction and enzyme engineering to provide a galactose oxidase (GOase) variant with remarkably high activity toward HMF, improved O2 binding and excellent productivity (>1,000,000 TTN). The biocatalyst and reaction conditions presented here for GOase catalysed selective oxidation of HMF to 2,5-diformylfuran offers a productive blueprint for further development, giving hope for the creation of a biocatalytic route to scalable production of furan-based chemical building blocks from sustainable feedstocks. |
| References: | [1] Birmingham, W. R., Toftgaard Pedersen, A., Dias Gomes, M., Bøje Madsen, M., Breuer, M., Woodley, J. M., Turner, N. J. Nat. Commun. 2021, 12 (1), 10. |
| Figures: | 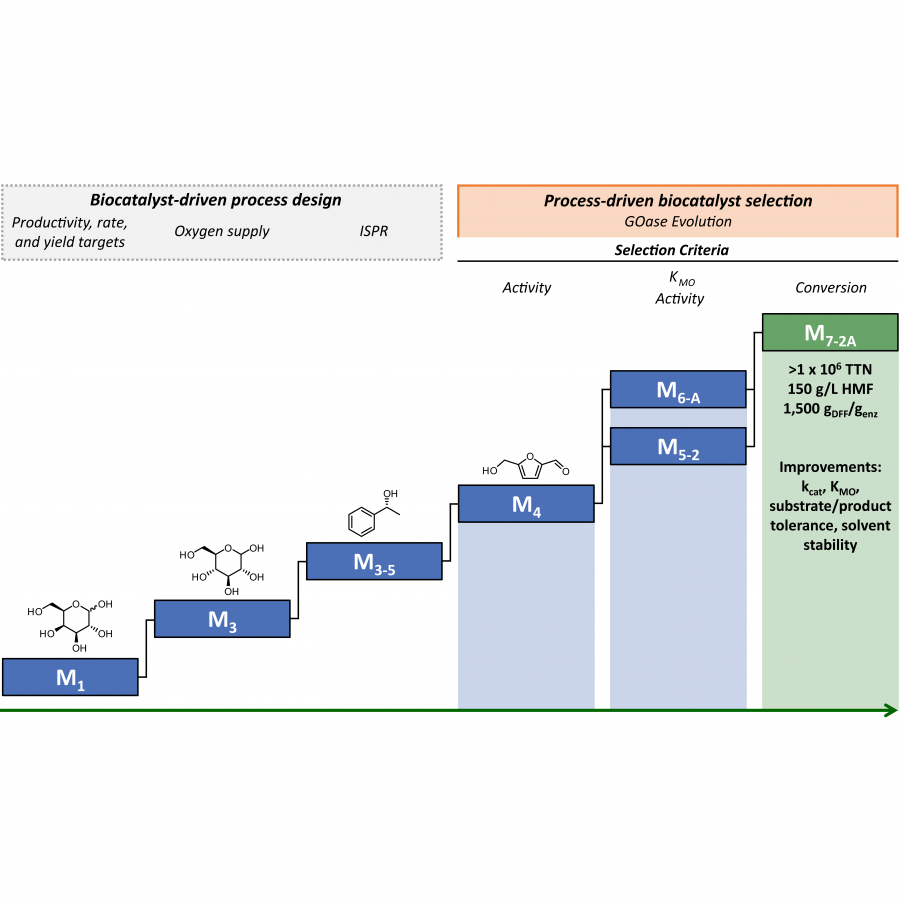 Abstract Figure |
#267 | The development of novel bio-catalytic strategies to construct enantiomerically pure sulfoxides |
|
| Presenting author: | Jingyue WU of UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Daniele CASTAGNOLO of UNIVERSITY COLLEGE LONDON |
| Other authors: | Silvia ANSELMI of UNIVERSITY COLLEGE LONDON Thomas MOODY of DEPARTMENT OF BIOCATALYSIS AND ISOTOPE CHEMISTRY, ALMAC |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | BVMO / Sulfoxide / / |
| Purpose: | Flavoenzymes have the potency to catalyse a vast number of monooxygenation reactions owing to the chemical versatility of cofactor flavin.[1] In BVMO-catalysed oxidation reactions, one atom of molecular oxygen is introduced to the organic substrate, and the other oxygen atom undergoes reduction to water. Having water as a by-product, BVMO attracts interest to be explored as a synthetic tool to address green chemistry. This enzyme has not only been used for performing Baeyer-Villiger oxidations[2] but also for other oxidation reactions including sulfoxidations, oxidations of boron and selenium-containing compounds, epoxidations, and N-oxidations.[3] Chemo- and stereo-selectivity are the major focus of previous studies. Only a few works conducted on substrates presented more than one group susceptible to BVMO-catalysed oxidation.[4,5] This work aims to set up a biocatalytic methodology for constructing enantiopure sulfoxides bearing multiple oxidative sites. A series of sulfides were synthesised using standard synthetic techniques as substrates and screened by a panel of BVMO enzymes. Selected enzymes were investigated and used for the methodology development. |
| References: | [1] M. Hall, Enzymes (Essen), 2020, 47, 37-62. [2] H. Leisch, K. Morley and P. C. K. Lau, Chem Rev, 2011, 111, 4165-4222. [3] G. de Gonzalo, M. D. Mihovilovic and M. W. Fraaije, ChemBioChem, 2010, 11, 2208-2231. [4] M. D. Mihovilovic, B. Müller, M. M. Kayser, J. D. Stewart, J. Fröhlich, P. Stanetty and H. Spreitzer, J Mol Catal B Enzym, 2001, 11, 349-353. [5] F. Zambianchi, S. Raimondi, P. Pasta, G. Carrea, N. Gaggero and J. M. Woodley, J Mol Catal B Enzym, 2004, 31, 165-171. |
#268 | Synthesis of 2' functionalised nucleosides via salvage pathway enzymes |
|
| Presenting author: | Matthew WILLMOTT of UNIVERSITY OF MANCHESTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Biocatalytic Cascades / Nucleosides / Nucleoside Analogues |
| Purpose: | Recent work on the synthesis of islatravir has highlighted the possibility to use aldolases and other enzymes from the nucleoside salvage pathway to generate nucleoside analogues. However this approach has currently been limited to 2' deoxy nucleotides. 2' functionalised nucleosides show broad use in therapeutic oligonucleotides due to their ability to increase binding to DNA and increase resistance of the oligonucleotides to endonucleases. If the enzymes in this pathway could be expanded to improve their substrate scope at the 2’ position this pathway could be used to generate these 2’ functionalised nucleosides biocatalytically The first step to this process will be improving the donor substrate scope of the aldolase enzyme. While aldolases are able to accept a broad range of acceptor (electrophiles) substrates they are generally considered to be quite restricted when it comes to the donor (nucleophile) substrate. Using bioinformatics several active site residues were identified as targets for mutagenesis with the potential to broaden the donor substrate scope of the enzyme. Single point mutations at several of these positions generated aldolases with a greatly increased activity to a range of more complex donor molecules towards which the WT enzyme showed no activity. This was further expanded into a two-step cascade include the synthesis of the aldol acceptor (D-glyceraldehyde-3-phophate) in situ via a kinase allowing a diverse range of 2' functionalised D-Ribose-5-Phosphates sugars to be synthesised stereoselectively. As demonstrated by previous work these pentose-5-phosphate sugars are key intermediates in the biocatalytic synthesis of nucleosides. To demonstrate the potential of the nucleoside salvage pathway enzymes are able to synthesise 2' functionalised nucleosides in addition to 2' deoxynucleosides the two step aldolase/kinase cascade was combined with phosphopentomutase (PPM) and nucleoside phosphorylases (NPs) to synthesis 2' OH , 2’ Me and 2’ F adenosine nucleotides from simple starting materials. Work to expand the substrate scope of these remaining two enzymes to include a broader range of functional groups is ongoing in our lab. This will hopefully allow the generation of a much broader range of 2’ functionalised nucleosides. |
| References: | (1) Huffman, M. A.; Fryszkowska, A.; Alvizo, O.; Borra-Garske, M.; Campos, K. R.; Canada, K. A.; Devine, P. N.; Duan, D.; Forstater, J. H.; Grosser, S. T.; Halsey, H. M.; Hughes, G. J.; Jo, J.; Joyce, L. A.; Kolev, J. N.; Liang, J.; Maloney, K. M.; Mann, B. F.; Marshall, N. M.; McLaughlin, M.; Moore, J. C.; Murphy, G. S.; Nawrat, C. C.; Nazor, J.; Novick, S.; Patel, N. R.; Rodriguez-Granillo, A.; Robaire, S. A.; Sherer, E. C.; Truppo, M. D.; Whittaker, A. M.; Verma, D.; Xiao, L.; Xu, Y.; Yang, H. Design of an in Vitro Biocatalytic Cascade for the Manufacture of Islatravir. Science (80-. ). 2019, 366 (6470), 1255-1259. https://doi.org/10.1126/science.aay8484. (2) Smith, C. I. E.; Zain, R. Therapeutic Oligonucleotides: State of the Art. Annu. Rev. Pharmacol. Toxicol. 2019, 59 (1), 605-630. https://doi.org/10.1146/annurev-pharmtox-010818-021050. (3) Haridas, M.; Abdelraheem, E. M. M.; Hanefeld, U. 2-Deoxy-d-Ribose-5-Phosphate Aldolase (DERA): Applications and Modifications. Applied Microbiology and Biotechnology. 2018. https://doi.org/10.1007/s00253-018-9392-8. |
#280 | Cry3Aa-formate dehydrogenase as a platform for regeneration of NADH for coupling reactions |
|
| Presenting author: | Reza YEKTA SAADABAD of THE CHINESE UNIVERSITY OF HONG KONG |
| Corresponding author: | Michael KENNETH CHAN of THE CHINESE UNIVERSITY OF HONG KONG |
| Other authors: | Marianne M LEE of THE CHINESE UNIVERSITY OF HONG KONG Xu XIONG of THE CHINESE UNIVERSITY OF HONG KONG |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cry3Aa / FDH / NADH regeneration / Genetically immobilization |
| Purpose: | NADH-dependent enzymes play an important role in a wide range of biological oxidation-reduction reactions. Their use in commercial biocatalysis is limited, however, due to the high cost of the required NADH cofactor. One attractive solution is the development of NADH regenerations systems which reduce the NADH required. Formate dehydrogenase (FDH) is a commonly used enzyme for this purpose since it can efficiently regenerate NADH by oxidizing formic acid into CO2 gas. However, it has low stability, cannot be recycled for multiple reaction cycles, and requires time-consuming and costly purification. Here, we present a strategy to stabilize FDH by fusion to Cry3Aa, a protein that naturally produces micrometer sized crystals within the bacterium Bacillus thuringiensis (Bt). The resulting Cry3Aa-FDH fusion crystals can be directly isolated from Bt by density centrifugation, greatly simplying its production costs. Moreover it can be co-immobilized with a second NADH-dependent enzyme allowing for direct coupling of the NADH produced for catalysis within same crystal. Two different enzymes, leucine dehydrogenase and alcohol dehydrogenase, were independently co-immobilized in Cry3Aa-FDH crystals as a coupling reaction to produce chiral intermediates to validate this system. According to the findings of this study, Cry3A-FDH/Cry3A-LDH and Cry3A-FDH/Cry3A-ADH can efficiently synthesize L-tert-leucine and ethyl (S)-3-hydroxybutyrate products without the minimal addition of exogenous NADH. This biosystem is reusable and can be used for multiple reactions, which is essential for industrial applications. We hope to investigate the platform's ability to simplify reactions and reduce associated costs for other industrial enzymes. |
| References: | [1] Q. SUN, Bioconjugate Chem. 2022, 33, 2, 386-396 [2] B. Heater, J. Am. Chem. Soc. 2020, 142, 22, 9879-9883 |
| Figures: | 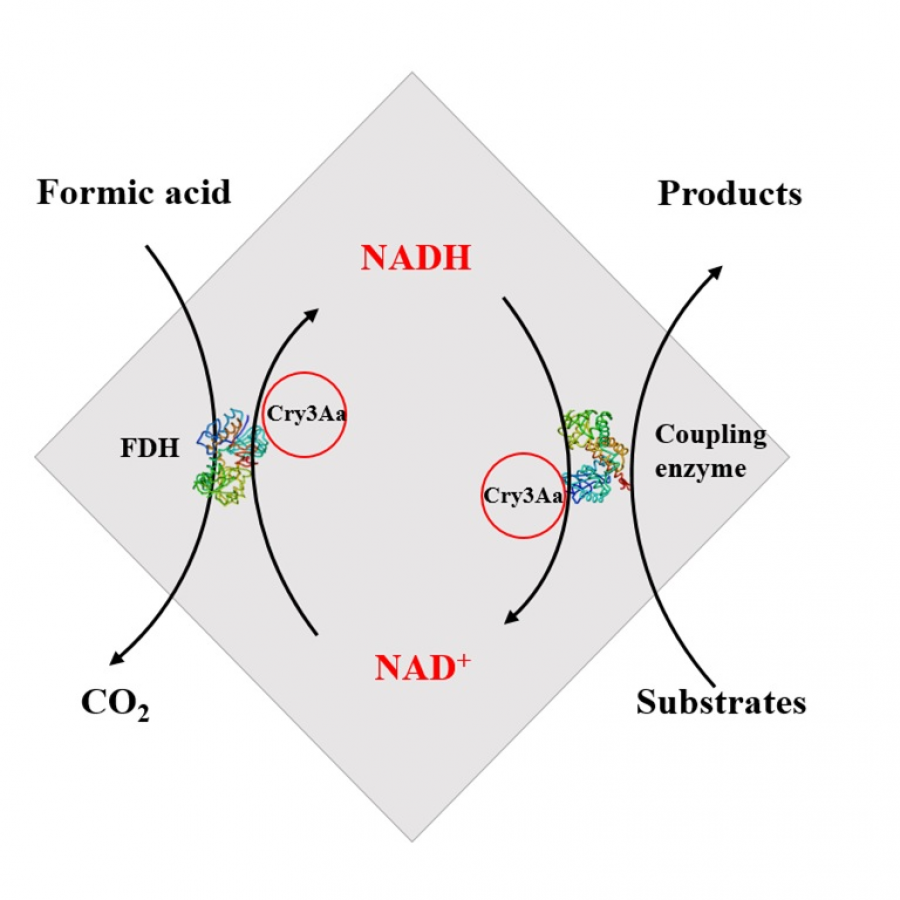 Graphical Abstract Cry3Aa -formate dehydrogenase (FDH) that is coupled with another enzyme (leucine dehydrogenase or alcohol dehydrogenase) to continuously regenerate NADH cofactors by oxidation of formic acid into CO2 gas, which is then utilized by the second enzyme. |
#281 | Cry1Ab-mediated In Vivo Co-immobilization of Enzymes for the Biosynthesis of Putrescine from L-Arginine |
|
| Presenting author: | XU XIONG of THE CHINESE UNIVERSITY OF HONG KONG |
| Corresponding author: | Michael Kenneth CHAN of THE CHINESE UNIVERSITY OF HONG KONG |
| Other authors: | Marianne Ming Ming LEE of THE CHINESE UNIVERSITY OF HONG KONG |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cry1Ab / putrescine / co-immobilization / |
| Purpose: | Putrescine is an important industrial chemical used in the production of polymers. It can be chemically synthesized from propene and methane, but this route is neither environmental-friendly nor sustainable due to its production of the toxic intermediate hydrogen cyanide, and its use of the non-renewable raw material propene. Another route that has attracted intense academic interest is the biological synthesis of putrescine due to its being renewable, environmentally friendly, and green. The biosynthesis of putrescine in cells has gained great attention, but this route is challenging for large-scale production due to the limited tolerance of cells to putrescine products. One route that avoids this limitation is the in vitro biosynthesis of putrescine, but a platform for immobilizing the enzymes is required. Here we report the direct co-immobilization of arginase and ornithine decarboxylase as genetic fusions to the crystal-forming protein Cry1Ab. Here arginase promotes the conversion of arginine to ornithine, which is further converted to putrescine by the ornithine decarboxylase. We show that Cry1Ab-mediated direct co-immobilization could produce active particles, which exhibited higher stability than their free enzyme counterparts and improved catalytic efficiency than the mixture of individually immobilized particles. Furthermore, the co-immobilized biocatalyst was shown to promote the conversion of arginine to putrescine for multiple reaction cycles. |
| References: | [1] B. Heater, J. Am. Chem. Soc. 2020, 142, 22, 9879-9883. [2] Q. SUN, Bioconjugate Chem. 2022, 33, 2, 386-396. |
| Figures: | 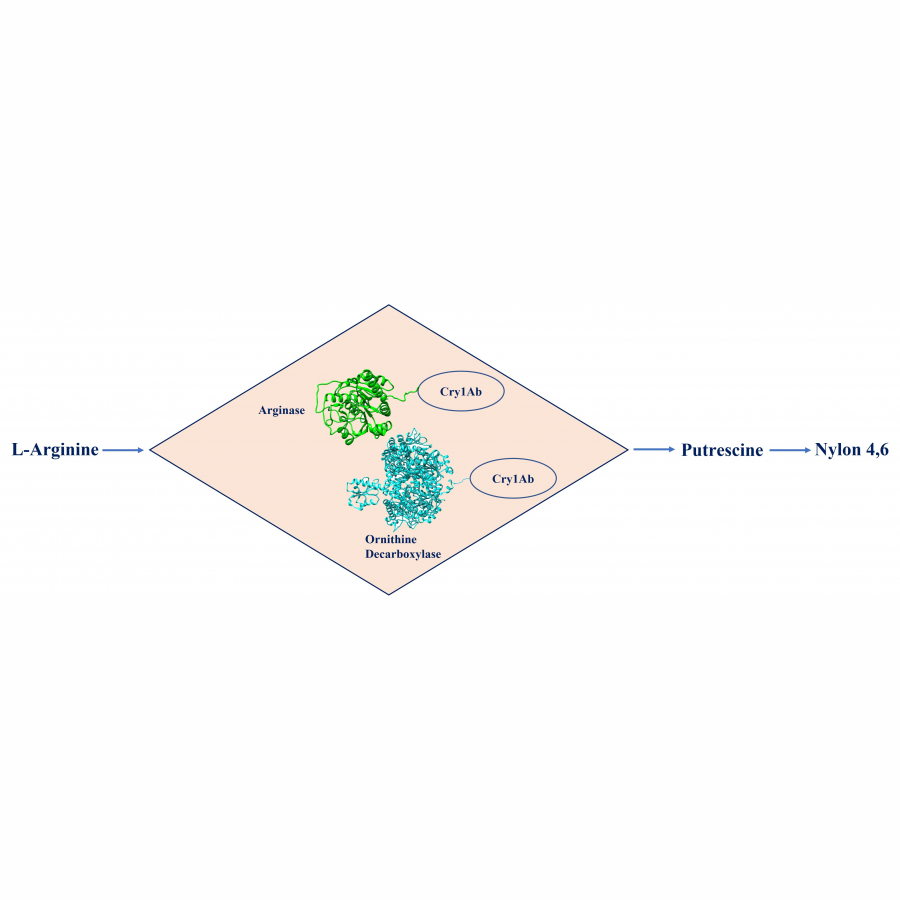 Graphic abstract Arginase and ornithine decarboxylase was directly co-immobilized by Cry1Ab protein in vivo. The active particles could be used to promote the conversion of L-arginine to putrescine and its subsequent conversion to nylon 4,6 was also demonstrated. |
#285 | Structure and Mutation of Deoxypodophyllotoxin Synthase (DPS), a C-C Bond Forming Enzyme |
|
| Presenting author: | Zoe INGOLD of UNIVERSITY OF YORK |
| Other authors: | Benjamin LICHMAN of UNIVERSITY OF YORK Gideon GROGAN of UNIVERSITY OF YORK |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | oxygenase / ring-closing / deoxypodophyllotoxin / iron-dependent |
| Purpose: | Deoxypodophyllotoxin synthase (DPS) is a member of the 2-oxoglutarate dependent dioxygenase (2-ODD) superfamily and catalyses a ring closing, C-C bond forming reaction. The product of this reaction, deoxypodophyllotoxin, is a polyphenol lignin ultimately derived from tyrosine via the phenylpropanoid pathway. The enzyme was discovered in 2015 during an investigation into the biosynthesis of podophyllotoxin in Podophyllum hexandrum (mayapple). (Scheme 1)[1]. Deoxypodophyllotoxin is the natural product precursor to several topoisomerase II inhibitors, which are on the World Health Organisation (WHO)’s list of essential medicines. Currently it is necessary to isolate this precursor directly from mayapple, which is slow-growing and has a limited environmental range, making it less than ideal as a source for the large-scale synthesis of topoisomerase II inhibitors[2]. Previous work has focused on developing synthetic biology platforms for the production of podophyllotoxin through heterologous gene expression, including DPS[2, 3, 4]. It has also been shown that DPS can accept a range of substrates, indicating it has potential in biocatalytic processes for the formation of diverse polycyclic aryllignans[5, 6, 7]. Here we present the structure of DPS, solved using X-ray crystallography to a resolution of 1.41 Å. The crystals were obtained in the space group P21 with a single monomer in the asymmetric unit displaying the squashed ß-barrel fold common to the 2-ODD superfamily. The open end of this supports the active site with the iron centre coordinated by the “facial triad” of His184, D186 and H239. The structure was used to inform a mutational analysis of DPS, which suggests a role for a D224-K187 salt bridge in maintaining substrate interactions and a catalytic role for H165, perhaps as the base for the proton abstraction at the final rearomatisation step. This work improves our understanding of specific residues‘ contributions to the DPS mechanism and can inform future engineering of the enzyme mechanism and substrate scope for the development of a versatile biocatalyst. |
| References: | 1. Lau, W., Sattely, E.S., Science, 2015, 349(6253), 1224-1228. 2. Decembrino, D., Raffaele, A., Knöfel, R., Girhard, M., Urlacher, V.B., Microbial Cell Factories, 2021, 20, 183. 3. Schultz, B.J., Kim, S.Y., Lau, W., Sattely, E.S., J.Am. Chem. Soc., 2019, 141(49), 19231-19235. 4. Kim, S.S., Wengier, D.L., Ragland, C.J., Sattely, E.S., ACS Synth. Biol., 2022, 11(10), 3379-3387. 5. Li, J., Zhang, X., Renata, H., Angew. Chem. Int. Ed., 2019, 58(34), 11657-11660. 6. Chang, W.C., Yang, Z.J., Tu, Y.H., Chien, T.C., Org. Letters, 2019, 21, 228-232. 7. Lazzarotto, M., Hammerer, L., Hetmann, M., Borg, A., Schmermund, L., Steiner, L. et al., Angew. Chem. Int. Ed., 2019, 58(24), 8226-8230. |
| Figures: | 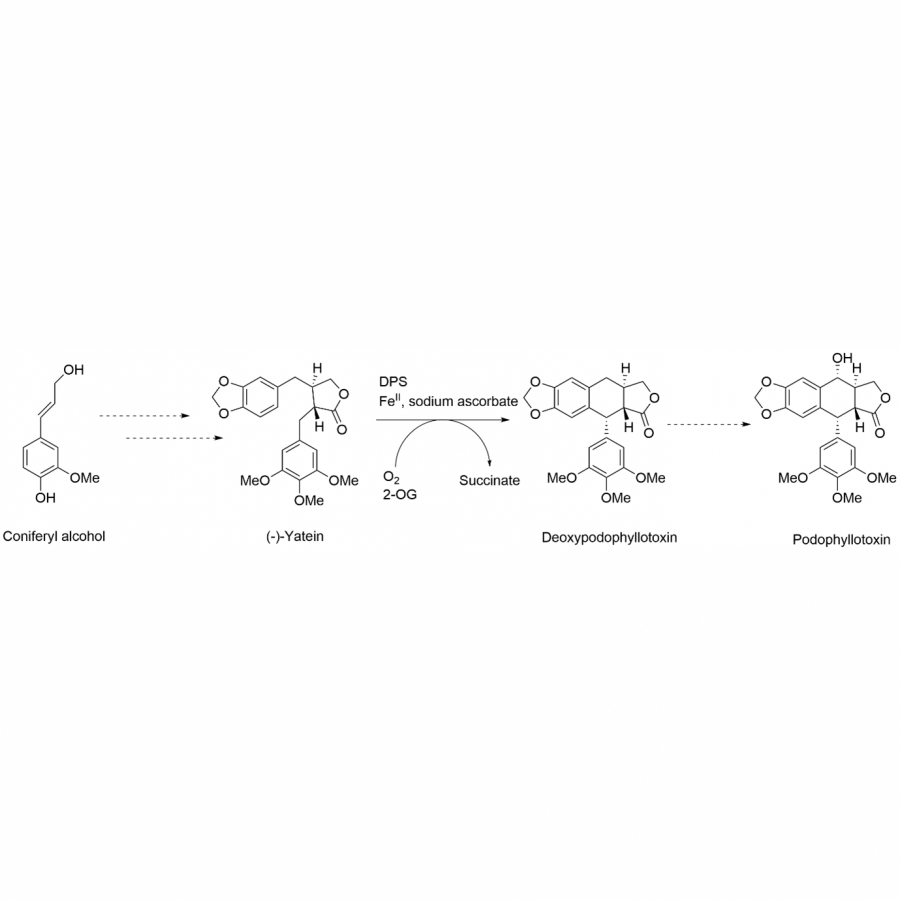 Figure 1: The biosynthetic route to podophyllotoxin in mayapple, highlighting the penultimate step that is catalysed by DPS |
#292 | Process engineering strategies for biocatalytic (cascade) reactions |
|
| Presenting author: | Selin KARA of AARHUS UNIVERSTY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Decarboxylase / Oxygenase / Non-conventional media / Flow biocatalysis |
| Purpose: | The application of nature’s catalysts enzymes for the synthesis of chemicals is a key emerging field of industrial biotechnology to meet current and future needs of our society for sustainable manufacturing of chemicals. Nature uses an elegant and efficient synthetic strategy: Coupling enzymes in multi-step pathways without intermediate isolation and purification steps with a precise spatial control of catalysis. Inspired by nature, the design of multi-step biotransformations has been attracting great attention within the biocatalysis community. The talk covers enzymatic (cascade) reactions and demonstration of those at the industrially relevant conditions with the help of process engineering. In particular, two use cases will be introduced covering decarboxylases and peroxygenases in cascading systems exploring the use of non-conventional media and different operational mode for enhancing the efficiency of these enzymatic applications. |
| References: | (1) M. Hobisch, P. De Santis, S. Serban, A. Basso, E. Byström, S. Kara, Peroxygenase-driven ethylbenzene hydroxylation in a rotating bed reactor, Organic Process Research & Development 2022, 26, 9, 2761-2765. DOI: 10.1021/acs.oprd.2c00211. (2) P. Petermeier, J.P. Bittner, S. Müller, E. Byström, S. Kara, Design of a green chemoenzymatic cascade for scalable synthesis of bio-based styrene alternatives, Green Chemistry 2022, 24, 6889-6899. DOI: 10.1039/d2gc01629j. (3) P. De Santis, N. Petrovai, L.-E. Meyer, M. Hobisch, S. Kara, A holistic carrier-bound immobilization approach for unspecific peroxygenase, Frontiers in Chemistry 2022, 10:985997. DOI: 10.3389/fchem.2022.985997. (4) L.-E. Meyer, B. Fogtmann Hauge, T. Müller Kvorning, P. De Santis, S. Kara, Continuous oxyfunctionalizations catalyzed by unspecific peroxygenase, Catalysis Science & Technology 2022, 12, 6473-6485. DOI: 10.1039/D2CY00650B. (5) L.-E. Meyer, D. Horváth, S. Vaupel, J. Meyer, M. Alcalde, S. Kara. A 3D printable synthetic hydrogel as an immobilization matrix for continuous synthesis with fungal peroxygenases, under revision. |
#293 | Sequential batches strategy for the enhancement of protein recovery from salmon frames by proteolysis |
|
| Presenting author: | Pedro VALENCIA of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA |
| Other authors: | Suleivys NUÑEZ of PONTIFICIA UNIVERSIDAD CATOLICA DE VALPARAISO Silvana VALDIVIA of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Cristian RAMIREZ of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Marlene PINTO of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Sergio ALMONACID of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | protein hydrolysis / byproduct proteolysis / salmon frames / sequential batch |
| Purpose: | The hydrolysis of proteins by proteases is characterized by a strong inhibition exerted by peptides formed during the reaction. Based on this fact, the efficiency should increase when the reaction products are withdrawn from the reaction media. The aim of this study was to test a new operational strategy consisting in a sequential batch operation where the aqueous phase containing the soluble peptides is withdrawn and the remaining solid phase is submitted to a second batch. The strategy was tested for the hydrolysis of salmon frame proteins by 13 AU subtilisin per kg at 55°C and pH 6.5 (native) during 2 h in a regular batch. Two sequential batches were operated during 1 h each at the same conditions. After 1 h the reaction mixture was centrifuged, and the different phases weighted and analyzed for nitrogen content. The solid phase was hydrolyzed in a second batch during 1 h at the same operating conditions. The nitrogen extraction was 26.6% ± 0.4 after 2 h of hydrolysis in a regular batch operation. Two sequential batches were operated during 1 h each with the same total protease dose (13 AU/kg) distributed as 75/25, 50/50 and 25/75 percentage in the first/second batch. The nitrogen extraction resulted in 49.0% ± 1.6, 45.9% ± 0.6 and 48.7% ± 0.8 for each protease dose distribution, respectively. These results showed that an increase in nitrogen extraction can be achieved without increasing operation time and protease dose. The sequential batches were also tested without the addition of protease in the second batch. The nitrogen extraction was 43.8% ± 1.6, 42.4% ± 0.6 and 39.8% ± 0.8 for protease dose of 75%, 50% and 25%, added to the first batch and without addition in the second batch, respectively. The adsorption of subtilisin was inferred from results as an explanation for the hydrolysis reaction observed in the second batch. The nitrogen extraction was significantly increased with the sequential batches strategy without increasing the operating time and protease dose compared to a one batch operation. A higher nitrogen extraction was obtained even without addition of protease in the second batch. The sequential batch is a promising strategy to enhance the efficiency of the enzymatic hydrolysis of byproduct proteins. |
| Figures: | 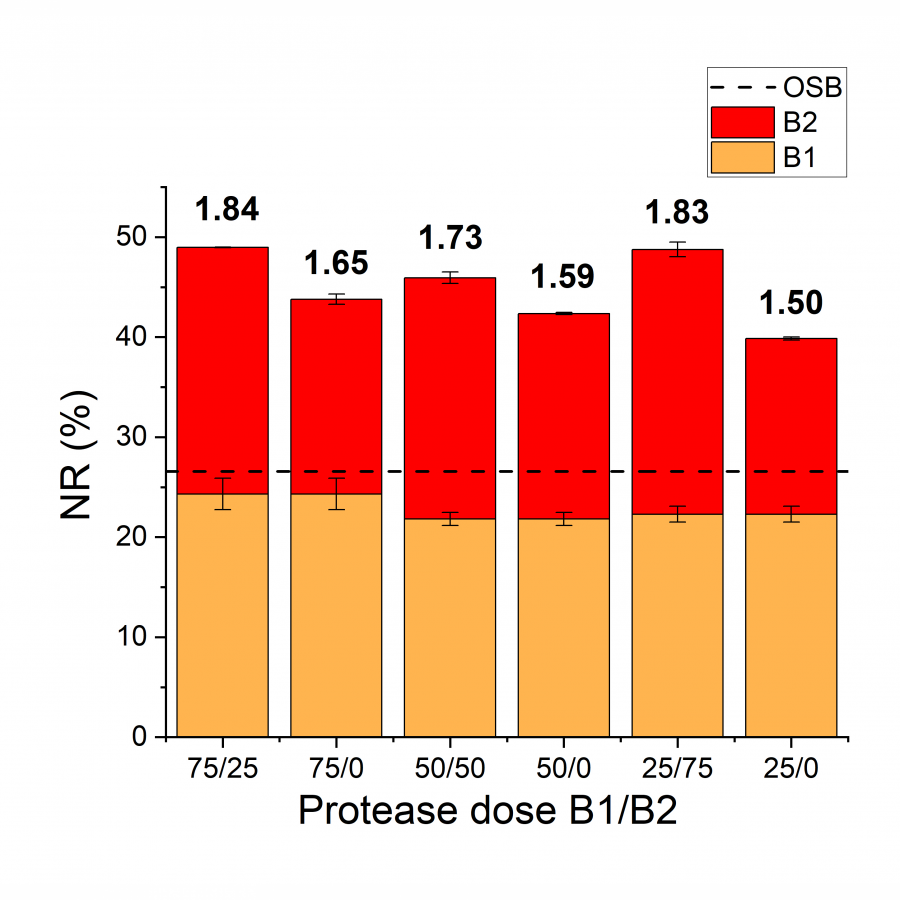 Nitrogen recovery and increment after the hydrolysis of SF by subtilisin at 55 °C for different protease doses. NR: nitrogen recovery.
OSB: one-stage batch.
B1: sequential batch 1.
B2: sequential batch 2. |
#294 | Combinatorial assembly and design of enzymes |
|
| Presenting author: | Rosalie LIPSH-SOKOLIK of WEIZMANN INSTITUTE |
| Other authors: | Sarel FLEISHMAN of WEIZMANN INSTITUTE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme design / xylanase / / |
| Purpose: | The design of structurally diverse enzymes is constrained by long-range interactions that are necessary for accurate folding. We introduce an atomistic and machine learning strategy for the combinatorial assembly and design of enzymes (CADENZ) to design fragments that combine with one another to generate diverse, low-energy structures with stable catalytic constellations. We applied CADENZ to endoxylanases and used activity-based protein profiling to recover thousands of structurally diverse enzymes. Functional designs exhibit high active-site preorganization and more stable and compact packing outside the active site. Implementing these lessons into CADENZ led to a 10-fold improved hit rate and more than 10,000 recovered enzymes. This design-test-learn loop can be applied, in principle, to any modular protein family, yielding huge diversity and general lessons on protein design principles. |
#295 | Mitigating product inhibition via an aqueous two-phase system in a biocatalytic process for p-coumaric acid production |
|
| Presenting author: | ALEXANDER VIRKLUND of TECHNICAL UNIVERSITY OF DENMARK |
| Other authors: | JOHN M. WOODLEY of TECHNICAL UNIVERSITY OF DENMARK ALEX T. NIELSEN of TECHNICAL UNIVERSITY OF DENMARK |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Aqueous two-phase system / In-situ product removal / Immobilization |
| Purpose: | p-Coumaric acid (pCA) is a promising chemical precursor to making thin, flexible, and transparent organic semiconductors, with applications in wearable electronics. pCA can be produced via biocatalysis, but the required tyrosine ammonia lyase (TAL) enzyme suffers from competitive product inhibition, which reduces production titers, rates, and yields [1]. We considered and tested several strategies to mitigate this problem, and we developed a method of in-situ product removal using an aqueous two-phase system (ATPS) consisting of a polymer and salt phase. pCA has a partition coefficient of around 1:20 in the salt:polymer phases, effectively removing pCA into the polymer phase. The implementation of the ATPS into a biocatalytic process still faced a challenge, as the whole cell biocatalyst also partitioned into the polymer phase due to the high density of the salt phase. In order to keep the biocatalyst in the salt phase, we tried immobilized the biocatalyst in a hydrogel and loading the beads into a catalyst basket. However, the common calcium-alginate hydrogel was unstable in the salts used for salt:PEG ATPS, but we found K-carrageenan to be stable in potassium salts. After making further optimizations to the composition of the ATPS, we finally tested the effectiveness of the immobilization procedure and the ATPS at improving pCA production in a rotating bed bioreactor. This work may help in the development of a viable pCA production bioprocess, and it is a case study in using bioprocess engineering to overcome limitations with a biocatalyst. |
| References: | [1] sariaslani, f. s., ann. rev. microb. 2007, 61(1), 51-69. |
| Figures: | 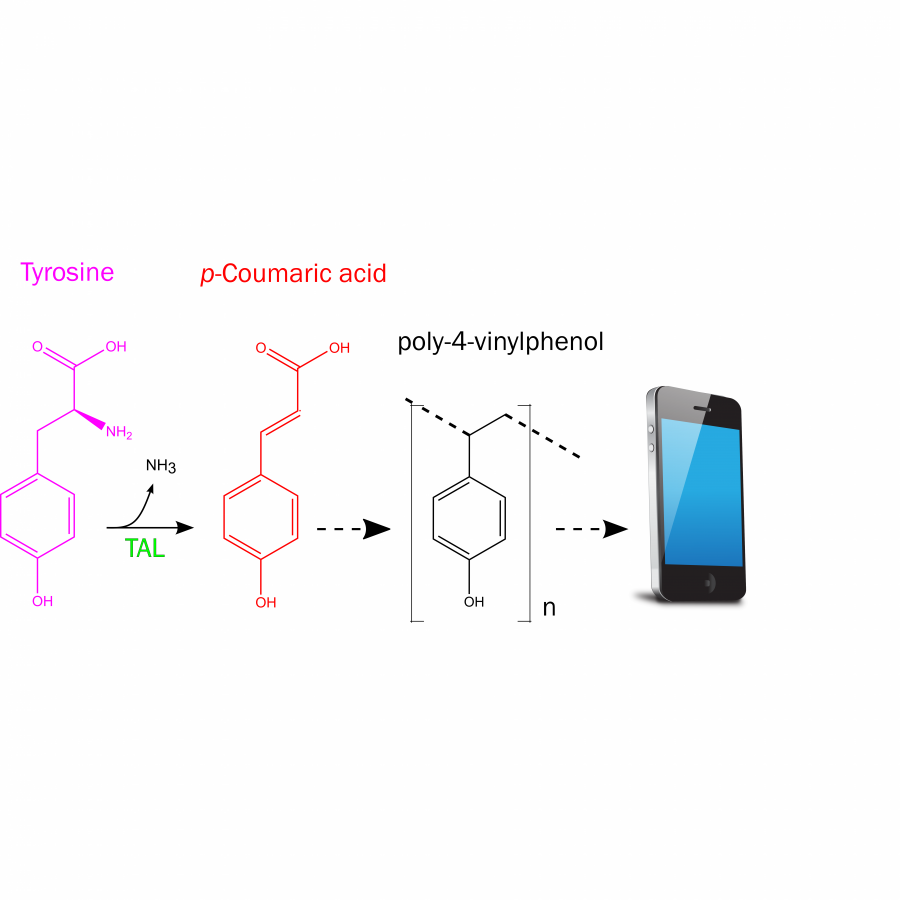 Biocatalysis for p-coumaric acid production TAL, tyrosine ammonia lyase 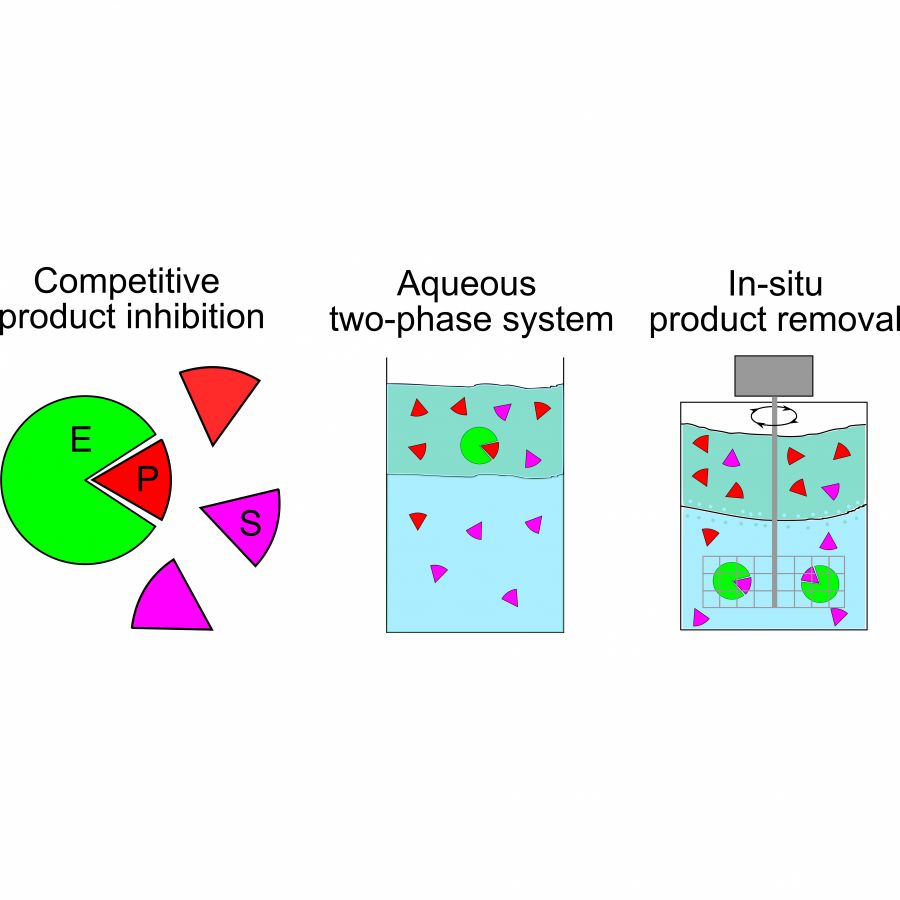 Mitigating product inhibition via in-situ product removal E, enzyme; P, product; S, substrate. |
#298 | Mutations increasing cofactor affinity improve stability and activity of a Baeyer-Villiger monooxygenase |
|
| Presenting author: | Florian RUDROFF of TU WIEN, INSTITUTE OF APPLIED SYNTHETIC CHEMISTRY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Baeyer-Villiger monooxygenase / protein engineering / structure-guided consensus approach / |
| Purpose: | The typically low thermodynamic and kinetic stability of enzymes is a bottleneck for their application in industrial synthesis. Baeyer-Villiger monooxygenases, which oxidize ketones to lactones using aerial oxygen, among other activities, suffer particularly from these instabilities. Previous efforts in protein engineering have increased thermodynamic stability but at the price of decreased activity. Here, we solved this tradeoff by introducing mutations in a cyclohexanone monooxygenase from Acinetobacter sp., guided by a combination of rational and structure-guided consensus approaches. We developed variants with improved activity (1.5 to 2.5-fold) and increased thermodynamic (+5 °C Tm) and kinetic stability (8-fold). Our analysis revealed a crucial position in the cofactor binding domain, responsible for an 11-fold increase in affinity to the flavin cofactor, and explained using MD simulations. This gain in affinity was compatible with other mutations. While our study focused on a particular model enzyme, previous studies indicate that these findings are plausibly applicable to other BVMOs, and possibly to other flavin-dependent monooxygenases. These new design principles can inform the development of industrially robust, flavin-dependent biocatalysts for various oxidations. |
| References: | [1] Mansouri, H. R.; Gracia Carmona, O.; Jodlbauer, J.; Schweiger, L.; Fink, M. J.; Breslmayr, E.; Laurent, C.; Feroz, S.; P. Goncalves, L. C.; Rial, D. V.; Mihovilovic, M. D.; Bommarius, A. S.; Ludwig, R.; Oostenbrink, C.; Rudroff, F., Mutations Increasing Cofactor Affinity, Improve Stability and Activity of a Baeyer-Villiger Monooxygenase. ACS Catal. 2022, 12 (19), 11761-11766. |
| Figures: | 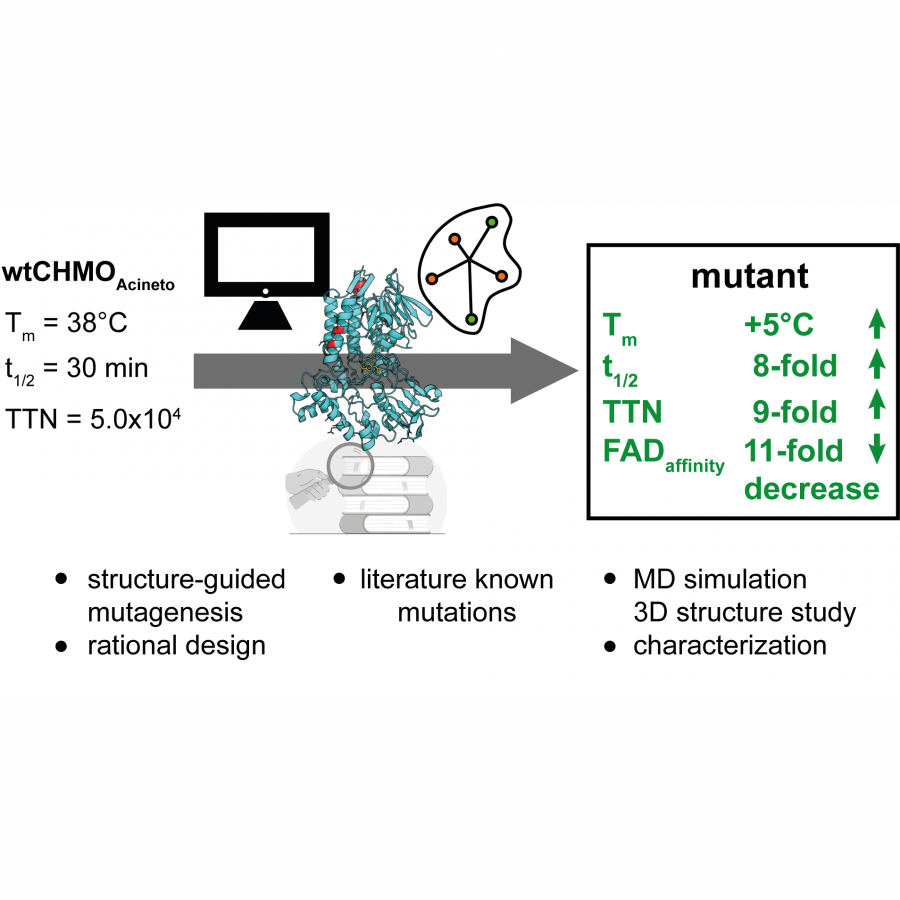 Figure 1. Protein engineering approach towards a highly improved BVMO variant. |
#299 | Enzymatic hydrolysis of salmon frame proteins at different byproduct/water ratios and pH regimes |
|
| Presenting author: | Pedro VALENCIA of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA |
| Other authors: | Silvana VALDIVIA of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Suleivys NUÑEZ of PONTIFICIA UNIVERSIDAD CATOLICA DE VALPARAISO Cristian RAMIREZ of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Marlene PINTO of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA Sergio ALMONACID of UNIVERSIDAD TECNICA FEDERICO SANTA MARIA |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | protein hydrolysis / byproduct proteolysis / salmon frame / high solid concentration |
| Purpose: | The enzymatic hydrolysis of proteins is an interesting alternative to add value to the salmon frames by converting this by-product into protein hydrolysate and bones. The protein hydrolysates are recognized sources of bioactive peptides. In adittion, bones can be transformed into a viable calcium source through nano milling. During the hydrolysis the mixing is favored by the addition of water, meanwhile, the cost is highly increased during the drying stage. The addition of alkali avoids the pH drop causing enzyme activity to decrease, meanwhile, the operation cost is increased. The evaluation of different by-product/water ratios was assessed using 50%, 75%, and 100% of ground salmon frames. The pH regimes were set up at controlled pH 8, initial pH 8 without control, and native initial pH 6.5 without control. The hydrolysis reactions were carried out at 55 °C in an agitated batch reactor using 13 AU subtilisin per kg salmon frame. Response variables were released alpha-amino groups, mass of soluble/insoluble fraction, and nitrogen extraction. The results showed that the released alpha-amino groups after 60 min of reaction decreased with the salmon frame concentration (Fig. 1). The hydrolysis without pH control allowed to avoid the use of alkali and control system but decreasing the production of amino groups at 68% of the controlled condition. The hydrolysis at high salmon frame concentration allowed to avoid the addition of water. However, the nitrogen recovery was 27.2, 45.8 and 49.1% at 60 min for 100%, 75%, and 50% of salmon frame for the controlled pH condition (Fig. 2). These results can be used to estimate the profitability of the process after considering the decrease in the operational cost and the effects on the product yield. |
| Figures: | 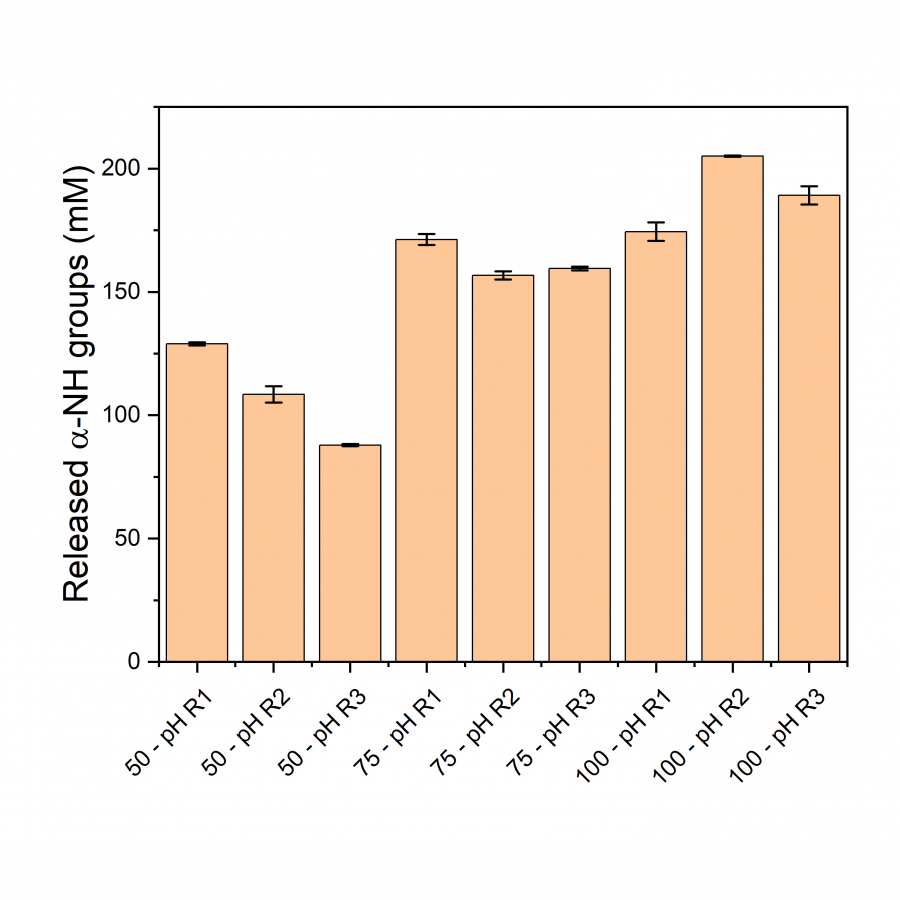 Released amino groups concentrations after 60 min of reaction for different byproduct/water ratios and pH regimes for the hydrolysis of salmon frame protein by subtilisine pH regimes
R1: initial pH 8.0 controlled.
R2: initial pH 8.0 uncontrolled.
R3: initial pH 6.5 (native) uncontrolled. 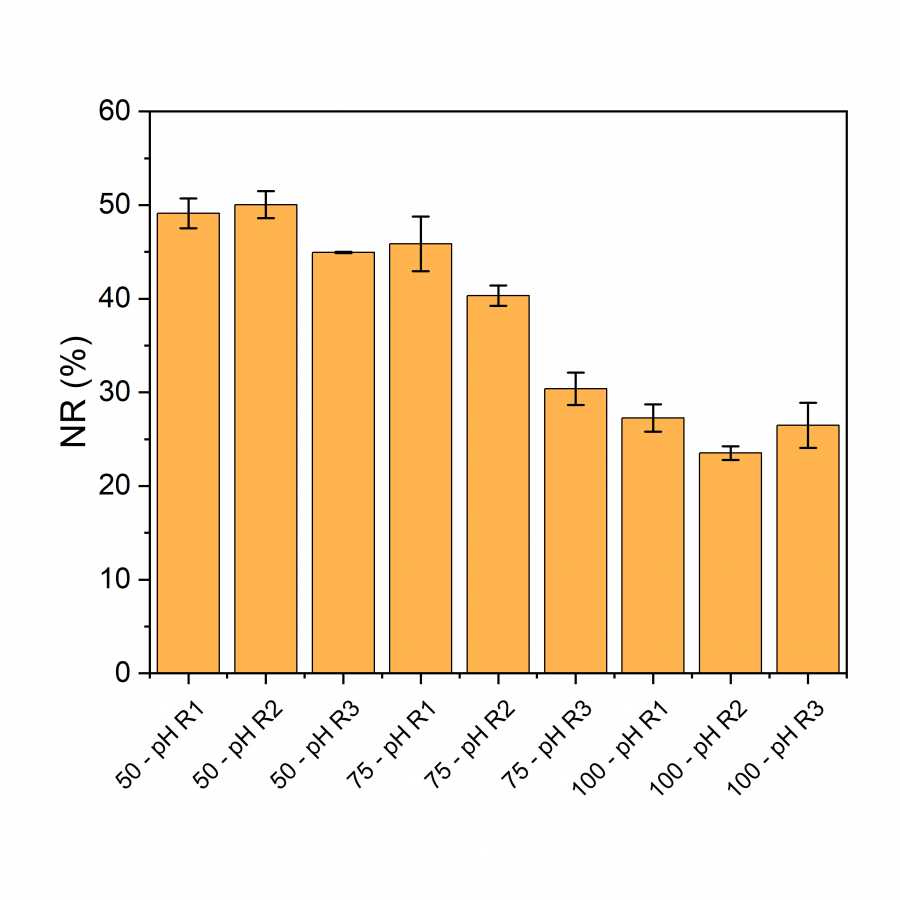 Nitrogen recovery of salmon frame protein at different byproduct/water ratior and pH regimes for the hydrolysis of salmon fram protein by subtilisin at 55°C. pH regimes:
R1: initial pH 8.0 controlled.
R2: initial pH 8.0 uncontrolled.
R3: initial pH 6.5 (native) uncontrolled. |
#303 | Full biocatalytic synthesis of D-tagatose from whey permeate as a raw material |
|
| Presenting author: | Fadia CERVANTES of CATALYSIS INSTITUTE CSIC |
| Corresponding author: | Fadia CERVANTES of CATALYSIS INSTITUTE CSIC |
| Other authors: | Sawssan NEIFAR of CENTRE OF BIOTECHNOLOGY OF SFAX Zoran MERDZO of MOLECULAR BIOLOGY CENTER SEVERO OCHOA Antonio O. BALLESTEROS of CATALYSIS INSTITUTE CSIC María FERNÁNDEZ-LOBATO of MOLECULAR BIOLOGY CENTER SEVERO OCHOA Samir BEJAR of CENTRE OF BIOTECHNOLOGY OF SFAX Francisco J. PLOU of CATALYSIS INSTITUTE CSIC |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | D-tagatose / Biocatalysis / immobilization / bioreactors |
| Purpose: | D-Tagatose is an isomer of D-galactose and the epimer at C-4 site of D-fructose. It is one of the most promising functional sweeteners, similar to the polyols in having a low caloric value and tooth-friendly property; however, it has no laxative effect and it is able to modulate lipid metabolism, and reduce the symptoms associated with type 2 diabetes. Tagatose has the GRAS (Generally Recognized as Safe) status granted by the US Food and Drug Administration (FDA) since 2001, so it can be used in confectionery, beverages, health foods, and dietary products as a low-calorie, full-bulk sweetener. Elsewhere, it has entered in a phase 3 clinical trial, as a good candidate to treat type 2 diabetes. Tagatose does not increase insulin levels for glucose control. This work was focused on the development of a totally biocatalytic methodology for obtaining D-tagatose, the best reaction conditions were studied for each step [1]. First step was the hydrolysis of whey permeate, and we chose the novel commercial β-galactosidase from Bifidobacterium bifidum (Saphera), because this enzyme with lactose concentrations lower than 200 g/L has hydrolysis as its main reaction and shows negligible transgalactosylation [2]. This reaction was performed with concentrated whey permeate at 45 °C and 3.75 units/mL of enzyme for 3 h. For second step we searched a microorganism that was capable of consuming D-glucose without consuming D-galactose from the hydrolyzed whey permeate; and the winner was the crabtree negative microorganism Komagataella phaffii (Pichia pastoris). Thus, 350 mg of wet weight cells were immobilized in alginate gel beads, and the resulting biocatalyst was able of completely eliminating D-glucose and efficiently reused for 13 cycles [3]. The third step was the isomerization of D-galactose with a L-arabinose isomerase from Bacillus stearothermophilus. The enzyme was successfully immobilized onto an amino polymethacrylate support and a packed bed bioreactor was developed and operated continuously for 9 days at 50 °C, with an initial productivity of D-tagatose of 77 g L-1 day-1. The fourth step was the search of a microorganism able to consume D-galactose without consuming D-tagatose. From 19 microorganisms tested, the best was Schwanniomyces occidentalis. This microorganism was immobilized with alginate and it could be reused for 3 cycles of 5 h each. As a conclusion, we have demonstrated that it is possible to obtain D-tagatose using an industrial waste as starting reagent, following a completely biocatalytic methodology. |
| References: | [1] FV-Cervantes, Cat. 10, 2020, 1-14. [2] V-Füreder, J. Agric. Food Chem. 68, 2020, 4930-4938. [3] FV-Cervantes, ACS Food Sci. Technol. 2, 2022, 682-690. |
| Figures: |  cycles with P. pastoris Percentage of eliminated D-glucose in different cycles with P. pastoris immobilized in calcium alginate beads, using a mixture of D-galactose and D-glucose (50 mg/mL of each sugar) as substrate. 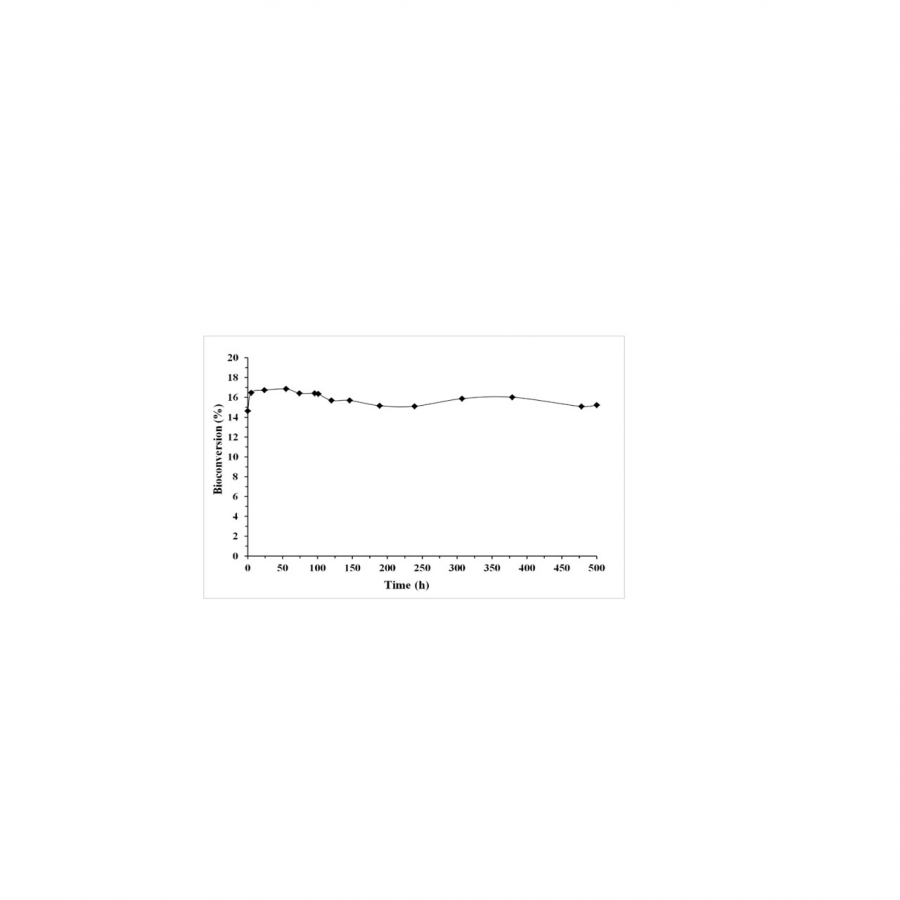 Bioreactor of L-arabinose isomerase Continuous packed bed reactor of L-AI US100 immobilized on Sepabeads EC-EA at 50 °C during 9 days of operation. The flow of the feeding solution (90 g/L galactose, 0.5 mM MnSO4 in 0.1 M MOPS buffer pH 7.5) was maintained at 0.03 mL/min. |
#304 | Engineering of an EneIRED for direct synthesis of asymmetric conjugate reduction-reductive amination products |
|
| Presenting author: | Anya MILETIC of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | EneIRED / Engineering / Directed Evolution / |
| Purpose: | Due to their ability to catalyse the synthesis of asymmetric amines, IREDs have many potentially useful applications in synthesis of agrochemicals and APIs, where enantiopure products are often desirable. Chemically synthesising enantiomerically pure compounds can be challenging, often requiring costly resolution processes of racemic mixtures, therefore utilising enantioselective biocatalysts to avoid these extra steps is favourable. Following the discovery of an IRED capable of catalysing conjugate reduction in tandem with reductive amination in a two-step mechanism, the wild-type substrate scope was investigated and was found to favour reactions between unhindered substrates. Rational design principles were applied alongside mutagenesis and directed evolution in an attempt to broaden the existing substrate scope and facilitate acceptance of sterically bulky substrates that were not previously accepted by the wild-type. |
#305 | In search of the perfect environment: The quest to maximize macromolecule stability |
|
| Presenting author: | Radović Mia of Faculty of Food Technology and Biotechnology |
| Corresponding author: | Marina CVJETKO BUBALO of FACULTY OF FOOD TECHNOLOGY AND BIOTECHNOLOGY |
| Other authors: | Mia RADOVIĆ of FACULTY OF FOOD TECHNOLOGY AND BIOTECHNOLOGY Wolfgang KROUTIL of INSTITUTE OF CHEMISTRY Tamara REITER of INSTITUTE OF CHEMISTRY Ivana RADOJČIĆ REDOVNIKOVIĆ of FACULTY OF FOOD TECHNOLOGY AND BIOTECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | macromolecules / deep eutectic solvents / stabilization / |
| Purpose: | Current literature unquestionably implies that deep eutectic solvents (DES)- mediated biocatalytic approach is an exciting new field with enormous possibilities for improving reaction efficiency and sustainability through better substrate solubility/loading, improved enzyme activity and stability. [1] Except for enzymes, other macromolecules used in biocatalysis, such as cofactors, struggle with long-term stabilization in liquid form. Due to their complex structure, macromolecules can be unstable and prone to denaturation, especially in harsh environments like extreme temperatures or pH conditions. Therefore, their stabilization is critical in many scientific fields, including biotechnology, pharmaceuticals, and medical research. This study aims to show that DES can be effective in stabilizing enzymes and other macromolecules by providing a suitable environment for their function and stability. DES can help to maintain their structure and reduce denaturation in harsh conditions. Moreover, in some cases, DES can also enhance the activity and selectivity of enzymes. [2] Implementation of green chemistry principles through the use of DES represents an up-to-date solution fighting waste generation. These non-toxic and non-flammable solvents are mixtures of cheap, natural and readily available components prepared by mixing hydrogen bond acceptors (HBA) such as quaternary ammonium salts and hydrogen bond donors (HBD) based on natural products, in a specific molar ratio resulting homogenous solution based on hydrogen bonds between DES components. One of the major attractions of making DES an alternative to organic solvents lies in the tremendous number of structural combinations, thus it is possible to rationally design an optimal one for each specific application. [3] The vast number of possible chemical structures, in addition to unique physical and chemical properties, and low environmental impact make DES interesting for industrial use. [4] Therefore, a new window of opportunities for DES application is presented in the stabilization of macromolecules. Stabilization of macromolecules refers to the methods that prevent or minimize the breakdown of these molecules, thereby maintaining their structure and function. So far, techniques for stabilizing macromolecules include chemical modifications (use of cross-linkers or protease inhibitors), physical methods (freeze-drying or lyophilization), use of chaperones or pH adjustment. Finding the right environment for an optimal desired state of a macromolecule with minimized degradation can be quite challenging. Therefore, due to their natural origin, DES can mimic the macromolecule’s natural environment more effectively. To sum up, this study presents the use of betaine and choline chloride-based DES as a medium for several alcohol dehydrogenases and nicotinamide cofactors long-term stabilization. In the end, we can conclude that macromolecule stabilization is a promising area of research with potential applications in various fields, including biotechnology and pharmaceuticals. |
| References: | [1] A. Paiva, R. Craveiro, I. Aroso, M. Martins, R. L. Reis and A. R. C. Duarte, ACS Sustain. Chem. Eng., 2014, 2, 1063-1071. [2] M. Panić et al., J. Chem. Technol. Biotechnol., 2021, 96, 14-30. [3] Y. Liu et al., Journal of Natural Products, 2018, 81, 679-690. [4] 1 M. A. R. Martins, S. P. Pinho and J. A. P. Coutinho, J. Solution Chem., 2019, 48, 962-982. |
| Figures: |  Graphical summary of the research DES (deep eutectic solvents) |
#313 | Investigating the Potential of Phenolic Compounds as Acceptor Substrates for Levansucrase-catalysed Transfructosylation Reactions |
|
| Presenting author: | Muriel Y K WONG MIN of MCGILL UNIVERSITY/ AFFILIATED TO INSTITUTE OF NUTRITION AND FUNCTIONAL FOODS (INAF) |
| Corresponding author: | Salwa KARBOUNE of MCGILL UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Levansucrase / Transfructosylation / Phenolic Compounds / Acceptor Specificity |
| Purpose: | Due to the promising physiological effects of levan and levan-type fructooligosaccharides, levansucrase (EC 2.4.1.10) has garnered much interest in the pharmaceutical and food industry. Levansucrase is a fructosyl-transferase that can catalyze the synthesis of complex oligosaccharides, by acquiring a fructosyl residue from a donor molecule and performing a non-Lenoir transfer to an acceptor molecule. The mechanism of action of levansucrase toward various carbohydrates has been well documented. However, in-depth studies are still needed to investigate and modulate the ability of phenolic compounds to act as acceptor substrates for levansucrase-catalyzed transfructosylation reactions. The enzymatic glycosylation of phenolic compounds is indeed seen as an attractive means to change their aqueous solubility, stability, and bioavailability. It is an interesting alternative to chemical glycosylation that requires multistep synthetic routes and results in low overall yields. This study characterized the acceptor specificity of levansucrases from Gluconobacter oxydans, Vibrio natriegens, Novosphingobium aromaticivorans and Burkholderia graminis towards various phenolic compounds using sucrose as a fructosyl donor. Their ability to catalyze the synthesis of fructosylated phenolic compounds were investigated via LCMS. The results showed that overall, levansucrase from V. natriegens had the highest catalytic efficiency and activity for the transfructosylation of phenolic compounds, while levansucrase from G. oxydans favored polymerization, oligomerization, or hydrolysis. LCMS analysis further confirmed that more than one fructosyl unit could be attached to the glycosylated phenolic compounds. The transfructosylation of Epicatechin by levansucrase from B. graminis had the most diversified products, with up to five fructosyl units transferred. This study suggests the high potential for application of levansucrase in the glycosylation of phenolic compounds. |
| References: | 1. Hill, A., Chen, L., Mariage, A., Petit, J.-L., De-Berardinis, V., Karboune, S. Catal. Sci. Technol., 9(2019), pp. 2931-2944. 2. Hill, A., Karboune, S., Narwani, T. J., & de Brevern, A. G. (2020). International journal of molecular sciences, 21(15). 3. Núñez-López, G., Herrera-González, A., Hernández, L., Amaya-Delgado, L., Sandoval, G., Gschaedler, A., Morel, S. (2019). Enzyme and Microbial Technology, 122, 19-25. |
| Figures: | 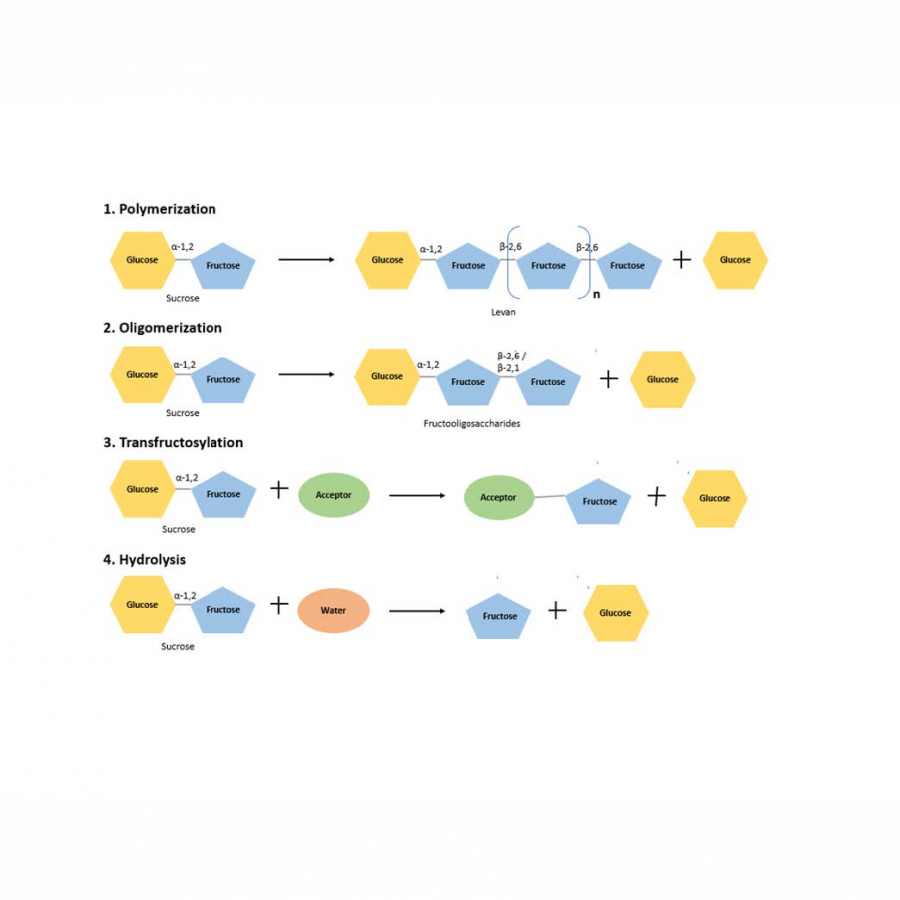 Reactions Catalyzed by Levansucrase no legend |
#319 | Bacterial peroxygenases - expanding the repertoire of oxyfunctionalization enzymes |
|
| Presenting author: | Nikola LONCAR of GECCO BIOTECH B.V. |
| Other authors: | Lur ALONSO COTCHICO of ZYMVOL BIOMODELING S.L. Davide CARRARETTO of UNIVERSITY OF PAVIA Andrea MATTEVI of UNIVERSITY OF PAVIA Marco W. FRAAIJE of UNIVERSITY OF GRONINGEN Maria Fatima LUCAS of ZYMVOL BIOMODELING S.L. |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | peroxygenase / enzyme discovery / industrial biocatalysis / melanin |
| Purpose: | Due to the extremely attractive biocatalytic potential of unspecific peroxygenases (UPOs), interest in the exploitation of UPOs for biocatalytic applications is booming. This can be seen from the number of scientific publications, patents, and biotech companies that started to offer fungal UPOs. Almost without exception, all the focus concerning biotechnological exploitation of peroxygenases is indeed on already known fungal UPOs. Yet, it has been shown that also bacteria contain peroxygenases (BUPOs), carrying out the same type of reactions. Nevertheless, these bacterial counterparts have received very little attention, while they show great promise as oxidative biocatalysts. Herein, we present a discovery of a set of BUPOs that can be easily produced and therefore it is possible to study and engineer them for diverse biocatalytic applications. |
#387 | Controllable iterative β-glucosylation from UDP-glucose by Bacillus cereus glycosyltransferase GT1: application for the synthesis of disaccharide-modified xenobiotics |
|
| Presenting author: | Jihye JUNG of INSTITUTE OF BIOTECHNOLOGY AND BIOCHEMICAL ENGINEERING |
| Corresponding author: | Bernd NIDETZKY of INSTITUTE OF BIOTECHNOLOGY AND BIOCHEMICAL ENGINEERING |
| Other authors: | Doreen SCHACHTSCHABEL of BASF SE Michael SPEITLING of BASF SE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Iterative glycosylation / Disaccharide modification / Leloir glycosyltransferases / Xenobiotics |
| Purpose: | Glycosylation in natural product metabolism and xenobiotic detoxification often leads to disaccharide-modified metabolites. The chemical synthesis of such glycosides typically separates the glycosylation steps in space and time. The option to perform the two-step glycosylation in one pot, and catalyzed by a single permissive enzyme, is interesting for a facile access to disaccharide-modified products. Here, we reveal the glycosyltransferase GT1 from Bacillus cereus (BcGT1) for iterative O-β-glucosylation from uridine 5'-diphosphate (UDP)-glucose to form a β-linked disaccharide of different metabolites, including C15-hydroxylated cinmethylin (15HCM). 15HCM is a phase I detoxification intermediate of the agricultural herbicide cinmethylin (CM), which is a benzyl ether derivative of the natural terpene 1,4-cineole. Screening studies of glycosyltransferase for β-D-mono-glucosylation of 15HCM from UDP-glucose revealed the product mixtures of the monosaccharide- and disaccharide-modified 15HCM formed from BcGT1 reaction. The disaccharide-modified products of 15HCM were determined in detailed product characterization with NMR and MS analysis. Combined with the comprehensive analysis of time courses for the enzymatic glucosylation of 15HCM, we identify thermodynamic and kinetic requirements for the selective formation of the disaccharide compared to the monosaccharide-modified 15HCM. Glycosylation reactions on methylumbelliferone and 4-nitrophenol involve reversible glycosyl transfer from and to UDP as well as UDP-glucose hydrolysis, both catalyzed by BcGT1. Collectively, this study delineates the iterative β-D-glucosylation of aglycones by BcGT1 and demonstrates applicability for the programmable one-pot synthesis of disaccharide-modified 15HCM. |
| References: | [1] J. Jung, D. Schachtschabel, M. Speitling, B. Nidetzky, J. Agric. Food Chem. 2021, 69, 14630-14642 [2] J. Jung, K. Schmoelzer, D. Schachtschabel, M. Speitling, B. Nidetzky, J. Agric. Food Chem. 2021, 69, 5491-5499 |
| Figures: | 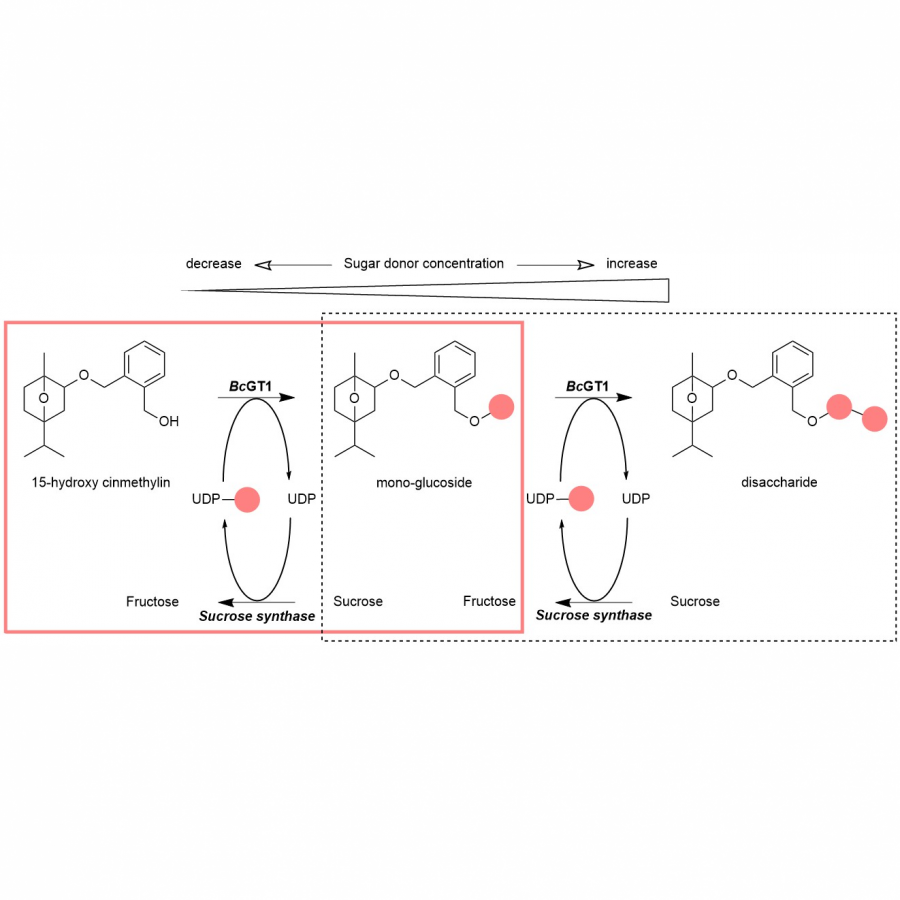 Controllable iterative glycosylation of 15-hydroxy cinmethylin by BcGT1 BcGT1 catalyzes the transfer of glucosyl
residue (red balls) from UDP-glucose to 15-hydroxy cinmethylin (15HCM), releasing 15HCM-β-D-glucoside, and to 15HCM-β-D-glucoside, forming 15HCM β-D-glucosyl-β-D-glucoside (disaccharides). |
#390 | Biosynthesis of the non-canonical amino acid piperazic acid by an enzyme cascade |
|
| Presenting author: | Dirk TISCHLER of RUHR UNIVERSITY BOCHUM |
| Other authors: | Simon SCHRÖDER of RUHR UNIVERSITY BOCHUM Artur MAIER of RUHR UNIVERSITY BOCHUM Sandy SCHMIDT of UNIVERSITY OF GRONINGEN Carolin MÜGGE of RUHR UNIVERSITY BOCHUM |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | flavoprotein monooxygenase / N-N bond formation / natural product / gene expression |
| Purpose: | Nature provides a large repertoire of non-ribosomal peptides with potent properties for various applications. The underlying bioactivity is especially interesting for the biosynthesis of drugs and related compounds. Respective (depsi)peptides are derived from proteogenic and non-canonical amino acids as well as carboxyclic acids and various combinations thereof. Here, we became intrigued by the biosynthetic machinery of some bacteria to form natural products such as metallophores and antimicrobial agents comprising N-N bonds.[1] The required precursors can be obtained from central metabolic pathways. Interestingly, some bacteria can N-hydroxylate amino acids selectively, such as L-ornithine towards N5-hydroxy-L-ornithine which can be further converted to L-piperazic acid. The latter represents one of those potent non-canonical amino acids. The Actinobacterium Kutzneria sp. 744 encodes the genetic information for the enzymes leading to L-piperazic acid, namely the flavoprotein N-hydroxylase (NMO) and piperazate synthase (PipS).[2] Especially, the latter enzyme is not well described with respect to biochemical properties nor biotechnological application. A phylogenetic investigation revealed that those enzymes can frequently be identified from Actinobacteria while only one was studied in somewhat detail up to now. This enzyme from Kutzneria sp. 744 (also designated KtzT) was reported to be a heme-protein with a narrow substrate scope. It is part of the biosynthetic machinery towards kutzneride compounds which are active against fungal and bacterial strains.[3] To date, no detailed data on enzyme production, heme-loading, enzyme kinetics, stability, structural information etc. were available. Hence, we cloned and produced the enzyme, reaching mediocre yields (0.4 mg protein per L broth with a heme loading of about 12%) in first attempts. A series of experiments to improve gene expression led to an almost 40-fold improved production of heme loaded and active catalyst. Another problem studying this enzyme was the supply of N-OH comprising substrates. This could be solved by our expertise on N-hydroxylating enzymes.[4,5] Here, we employed the NMOs from Thermocrispum agreste (TheA) to produce N5-hydroxy-L-ornithine or from Gordonia rubripertincta (GorA) to provide other N-OH molecules in situ. As this enzyme class is NADPH-dependent, we had to recycle the liberated NADP+. This was achieved with an FDH variant from Candida boidinii. All in all, we optimized the cascade and were able to use it to study KtzT and related PipS enzymes. Activity was followed by an HILIC-MS/MS approach which allowed to separate substrates and products and to detect compounds by tandem mass spectrometry. For the first time, we were able to demonstrate that the here employed NMOs can produce a series of N-hydroxylated intermediates of which some can be used by PipS. Our studies were corroborated by iterative bioinformatics approach comprising ligand docking and refinement by molecular dynamics simulations to predict heme positioning and substrate binding in KtzT. This resulted in a trustworthy model of KtzT containing a docked heme and substrate. This information was then employed to conduct mutagenesis experiments as well as to explain substrate promiscuity of this enzyme family for the first time. In conclusion, we found the family of PipS enzymes is very promising to produce various N-N containing molecules, especially in artificial enzyme cascades, where further optimization might lead to interesting future applications. |
| References: | [1] Mügge, C., Heine, T., Baraibar, A.G., van Berkel, W.J.H., Paul, C.E., Tischler, D., Flavin-dependent N-hydroxylating enzymes: distribution and application. Appl. Microbiol. Biotechnol. 2020, 104, 6481-6499. [2] Du, Y.-L., He, H.-Y., Higgins, M.A., Ryan, K. S., A heme-dependent enzyme forms the nitrogen-nitrogen bond in piperazate. Nat. Chem. Biol. 2017, 13, 836-838 [3] Fujimori, D.G., Hrvatin, S., Neumann, C.S., Strieker, M., Marahiel, M.A., Walsh, C.T., Cloning and characterization of the biosynthetic gene cluster for kutznerides. Proc. Natl. Acad. Sci. USA 2007, 104, 16498-16503. 262:501-504. [4] Heine, T., Mehnert, M., Schwabe, R., Tischler, D., Thermochelin, a hydroxamate siderophore from Thermocrispum agreste DSM 44070. Solid State Phenom. 2017, 262, 501-504. [5] Esuola, C.O., Babalola, O.O., Heine, T., Schwabe, R., Schlömann, M., Tischler, D., Identification and characterization of a FAD-dependent putrescine N-hydroxylase (GorA) from Gordonia rubripertincta CWB2. J. Mol. Catal. B Enzym. 2016, 134, 378-389. |
| Figures: | 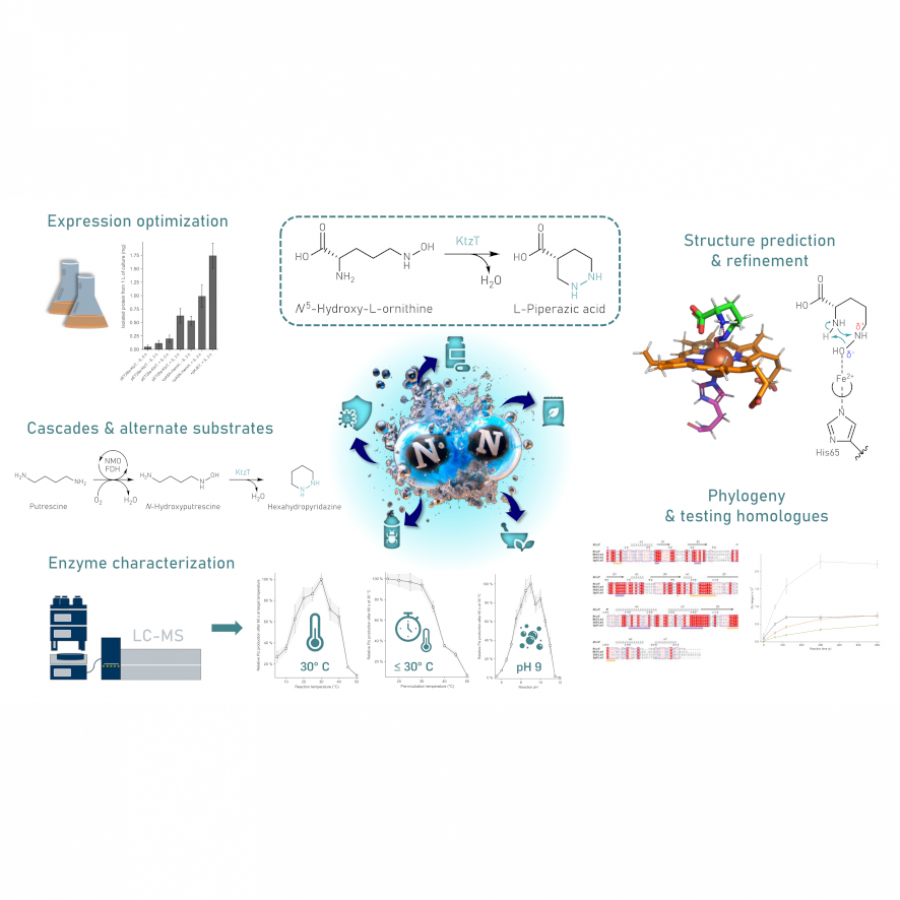 KtzT A biochemical study of KtzT including expression optimization, assay development, activity determinants and structural probing. |
#393 | Glycoside-phosphorylases: in vitro and in cellulo implementation for the production of valuable glycosides |
|
| Presenting author: | Maxant VIVIER of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Corresponding author: | Gabrielle POTOCKI-VERONESE of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Other authors: | Julien DURAND of SWEETECH Pietro TEDESCO of TOULOUSE BIOTECHNOLOGY INSTITUTE Fabien LETISSE of INSTITUT DE PHARMACOLOGIE ET DE BIOLOGIE STRUCTURALE Yannick MALBERT of SWEETECH Gabrielle POTOCKI-VERONESE of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Glycoside Phosphorylases / Mannosides / Metabolic engineering / |
| Purpose: | Glycoside Phosphorylases (GPs) are reversible Carbohydrate Active Enzymes enzymes able to degrade glycosides thanks to inorganic phosphate (phosphorolysis) or to synthesize glycosides thanks to sugar-phosphates and adequate acceptors (reverse phosphorolysis) [1]. Without the need of costly activated-sugars, they can be used to produce valuable glycosides. Such biocatalysts represent a solid alternative to natural extraction and chemical synthesis, which suffer from low yield, lack of structural purity, high energy-use and sustainability issues [2]. However, the reverse phosphorolysis reaction is difficult to exploit at industrial scale because of the high cost and limited commercial availability of sugar-phosphates, as well as unfavourable reaction equilibria. Those limitations can be overcome by implementing GPs in engineered microbial cells. The cells can provide substrates for reverse phosphorolysis, and promote GP synthesis by displacing the reaction equilibrium with secretion of the products of interest. For that, the microbial chassis must be able to accumulate cytoplasmic sugar-phosphates, which can be achieved by metabolic engineering. The proof of concept of this technology was made in an engineered Escherichia coli K12 strain [3] that ultimately was able to synthesize β-mannosides (β-MOS) of pharmaceutical interest once transformed with mannoside phosphorylases from the GH130 family. Notably, mannosides linked with β-1,2 linkages (β-1,2-MOS) are antigenic patterns of the pathogenic yeast Candida [4]. They can be used to prevent and cure candidiasis, an infectious disease affecting more than 300 million patients per year, and possibly lethal in its most serious form [5]. Implemented in engineered microbial cells or used in vitro, GPs are powerful tools to access a wide range of glycosides previously unreachable, and will undoubtedly be significant players in the coming years. |
| References: | [1] Li, A. et al. Discovery and Biotechnological Exploitation of Glycoside-Phosphorylases. International Journal of Molecular Sciences 23, 3043 (2022) [2] Shivatare, S. S. & Wong, C.-H. Synthetic Carbohydrate Chemistry and Translational Medicine. J. Org. Chem. 85, 15780-15800 (2020) [3] Durand, J., Letisse, F., Tedesco, P. & Veronese, G. Bacterial Strains and Method for Producing Oligosaccharides. (2021) [4] Shibata, N., Kobayashi, H. & Suzuki, S. Immunochemistry of pathogenic yeast, Candida species, focusing on mannan. Proceedings of the Japan Academy, Series B 88, 250-265 (2012) [5] Lipinski, T. et al. A β-mannan trisaccharide conjugate vaccine aids clearance of Candida albicans in immunocompromised rabbits. Vaccine 30, 6263-6269 (2012). |
#402 | Primary determinants for substrate preference and regioselectivity of Burkholderia thailandensis lipoxygenase |
|
| Presenting author: | RUTH CHRISNASARI of WAGENINGEN UNIVERSITY & RESEARCH |
| Other authors: | Marie HENNEBELLE of WAGENINGEN UNIVERSITY & RESEARCH Willem J. H VAN BERKEL of WAGENINGEN UNIVERSITY & RESEARCH Jean-Paul VINCKEN of WAGENINGEN UNIVERSITY & RESEARCH Tom A. EWING of WAGENINGEN UNIVERSITY & RESEARCH |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lipoxygenase / regioselectivity / substrate preference / mutagenesis |
| Purpose: | Lipoxygenases (LOXs) are enzymes that catalyze regioselective dioxygenation of polyunsaturated fatty acids (PUFAs) into fatty acid hydroperoxides (FAHPs). The regioselective dioxygenation of PUFAs opens up a number of interesting applications in the food and chemical industries as the position of the hydroperoxide group in FAHPs will determine which products, with which functionalities can be derived from them. The regioselectivity of LOXs is regulated by certain structural features, including the depth of the substrate-binding pocket [1] and the position of the migration channel that shuttles molecular oxygen to the active site [2]. LOXs produced by bacteria have gained more attention in the past few years, because they have been reported to be active towards a broad range of PUFAs [3]. However, their specific activity towards each of these PUFAs can vary. Currently, there is no report on the structural features that determine the substrate preference of bacterial LOXs. In this study, we focused on understanding the structural determinants of substrate specificity in Burkholderia thailandensis LOX which has shown a high activity at pH 6 at 20-30 °C. This enzyme preferentially used ω-6 PUFAs as substrates and catalyzed the dioxygenation regioselectivity at ω-5 position. Mutations were performed on specific residues, that were proposed to play a role in the substrate preference and regioselectivity, i.e. Leu445, Phe446 and Ala431. Mutation Leu445Ala changed the substrate specificity of the enzyme from ω-6 to ω-3 PUFAs, while mutations Phe446Val and Ala431Gly enabled the enzyme to utilize both ω-6 and ω-3 PUFAs equally. Mutation Leu445Ala changed the regioselectivity of the enzyme from ω-5 to ω-2 carbon atom on ω-3 PUFAs as the substrate, while mutations Phe446Val and Ala431Gly made the enzyme less regioselective. These findings provide insights into specific residues involved in the substrate preference and regioselectivity of Burkholderia thailandensis LOX. Manipulating them may allow the synthesis of different FAHPs and therefore stimulate the exploitation of bacterial LOXs as a green alternative in a wide range of applications. |
| References: | [1] R. Vogel et al. J. Biol Chem. 2010, 285(8), 5369-5376. [2] M. E. Newcomer and A. R. Brash. Protein Sci. 2015, 24(3), 298-309. [3] R. Chrisnasari, et al. Biotechnol Adv. 2022, 61, 108046. |
| Figures: | 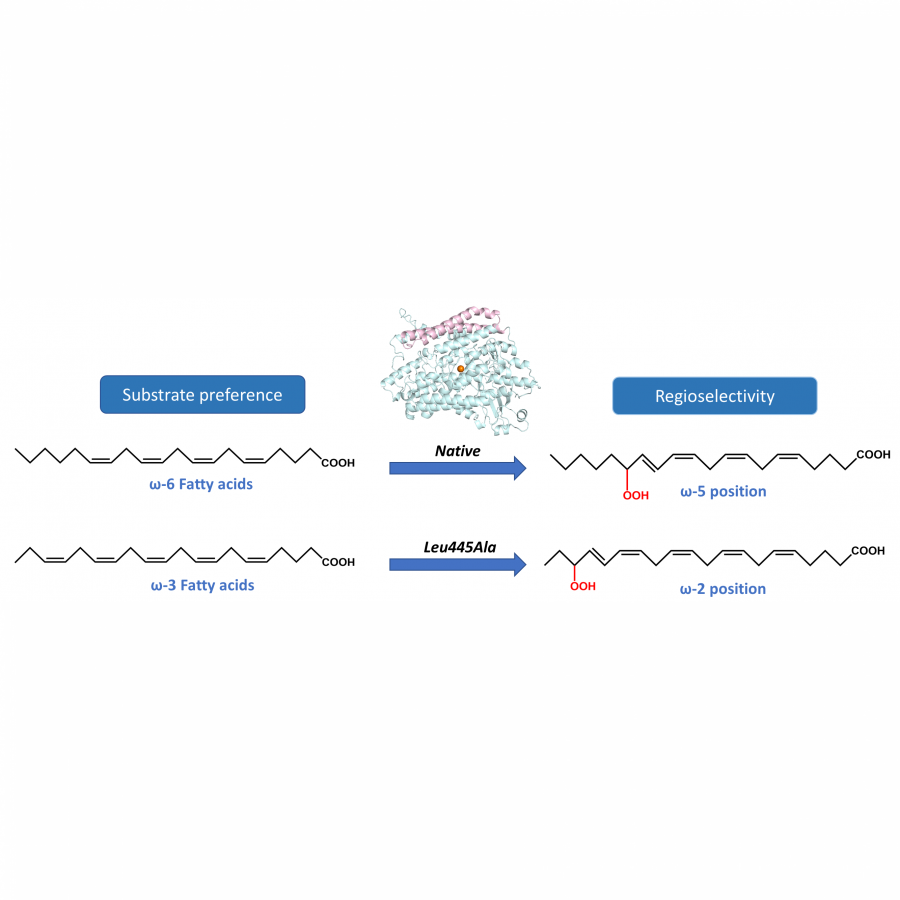 The effect of mutation on substrate preference and regioselectivity of B. thailandensis LOX Mutation Leu445Ala changes the substrate preference of the enzyme from ω-6 to ω-3 PUFAs and changes the dioxygenation regioselectivity from ω-5 to ω-2 carbon atom on ω-3 PUFAs. |
#403 | Expanding the one-carbon chemistry of thiamine diphosphate-dependent enzymes |
|
| Presenting author: | Simon BURGENER of UNIVERSITY OF BASEL |
| Other authors: | Maren NATTERMANN of MAX PLANCK INSTITUTE FOR TERRESTRIAL MICROBIOLOGY Tobias ERB of MAX PLANCK INSTITUTE FOR TERRESTRIAL MICROBIOLOGY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | One-carbon chemistry / Thiamine diphosphate-dependent enzymes / Enzyme engineering / Synthetic biochemistry |
| Purpose: | The synthesis of complex molecules from simple, renewable carbon units (and eventually from CO2) is essential to achieve a circular economy. A significant challenge in this regard is the direct condensation of one-carbon molecules, for which only highly complex enzymes are known so far. Thiamine diphosphate-dependent (ThDP) enzymes offer a promising solution because they are versatile C-C bond forming catalysts and can be readily engineered. In this work, we explored and expanded the biocatalytic potential of the oxalyl-CoA decarboxylase (OXC)/2-hydroxyacyl-CoA lyase (HACL) superfamily, which catalyzes the shortening of acyl-CoA thioester substrates through the release of the one-carbon unit formyl-CoA. We demonstrated that members of this superfamily can operate in reverse to extend various aldehydes with one-carbon units, yielding the corresponding 2-hydroxyacyl-CoA thioesters. We improved the catalytic properties of OXC by rational enzyme engineering and combined it with two newly described enzymes to create an enzymatic cascade that enabled the continuous conversion of oxalate and aromatic aldehydes into valuable (S)-α-hydroxy acids with up to 99% enantiomeric excess. Furthermore, we employed directed evolution to convert OXC into a glycolyl-CoA synthase (GCS) that condenses the two one-carbon units formyl-CoA and formaldehyde. The quadruple variant MeOXC4 showed a 100,000-fold switch between OXC and GCS activities, a 200-fold increase in GCS activity compared to the wild type, and formaldehyde affinity comparable to natural formaldehyde-converting enzymes. Our work highlights the potential of ThDP-dependent enzymes in biocatalytic one-carbon conversion and offers a promising strategy for the sustainable production of valuable products from renewable feedstocks. |
| References: | [1] Burgener, S., Cortina, N.S., Erb, T.J., Angew. Chem. Int. Ed. 2020, 59, 5526-5530. https://doi.org/10.1002/anie.201915155 [2] Nattermann, M., Burgener, S., Pfister, P., Chou, A., Schulz, L., Lee, S.H., Paczia, N., Zarzycki, J., Gonzalez, R., Erb, T.J., ACS Catal. 2021, 11, 5396-5404. https://doi.org/10.1021/acscatal.1c01237 |
| Figures: |  Oxalyl-CoA Decarboxylase Enables Nucleophilic One-Carbon Extension of Aldehydes to Chiral 2-Hydroxy Acids . 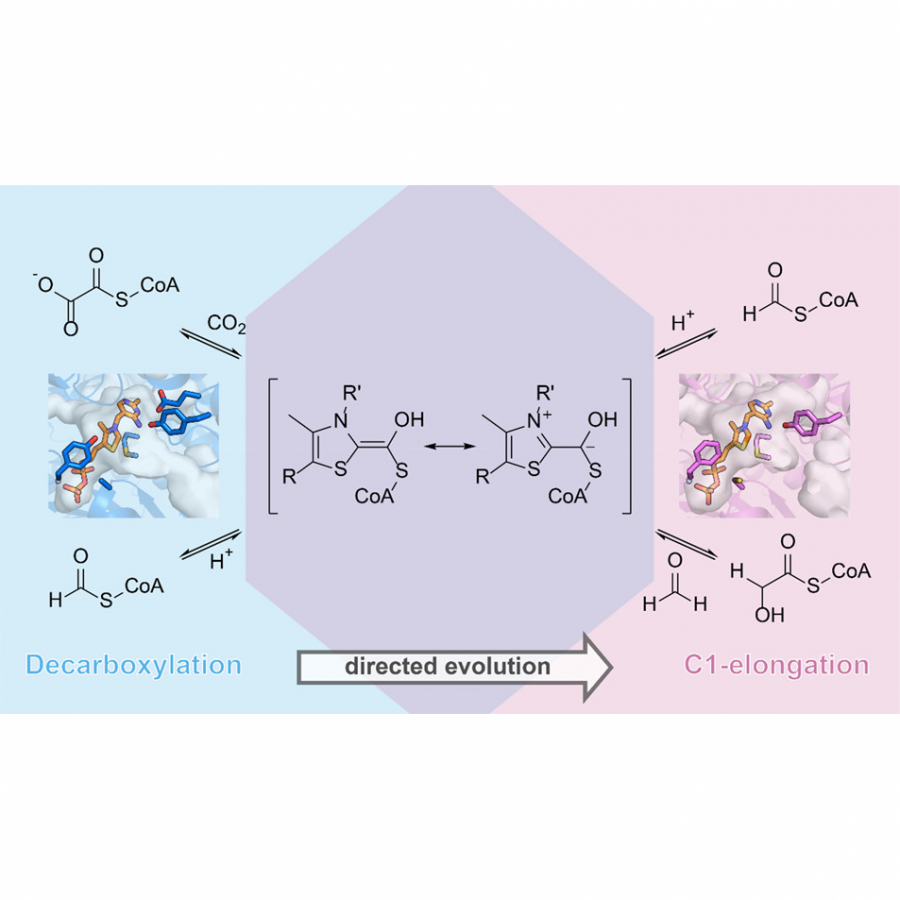 Engineering a Highly Efficient Carboligase for Synthetic One-Carbon Metabolism . |
#406 | EziG®: a universal enzyme immobilisation matrix enabling access to novel chemicals and process solutions |
|
| Presenting author: | Ashley MATTEY of ENGINZYME |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme immobilisation / Continuous processes / Industrial biocatalysis / |
| Purpose: | Immobilised enzymes are essential tools for industrial biocatalysis, combining the selectivity of enzymes with the operational stability and versatility of heterogeneous catalysts. However, the choice of immobilisation support can greatly affect the performance and stability of such enzymes, and finding the optimal material for immobilisation can be a time-consuming and costly process. A fundamental component of our platform is EziG®, a universal immobilisation matrix which offers a robust solution for a wide range of biocatalytic applications. EziG® is designed to provide superior enzyme loading, activity, and stability, and is compatible with a wide range of enzymes and substrates. Using EziG®, we have successfully produced a variety of novel chemicals and process solutions, including pharmaceutical intermediates, flavors, and fragrances. Here, we will present data on the optimisation and scale-up of EziG®-based biocatalytic processes, as well as a case study demonstrating the versatility and efficacy of the platform. |
#411 | Coupled molecular dynamics mediates interaction between long-range mutations and its application in enzyme engineering |
|
| Presenting author: | Haoran YU of INSTITUTE OF BIOENGINEERING, COLLEGE OF CHEMICAL AND BIOLOGICAL ENGINEERING, ZHEJIANG UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Dynamics correlation / Epistasis / Long-range mutations / Stability-activity tradeoff |
| Purpose: | When several substitutions are made in a single protein, the mutations can potentially interact in a non-additive manner, resulting in epistatic effects, which can hamper protein engineering strategies to improve enzyme properteis. We examined the role of protein dynamics in mediating epistasis between pairs of mutations for E. coli transketolase (TK) [1]. Epistasis was determined for conformational protein stability, and also for kinetic inactivation by heat-induced aggregation, and observed between both neighbouring and distant mutations. Molecular dynamics simulations and a pairwise cross-correlation analysis revealed how mutations influence their dynamics both locally, and also in specific regions distant in the structure. This effect was found to mediate epistatic interactions between distant mutations, and was subsequently exploited to improve the stability of a TK variant 3M [2] and the activity of 2,3-butanediol dehydrogenase from Corynebacterium glutamicum (CgBDH) [3]. The TK 3M variant was evolved to accept novel aromatic substrates, but suffered a trade-off in stability through a loss in unfolding cooperativity. Molecular dynamics simulations revealed increased flexibility in several interconnected active-site regions, that also form part of the dimer interface (Figure 1). Mutating the newly flexible active-site residues to regain stability risked losing the new activity. We therefore targeted stabilising mutations to residues outside of the active site, whose dynamics were correlated with the newly flexible active-site residues. This re-established the WT-level of stability and unfolding cooperativity, giving a 10.8-fold improved half-life at 55 °C (Figure 1), and increased Tm and Tagg by 3 °C and 4.3 °C, respectively. Molecular dynamics simulations confirmed that the mutations rigidified the active-site via the correlated network [2]. CgBDH is a homotetramer with its last amino acid residue Asn258 converging at the center of the tetramer. The last amino acid is located distal from the active center but in the hydrogen bond network involved with active sites. Specifically, Asn258 is located 14 Å away from the active sites Lys158 and Tyr154, but forms a hydrogen bond with Arg162, which then has a connection with the two catalytic residues through two hydrogen bonds (Figure 2). We hence assumed that introduction of interchain disulfide bonds by mutation N258C might improve the enzyme stability and impact the enzyme activity. In the results, the mutant showed a 14.8-fold improved half-life, a 7.9-fold improved catalytic efficiency (kcat/Km) toward diacetyl. MD simulations confirmed that a dynamics cross correlation network involved with the catalytic sites was reconstructed in the variant and the dynamics change caused by the distal disulfide bond was propagated through the interactions network (Figure 2). This improved the enzyme stability and activity by decreasing the flexibility and locking more “reactive” pose, respectively [3]. This work provides new insights into the mechanism of the interaction between long-range mutations and point outs the importance of long-range mutations in protein engineering. |
| References: | [1] Yu H., Dalby P. A.. Coupled molecular dynamics mediate long-and short-range epistasis between mutations that affect stability and aggregation kinetics. Proceedings of the National Academy of Sciences, 2018, 115(47): E11043-E11052. [2] Yu H., Dalby. P. A.. Exploiting correlated molecular-dynamics networks to counteract enzyme activity-stability trade-off. Proceedings of the National Academy of Sciences, 2018, 115(52): E12192-E12200. [3] Pu Z., Yang L., Yu H. Reconstructing dynamics correlation network to simultaneously improve enzyme activity and stability of 2,3-butanediol dehydrogenase by design of distal interchain disulfide bonds. Submitted |
| Figures: | 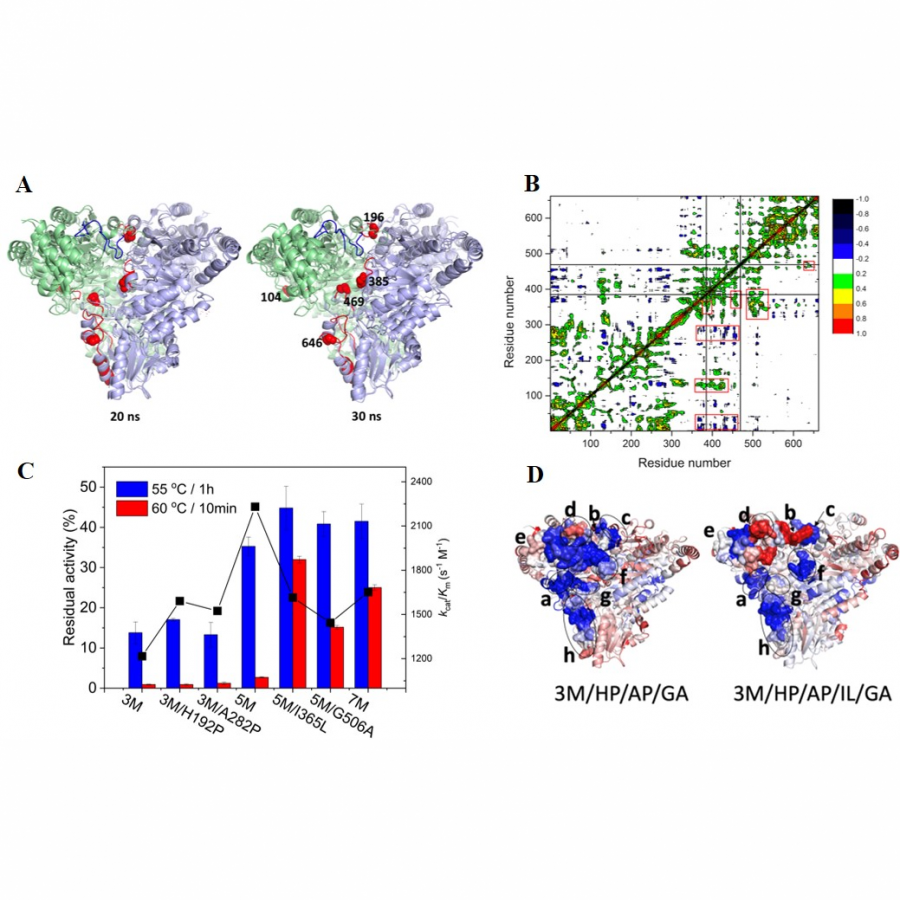 Long-range mutations counteract the activity-stability trade-off of TK 3M A. The TK 3M mutant shows a higher flexibility in the active center. B. Dynamics correlation analysis to identify the long=range mutation sites. C. The mutants designed improved the stability of TK 3M without compromise of activity. D. Long-range mutants 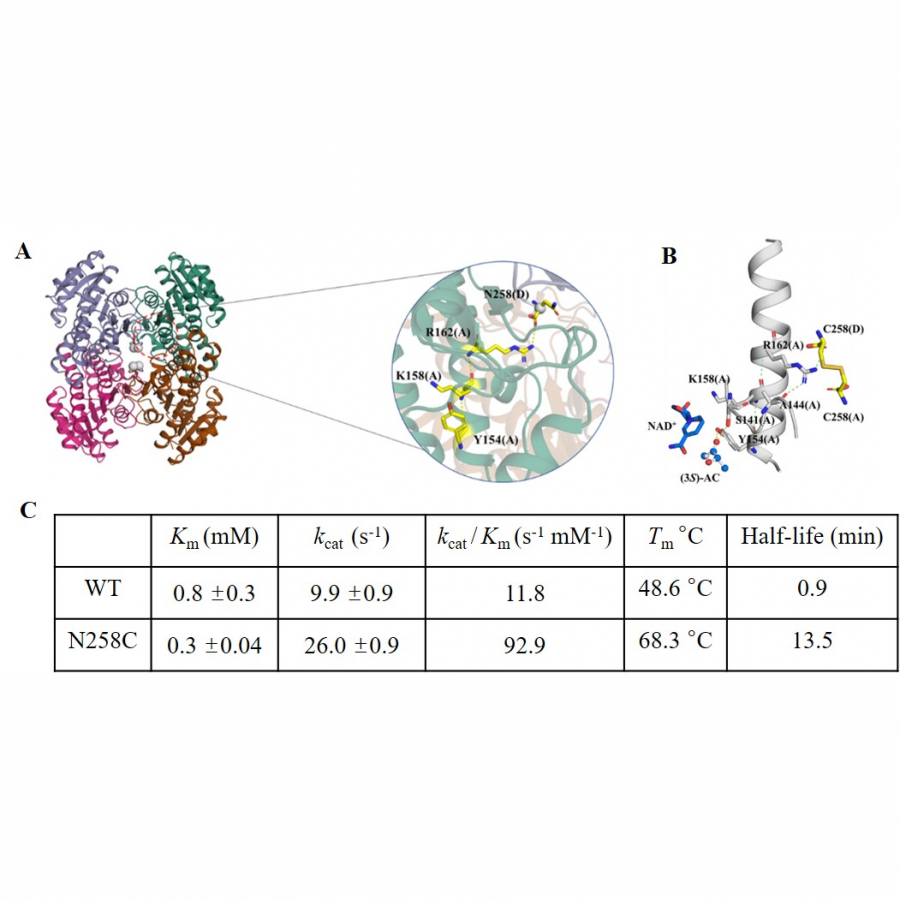 Long-range mutant at C-terminal of CgBDH simultaneously improved the enzyme stability and activity A, The C-terminal residue of CgBDH converges at the center of tetramer. B, N258C mutant reconstructed an interaction network involved with active sites. C, Characteristics of wild-type and mutant CgBDH |
#413 | Rutinosidase and other diglycosidases: Rising stars in biotechnology |
|
| Presenting author: | Vladimír KREN of INSTITUTE OF MICROBIOLOGY, CZECH ACADEMY OF SCIENC |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme synhesis / Glycosidases / Rutinosidase / Anti-aging agents |
| Purpose: | Vladimír KREN, Katerina BRODSKY, Michal KOTIK, Pavla BOJAROVA Institute of Microbiology of the Czech Academy of Sciences, Laboratory of Biotransformation, Videnska 1083, CZ-142 00, Prague 4, Czech Republic; kren@biomed.cas.cz Diglycosidases are glycosidases catalyzing the cleavage of entire disaccharide moieties from the aglycone. Rutinosidases, main diglycosidase representatives, cleave rutinose (α-L-Rha-(1-6)-β-D-Glc) from rutin or other rutinosides (Fig. 1A). Some diglycosidases can be classified as monoglucosidases with extended substrate specificity. They also have distinct synthetic (transglycosylating) abilities. Rutinosidase from A. niger [1] and A. oryzae (GH5-23) can glycosylate various acceptors, including phenols, in a good yield using priceworthy rutin as a glycosyl donor. Surprisingly, they are even able to glycosylate species such as inorganic azide to form β-rutinosyl azide [2] or carboxylic acids forming (anomeric) glycosyl esters [3], which is a unique property in the glycosidase family. The variant of A. niger rutinosidase mutated at the catalytic nucleophile residue E319A is capable of generating α-rutinosyl azide [2]. It was found that rutinosidase is able to accept quercetin 3-β-glucopyranoside as a substrate and therefore it is also able to transfer a β-glucosyl moiety [4]. Thus, this enzyme has a dual glycosylation activity, generating either rutinosides or glucopyranosides. Its broad substrate specificity has also been demonstrated in the enzymatic cleavage of various 6"-acylated quercetin-3-O-β-glucopyranosides (Fig. 1B). Rhamnose-containing compounds (such as rutinose) are attracting attention due to their anti-cancer activity and as skin anti-aging agents in dermatology [3]. Their easy availability through the action of rutinosidase opens a whole new avenue in cancer therapy, biomedicine, dermatology, and other fields. Acknowledgment: We acknowledge the support by the Czech Science Foundation project No. 22-00197K and by the COST Action CA18132. |
| References: | References [1] P. Pachl, J. Kapešová, J. Brynda, L. Biedermannová, H. Pelantová, P. Bojarová, V. Kren, P. Rezácová, M. Kotik (2020) FEBS J. (287), 3315-3327 [2] M. Kotik, K. Brodsky, P. Halada, H. Javurková, H. Pelantová, D. Konvalinková, P. Bojarová, V. Kren (2021), Cat. Commun. (149), 106193 [3] I. Bassanini, J. Kapesová, L. Petrásková, H. Pelantová, K. Markosová, M. Rebros, K. Valentová, M. Kotik, K. Kánová, P. Bojarová, J. Cvacka, L. Turková, E. E. Ferrandi, I. Bayout, S. Riva, V. Kren (2019) Adv. Synth. Catal. (361), 2627-2637 [4] K. Brodsky, M. Kutý, H. Pelantová, J. Cvacka, M. Rebros, M. Kotik, I. Kutá Smatanová, V. Kren, P. Bojarová (2020) Int. J. Mol. Sci. (21), 5671 [5] R. Novotná, D. Skarupová, J. Hanyk, J. Ulrichová, V. Kren, P. Bojarová, K. Brodsky, J. Vostálová, J. Franková (2023) Molecules (28), 1728 |
| Figures: | 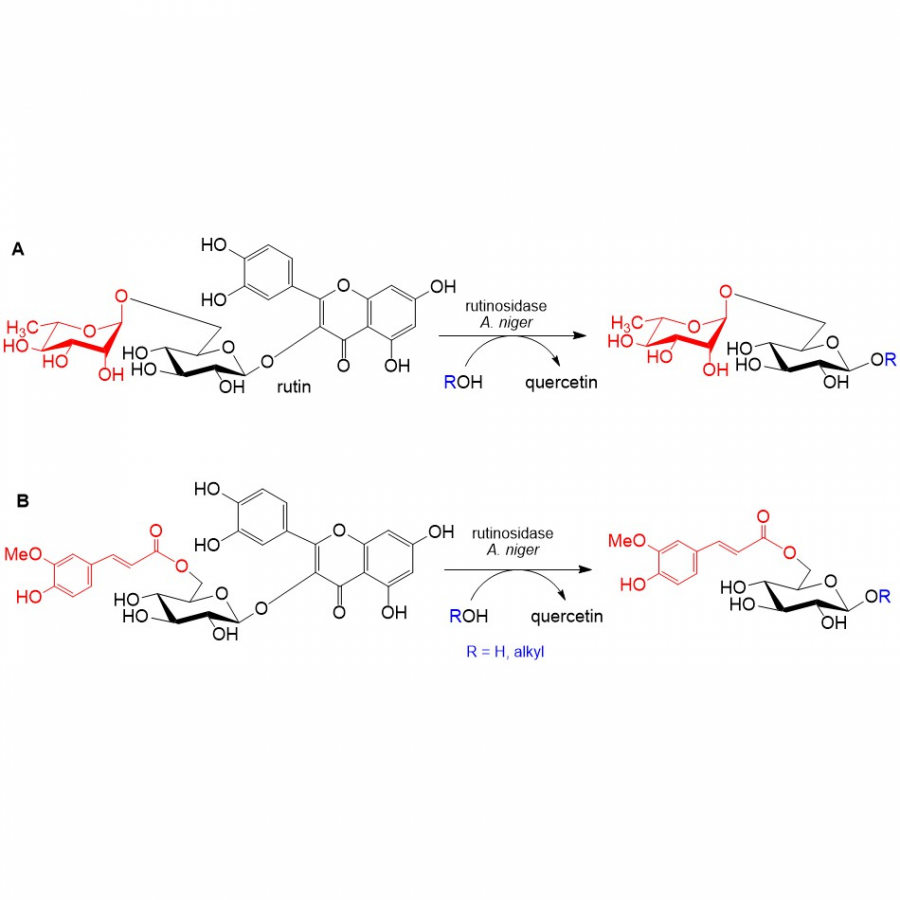 Figure 1. A – Hydrolysis or transglycosylation catalyzed by rutinosidase using the natural substrate rutin; B – rutinose acting upon a non-natural substrate – 6”-acylated (feruloyl) quercetin 3-O-β-glucopyranoside, demonstrating its broad substrate specificity. |
#418 | Exploring diastereoselectivity mechanism of L-threonine aldolase |
|
| Presenting author: | WU Jianping of ZHEJIANG UNIVERSITY |
| Corresponding author: | Wenlong ZHENG of ZHEJIANG UNIVERSITY |
| Other authors: | Jianping WU of ZHEJIANG UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | L-threonine aldolase / directed evolution / diastereoselectivity / protein engineering |
| Purpose: | L-Threonine aldolase (LTA), a PLP-dependent enzyme, is an attractive tool in organic chemistry for catalyzing the asymmetric formation of β-hydroxy-α-amino acids with two chiral centers from aldehyde and glycine. β-Hydroxyl-α-amino acids are widely used in the fields of medicine, food and agriculture as a kind of chiral building blocks. The wild enzyme has a strict selectivity for Cα of β-hydroxy-α-amino acids but a moderate selectivity for Cβ, limiting its wide application in stereospecific carbon-carbon bond synthesis. In this work, diastereoselectivity mechanism of LTA was explored using molecular dynamics simulations. And then, the diastereoselectivity of CpLTA from Cellulosilyticum sp was engineered based on the insights. Guided by the molecular dynamics simulations, “path hypothesis” and “Prelog rule” were proposed to elucidate diastereoselectivity mechanism of LTA. We assumed that the active pocket of LTA has two substrate access paths named syn path and anti path. L-syn configuration products were formed by the substrate aldehyde entering the active center from syn path, as the electron of Cα anion of PLP-Gly (quinonoid intermediate form) transferred to carbonyl carbon atom of aldehyde from si-face. On the contrary, L-anti configuration products were formed by the substrate aldehyde entering the active center from anti path, as the electron of Cα anion transferred to carbonyl carbon atom of aldehyde from re-face. Furthermore, with CpLTA as an object, a mutability landscape was first constructed by performing saturation mutagenesis at substrate access tunnel amino acids. CAST/ISM strategy was then performed to tune diastereoselectivity. As a result, diastereoselectivity of mutant H305L/Y8H/V143R was improved from 37.2%syn to 99.4%syn. Besides, diastereoselectivity of mutant H305Y/Y8I/W307E was inverted to 97.2%anti. The study would be useful to expand LTA applications and guide the engineering of other C-C bond formation enzymes. The work has been published online in Angew. Chem. Int. Ed. (doi.org/10.1002/anie.202213855) |
| References: | Wenlong Zheng; Zhongji Pu; Lanxin Xiao; Gang Xu; Lirong Yang; Haoran Yu; Jianping Wu ; Mutability-Landscape-Guided Engineering of l-Threonine Aldolase Revealing the Prelog Rule in Mediating Diastereoselectivity of C-C Bond Formation, Angewandte Chemie International Edition, 2022, 62(2): e202213855 |
| Figures: | 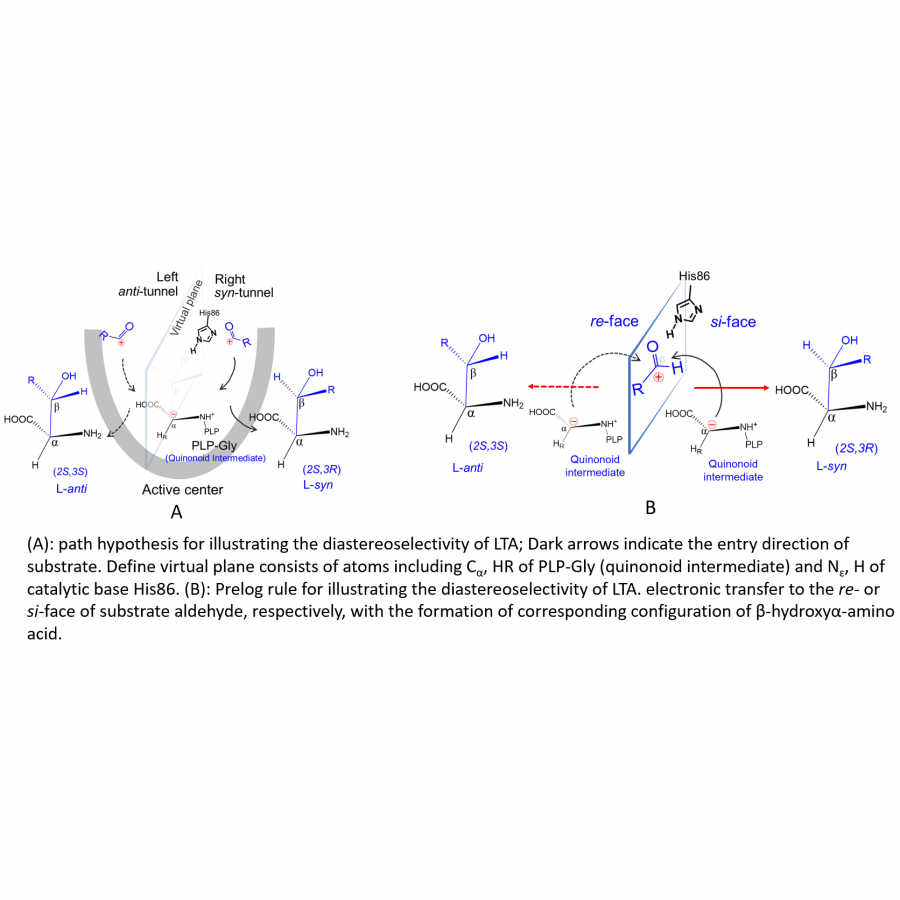 diastereoselectivity mechanism of LTA (A): path hypothesis for illustrating the diastereoselectivity of LTA; Dark arrows indicate the entry direction of substrate. Define virtual plane consists of atoms including Cα, HR of PLP-Gly (quinonoid intermediate) and Nɛ, H of catalytic base His86. (B  Directed evolution (A) Models of CpLTA with the docked ligands PLP-Gly.(B): The evolutionary process of CpLTA in diastereoselectivity. syn: mutant with Lsyn-MTPS preference. anti: mutant with L-anti-MTPS preference. conv: conversion rate.
|
#421 | Overcoming enzyme inactivation in biocatalysis by co-entrapment in ionic liquid gel materials |
|
| Presenting author: | Jose Angel PEREZ TOMAS of QUEEN'S UNIVERSITY BELFAST |
| Other authors: | Andrew MARR of QUEEN'S UNIVERSITY BELFAST Patricia MARR of QUEEN'S UNIVERSITY BELFAST Chris ALLEN of QUEEN'S UNIVERSITY BELFAST Stefan MIX of ALMAC GROUP Gareth BROWN of ALMAC GROUP |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ionic liquids / Entrapment / Polyacrylamide / Lipase |
| Purpose: | A roadmap of biochemical routes lies at the heart of every living organism, enabled and maintained by nature’s signature catalysts, enzymes. Enzymes embody the highest ideals of green chemical catalysts: They are derived from renewable sources and are biodegradable and biocompatible [1]. Although the use of these biocatalysts in industrial processes is not new, their widespread implementation can be expensive and thus severely limited [2]. Commercially available epoxy resins can be used to reduce costs by covalently immobilising enzymes onto their surface, enabling their recovery and reuse. However, these materials do not protect the enzyme from inhibitors present in the reaction mixture and could disrupt the protein structure as a result of the physical linkage to the support [3]. In this regard, entrapment methods offer a more sophisticated approach, creating a porous network that physically entraps the enzyme along with a solvent of choice. Taking the well-studied polyacrylamide hydrogels as a starting point, the functional groups of the polymeric matrix can be altered and tuned through monomer co-polymerisation. Additionally, the use of ionic liquids as co-solvents in the entrapment process renders polymeric materials with superior mechanical properties [4], an important consideration in industrial operations. Ionic liquids are highly tuneable, non-volatile solvents that have been used in the past to assist biocatalysis in various ways [5]. Flexibility in both monomer and ionic liquid design provides a vast toolkit for microenvironment engineering. The ideal microenvironment would have minimum leach into the reaction solvent, would preserve enzymatic activity and would attract substrates into the matrix while allowing products to diffuse back into the bulk phase. Entrapment offers the benefits of biphasic reaction systems while keeping the biocatalyst in a conveniently retrievable solid form. At the end of its useful life, the ionic liquid can be extracted and re-used in further material syntheses, while the polymeric material can be degraded and disposed of [6]. In this work, co-entrapment of enzymes and green ionic liquids has been employed to assist in biocatalytic reactions, such as transesterification and hydrolysis; and the effects of altering the polymer matrix, and adding ionic liquids, assessed. |
| References: | [1]. Sheldon, R. A. Biocatalysis in ionic liquids: State-of-the-union. Green Chem. 2021, 23, 8406-8427. [2]. Sheldon, R. A. & van Pelt, S. Enzyme immobilisation in biocatalysis: Why, what and how. Chem. Soc. Rev. 2013, 42, 6223-6235. [3]. Imam. H. T., Marr, P. C. & Marr, A. C. Enzyme entrapment, biocatalyst immobilization without covalent attachment. Green Chem. 2021, 23, 4980. [4]. Wang, M., Zhang, P., Shamsi, M., Thelen, J. L., Qian, W., Truong, V. K., Ma, J., Hu, J. & Dickey, M. D. Tough and stretchable ionogels by in-situ phase separation. Nat. Mater. 2022, 21, 359-365. [5]. Imam, H. T., Krashnan, V., Rebros, M. & Marr, A. C. Applications of ionic liquids in whole-cell and isolated enzyme biocatalysis. Molecules 2021, 26, 4701. [6]. Vijayalakshmi, S.P., Raichur, A. & Madras, G. Thermal degradation of poly(ethylene oxide) and polyacrylamide with ascorbic acid. J. Appl. Polym. Sci. 2006, 101, 3067-3072. |
| Figures: | 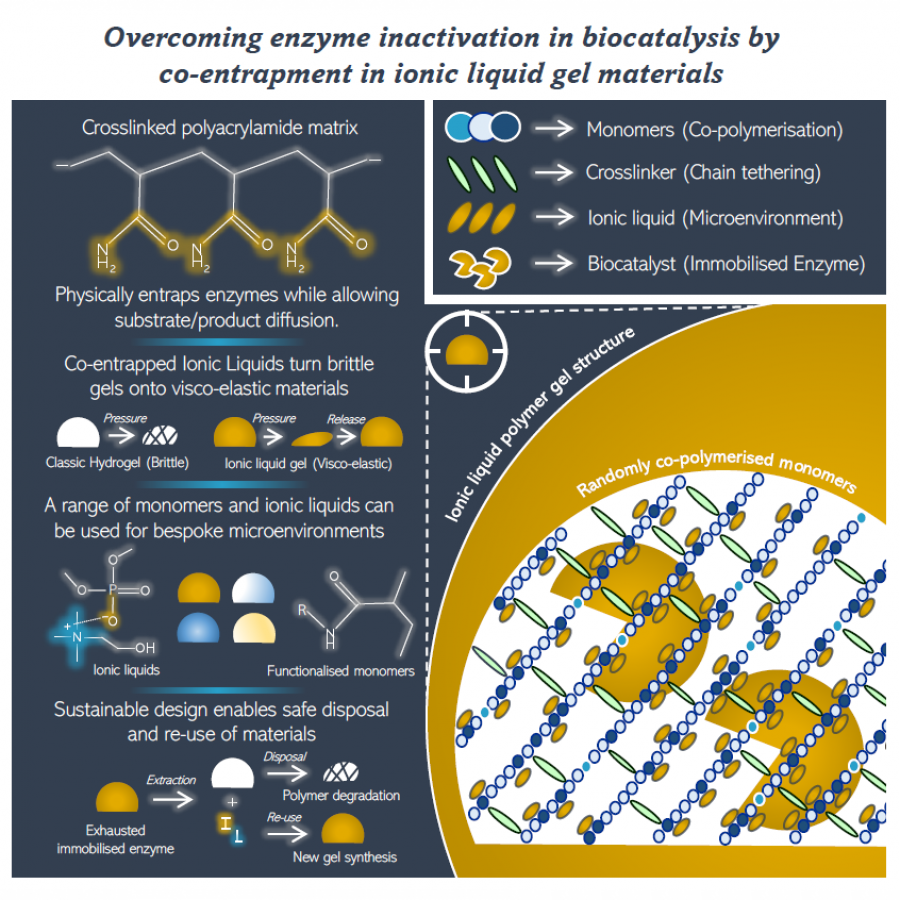 Graphical abstract Visual description of ionic liquid polymer gel materials for enzyme entrapment: Structure, properties, versatility and degradation. |
#425 | Computational-aided engineering of a selective unspecific peroxygenase toward enantiodivergent beta-ionone hydroxylation |
|
| Presenting author: | Judith MUENCH of MARTIN-LUTHER UNIVERSITY HALLE-WITTENBERG |
| Other authors: | Jordi SOLER of UNIVERSITAT DE GIRONA Marc GARCIA-BORRAS of UNIVERSITAT DE GIRONA Martin J. WEISSENBORN of MARTIN LUTHER-UNIVERSITY HALLE-WITTENBERG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | unspecific peroxygenase / enantioselectivity / directed evolution / computational-guided protein engineering |
| Purpose: | Unspecific peroxygenases (UPOs) are fungal, secreted, heme containing enzymes. They perform oxyfunctionalization reactions within a broad substrates scope utilizing H2O2 without additional reductive equivalents or electron transfer chains.[1] The development of these enzymes for industrial applications has been a focus of research over the last decade, with engineering efforts targeting heterologous expression, activity, stability, and improvements in chemo- and regioselectivity.[2, 3]However, the targeted engineering of enantioselectivity for specific substrates with poor starting enantioselectivity remained a missing integral piece until now. We pursued this endeavor using the terpene β-ionone as model substrate. Ionones are valuable substrates used in the fragrance industry and in the synthesis of carotenoids and Vitamin A.[4, 5] The conversion of α- and β-ionone has already been shown with several UPOs, leading to a diverse range of hydroxylation and epoxidation products.[6] It also has been pursued using various P450s.[7-9] P450 engineering efforts led to a 280-fold increase in product formation rate toward α- and β-ionone hydroxylations. Enhancing the enantioselectivity, however, has proved challenging.[7] Enantioselective 4 hydroxy-β-ionone formation has been achieved solely through enzymatic kinetic resolution[10] and by recombinantly in T. ni cells expressed CYP2B6.[9] We engineered MthUPO derived from Myceliophthora thermophila to enantioselectively access C4 hydroxylated stereoisomers of β-ionone. In this study, a computational-aided engineering approach based on a combination of DFT model calculations and MD simulations has been applied. These simulations were used to characterize near-attack conformations of the selective hydroxylation which revealed relevant binding modes of the model substrate β-ionone (Figure 1). The identification of the relevant residues for substrate positioning facilitated the design of a small smart library to modify the active site pocket of MthUPO. In this way, we could direct the selectivity of the oxyfunctionalization toward enantioselective R/S C4 hydroxylation. Enzyme variants were expressed in Saccharomyces cerevisiae in a 96-well microtiter plate. The screening was performed by the previously developed Multiple Injection in a Single Experimental Run (MISER) GC-MS method [11, 12] focusing on activity increase. The MISER setup involves injecting 96 samples into the GC in a single experimental run, with product quantifications performed exclusively in the MS through different m/z ratios, eliminating the need for substrate/product separation. This setup enables an injection frequency of up to 30 s, allowing for GC analysis of one microtiter plate within 48 minutes. Rescreening of the best variants with a chiral GC-MS led to the determination of the enantioselectivities. After two rounds of iterative enzyme evolution, the activity increased up to 17-fold and the regioselectivity reached up to 99.6 % for the 4-hydroxy-β-ionone. Enantiodivergent variants were identified with enantiomeric ratios of 96.6:3.4 (R) and 0.3:99.7 (S), respectively (Figure 2). Finally, in silico analysis of the best performing, highly enantioselective variants revealed the molecular basis of the selectivity, which was achieved by only two (R-selectivity) and four (S-selectivity) mutations, respectively. |
| References: | [1] Muench, J., et al. ACS Catal., 2021. 11(15), 9168-9203. [2] Beltran-Nogal, A., et al. Curr. Opin. Struct. Biol., 2022. 73, 102342. [3] Monterrey, D.T., et al. Current Opinion in Green and Sustainable Chemistry, 2023, 100786. [4] Beekwilder, J., et al. J. Biotechnol., 2014. 192, 383-392. [5] Parker, G.L., et al. Tetrahedron, 2016. 72(13), 1645-1652. [6] Babot, E.D., et al. J. Agric. Food Chem., 2020. 68(19), 5375-5383. [7] Urlacher, V.B., et al. Appl. Microbiol. Biotechnol., 2006. 70(1), 53-59. [8] Celik, A., et al. Org. Biomol. Chem., 2005. 3(16), 2930-2934. [9] Marumoto, S., et al. Planta Medica, 2017. 83(03/04), 292-299. [10] Kakeya, H., et al. Agric. Biol. Chem., 1991. 55(7), 1873-1876. [11] Knorrscheidt, A., et al. ChemCatChem, 2020. 12(19), 4788-4795. [12] Knorrscheidt, A., et al. ACS Catal., 2021. 11(12), 7327-7338. |
| Figures: | 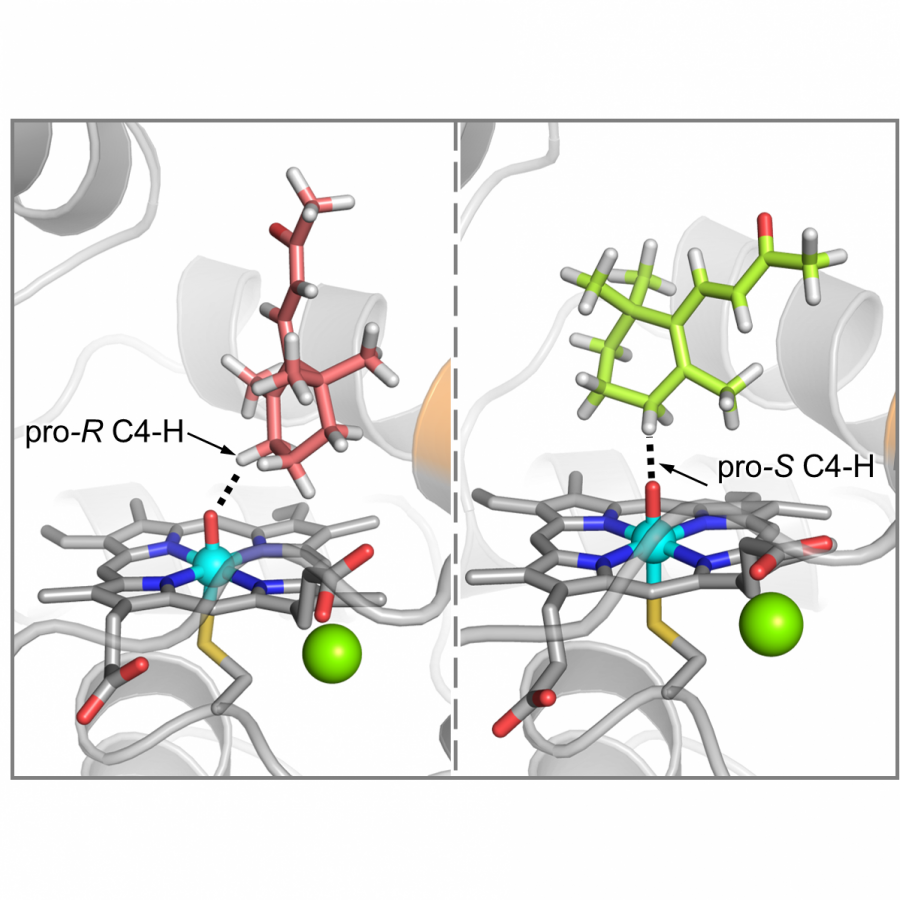 Analysis of beta-ionone near-attack conformations. Restrained MD-simulations are performed to explore near-attack conformations for selective C4-hydroxylation and characterize relevant binding modes of the model substrate beta-ionone.
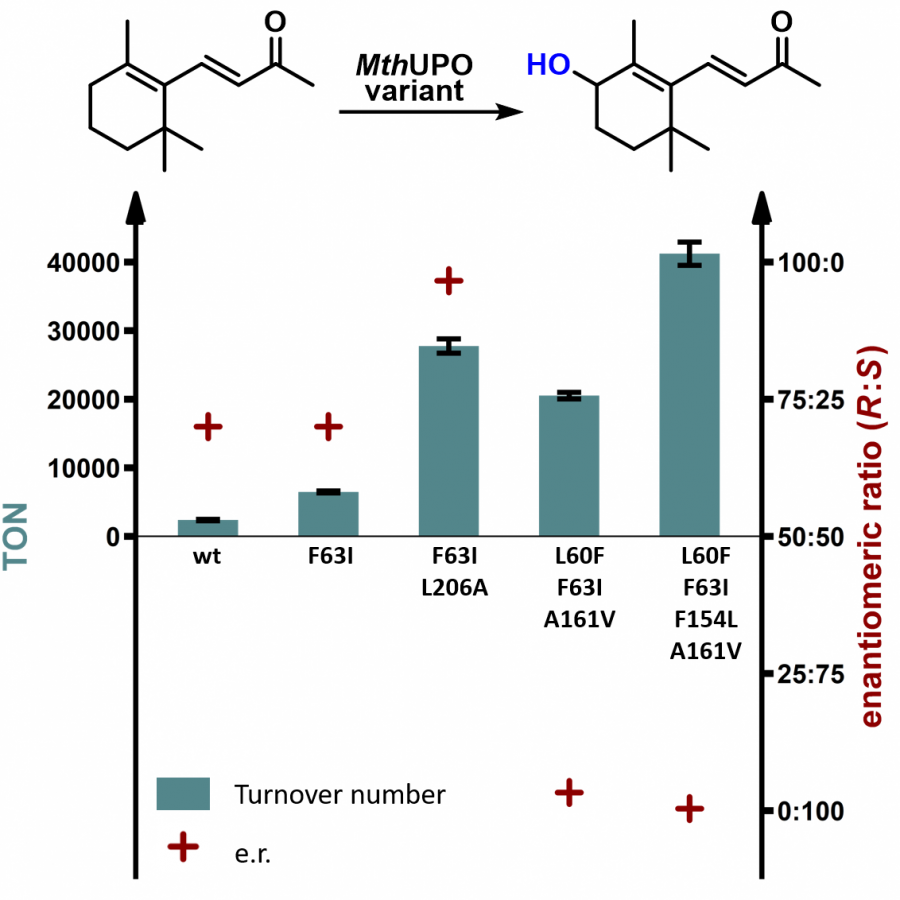 Turnover number and enantioselectivity of different engineered MthUPO variants. Turnover data are mean +/- s.d. of measurements in triplicates. TON (teal bars) determined by GC-MS and enantiomeric excess (red cross) by chiral GC-MS.
|
#428 | Exploring the Scope of Methyltransferase Biocatalysis |
|
| Presenting author: | Jiaming PENG of UNIVERSITY OF BASEL |
| Corresponding author: | Florian SEEBECK of UNIVERSITY OF BASEL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Halide methyltransferase / Fluoromethylation / SAM recycling |
| Purpose: | S-adenosylmethionine (SAM)-dependent methyltransferases constitute a large family of enzymes that catalyze regio-, chemo- and stereospecific methylation of complex natural products. [1] These enzymes could be very useful tools for chemoenzymatic reaction and diversification of natural or synthetic compounds. Until recently, preparative applications of methyltransferases (MT) in vitro were limited because of the requirement for SAM as a stoichiometric methyl donor. Introduction of a simple SAM-regeneration process based on the ability of halide methyltransferases (HMT) to transfer methyl-groups from methyl iodide to S-adenosylhomocysteine (SAH) has highlighted a general strategy to harness enzyme-catalyzed alkylation in biocatalysis. [2 - 4] This strategy extends beyond methyl groups and applies to more complex alkyl groups. [5, 6] Fluoromethyl groups may be of particular interest in this regard. Strategic fluorination can optimize the pharmacological properties of drugs by modulating their membrane permeability and metabolic stability. [7] Hence, the development of organic fluorides and the methodologies towards fluorination are of great interest. [8] We demonstrated successful enzyme-catalyzed attachment of fluoromethyl group onto C-, N- and O- and S-nucleophiles using fluormethyl iodide as a reagent. [9] In this presentation we will discuss our latest efforts to exploit the remarkable reactivity of some fluoromethylated products for the development of novel biocatalytic processes. |
| References: | [1] A. W. Struck, M. L. Thompson, L. S. Wong, J. Micklefield. ChemBioChem, 2012, 13, 2642-2655. [2] C. Liao, F.P. Seebeck. Nat. Catal, 2019, 2, 696-701. [3] C. Liao, F.P. Seebeck. Angew. Chemie. Int. Ed., 2020, 59, 7184-7187. [4] L. L. Bengel, B. Aberle, A. N. Egler Kemmerer, S. Kienzle, B. Hauer, S. C. Hammer. Angew. Chemie. Int.Ed., 2021, 60, 5554-5560. [5] Q. Tang, C. W. Grathwol, A. S. Aslan Uezel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst, U. T. Bornscheuer. Angew. Chemie. Int. Ed., 2021, 60, 1524-1527. [6] K. H. Schuelke, F. Ospina, K. Hoernschemeyer, S. Gergel, S. C. Hammer. ChemBioChem., 2021, 23, e2021006. [7] A. Rentmeister, F. Arnold, R. Fasan. Nat. Chem. Biol., 2009, 5, 26-28. [8] R. Senatore, M. Malik, M. Spreitzer, W. Holzer, V. Pace. Org. Lett., 2020, 22, 1345-1349. [9] J. Peng, C. Liao; C. Bauer, F.P. Seebeck. Angew. Chemie. Int. Ed., 2021, 60, 27178-27183. |
| Figures: | 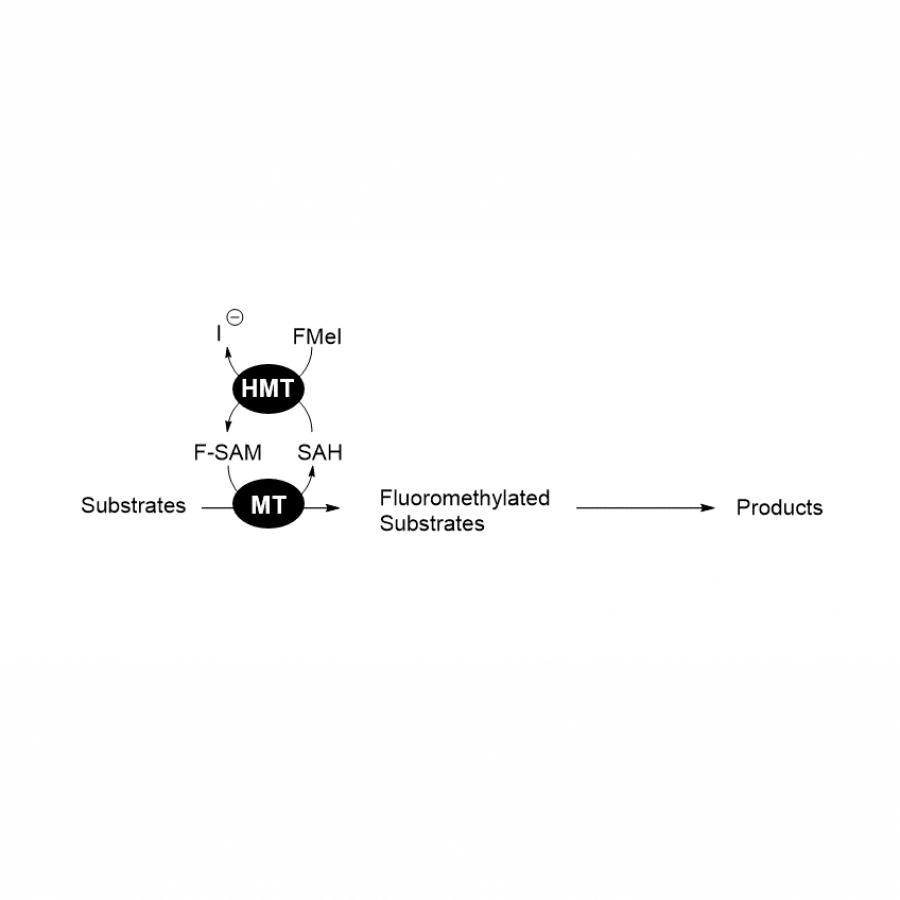 HMT-MT Cascade HMT-MT cascade transfers fluoromethyl group to small and macromolecules that are subject to further reaction. |
#429 | Enzymatic acylation of peptides |
|
| Presenting author: | Laura RODRIGUEZ PEREZ of UNIVERSITY OF MANCHESTER |
| Other authors: | Christian SCHNEPEL of UNIVERSITY OF MANCHESTER William GOUNDRY of EARLY CHEMICAL DEVELOPMENT,PHARMACEUTICAL SCIENCES, R&D, ASTRAZENECA Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / click chemistry / peptide functionalization / lysine acetyltransferases |
| Purpose: | Peptide modification is a field of interest due to its potential applications in the biomedical area, for example, in the manufacture of peptide-drug conjugates. Nevertheless, production of peptide conjugates is non-trivial, and chemical methods tend to display an array of shortcomings such as poor thermostability, low potency, or off-target toxicity.[1] Biocatalysis is an attractive alternative to chemical bioconjugation, as enzymes exhibit exceptional selectivity, fast kinetics, high yields and require mild conditions.[2] As a result, they can be considered to be superior to chemical methods in many aspects. In this work, we developed an enzymatic method to acylate peptides, allowing the introduction of a range of bioorthogonal probes. The adenylation domain of the carboxylic acid reductase from Segniliparus rugosus was used to obtain in-situ the CoA derivative of the respective acids. The CoA substrates were employed by lysine acetyltransferase p300 for acylation, functionalizing a 20-mer peptide with bioorthogonal handles. Following the one-pot reaction, modification of up to five residues in peptide H4(1-20) was observed. Furthermore, peptide labelling with stable isotopes was also achieved via acetylation using the same enzymatic system. |
| References: | [1] P. Hoppenz, S. Els-Heindl and A. G. Beck-Sickinger, Frontiers in chemistry, 2020, 8, 571. [2] Y. Zhang, K.-Y. Park, K. F. Suazo, and M. D. Distefano, Chemical Society Reviews, 2018, 47, 9106. |
| Figures: | 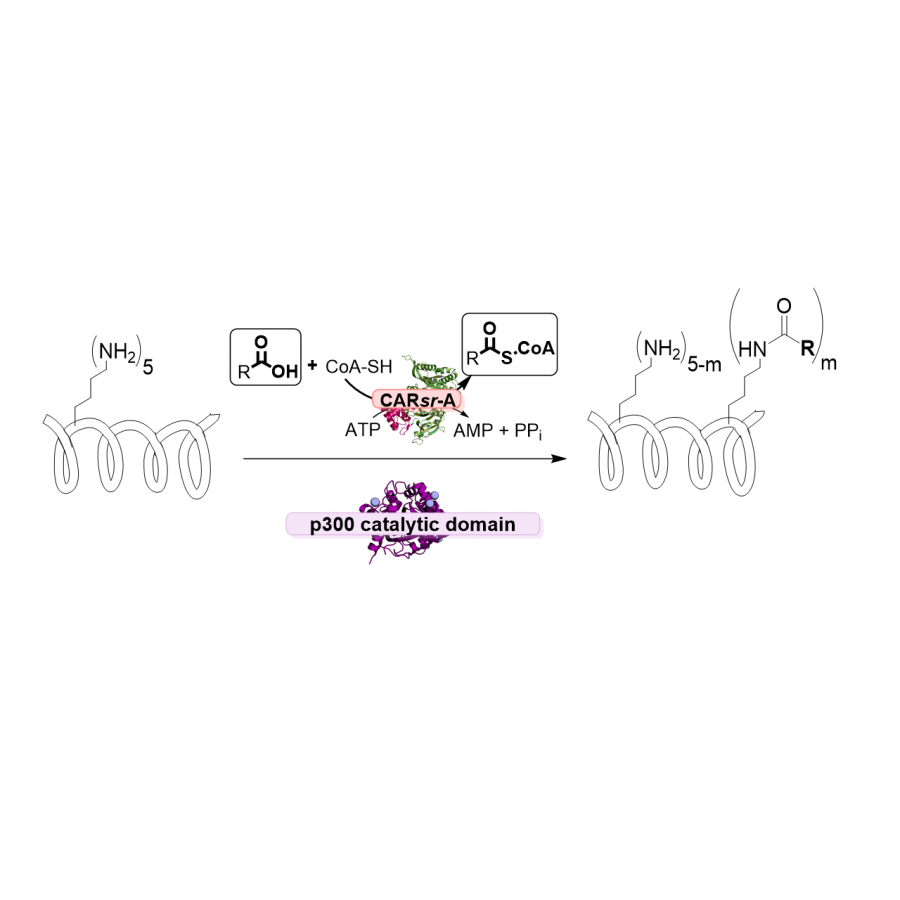 Scheme of the enzymatic modification of peptide H4(1-20) CARsr-A was employed to form the CoA derivatives of the respective acids, which were used by enzyme p300 to functionalize peptide H4(1-20) |
#431 | A sustainable end-of-life solution for thermoset composites: Directed enzyme evolution for the degradation of vinyl-ester resins. |
|
| Presenting author: | Mikel DOLZ of INSTITUTE OF CATALYSIS & PETROCHEMISTRY (ICP-CSIC) |
| Other authors: | Javier VIÑA-GONZALEZ of EVOENZYME Miguel ALCALDE of INSTITUTE OF CATALYSIS & PETROCHEMISTRY (ICP-CSIC) |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Thermoset composites / Directed Evolution / Cutinases / polyester resins |
| Purpose: | The handling and manage of the end-of life of complex artificial materials, such as thermoset composites, is a rather significant challenge for nowadays sustainability. These materials are known for their durability due their exceptional mechanical, thermal and chemical resistance. As landfill storage and incineration are the most typical destinations – thus harming the surrounding ecosystems – environmentally-friendlier approaches need to be developed and implemented (1). We are currently working in engineering improved biocatalysts for degradation of these materials. Cutinases (EC 3.1.1.74) exhibit many of the necessary qualities for a promising degrading biocatalyst, due to their natural activity to hydrolase ester bonds – a motif with a widespread of magnitude in several thermoset composites (1). In our study, the cutinase from Fusarium solani (FsC) was chosen as departure point for directed evolution towards the degradation of vinyl-ester and poly-ester resins. As these polymeric matrix show extreme complexity and high hydrophobicity, their direct use as a substrate in a high-throughput manner is precluded. Instead, a colorimetric HTS assay was developed based on p-nitrophenyl trimethylacetate (3-MA), which represents a structural soluble scaffold molecule present both in vinyl- and poly-ester resins. Using this assay, we performed focused directed evolution by MORPHING (2) targeting the active site and surroundings to random mutagenesis and recombination, which rendered 5 different first-generation mutant winners. In a wise-step approach, beneficial mutations were recombined by Site-Directed Recombination (SDR) (3) while saturating hot spot-residues identified during the evolution campaign. As a result, we obtained a final mutant variant, referred to as RancoR, which displayed a 30-fold total activity improvement with respect to parental type FsC. Both FsC and RancoR were cloned into the overexpressing host Pichia pastoris for upscale production and ulterior purification and characterization, which revealed the improved biochemical characteristics of the evolved biocatalyst. |
| References: | 1. Pickering SJ. Recycling technologies for thermoset composite materials—current status. Compos. Part A Appl. Sci. 2006 Aug 1;37(8):1206-15. 2. Gonzalez-Perez D, Molina-Espeja P, Garcia-Ruiz E, Alcalde M. Mutagenic organized recombination process by homologous in vivo grouping (MORPHING) for directed enzyme evolution. PLoS One. 2014;9(3):e90919. 3. Viña-Gonzalez J, Alcalde M. In vivo site-directed recombination (SDR): An efficient tool to reveal beneficial epistasis. Methods Enzymol. 2020;643:1-13. |
#442 | Enzymatic oxidation of plant oils to C18 trihydroxy fatty acids at high concentrations by lipoxygenase and epoxide hydrolase |
|
| Presenting author: | LEE JIN of KONKUK UNIVERSITY |
| Other authors: | SHIN KYUNG CHUL of KONKUK UNIVERSITY OH DEOK KUN of KONKUK UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biotransformation / C18 trihydroxy fatty acid / lipoxygenase / epoxide hydrolase |
| Purpose: | Plant oxylipins, including C18 Trihydroxy fatty acids (THFAs), use as antifungal agents and adjuvants for vaccine. They have been synthesized by chemical methods, which have disadvantages such as low yields by multi-step reactions and cause of environmental pollution. However, the synthesis of THFAs by microorganisms and plants show still too low concentrations and productivities for industrial synthesis. Here, recombinant Escherichia coli cells co-expressing bacterial linoleate (LA) 13-lipoxygenase with high isomerization activity and epoxide hydrolase converted 200 mM of LA, α-linolenic acid (ALA), and γ-linolenic acid (GLA), into 11R,12R,13S-THFAs via epoxy hydroxy fatty acids with high molar yields (>60%) in a baffled flask. In the conversion, GLA-derived 12S,13S-epoxy-11R-hydroxyoctadecadienoic acid and 11R,12R,13S-trihydroxyoctadecadienoic acid were identified as new compounds by NMR analysis. For the production of THFA from safflower oil as LA source, the content of LA in safflower oil hydrolyzate was increased by adding adsorbent resin SP207 to the hydrolysis reaction of safflower oil by lipase because palmitic acid and glycerol were removed through selective binding of the resin. The resin-treated safflower oil hydrolyzate containing 250 mM LA, which was obtained from 93 g L−1 safflower oil, was converted into 230 mM 11R,12R,13S-trihydroxyoctadecenoic acid in 24 h, with a productivity of 9.6 mM h−1 and a molar yield of 92% by the recombinant cells in a bioreactor. Therefore, we succeeded in the cost-effective, efficient, and environmentally friendly biotransformation of safflower oil into THFAs. |
| Figures: | 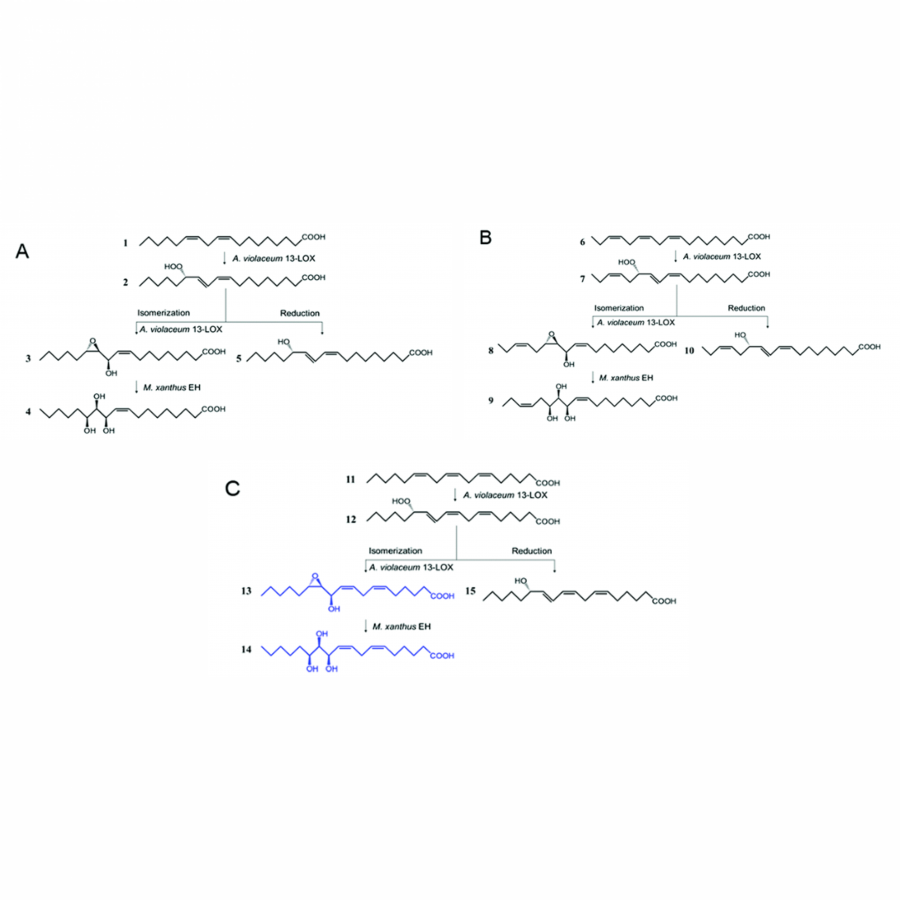 Fig. 1. Production of trihydroxy fatty acids by whole recombinant E. coli co-expressing A. violaceum LA 13S-LOX and M. xanthus EH. (A) Production of 11R,12R,13S-trihydroxy-9Z-octadecenoic acid (11R,12R,13S-TriHOME) from linoleic acid (LA) via 12S,13S-epoxy-11R-hydroxy-9Z-octadecenoic acid (12S,13S-EHOME). (B) Production of 11R,12R,13S-trihydroxy-9Z,15Z-octadecadienoic acid (11R,12R,1 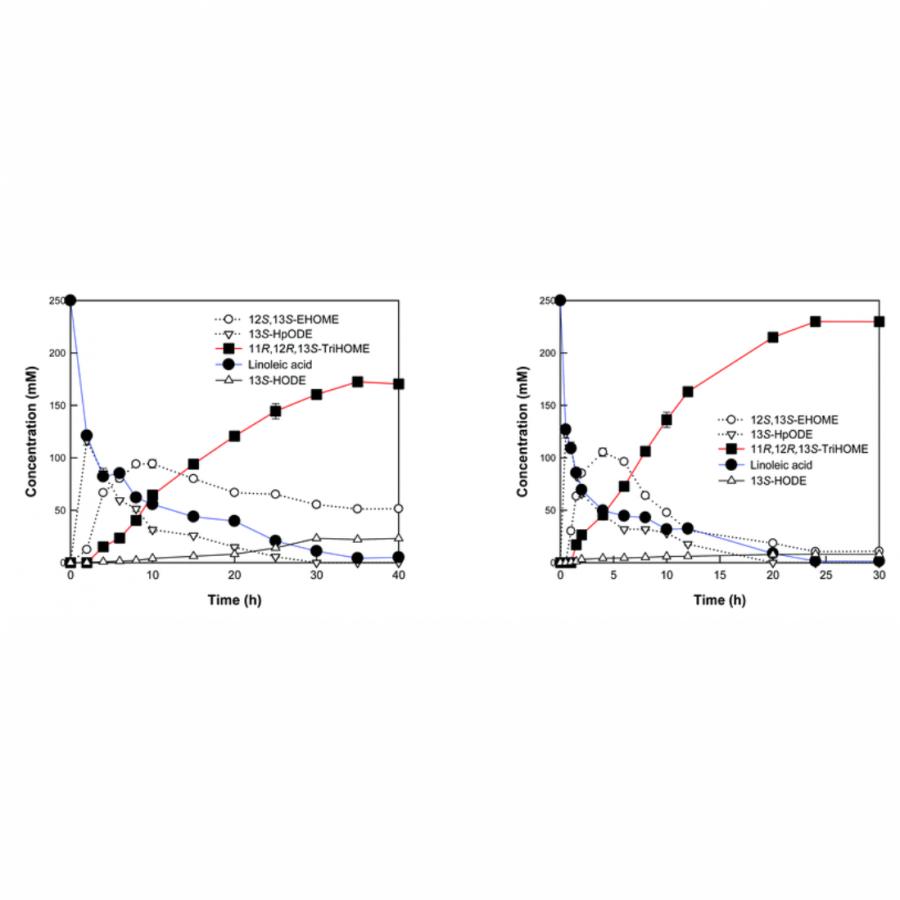 Fig. 5. Time-course reactions for the production of 11R,12R,13S-TriHOME from LA in safflower oil hydrolyzate by whole recombinant E. coli cells co-expressing A. violaceum LA 13S-LOX and M. xanthus EH under the optimized reaction conditions. (A) Biotransformation of LA in safflower oil hydrolyzate obtained without absorbent resin into 11R,12R,13S-TriHOME. (B) Biotransformation of LA in safflower oil hydrolyzate obtained from absorbent resin treatment into 11R,12R,13S-TriHOME. |
#447 | Holistic understanding of alcohol dehydrogenase catalysis in deep eutectic solvents through experimental and computational approaches |
|
| Presenting author: | Ningning ZHANG of LEIBNIZ UNIVERSITY HANNOVER |
| Other authors: | Jan Philipp BITTNER of HAMBURG UNIVERSITY OF TECHNOLOGY Pablo DOMÍNGUEZ DE MARÍA of SUSTAINABLE MOMENTUM Sven JAKOBTORWEIHEN of HAMBURG UNIVERSITY OF TECHNOLOGY Irina SMIRNOVA of HAMBURG UNIVERSITY OF TECHNOLOGY Selin KARA of LEIBNIZ UNIVERSITY HANNOVER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | redox biocatalysis / non-aqueous media / deep eutectic solvents / alcohol dehydrogenase |
| Purpose: | Biocatalysis starts a transition phase from default aqueous media to non-aqueous media in the spirit of Green Chemistry. The use of oxidoreductases (EC1) in non-conventional media is particularly vital for the synthesis of value-added chiral chemicals.[1] Deep eutectic solvents (DES) have emerged as a new class of sustainable solvents with tremendous tunability and biocompatibility.[2,3] Redox biocatalysis in DESs combines the best of two worlds: enzymes’ selectivity and DESs’ designability.[4] Design redox biocatalysis in DESs requires an in-depth understanding of DES’s impact on enzymes. This stimulates us to study DESs’ effects on oxidoreductases by assessing the catalytic performance of alcohol dehydrogenases (ADHs) in DES-water mixtures with the aid of molecular dynamics (MD) simulations.[5–7] Enzymes’ activity was found positively correlated to water activity (αW) due to the changes in solvation layers surrounding enzymes.[6–7] Individual DES components were first revealed to have discrepant effects on enzymes, e.g., positive (Gly) or negative (ChCl), promoting the knowledge-oriented design of a new enzyme-compatible eutectic mixture (ChCl-Gly, 1:9).[7] We will deepen the study with experimental analyses and in silico simulations (Fig. 1). Acknowledgment: The authors thank the Deutsche Forschungsgemeinschaft (DFG) (grant numbers: KA 4399/3-2, SM82/25-2) for the financial support. |
| References: | [1] L. Huang, P. Dominguez De María, S. Kara, Chim. Oggi. 2018, 36, 48-56. [2] M. Pätzold, S. Siebenhaller, S. Kara, A. Liese, C. Syldatk, D. Holtmann, Trends Biotechnol. 2019, 37, 943-959. [3] N. Zhang, F. Steininger, L.-E. Meyer, K. Koren, S. Kara, ACS Sustainable Chem. Eng. 2021, 9, 8347-8353. [4] V. Gotor-Fernández, C. E. Paul, J. Biotechnol. 2019, 293, 24-35. [5] N. Zhang, J. P. Bittner, M. Fiedler, T. Beretta, P. D. de María, S. Jakobtorweihen, S. Kara, ACS Catal. 2022, 12, 9171-9180. [6] L. Huang#, J.P. Bittner#, P. Dominguez de María, S. Jakobtorweihen, S. Kara, ChemBioChem 2020, 21, 811-817. (#shared first author) [7] J. P. Bittner#, N. Zhang#, L. Huang, P. Dominguez de María, S. Jakobtorweihen, S. Kara, Green Chem. 2022, 24, 1120-1131. (#shared first author) |
| Figures: | 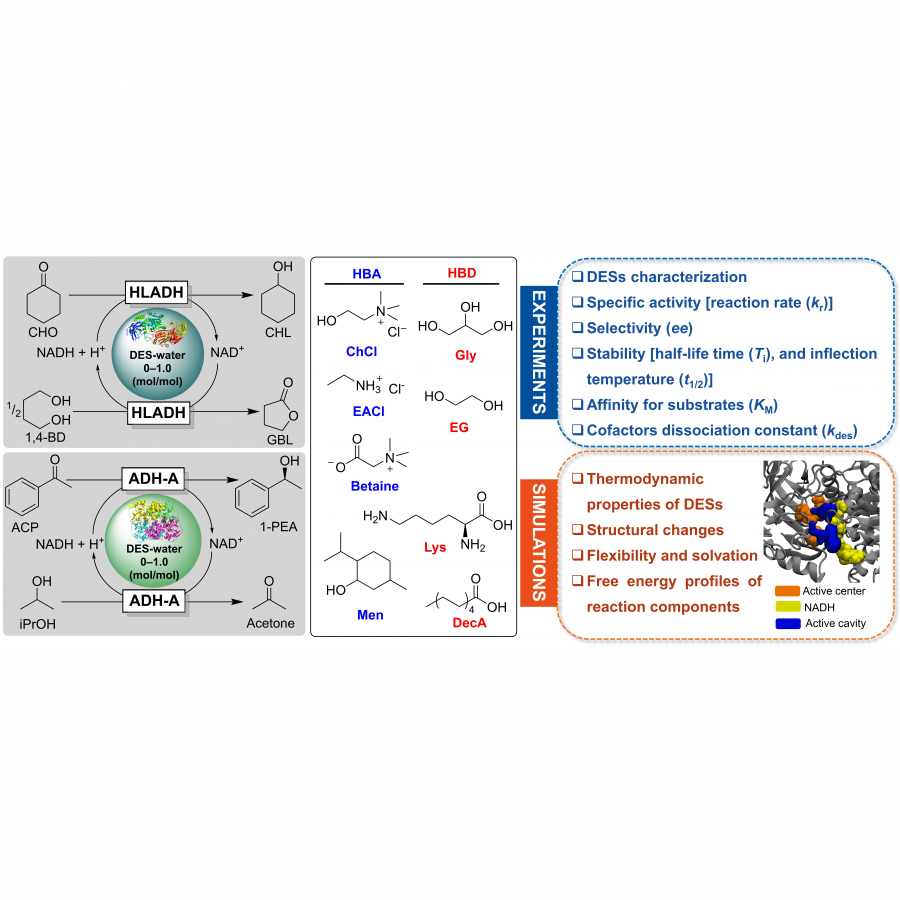 Fig.1 Reduction of cyclohexanone (CHO) and acetophenone (ACP) catalyzed by horse live ADH (HLADH) and ADH-A in various DES-water mixtures. HBA: hydrogen bond acceptor, HBD: hydrogen bond donor. |
#448 | Biochemical characterization of a new unspecific peroxygenases from Botryobasidium botryosum |
|
| Presenting author: | Margarita SEEGER of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG |
| Corresponding author: | Anett SCHALLMEY of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Unspecific peroxygenases / Enzyme characterization / / |
| Purpose: | Unspecific peroxygenases (UPOs; EC 1.11.2.1) represent a recently discovered subfamily of predominantly fungal heme-thiolate enzymes. They catalyze a broad range of oxidative transformations including the selective oxyfunctionalization of unactivated hydrocarbons by transferring peroxide-borne oxygen. The reaction mechanism of UPOs is similar to the peroxide shunt pathway of cytochrome P450 monooxogenases but without the requirement of complex cofactors such as NAD(P)H or electron-transport systems [1]. Additionally, hydrogen peroxide-driven biocatalysis combines the high oxidation power of H2O2 and its environmentally friendly properties with the high efficiency and selectivity of enzymatic reactions, making UPOs of high interest for biotechnological applications [2]. One major bottleneck so far remains the lack of recombinant functional expression of UPO genes with satisfactory expression levels [3]. Only recently, it has become possible to produce small amounts of active UPOs using an Escherichia coli expression system, although this does not work in the case of all UPO constructs and always occurs without glycosylation [4]. In this study, we report one new peroxygenases from the Basidiomycete Botryobasidium botryosum (BboUPO), which was successfully obtained, purified, and initially characterized with respect to its basic biochemical features, utilizing E. coli as expression host. BboUPO displays oxidation activity in presence of 10% acetonitrile, acetone, and methanol without exceedingly large decreases in the enzymatic activities. |
| References: | [1] M. Hofrichter, et al. Monooxygenase, peroxidase and peroxygenase properties and mechanisms of cytochrome P450, 2015, 341-368. [2] B. Burek, et al. Green Chemistry 2019, 21(12), 3232-3249. [3] Y. Wang, et al. Curr.Opin Chem. Biol. 2017, 37, 1-9. [4] D. Linde, et al. Appl. Environ. Microbiol. 2020, 86(7), e02899-19. |
#458 | Enhancing ferredoxin NADP+ reductase stability by heterologous expression in Pichia pastoris |
|
| Presenting author: | Cristina ALIERTA VIÑAO of UNIVERSIDAD DE ZARAGOZA |
| Corresponding author: | Susana VELASCO-LOZANO of UNIVERSIDAD DE ZARAGOZA |
| Other authors: | Juan C. ALMADA of UNIVERSIDAD DE ZARAGOZA Ana Daniela VEGA-RODRIGEZ of CENTRO DE INVESTIGACIÓN Y ASISTENCIA EN TECNOLOGÍA Y DISEÑO DEL ESTADO DE JALISCO Juan Carlos MATEOS-DIAZ of CENTRO DE INVESTIGACIÓN Y ASISTENCIA EN TECNOLOGÍA Y DISEÑO DEL ESTADO DE JALISCO Jorge Alberto RODRIGUEZ-GONZALEZ of CENTRO DE INVESTIGACIÓN Y ASISTENCIA EN TECNOLOGÍA Y DISEÑO DEL ESTADO DE JALISCO |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | stability / ferredoxin NADP+ reductase / glycosylations / P. pastoris |
| Purpose: | In nature, ferredoxin NADP+ reductase (FNR) is the main flavoenzyme which regenerates NADPH by receiving the electrons dispensed from photosystem I and catalyzing the two-electron reduction of NADP+ to NADPH. This catalytic capacity has been harnessed outside the cell in the so called “electrochemical leaf”.1 Under these conditions, FNR catalyzes the reversible interconversion of NADPH into NADP+ when it is confined into an electrode surface. Moreover, this bioelectrocatalytic concept has been exploited as a cofactor regenerating tool when assembled with cofactor-dependent multi-enzyme systems.2 Among the reported FNRs, the one from the cyanobacterium Anabaena sp. (AsFNR) has been well recombinantly expressed in E. coli with relatively high protein yields, thus allowing been widely studied and kinetically characterized.3 Despite these advantages, AsFNR displays very poor stability (it is completely inactivated after spending one night stored at 4 ºC). This drawback hampers its application as a robust cofactor-regenerating system in large scale biotransformations. Several strategies have been focused on increasing the stability of this enzyme mainly by immobilization and rational mutations.4 Nowadays, the yeast expression system P. pastoris is one of the most popular and standard tool for the production of recombinant proteins in molecular biology. Overall the benefits of this strategy include appropriate folding and secretion of recombinant proteins to the external environment of the yeast cell.5 Moreover, N-glycosylation is one of the most common forms of protein post-translational modification in this yeast expression system; which is closely related with the increase in thermal stability of the resulting enzymes.6, 7 Based on these previous findings, herein we report for the first time the heterologous expression of AsFNR in P. pastoris aiming at increasing the operational stability of this enzyme through the natural N-glycosylation process. We have cloned a his-tagged AsFNR construct extracted from a pET28a vector and inserted it into a pGAPZαA vector. After the successful clone construction, we optimized the heterologous expression of rAsFNR in P. pastoris in defined and enriched mediums. Once rAsFNR is secreted to the culture medium, we have purified it by IMAC chromatography and kinetically characterized. The obtained pure rAsFNR displays higher melting temperature and higher operational stability than the one expressed in E. coli expression system. |
| References: | 1 Siritanaratkul, B. et al. Chem. Sci. J. (2017) 8, 4579-4586 2 Armstrong, F. A., Cheng, B., Herold, R. A., Megarity, C. F. & Siritanaratkul, B. Chem. Rev. (2022) 3 Hermoso, J. A. et al. Journal of Molecular Biology (2002) 319, 1133-1142 4 Bes, M. T., Gomez-Moreno, C., Guisan, J. M. & Fernandez-Lafuente, R. Journal of Molecular Catalysis A: Chemical (1995) 98, 161-169 5 Yang, Z. & Zhang, Z. Biotechnol. Adv. (2018) 36, 182-195 6 Han, M. et al. Enzyme Microb. Technol. (2014) 54, 32-37 7 Skropeta, D. Bioorganic & Medicinal Chemistry (2009) 17, 2645-2653 |
#464 | New hyaluronic acid degrading enzymes from fungi |
|
| Presenting author: | MARINA MINGUET-LOBATO of CATALYSIS INSTITUTE CSIC |
| Other authors: | FADIA V. CERVANTES DOMINGUEZ of CATALYSIS INSTITUTE CSIC DAVID FERNANDEZ-POLO of CATALYSIS INSTITUTE MARIA FERNANDEZ-LOBATO of MOLECULAR BIOLOGY SEVERO OCHOA CENTER UAM-CSIC FRANCISCO J. PLOU of CATALYSIS INSTITUTE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | hyaluronic acid / lyases / glycosidases / hyaluronidases |
| Purpose: | Hyaluronic acid or hyaluronan (HA) is a linear anionic non-ramified and non-sulphated glycosaminoglycan (GAG) composed by repeat units of β-1,4-D-glucuronic acid-β-1,3-N-acetyl-D-glucosamine (Fig. 1). HA plays numerous roles in biological processes due to its unique structural properties, as mechanical support of cells being the major component of extracellular matrix, virulence and cell signalling, migration and proliferation. Hyaluronidases (HAases) comprise a group of enzymes, widely distributed in all kingdoms of life, that primordially degrades HA, although possess activity towards other GAGs (chondroitin and its derivatives), thus participating in the referred processes as well. These enzymes can be divided in three types according to their enzymatic activities: (1) HA endo-β-1,4-acetyl-hexosaminidases, which cleave β-1,4 glycosidic linkages to form oligomers with tetrasaccharides as final products and with GlcNAc at the reducing end (mainly found in mammals); (2) HA endo-β-1,3-glucuronidases, which cleave β-1,3 glycosidic linkages yielding tetra- or hexa-saccharides with a GlcNAc at the non-reducing end and GlcA at the reducing end (found in leeches); (3) HA lyases, which catalyse β-elimination reactions yielding unsaturated carbon-carbon bond products, with unsaturated disaccharides as final products (mainly found in bacteria). In recent years, these enzymes have aroused enormous interest in the scientific community due to the increasing number of biotechnological applications as drug delivery, cosmetic surgeries, cancer therapies, HA processing and hyalo-oligosaccharides (HAOS) preparation [1, 2]. Among microbial sources, fungi hyaluronidases are less studied in comparison with bacterial ones. The aim of this work was to find new microbial sources of hyaluronidases focusing on that coming from fungi and the characterisation of their ability to degrade HA. A mycelial microorganism not yet characterized was isolated from colloidal chitin containing minimal media and was screened for the discovery of enzymatic degrading activities towards different polysaccharides (basically chitin and HA) with positive result. This positive HA-degrading, apparently fungus, was cultivated in different developed screening media for HAase activity. Extracellular HAase expression was achieved to its maximum in a minimal medium containing HA as carbon source. The optimal pH and temperature of crude enzymatic extract for the degradation of high molecular weight HA were set at 4.5 and 50 °C. Time-course HA degradation was monitored by HPAEC-PAD and SEC-ELSD developed methods from cultures and reactions with the protein extracts. Maximal activity was achieved after 48 h of expression, although activity was observed after only 6 h of expression on cultures. Main HAOS formed upon prolonged times of reaction were characterized by MALDI-TOF MS. Product analysis confirmed that main positive mode detected oligomers contained an unsaturation and, therefore, were produced in a β-elimination manner. However, oligosaccharides without unsaturation were also detected in negative mode, being the minimal polymerization degree a tetrasaccharide, thus suggesting the presence of an HA-4-glycanohydrolase enzyme. To our knowledge, this is the first time that two different HA degradation modes were detected from an unique fungal organism. Our results suggest a multi-enzymatic attack to high molecular weight degradation and we are currently performing transcriptomic analysis of this microorganism in order to characterise the responsible genes of the observed enzymatic activities. |
| References: | [1] yue-sheng z., biotch. adv. 2022, 60, 108018. [1] sliadovskii d., sci. rep. 2021, 11, 22600. |
| Figures: |  Hyaluronic acid chemical structure representation. Structural unit of hyaluronan polymer consisted of N-acetyl-D-glucosamine and D-glucuronic acid. Glycosydic linkage types between sugar units are indicated. |
#475 | Investigating expression of urethane bond hydrolyzing enzyme |
|
| Presenting author: | Jokūbas KRUTKEVIČIUS of VILNIUS UNIVERSITY, LIFE SCIENCES CENTER, INSTITUTE OF BIOTECHNOLOGY, SECTOR OF APPLIED BIOCATALYSIS |
| Other authors: | Inga MATIJOŠYTĖ of VILNIUS UNIVERSITY, LIFE SCIENCES CENTER, INSTITUTE OF BIOTECHNOLOGY, SECTOR OF APPLIED BIOCATALYSIS |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Polyurethane / Recombinant expression / Urethanase / Circular Economy |
| Purpose: | The abundance of industrial and domestic wastes is still neither recycled, nor used in any other profitable way. Currently, intensively promoted the Circular Economy model implicates that the generated waste has to re-enter the production line and save valuable recourses. Each waste material requires specific treatment, and a unified process may not be applied. For example, polyurethane (PU) is an excellent material that finds its way to be used in many industrial and domestic products—from insulation to sleeping mattress. Also, nearly half of the global unrecycled PU waste finds its way into landfills. PU represent 8 % of all plastics produced annually. The treatment of polyurethane waste by hydrolysis, glycolysis, pyrolysis demands a substantial amount of energy and has not been applied on a commercial level because it is economically not favourable. Biocatalysis can be foreseen as one of the alternative treatment methods to for such type of waste [1,2]. Currently, there are no biocatalytic or green chemical PU utilization technologies or methods. Some PU are susceptible to enzymatic and microbial degradation [3]. The aim of this research is to investigate enzymes that could hydrolyse urethane bond in PU. Various environmental samples have been screened and PU degrading microorganisms were isolated. Urethane bond hydrolysing enzyme urethanase (GenBank: MK757456.1) was found in Lysinibacillus sp. strain TBS 101. This bacterium was isolated from Lithuanian soil samples. Recombinant urethanase was successfully cloned and synthesized. Its ability to hydrolyse a model substrate—ethyl carbamate (urethane)—has been confirmed by Berthelot reaction. Specific enzyme activity reached up to 4.4 U/mg in E.coli. Further research is directed towards increasing the amount of urethanase by using heterologous expression systems, such as Pichia pastoris yeast, solubility enhancing partner proteins, such as maltose-binding protein (MBP), and codon optimisation. The attained results in more detail will be presented during the poster session. |
| References: | [1] A. Kemona et al. Polymers, 2020, 12(8), 1752. [2] D. Simón et al. Waste Management, 2018, 76, 147-171. [3] A. Magnin et al. Biotechnology Advances, 2020, 39, 107457. |
#477 | The Discovery and Characterization of a Fungal Aldolase with High Formaldehyde Affinity |
|
| Presenting author: | Yonghui XIE of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY,CHINESE ACADEMY OF SCIENCES |
| Corresponding author: | Xiang SHENG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY,CHINESE ACADEMY OF SCIENCES |
| Other authors: | Hongjun DONG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY,CHINESE ACADEMY OF SCIENCES Xiang SHENG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY,CHINESE ACADEMY OF SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Formaldehyde assimilation / Class II pyruvate aldolase / Homoserine cycle / Enzyme discovery |
| Purpose: | The homoserine cycle is an artificially designed methanol assimilation pathway, relying on two promiscuous formaldehyde-condensing aldolase reactions [1]. It is expected to outperform the generally used natural pathways such as the RuMP cycle and the serine cycle. However, previous study revealed that the low activity of pyruvate aldolase, more specifically the binding affinity of formaldehyde, represents a limiting factor for the efficiency of the homoserine cycle [1]. In the present study, we used a genomic enzymology web tool [2], called Enzyme Function Initiative-Enzyme Similarity Tool (EFI-EST), to mine new aldolases for the reversible aldol condensation of pyruvate with formaldehyde among the Class II pyruvate aldolase. On the basis of the generated sequence similarity networks, we segregated the family into different functional clusters and selected one from each cluster for the characterization of formaldehyde-condensing activity by the colorimetric method developed by ourselves. A number of active aldolases were identified. The one with the highest affinity of formaldehyde was from fungi and the measured Km value was 0.34mM, which is two orders of magnitude lower than those precedingly reported [3,4]. The multiple sequence and structure alignment of aldolases indicate that a hydrophobic flexible loop of the adjacent subunit may play an important role in the improved binding affinity of pyruvate aldolase toward formaldehyde. We thus identified a novel aldolase, whose condensation rate and binding affinity of formaldehyde are high enough to be physiologically applicable, which opens the way for the application of this unnatural condensation reaction in vivo. |
| References: | [1] He, H.; Hoper, R.; Dodenhoft, M.; Marliere, P.; Bar-Even, A., Metab. Eng. 2020, 60, 1-13. [2] Gerlt, J. A., Biochemistry. 2017, 56, 4293-4308. [3] Bosch, S.; Sanchez-Freire, E.; del Pozo, M. L.; Cesnik, M.; Quesada, J.; Mate, D. M.; Hernandez, K.; Qi, Y.; Clapes, P.; Vasic-Racki, D.; Findrik Blazevic, Z.; Berenguer, J.; Hidalgo, A., ACS Sustain. Chem. Eng. 2021, 9, 5430-5436. [4] Hernandez, K.; Bujons, J.; Joglar, J.; Charnock, S. J.; Dominguez de Maria, P.; Fessner, W. D.; Clapes, P., ACS Catalysis. 2017, 7, 1707-1711. |
| Figures: |  Sequence Similarity Networks (SSNs) for sequences from the Class II pyruvate aldolase generated with EFI-EST. Alignment score is 60, 45% sequence identity. we selected one from each cluster for the characterization of formaldehyde-condensing activity.  The Kinetic Properties of Aldolases for the condensation of pyruvate with formaldehyde. Aldolase activity was detected by the colorimetric method developed by ourselves, relying on monitoring the consumption of formaldehyde. |
#478 | VAO-mediated oxidation of C-N bond for thiazoline synthesis |
|
| Presenting author: | Lei GAO of PEKING UNIVERSITY |
| Other authors: | Haowen ZHANG of PEKING UNIVERSITY Xiaoguang LEI of PEKING UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | thiazoline / vanillyl alcohol oxidases / chemo-enzymatic synthesis / heterocycles |
| Purpose: | Heterocycles prevalently exist in the structures of drugs, natural products, and pharmacologically active synthetic molecules. Motivated by the significance of heterocycles in medicinal chemistry, chemists have made great effort to unveil the biosynthetic pathways of heterocycle-containing natural products, resulting in successful identification of enzymes catalyzing heterocycle formation in NRPs[1,2,3], polyketide-NRP hybrids[4,5], or RiPPs[6]. However, these enzymes typically catalyze intramolecular cyclization reactions on structurally complex substrates and are featured with limited substrate scope, thus dramatically reducing their synthetic application in medical chemistry. Thiazoline and thiazole are two representative heterocycles found in natural products and drugs such as desferrithiocin, SP-420, SPB01403 and Uloric (as shown in Figure 1A). Chemical synthesis of thiazoline rings is majorly dependent on the condensation reaction of 2-aminoethane-1-thiol (or its derivatives) with a cyanide group, which is typically derived from an aldehyde group.[7] Here, we report our discovery of a new enzymatic transformation from aldehydes to thiazolines. This unique transformation is catalyzed by vanillyl alcohol oxidases (VAOs), which was previously characterized as a subfamily of FAD dependent oxidase to catalyze oxidation of alcohols or enantioselective hydroxylation of 4-alkylated phenols (Figure 1B).[8] The VAO-mediated synthesis of thiazolines from aldehydes is achieved via a cascade reaction, composed of a spontaneous cyclization and enzymatic benzylic oxidation. We found that VAOs exhibited substrate promiscuity towards a wide range of 4-hydroxybenzaldehyde derivatives (Figure 1C). Notably, methyl cysteine can also be recognized as the surrogate of 2-aminoethane-1-thiol, offering new opportunity to achieve the green and efficient synthesis of the clinical drug SP-420. |
| References: | 1. Shen, Q. et al. Characterization of a cryptic NRPs gene cluster in bacillus velezensis fzb42 reveals a discrete oxidase involved in multithiazole biosynthesis. ACS Catal. 2022, 12, 3371. 2. Dan, Q., et al. Fungal indole alkaloid biogenesis through evolution of a bifunctional reductase/Diels-Alderase. Nat. Chem. 2019, 11, 972. 3. Chen, XW., et al. Unexpected assembly machinery for 4(3H)-quinazolinone scaffold synthesis. Nat. Commun. 2022, 13, 6522. 4. Yu, Z., et al. Discovery and biosynthesis of karnamicins as angiotensin converting enzyme inhibitors. Nat. Commun. 2023, 14, 209. 5. Ohashi, M. et al. An enzymatic Alder-ene reaction. Nature 2022, 586, 64. 6. Hudson, G. A. In Vitro biosynthesis of the core scaffold of the thiopeptide thiomuracin. J. Am. Chem. Soc. 2015, 137, 51, 16012. 7. Bergeron, R. J. et al. Desazadesmethyldesferrithiocin analogues as orally effective iron chelators. J. Med. Chem. 1999, 42, 1, 95. 8. Gygli G., et al. On the origin of vanillyl alcohol oxidases. Fungal Genet. Biol. 2018, 116, 24. |
| Figures: | 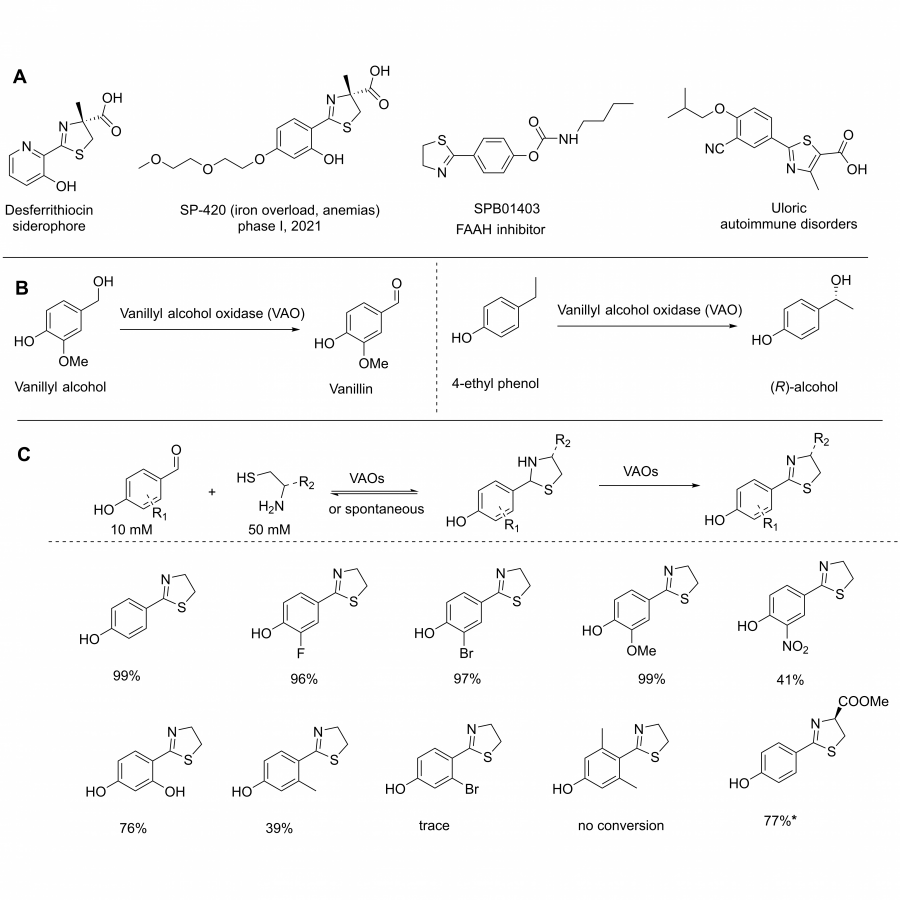 VAO-mediated oxidation of C-N bond for thiazoline synthesis (A) Representative bioactive thiazoline and thiazole derivatives. (B) Original function of vanillyl alcohol oxidases
(C) chemo-enzymatic synthesis of thiazoline using VAO-mediated oxidation. |
#482 | Ancestral sequence reconstruction enlightens the structural basis of vitamin C biosynthesis |
|
| Presenting author: | Alessandro BOVERIO of UNIVERSITY OF PAVIA |
| Corresponding author: | Marco W. FRAAIJE of UNIVERSITY OF GRONINGEN |
| Other authors: | Maria Laura MASCOTTI of UNIVERSITY OF GRONINGEN Andrea MATTEVI of UNIVERSITY OF PAVIA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | structural biology / Ancestral sequence reconstruction / oxidoreductases / |
| Purpose: | Ascorbic acid (Vitamin C) is a key element in eukaryotes involved in several roles in the cell1. Apart from being a successful redox system counteracting the deleterious action of Reacting Oxygen Species (ROS)2, Ascorbic acid is also used as a cofactor in several metal-dependent monooxygenases responsible, for example, of hydroxylation reactions during collagen biosynthesis3. In nature, the final step of vitamin C biosynthesis is catalyzed by a class of flavin dependent enzymes called aldonolactone oxidoreductase4. In animals, the enzyme is responsible for the oxidation of L-gulono 1,4 lactone to ascorbic acid. The enzyme uses molecular oxygen as electron acceptor with the flavin cofactor that is covalently bound to the enzyme with a histidyl linkage. On the other hand, in plants, the enzyme (L-galactono 1,4 lactone dehydrogenase) poorly reacts with oxygen using cytochrome c as electron acceptor, without having the flavin covalently bound. Despite an extensive biochemical characterization5,6, no structural data have been reported so far for aldonolactone oxidoreductases leaving an open question about their mechanism of action. By applying Ancestral Sequence Reconstruction (ASR) we were able to infer the evolutionary history of L-galactono 1,4 lactone dehydrogenase. The last common ancestors of embryophyta and viridiplantae were resurrected and characterized. The enzymes were recombinantly expressed in E. coli with a high yield. By deleting the mitochondrial target sequence at the N-terminus of the amino acid sequences the enzymes were purified as soluble cytoplasmatic proteins. Their substrate scope was investigated by testing a series of carbohydrates. Finally, the combination of site directed mutagenesis and x-ray crystallography allowed to highlight key features regarding the mechanism of action of this class of enzyme. |
| References: | (1) Linster, C. L., and Van Schaftingen, E. (2007) Vitamin C: Biosynthesis, recycling and degradation in mammals. FEBS Journal. (2) Wheeler, G., Ishikawa, T., Pornsaksit, V., and Smirnoff, N. (2015) Evolution of alternative biosynthetic pathways for vitamin C following plastid acquisition in photosynthetic eukaryotes. Elife 2015. (3) Mandl, J., Szarka, A., and Bánhegyi, G. (2009) Vitamin C: Update on physiology and pharmacology. Br J Pharmacol. (4) Aboobucker, S. I., and Lorence, A. (2016) Recent progress on the characterization of aldonolactone oxidoreductases. Plant Physiology and Biochemistry. (5) Leferink, N. G. H., Jose, M. D. F., van den Berg, W. A. M., and van Berkel, W. J. H. (2009) Functional assignment of Glu386 and Arg388 in the active site of l-galactono-γ-lactone dehydrogenase. FEBS Lett 583. (6) Leferink, N. G. H., Fraaije, M. W., Joosten, H. J., Schaap, P. J., Mattevi, A., and van Berkel, W. J. H. (2009) Identification of a gatekeeper residue that prevents dehydrogenases from acting as oxidases. Journal of Biological Chemistry 284. |
#484 | α-GLUCOSYLATION OF FLAVONOIDS: DIVERSITY OF ACCEPTORS AND PRODUCTS |
|
| Presenting author: | Eglantine DON SIMONI of GIVAUDAN |
| Other authors: | Daniel AURIOL of GIVAUDAN Carole LAMBERT of GIVAUDAN Jane HUBERT of NATEXPLORE Alexis KOTLAND of NATEXPLORE Perrine LEMAGNEN of GIVAUDAN Chantal PAULUS of GIVAUDAN Boris ELBAUM of GIVAUDAN Pascale AURIOL of GIVAUDAN Julie MARBOEUF of GIVAUDAN Cyrille JARRIN of GIVAUDAN Patrick ROBE of GIVAUDAN Romain REYNAUD of GIVAUDAN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alpha-glucosylation / Flavonoids / Cyclodextrin glucanotransferase / Dextransucrase |
| Purpose: | For many years phenolic compounds have had a strong success in the cosmetic industry due to their antioxidant, lightening, anti-inflammatory and UV-protection properties [1]. They are extracted from plants and some pure molecules like Arbutin, Hesperidin, Resveratrol, and more recently Bakuchiol, acquired a strong reputation. Flavonoids, including Hesperidin, are a subfamily of polyphenols, with a C6-C3-C6 carbon skeleton composed of two phenyl rings (A and B) and a heterocyclic ring (C) (formation of ring C is not achieved in the case of dihydrochalcones) as seen in Figure 1 [2]. This structure makes flavonoids have low water solubility and low stability depending on substituents of ring A and B. Glucosylation, the transfer of a glucose moiety from a donor to an acceptor by enzymatic reaction, is a way to increase water solubility and stability of phenolic compounds. In the 1990’s Hayashibara developed industrial processes for the alpha-glucosylation of Hesperidin, Rutin and Naringin using Cyclodextrin glucanotransferase (CGTase) that usually works to convert starch and dextrins into cyclodextrins. The three flavonoids are natural glucosides and are recognized as substrate of the CGTase as they are structurally similar to its natural acceptors. While enzymatic glycosylation requires a specific recognition of substrate by the enzyme [3] an exhaustive review of the literature revealed that CGTases are able to transfer one or several glucose units on phenolic aglycones. As example CGTase from Thermoanaerobacter sp. can modify the following compounds at very variable molar yield (%): Kaempferol (15%), Pterostilbene (3%), Epigallocatechin gallate (65%), Resveratrol (50%), Hesperetin (4%) [4, 5, 6, 7, 8]. It appeared that conducting a study on the specificity of a CGTase to a large panel of phenolic compounds would be of great interest to understand how the structure of acceptor could influence the efficacy of conversion. Thirty-one flavonoids, with varied chemical structures, were tested through glucosylation by CGTase from Thermoanaerobacter sp. As expected, natural glycosides were more prone to glucosylation and we found 7 aglycones exhibiting conversion yield above 45% (Figure 1). No absolute structural feature was identified as a predictive criterion for the success of conversion. However, molecular characteristics seemed to have a positive impact: the presence of glycosyl (Phloridzin vs Phloretin, Rutin vs Quercetin, Naringin vs Naringenin), the presence of glucuronyl (Baicalin vs Baicalein), the absence of methylation (Luteolin vs Hesperetin) and the hydroxylation in position 3 (Myricetin vs Tricetin, Taxifolin vs Eriodictyol). Then a comparison of epigallocatechin gallate (EGCG) alpha-glucosylation with two enzymes was conducted: Thermoanaerobacter sp. CGTase using alpha-cyclodextrin as donor and Dextransucrase (DS) from Leuconostoc mesenteroides NRRL B512F, using sucrose as donor [9,10]. Reaction medium was purified and fractionated by centrifugal partition chromatography to separate the different glucosides. Both enzymes mainly produced a diversity of glucosides of which the major one was a monoglucoside. NMR characterization of EGCG-G1 revealed that both enzymes generate two different structures: DS was more prone to create Epigallocatechin gallate 4’-O-glucoside as reported by [11], while CGTase mainly converts EGCG onto Epigallocatechin gallate 3'-O-glucoside as reported by [6]. Morevover, CGTase produced a diversity of glucosides where DS specifically synthesized mono- and diglucoside of EGCG (Figure 2). Our work provides new insights in the broad diversity of acceptors of CGTase from Thermoanaerobacter sp. It also underlines the interest of glucosylation with alpha-transglucosidases that efficiently and specifically modify acceptors from cost-efficient and renewable material. |
| References: | [1] De Lima Cherubim, D.J.; Buzanello Martins, C.V.; Oliveira Fariña, L.; da Silva de Lucca, RA. Polyphenols as natural antioxidants in cosmetics applications. J Cosmet Dermatol. 2020 Jan;19(1):33-37. doi: 10.1111/jocd.13093. [2] Barreca, D.; Mandalari, G.; Calderaro, A.; Smeriglio, A.; Trombetta, D.; Felice, MR.; Gattuso, G., Citrus Flavones: An update on sources, biological functions, and health promoting properties. Plants (Basel). 2020 Feb 26;9(3):288. doi: 10.3390/plants9030288. [3] Desmet, T.; Soetaert, W.; Bojarová, P.; Křen, V.; Dijkhuizen, L.; Eastwick-Field, V.; Schiller, A. Enzymatic glycosylation of small molecules: challenging substrates require tailored catalysts. Chem. Weinh. Bergstr. Ger. 2012, 18, 10786-10801. doi:10.1002/chem.201103069. [4] Choung, W.-J.; Hwang, S.H.; Ko, D.-S.; Kim, S.B.; Kim, S.H.; Jeon, S.H.; Choi, H.-D.; Lim, S.S.; Shim, J.-H. Enzymatic synthesis of a novel kaempferol-3-O-β-D-glucopyranosyl-(1→4)-O-α-D-glucopyranoside using cyclodextrin glucanotransferase and its inhibitory effects on aldose reductase, inflammation, and oxidative stress. J. Agric. Food Chem. 2017, 65, 2760-2767. doi:10.1021/acs.jafc.7b00501. [5] Gonzalez-Alfonso, J.L.; Rodrigo-Frutos, D.; Belmonte-Reche, E.; Penalver, P.; Poveda, A.; Jimenez-Barbero, J.; Ballesteros, A.O.; Hirose, Y.; Polaina, J.; Morales, J.C.; Fernandez-Lobato, M.; Plou, F.J. Enzymatic synthesis of novel pterostilbene alpha-glucoside by the combination of cyclodextrin glucanotransferase and amyloglucosidase. J. Agric. Food Chem. 2018, 66, 7402-7408. doi:10.1021/acs.jafc.8b02143. [6] Gonzalez-Alfonso, J.L.; Leemans, L.; Poveda, A.; Jimenez-Barbero, J.; Ballesteros, A.O.; Plou, F.J. Efficient α-glucosylation of epigallocatechin gallate catalyzed by cyclodextrin glucanotransferase from Thermoanaerobacter Species. J. Agric. Food Chem. 2018, 66, 7402-7408. doi:10.1021/acs.jafc.8b02143. [7] González-Alfonso, J.L.; Míguez, N.; Padilla, J.D.; Leemans, L.; Poveda, A.; Jimnez-Barbero, J.; Ballesteros, A.O.; Sandoval, G.; Plou, F.J. Optimization of regioselective α-glucosylation of Hesperetin catalyzed by cyclodextrin glucanotransferase. Mol. Basel Switz. 2018, 23, E2885. doi:10.3390/molecules23112885. [8] Torres, P.; Poveda, A.; Jimenez-Barbero, J.; Parra, J.L.; Comelles, F.; Ballesteros, A.O.; Plou, F.J. Enzymatic synthesis of α-glucosides of resveratrol with surfactant activity. Adv. Synth. Catal. 2011, 353, 1077-1086. doi:10.1002/adsc.201000968. [9] Patent EP2027279B1. Phenolic compounds with cosmetic and therapeutic applications. Publication 2017. [10] Nadim, M.; Auriol, D.; Lamerant-Fayel, L.; Lefèvre, F.; Dubanet, L.; Redziniak, G.; Kieda, C.; Grillon, C. Improvement of polyphenol properties upon glucosylation in a UV-induced skin ageing model. Int. J. Cosm. Sci. 2014, 36, 579-587. doi:10.1111/ics./12159. [11] Moon, Y.-H.; Lee, J.-H.; Ahn, J.-S.; Nam, S.-H.; Oh, D.-K.; Park, D.-H.; Chung, H.-J.; Kang, S.; Day, D. F.; Kim, D. Synthesis, structure analyses, and characterization of novel epigallocatechin gallate (EGCG) glycosides using the glucansucrase from Leuconostoc mesenteroides B-1299CB. J. Agric. Food Chem. 2006, 54, 1230-1237. doi.org/10.1021/jf052359i |
| Figures: | 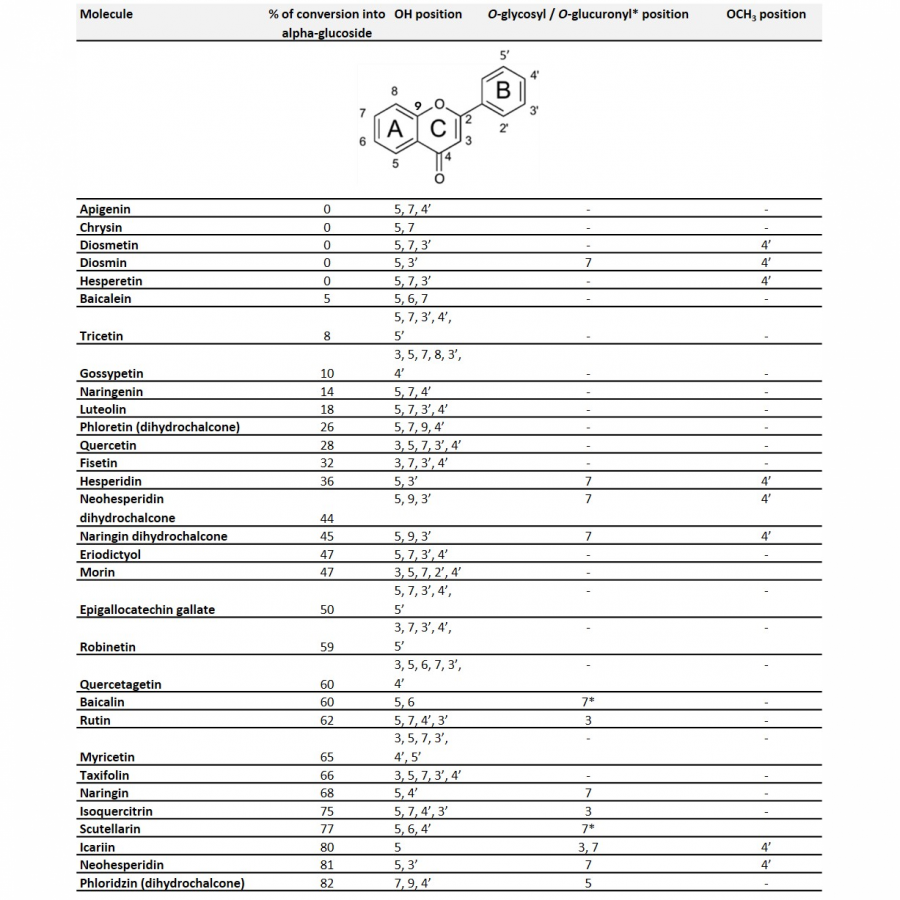 Characteristics of 31 flavonoids involved in transglucosylation with CGTase from Thermoanaerobacter sp. The table reports the molar conversion yield of total alpha-glucosides and presents the main structural features of the molecules. 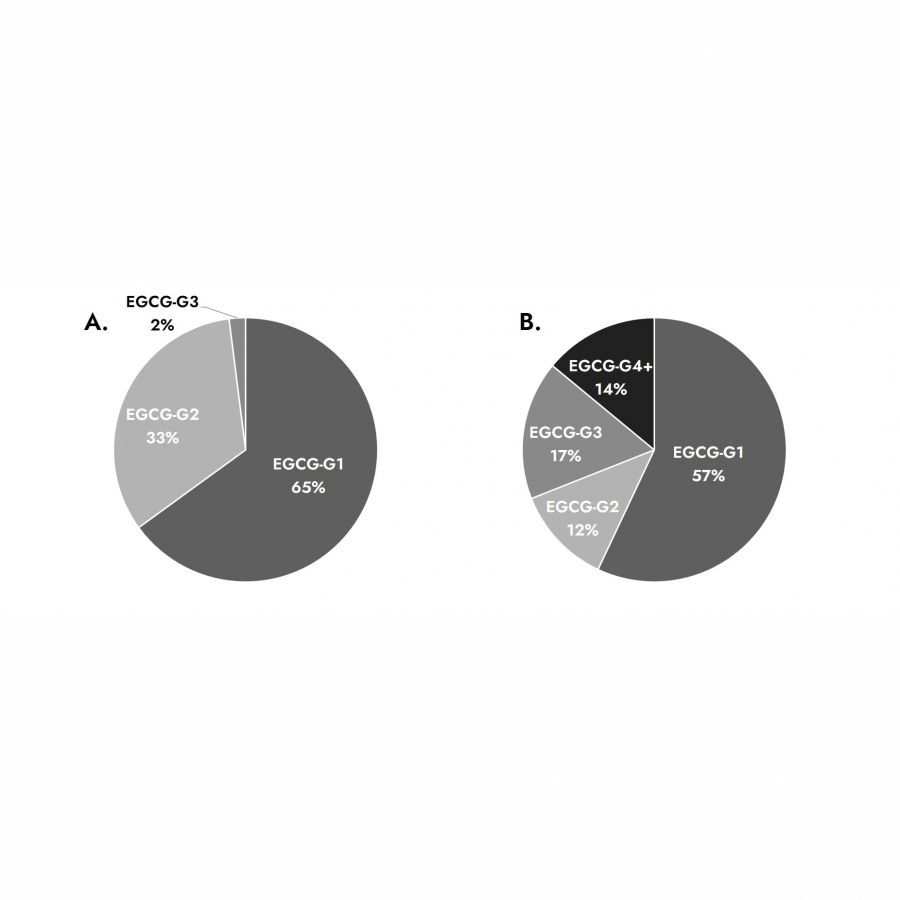 Distribution of Epigallocatechin gallate glucosides synthesized by Dextransucrase from Leuconostoc mesenteroides NRRL B512F (A) and Cyclodextrin glucanotransferase from Thermoanaerobacter sp (B) expressed as percentage of total glucosides. Epigallocatechin glucosides are grouped under monoglucosides (EGCG-G1), diglucosides (EGCG-G2), triglucosides (EGCG-G3) and tetra- and polyglucosides (EGCG-4+). |
#489 | Enhancement of the overexpression and stability of strictosidine synthase from Rauvolfia serpentina by protein engineering |
|
| Presenting author: | Isabel OROZ-GUINEA of UNIVERSITY OF GRAZ |
| Other authors: | Tasha TAMGUE-CHRAKO-OH of UNIVERSITY OF GRAZ Vinz VOM-HAGEN of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Strictosidine synthase / Pictet-Splenger reaction / Stability and solubility enhancement / Protein engineering |
| Purpose: | The Pictet−Spengler reaction offers a valuable route to 1,2,3,4-tetrahydro-β-carboline (THBC) and isoquinoline molecules. These scaffolds are found in important pharmaceuticals such as Tadalafil, a phosphodiesterase 5 (PDE5) inhibitor, administered to treat male erectile dysfunction.[1] Moreover, due to their pharmacological activities they are investigated for their potential use as antivirals, fungicides, antimalarials, antileishmanial, antithrombotic, analgesic and anticancer drugs.[2] However, one of the main challenges during the synthesis TBHCs derivatives is the introduction of chirality of C1. In this sense, a promising alternative to the traditional organic synthesis strategies is the application of biocalatalysis. Strictosidine synthases (STRs) catalyze the Pictet−Spengler condensation of tryptamine and the aldehyde secologanin to give (S) strictosidine as a key intermediate in indole alkaloid biosynthesis (Scheme 1).[3] Thus, STRs could be valuable asymmetric biocatalysts for applications in synthesis. In this sense, STRs also accept short-chain aliphatic aldehydes to give enantioenriched alkaloid products with up to 99% ee.[4] However, a drawback for the implementation of STRs in the synthesis of pharmaceutical compounds is their low heterologous overexpression, solubility and stability. Hence, to overcome this issue the work presented here describes the computer-aided design of novel enzyme variants based on the STR from Rauvolfia serpentina (RsSTR) with improved protein solubility and stability. Amino acid changes to increase the stability and/or the solubility of RsSTR were identified. For this purpose, different bioinformatic tools were used, thus, positions relevant for the stability were detected using the Protein Repair One-Stop Shop (PROSS)[5] server and via calculation of ‘stability hot spots’ using the HotSpot Wizard (FireProt),[6] while mutations reducing the aggregation tendency were predicted with the webserver SolubiS.[7] Overexpression levels of the different variants were studied in terms of RsSTR percentage present in the total soluble protein fraction, milligram of RsSTR per gram of E. coli cells and mg of purified RsSTR per gram of E. coli cells. Protein stability was tested by quantifying the melting temperature of the variants and the wt. Additionally, to determine the possible effects that the new introduced amino acid changes could have on the enzymatic activity, specific activity of the produced SrSTR variants was measured and compared with the activity displayed by the wt enzyme. |
| References: | [1] a).- A. Daugan, P. Grondin, C. Ruault, A. C. Le Monnier de Gouville, H. Coste, J. M. Linget, J. Kirilovsky, F. Hyafil and R. Labaudinièret, J. Med. Chem. 2003, 46, 4533-4542; b).- A. Daugan, P. Grondin, C. Ruault, A. C. Le Monnier De Gouville, H. Coste, J. Kirilovsky, F. Hyafil and R. Labaudinière, J. Med. Chem. 2003, 46, 4525-4532. [2] J. Wang, F. Gong, T. Liang, Z. Xie, Y. Yang, C. Cao, J. Gao, T. Lu and X. Chen, Eur. J. Med. Chem. 2021, 225, 113815. [3] R. Roddan, J. M. Ward, N. H. Keep and H. C. Hailes, Curr. Opin. Chem. Biol. 2020, 55, 69-76. [4] D. Pressnitz, E.-M. Fischereder, J. Pletz, C. Kofler, L. Hammerer, K. Hiebler, H. Lechner, N. Richter, E. Eger and W. Kroutil, Angewandte Chemie, International Edition 2018, 57, 10683-10687. [5] a).- A. Goldenzweig, M. Goldsmith, Shannon E. Hill, O. Gertman, P. Laurino, Y. Ashani, O. Dym, T. Unger, S. Albeck, J. Prilusky, Raquel L. Lieberman, A. Aharoni, I. Silman, Joel L. Sussman, Dan S. Tawfik and Sarel J. Fleishman, Mol. Cell 2016, 63, 337-346; b).- J. J. Weinstein, A. Goldenzweig, S. Hoch and S. J. Fleishman, Bioinformatics 2020, [6] D. Bednar, K. Beerens, E. Sebestova, J. Bendl, S. Khare, R. Chaloupkova, Z. Prokop, J. Brezovsky, D. Baker and J. Damborsky, PLoS Comput. Biol. 2015, 11, e1004556. [7] J. Van Durme, G. De Baets, R. Van Der Kant, M. Ramakers, A. Ganesan, H. Wilkinson, R. Gallardo, F. Rousseau and J. Schymkowitz, Protein Eng. Des. Sel. 2016, 29, 285-289. |
| Figures: | 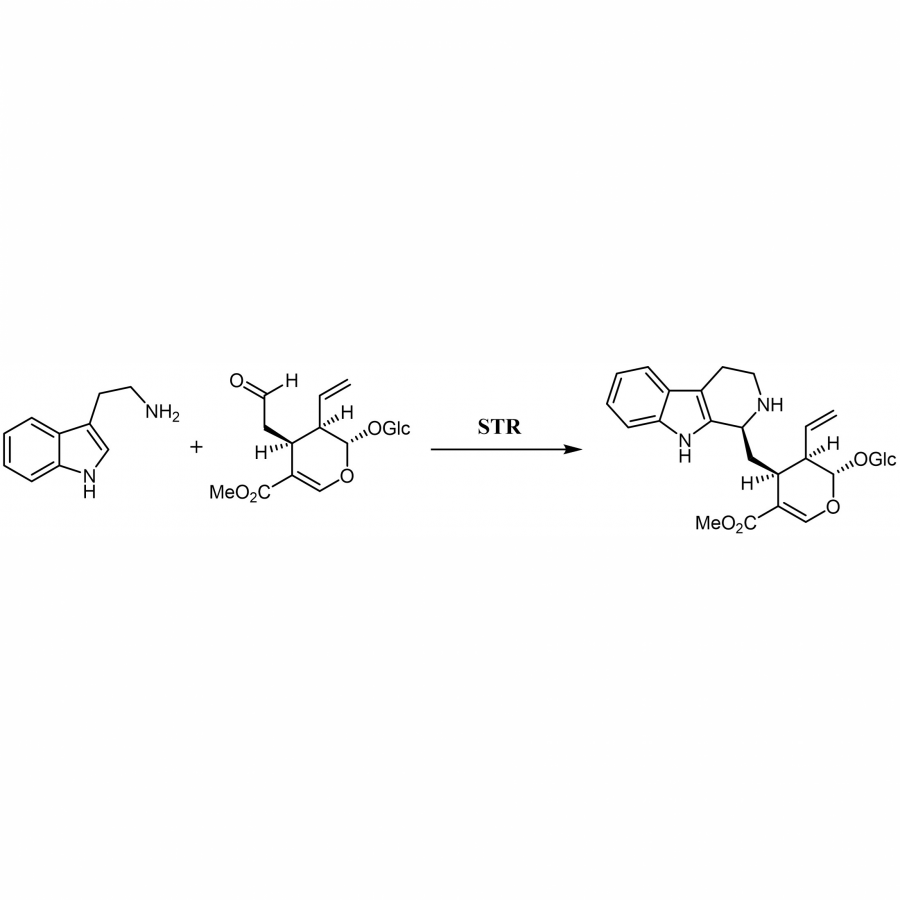 Biocatalytic Pictet-Splenger condensation Strictosidine synthase synthesis of (S)-strictosidine by Pictet-Splenger condensation of tryptamine and secologanine |
#490 | Co-encapsulation of P450 enzymes and their redox partner, in virus-like particles as nano-scale bioreactors |
|
| Presenting author: | Loic BOURDON of UNIVERSITY OF QUEENSLAND |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cytochromes P450 / Virus-Like-Particles / Co-encapsulation / |
| Purpose: | The superfamily of monooxygenase enzymes, the cytochromes P450, catalyze a large diversity of reactions making them highly interesting biocatalysts for diverse biotechnological industries. However, to be profitable, biocatalysts need to be both stable and easily recovered from biocatalytic reactions to reduce the costs involved in their use. The Gillam lab has used ancestral reconstruction (ASR) to engineer thermostable, yet catalytically promiscuous P450s, based on xenobiotic-metabolizing P450s. Ancestral P450s have shown increases of up to ~ 30 degrees in thermostability compared to their extant counterparts.[1-4] However, the need to recover biocatalysts from bioreactors, and re-use them if possible, remains a challenge, since P450s are embedded in a membrane or in cells for most biocatalytic applications, complicating the recovery and work-up of product. As a biotechnological solution, encapsulating P450s in virus-like particles (VLPs) would facilitate their recovery and re-use, since the separation of the biocatalyst from the process bioreactor could be achieved using centrifugation. At the same time, immobilizing one or multiple enzymes within a VLP has been shown to improve biocatalysts’ stability against harsh process conditions.[5] This presentation will focus on co-encapsulating ancestral P450s with their redox partner, cytochrome P450 reductase (CPR) into VLPs derived from the P22 bacteriophage. P22 VLP is composed of an assembly of ~420 coat proteins in a non-infectious spherical structure (~50nm diameter) that do not contain genetic material. With a simple linker between the native scaffold protein and the biocatalyst, co-expression in Escherichia.coli gives an effective delivery system where the VLPs encapsulate the biocatalysts of interest. In the current project, the activity of ancestral P450s towards a luminescent substrate (MultiCYP) was compared when encapsulated into VLPs compared to embedded in a membrane. The activity could be supported using different concentrations of oxygen surrogates, but activity was ~20% lower for P450s in VLPs compared to P450s in bacterial membranes, a difference that may be related to either greater sensitivity to the damaging effect of oxygen surrogates or limited transfer of substrate or product across the VLP capsid. The reusability of ancestral P450s encapsulated into VLPs was assessed using ultracentrifugation to separate the P450s in VLPs from the reaction mixture containing oxygen surrogates. While specific activity (i.e. activity per mg protein) was maintained, roughly 85% loss of total protein was observed over 5 recovery cycles and total activity declined in parallel. Co-encapsulation of the CPR that naturally supports the P450 activity would avoid the protein-degrading, use of oxygen surrogates. Two main methods to obtain VLPs co-encapsulating P450s and CPR have been pursued: firstly, in vitro assembly by reconstitution from pure proteins; and secondly, in vivo assembly realized directly within the transformed E.coli expressing the coat protein, P450 fused to the-scaffold protein and CPR fuse to the scaffold protein. Successful co-encapsulation of ancestral P450s and CPR will enable prolonged reactions at process-type temperatures with a high recycling capacity. |
| References: | [1] Y. Gumulya., t al., ChemCatChem. 2018, 11, 841-850. [2] A. G. Bart., et al., Journal of Biological Chemistry. 2020, 295, 5640-5653. [3] Y. Gumulya., t al., Biochemical journal. 2018, 474, 1-19. [4] Y. Gumulya., t al., Nature Catalysis. 2018, 1, 878-888. [5] S. Das., et al., Biochemistry. 2020, 59, 2870-2881. |
| Figures: | 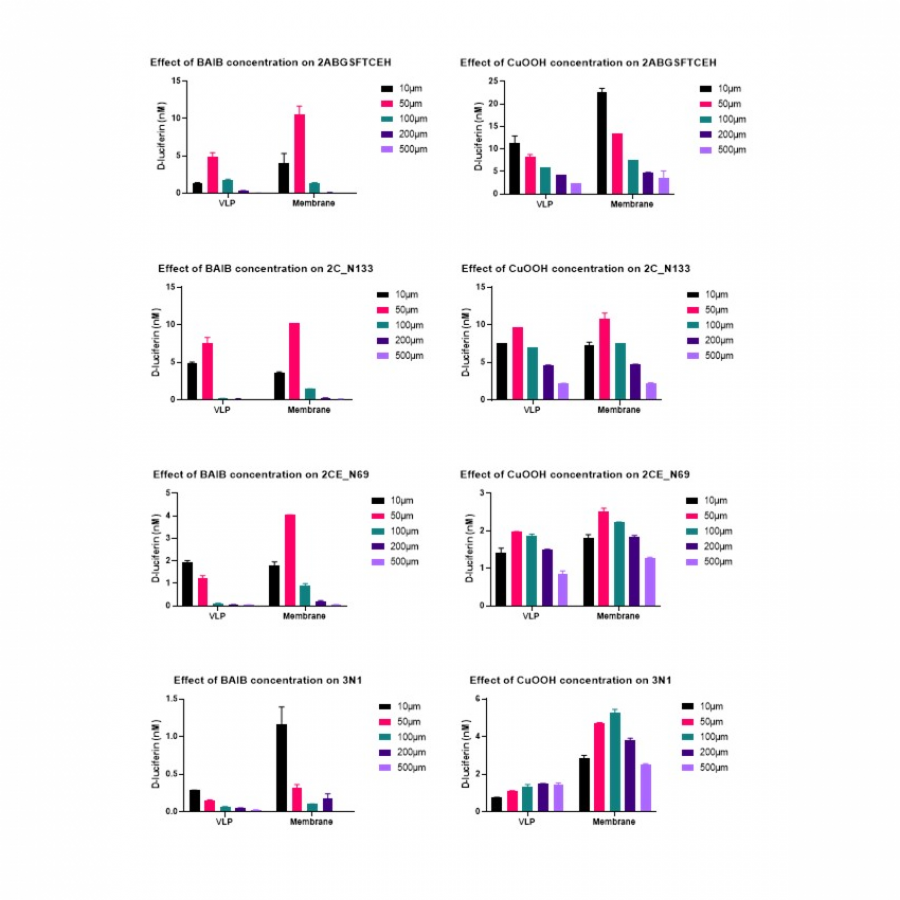 Effect of Oxygen surrogates on P450-Glo™ luciferin reactions P450-Glo™ luciferin reactions were carried out with 40nM P450 (Membrane: bacterial membrane preparation / VLP: Encapsulated in VLP (P22) by expression with long scaffold protein) and 50 µM luciferin-MultiCYP. Catalysis was supported using a range of oxyge 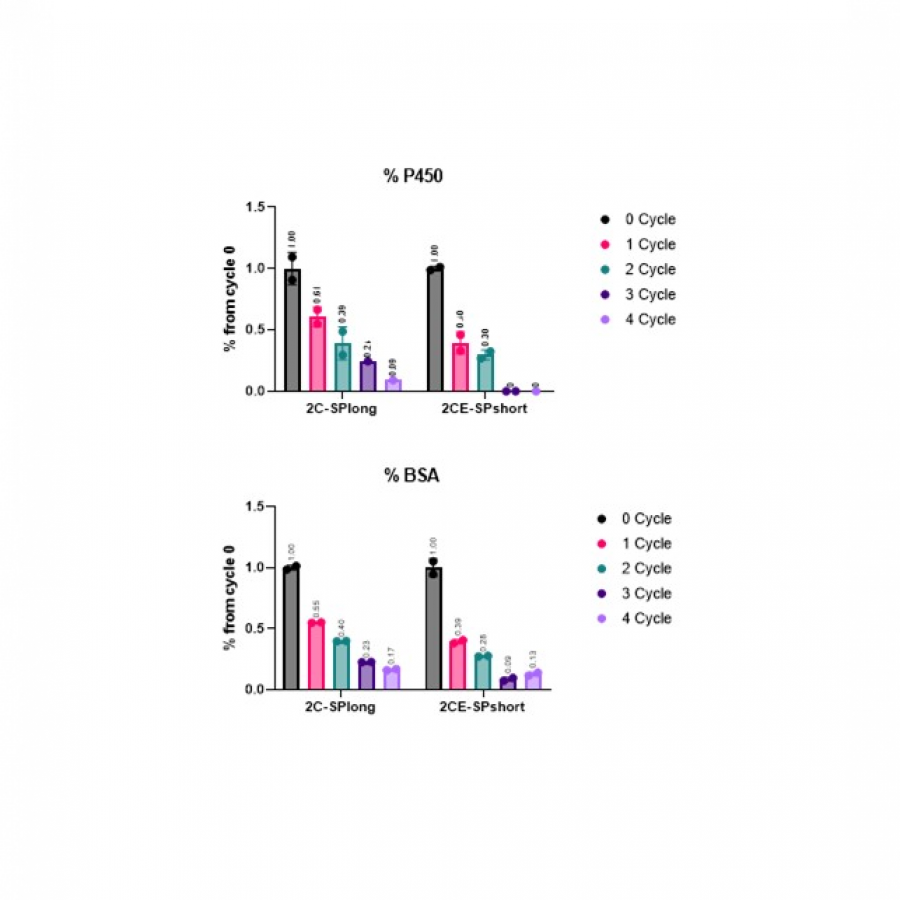 Recyclability of P450 encapsulated in VLP Measure of the relative concentration of P450 and total soluble protein during a cycle of recyclability assay, P450 sample incubate for 30min at 37°C in presence of 40 µM Cumene Hydroperoxide (to reproduce reactions conditions). Recyclability was done by |
#491 | Getting the most out of cheese whey: An integrated biotechnological platform for the synthesis of bio-based surfactants |
|
| Presenting author: | Marina Simona ROBESCU of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES |
| Other authors: | Riccardo SEMPROLI of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES Sara SANGIORGIO of UNIVERSITY OF MILAN, DEPARTMENT OF CHEMISTRY Silvia DONZELLA of UNIVERSITY OF MILAN, DEPARTMENT OF FOOD, ENVIRONMENTAL AND NUTRITIONAL SCIENCES Eleonora PARGOLETTI of UNIVERSITY OF MILAN, DEPARTMENT OF CHEMISTRY Marco RABUFFETTI of UNIVERSITY OF MILAN, DEPARTMENT OF CHEMISTRY Teodora BAVARO of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES Giuseppe CAPPELLETTI of UNIVERSITY OF MILAN, DEPARTMENT OF CHEMISTRY Concetta COMPAGNO of UNIVERSITY OF MILAN, DEPARTMENT OF FOOD, ENVIRONMENTAL AND NUTRITIONAL SCIENCES Francesco MOLINARI of UNIVERSITY OF MILAN, DEPARTMENT OF FOOD, ENVIRONMENTAL AND NUTRITIONAL SCIENCES Giovanna SPERANZA of UNIVERSITY OF MILAN, DEPARTMENT OF CHEMISTRY Daniela UBIALI of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sugar fatty acid esters / bio-based surfactants / cheese whey permeate / waste upcycling |
| Purpose: | Cheese whey (CW) is the main waste stream of dairy industry accounting for ~200 million tons worldwide with ~40 million tons being produced in the European Union. [1] After protein recovery, the resulting cheese whey permeate (CWP) contains mainly lactose, thus providing a pool of carbohydrates (lactose, glucose and galactose) that can be upcycled for the synthesis of fine and commodity chemicals. In this project, an integrated bioprocess was set-up for the synthesis of Sugar Fatty Acid Esters (SFAE) which are valuable non-ionic surfactants. [2] CWP was used both as the feedstock for enzymatic biotransformations and for growing oleaginous yeasts to produce the galactose-based “head” and the lipid “tail” of SFAE, respectively (Figure). As a proof-of-concept, 1-butyl β-D-galactopyranoside was synthesized by reacting commercial lactose (1 mmol) with 1-BuOH through a transglycosylation reaction catalyzed by the covalently immobilized β-galactosidase from Aspergillus oryzae in a ternary system composed of alcohol/acetone/McIlvane buffer pH 4.3 (50/30/20). Subsequently, 1-butyl β-D-galactopyranoside was submitted to the esterification step with commercial palmitic acid in a solvent-free system at 80 °C by using Novozym 435 as the biocatalyst, affording pure n-butyl 6-O-palmitoyl-β-D-galactopyranoside. Interfacial features and emulsifying properties of the synthesized SFAE were evaluated. This sugar ester showed promising interfacial features and was able to stabilize water-in-oil emulsions up to 48 h. [3] At this point, we moved to the use of the crude CWP as substrate. On the one hand, CWP was submitted directly to the transglycosylation reaction in 1-BuOH, giving 1-butyl β-D-galactopyranoside in a yield comparable to that of the reference process (40%). On the other hand, microbial lipids were produced by growing oleaginous yeasts on CWP and mango syrup obtaining cells with a 45% lipid content. Microbial lipids were extracted and their composition both in terms of lipid species and fatty acid profile was assessed by GC-MS. Triglycerides are the most abundant lipids (77%) containing almost 50% saturated fatty acids (palmitic and stearic acids) and mostly oleic acid as the unsaturated component. [4] The esterification of 1-butyl β-D-galactopyranoside with the microbial lipids is currently in progress. Acknowledgements This work was financially supported by Cariplo Foundation (Italy) (call: “Circular Economy for a sustainable future 2020”, project BioSurf, ID 2020-1094). M.S.R. is supported by the project NODES which has received funding from the MUR – M4C2 1.5 of PNRR with grant agreement no. ECS00000036. |
| References: | [1] F.J. Barba, Foods 2021, 10, 564. [2] B. Pérez, S. Anankanbil, Z. Guo, in Fatty Acids: Chemistry, Synthesis and Applications, 2017, Chp. 10, 329-354. [3] R. Semproli, M.S. Robescu, S. Sangiorgio, E. Pargoletti, T. Bavaro, M. Rabuffetti, G. Cappelletti, G. Speranza, D. Ubiali, ChemPlusChem 2023, 88, e202200331. [4] S. Donzella, A. Fumagalli, S. Arioli, L. Pellegrino, P. D’Incecco, F. Molinari, G. Speranza, D. Ubiali, M.S. Robescu, C. Compagno, Fermentation 2022, 8, 341. |
| Figures: | 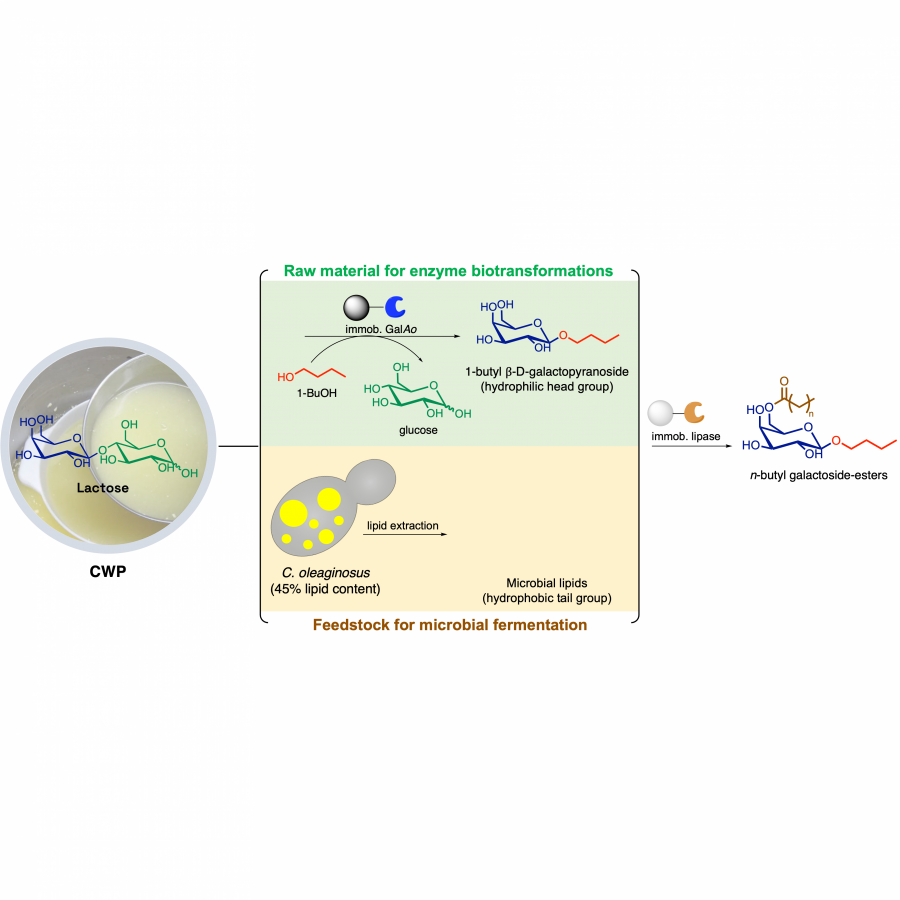 Figure Integrated bioprocess for the upcycling of cheese whey permeate into bio-based surfactants (www.biosurfproject.it). |
#495 | Enzymatic degradation of low molecular weight polyurethane model systems in ionic liquids |
|
| Presenting author: | Pedro LOZANO of UNIERSIDAD DE MURCIA (SPAIN) |
| Other authors: | Rebeca SALAS of UNIVERSIDAD DE MURCIA ROCIO VILLA of UNIVERSIDAD DE MURCIA JAIRTON DUPONT of UNIVERSIDAD DE MURCIA ANTONIO DONAIRE of UNIVERSIDAD DE MURCIA EDUARDO GARCIA-VERDUGO of UNIVERSIDAD JAUME I |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | applied biocatalysis / ionic liquids / sustainable chemistry / polyurethane |
| Purpose: | The high increase in the production of a wild range of manmade polymers has become a huge threat to the environment due to their unabated disposal. The polymeric materials have very stable chemical bonds, and their xenobiotic nature marks them a rising problem all around the globe. Among them, the polyurethane family of polymers account for the highest market share owing to their high resilience, durability, and versatility which ultimately leads to the generation of huge volumes of plastic waste, posing severe environmental problems. [1] Polyurethane (PU) is synthesized from two monomers, a diol and a highly toxic and reactive isocyanate in the presence of some additives. The resulting polymer is characterized for the urethane bond in its structure, being necessary the use of severe reaction conditions (i.e. >200 °C) to obtain the depolymerization of this chemical structure. [2] Moreover, state-of-the-art chemical methods for recycling are not adequate for handling the current volumes of waste. [3] In this work, it is proposed a new sustainable approach towards PU degradation by means of using Ionic Liquids (IL) and enzymes as catalysts. ILs are exceptional non-aqueous reaction media for carrying out both chemo- and biocatalytic processes and they also have been shown to be an excellent tool to integrate reaction and separation steps. [4] This study aimed at developing a green recycling pathway for PU by means of a PU-model system. Different enzyme families coupled with an IL/(bio)catalysts system have been assayed under mild reaction conditions. The results shown that the combination of suitable IL (e.g., [C4mim] [NTf2], [N4111] [NTf2], etc), and biocatalyst results in an excellent synergy able to break the intra-molecular urethane bond of this molecule. |
| References: | [1] Skleničková, K. Abbrent, S. Halecký, M. Kočí, V. and Beneš, H. Polyurethane Foam Residue Biodegradation through the Tenebrio molitor Digestive Tract: Microbial Communities and Enzymatic Activity. Crit. Rev. Environ. Sci. Technol., 2022, 52, 157-202. [2] Vanbergen, T. Verlent, I. De Geeter, J. Haelterman, B. Claes L. and De Vos, D. Recycling of Flexible Polyurethane Foam by Split-Phase Alcoholysis: Identification of Additives and Alcoholyzing Agents to Reach Higher Efficiencies. ChemSusChem, 2020, 13, 3835-3843. [3] Simón, D. Borreguero, A. M. de Lucas, A. and Rodríguez, J. F. Recycling of polyurethanes from laboratory to industry, a journey towards the sustainability. Waste Manag., 2018, 76, 147-171. [4] Villa, R. Alvarez, E. Porcar, R. García-Verdugo, E. Luis, S. V. Lozano, P. Ionic liquids as an enabling tool to integrate reaction and separation processes. Green Chem, 2019, 21, 6527-6544. Acknowledgements This work was partially supported by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021- 124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN -European Union Next Generation EU-PRTR (TED2021-129626B-C21/C22) and SENECA (21884/PI/22) for financial support. J. D. is fellow of the “Maria Zambrano” Program at the University of Murcia (Spain). R.V is fellow of the “Margarita Salas” Program” at the University of Murcia (Spain). |
| Figures: | 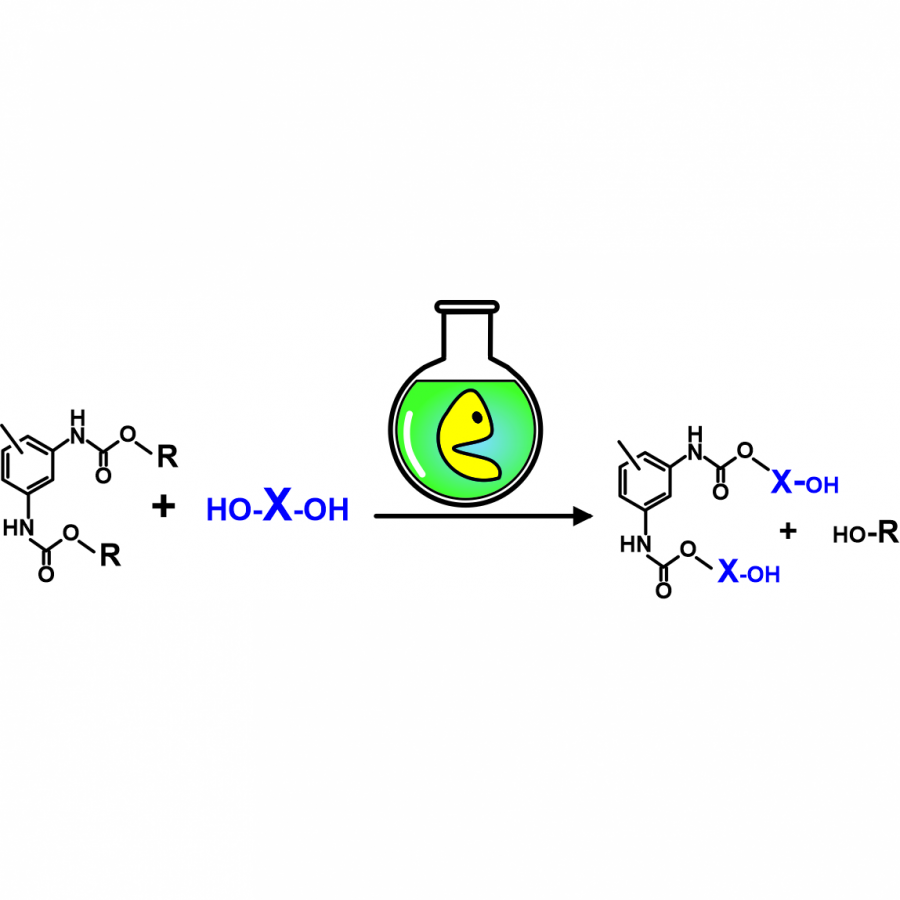 Figure 1 Schema of the degradation reaction of a polyurethane model system by using an IL/enzyme catalyst system |
#502 | Flow-Injection Mass Spectrometry: A Rapid and Versatile Method for Screening Enzymatic Reactions |
|
| Presenting author: | OROZ-GUINEA Isabel of UNIVERSITY OF GRAZ |
| Corresponding author: | Joerg SCHRITTWIESER of UNIVERSITY OF GRAZ |
| Other authors: | Sarah BERGER of UNIVERSITY OF GRAZ Christopher GRIMM of UNIVERSITY OF GRAZ Jonathan NYENHUIS of UNIVERSITY OF GRAZ Stefan PAYER of UNIVERSITY OF GRAZ Isabel OROZ-GUINEA of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flow Injection Analysis / Mass Spectrometry / High-Throughput Screening / |
| Purpose: | The screening of enzymatic reactions traditionally relies on photometric and fluorometric assays, which offer high throughput but are restricted to substrates with the required optical properties, or on chromatographic methods, which are very general but require analysis times of several minutes per sample. Mass spectrometry (MS) without chromatographic separation has the potential to bridge the gap between these two approaches, as it is broadly applicable and relatively fast (analysis times ≤1 min/sample). Powerful MS-based high-throughput screening methods have been reported in recent years,[1–3] but these rely on specialised or even custom-built equipment, which is accessible to only few research groups. We have found that flow-injection mass spectrometry (FIA–MS) performed on a common instrumental platform, single-quadrupole HPLC–MS, can be used for the qualitative and quantitative analysis of diverse biotransformations. Samples are injected into the eluent flow of a liquid chromatograph with no column installed and are thus introduced into the electrospray MS with a throughput of 1 sample per minute. Common organic buffers (e.g., bicine, tricine, MOPS) present in the biotransformations can fulfil the function of an internal standard, allowing a linear quantification of analytes over 1–2 orders of magnitude in concentration. We demonstrate the application of FIA–MS to the screening of reactions catalysed by imine reductases, transaminases, methyltransferases, and a strictosidine synthase, using crude biocatalyst preparations (lysates or whole cells) and straightforward, plate-based liquid handling workflows. In each case, FIA–MS rapidly generated actionable insights into enzyme activity and selectivity that were readily confirmed by chromatographic re-analysis. We are, therefore, convinced that FIA–MS will become a useful additional tool for the screening of enzymatic reactions. |
| References: | [1] E. E. Kempa, J. L. Galman, F. Parmeggiani, J. R. Marshall, J. Malassis, C. Q. Fontenelle, J.-B. Vendeville, B. Linclau, S. J. Charnock, S. L. Flitsch, N. J. Turner, P. E. Barran, JACS Au 2021, 1, 508-516. [2] D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore, S. Sun, R. T. Kennedy, Angew. Chem. Int. Ed. 2020, 59, 4470-4477. [3] I. Sinclair, M. Bachman, D. Addison, M. Rohman, D. C. Murray, G. Davies, E. Mouchet, M. E. Tonge, R. G. Stearns, L. Ghislain, S. S. Datwani, L. Majlof, E. Hall, G. R. Jones, E. Hoyes, J. Olechno, R. N. Ellson, P. E. Barran, S. D. Pringle, M. R. Morris, J. Wingfield, Anal. Chem. 2019, 91, 3790-3794. |
| Figures: | 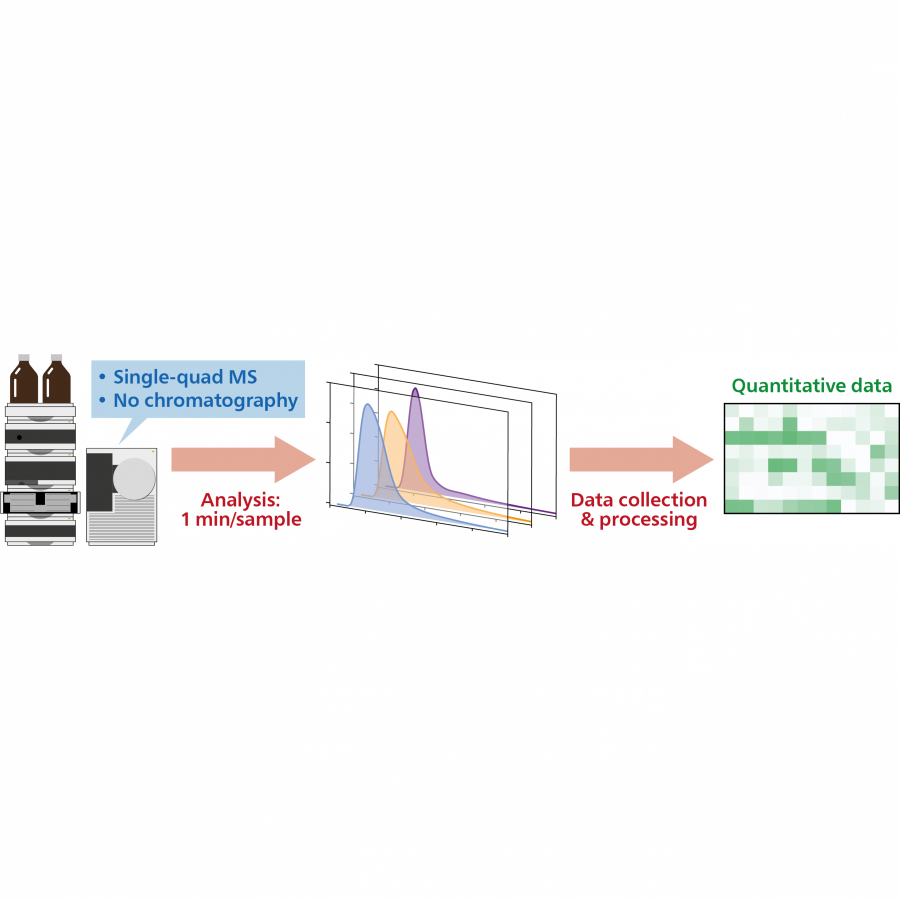 Figure 1 Screening of enzymatic reactions by flow injection analysis mass spectrometry |
#509 | A novel bacterial short-chain dehydrogenase/reductase (SDR) active on guaiacyl, p-hydroxyphenyl and syringyl monolignols |
|
| Presenting author: | Anna BRILHANTE of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY |
| Other authors: | Priscila GIUSEPPE of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY Lúcia WOLF of BRAZILIAN BIORENEWABLES NATIONAL LABORATORY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | alcohol dehydrogenase / aromatic compounds / lignin / biotransformation |
| Purpose: | The conversion of lignin-derived aromatics (LDA) into value-added chemicals is a renewable alternative to support the transition from a fossil-based industry to a sustainable economy based on renewable sources. Given the heterogeneity of lignin depolymerization products, convergent biocatalysis performed by microorganisms has emerged as a promising approach to funneling LDA into target molecules. Biological pathways and enzymes for the catabolism of aromatic compounds derived from the syringyl (S), guaiacyl (G), and p -hydroxyphenyl (H) subunits of lignin (e.g. syringate, vanilate, and p-coumarate, respectively) have been described for some bacterial strains [1] . Despite these advances, there is still little information regarding the enzymatic strategies for the catabolism of the monolignols coniferyl alcohol (G), p-coumaryl alcohol (H) and sinapyl alcohol (S). In this work, we identified an aryl-alcohol dehydrogenase (XarA) from Xanthomonas citri pv. citri 306 that belongs to the short-chain dehydrogenase/reductases (SDR) superfamily. Its gene (XAC0353) is located close to the operon of protocatechuate catabolism, a key intermediate in the metabolism of H and G lignin-derived aromatic compounds. XarA has about 60% sequence identity with a coniferyl alcohol dehydrogenase from Pseudomonas sp. HR199 (CalAHR199), the only bacterial SDR active on coniferyl alcohol with experimental data available so far (U.S. Patent Application Publication No. 2003/0228670 A1). However, the substrate range of CalAHR199 was poorly investigated and the structural properties defining substrate recognition mechanisms of bacterial SDRs able to oxidize monolignols are still elusive. Therefore, our group aims to investigate XarA substrate profile and structure-function relationships using as main approaches biochemical and biophysical assays in vitro, X-ray diffraction crystallography, cryo-EM and molecular dynamic simulations. Here, we highlight the main results obtained so far. Biochemical assays demonstrated that XarA is a NAD+-dependent dehydrogenase, with optimal activity at 30 °C (pH 10). XarA specific activity on several aryl-alcohols, evaluated by NADH quantification using HPLC, showed the following profile (relative activity): p-coumaryl alcohol (100%) > cinnamyl alcohol (83%) > coniferyl alcohol (48%) > sinapyl alcohol (34%) > 4-hydroxybenzyl alcohol (10%) > vanillyl alcohol (3%) > benzyl alcohol (2%), indicating that XarA has greater activity on aryl alcohols containing longer (propenol) side chains, with a decrease in specific activity as the degree of methoxylation in the aromatic ring increases, and only a residual activity on aryl alcohols with shorter (methanol) side chains. XarA formed crystals in complex with NAD+, which were soaked with coniferyl alcohol and diffracted to 2.9 Å resolution. The crystal structure was solved by Molecular Replacement, using an AlphaFold-generated model structure as template, and model refinement is in progress. SEC-RALS/LALS assays indicate that XarA forms dodecamers in solution that can dissociate into smaller oligomers depending on the medium composition. The mechanisms underlying this oligomeric state transition will be further investigated by cryo-EM analyses. In summary, to the best of our knowledge, this is the first report of a bacterial SDR family member able to convert the three main precursors of lignin (G, H and S monolignols) into their respective aldehydes, the first step required for their entry into catabolic funneling pathways. By combining structural analyses, molecular dynamics simulations and kinetic studies, we expect to provide a deeper understanding on the substrate recognition mechanisms that determine the substrate preference of XarA, thus contributing to the knowledge regarding the structure-function relationships of SDRs active on monolignols and supporting the development of biotechnologies for the sustainable production of industrially relevant bioproducts. |
| References: | [1] Ravi, K. et al, 2019, bioresour. technol., 285, 121327. |
#514 | What neutrons can do for you: The single crystal neutron diffractometer BIODIFF at the Heinz Maier-Leibnitz Zentrum (MLZ) and a short excursion to Small-angle neutron diffraction |
|
| Presenting author: | Tobias Erich SCHRADER of FORSCHUNGSZENTRUM JÜLICH, JUELICH CENTRE FOR NEUTRON SCIENCE |
| Other authors: | Andreas OSTERMANN of FRMII, TECHNISCHE UNIVERSITÄT MÜNCHEN |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | neutron / Small-angle / crystallography / scattering |
| Purpose: | Neutrons are scattered from the nuclei and x-rays are scattered from the electrons of the atoms in a protein crystal. This renders these two scattering probes as being complementary to each other. The neutrons can see the hydrogen atom positions in a protein crystal. This allows to determine protonation states of crucial amino acid residues in the active centre of an enzyme or one can detect water clusters and proton paths to the active centre by locating water molecules and their exact orientation and hydrogen bonding. Using the fact that hydrogen has a negative scattering length and deuterium has a positive one, proteins or DNA can be matched out at different solvent compositions of heavy and light water. This is heavily used in Small-angle neutron scattering where for example protein-DNA complexes can be disentangled in solution phase that way. In this contribution neutron protein crystallography is introduced using the example of alcohol dehydrogenase from the organism Lactobacillus brevis (LbADH), an enzyme which catalyzes the reduction of prochiral ketones to the corresponding secondary alcohols [1]. The data set for this project was taken with the instrument BIODIFF. The neutron single crystal diffractometer BIODIFF at the research reactor Heinz Maier-Leibnitz (FRM II) is especially designed to collect data from crystals with large unit cells. The main field of application is the structural analysis of proteins, especially the determination of hydrogen atom positions. BIODIFF is a joint project of the Jülich Centre for Neutron Science (JCNS) and the FRM II. BIODIFF is designed as a monochromatic instrument with a narrow wavelength spread of less than 3 %. To cover a large solid angle the main detector of BIODIFF consists of a neutron imaging plate in a cylindrical geometry with online read-out capability. BIODFF is equipped with a standard Oxford Cryosystem “Cryostream 700+” which allows measurements at 100 K. But in case of the alcohol dehydrogenase project, room temperature data collection was chosen. Here, the crystal was kept in a glass capillary. The resulting data led to a better understanding of the role of the Magensium ion in substrate binding and it showed a new hydrogen bonding network close to the active centre of the enzyme. It also showed nicely the complementary nature of x-ray and neutron protein crystallography. The metal ion in Figure 1 has not been detected by neutron scattering but it was easily seen by x-ray scattering. The reason for this lies in a cancellation effect between the negative scattering length of Manganese ions and the average positive scattering lengths of Magnesium ions which just cancel to zero in this position. The Magnesium ions were present in the crystallization condition, but the Manganese Ions must stem from the expression of the protein in the E.coli expression system. Time permitting, I will show some results on protein crystal growth based on Small-angle neutron scattering data where radiation damage did not play a role [2]. So, the complete time and length scale from the monomeric protein molecules to the final protein crystals could be followed without disturbing the growth of the protein crystals with radiation damage which would not have been possible using SAXS measurements. E-mail: t.schrader@fz-juelich.de |
| References: | [1] Hermann et al., Acta Cryst. (2018). F74, 754 [2] Heigl et al., Cryst. Growth Des. 2018, 18, 1483 |
| Figures: | 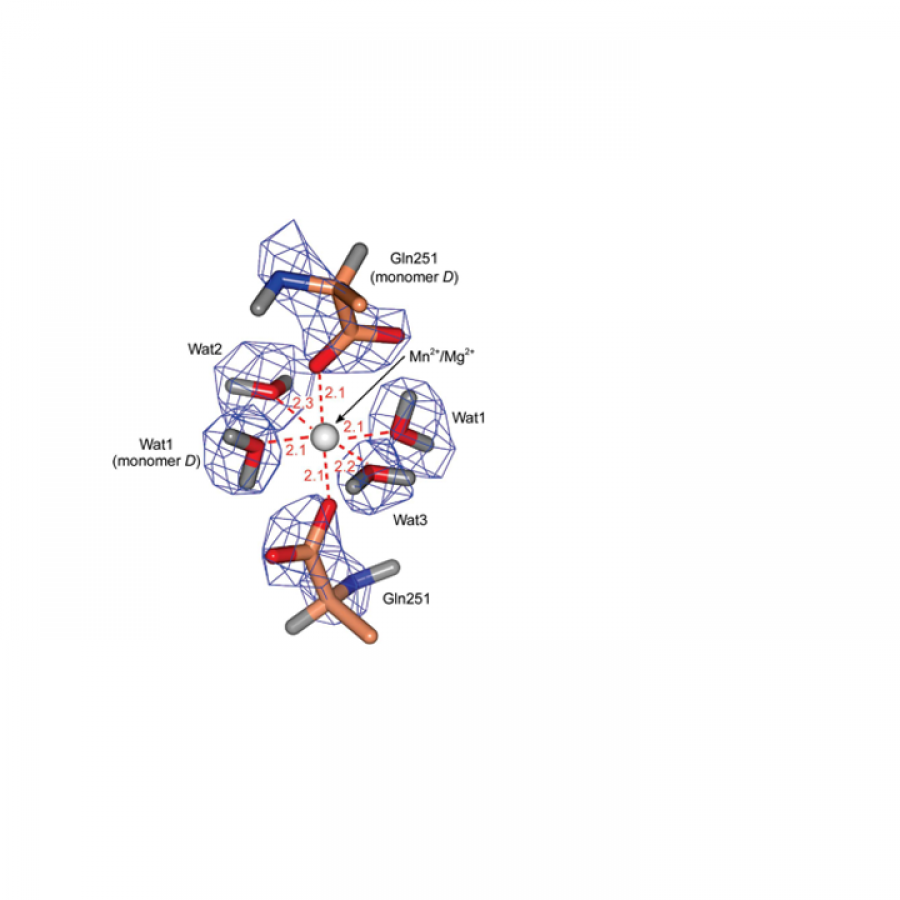 Figure 1 An neutron omit map of the involved amino acid residues from two copies of the protein Alcohol dehydrogenase and the water molecules. No density is observed for the central metal ion. 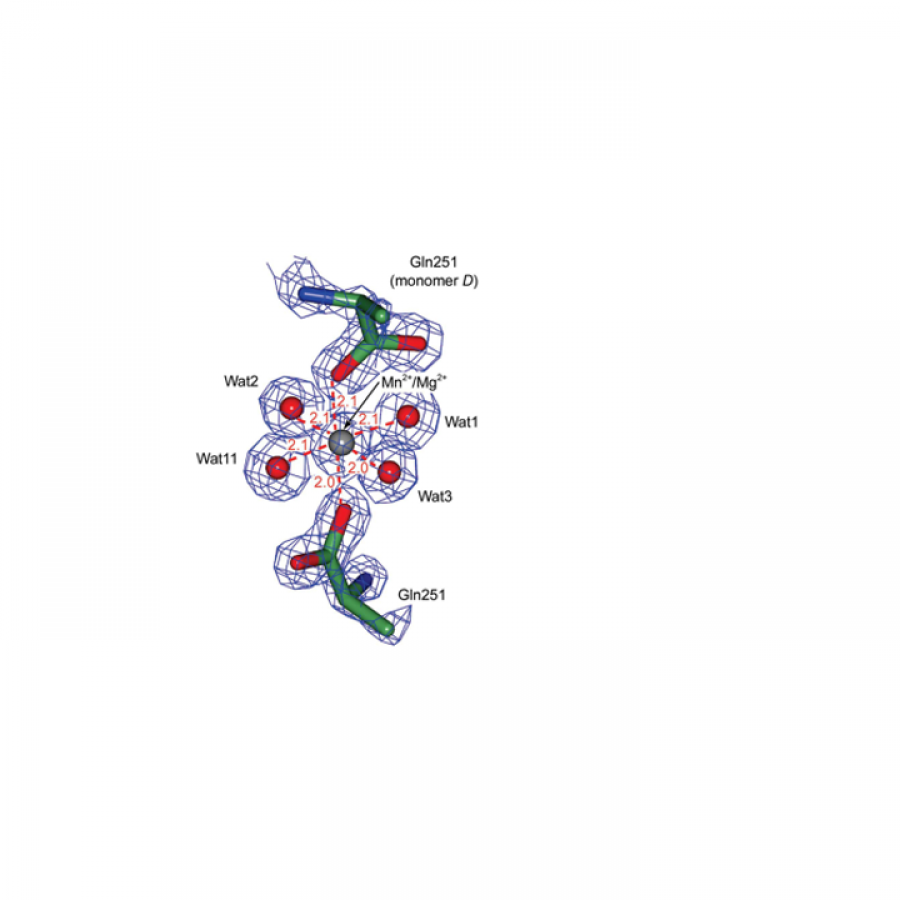 Figure 2 The same spot as in Figure 1 in the crystal as seen by x-rays: Here the metal atom is clearly observed. Figures taken from [1]. |
#518 | Peroxygenase-Promoted Enzymatic Cascades for the Valorisation of Fatty Acids |
|
| Presenting author: | Yinqi WU of DELFT UNIVERSITY OF TECHNOLOGY |
| Corresponding author: | Frank HOLLMANN of DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Caroline PAUL of DELFT UNIVERSITY OF TECHNOLOGY Thomas HILBERATH of DELFT UNIVERSITY OF TECHNOLOGY Ewald JONGKIND of DELFT UNIVERSITY OF TECHNOLOGY Wuyuan ZHANG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY, CHINESE ACADEMY OF SCIENCES Miguel ALCALDE of INSTITUTE OF CATALYSIS AND PETROCHEMISTRY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | fatty acid valorisation / peroxygenase / selective oxyfunctionalisation / multi-enzyme cascades |
| Purpose: | Fatty acids as renewable feedstock are used for the synthesis of a range of products such as polymer building blocks, paints, coatings, surfactants and lubricants.[1] Biocatalytic transformations of fatty acids generally relies on pre-existing functional groups, such as the carboxylate group or C=C-double bonds, enabling (trans)esterification, amidation, reduction reactions, epoxidation and hydration. Addition of new functionalities into the hydrocarbon part opens up new possibilities for fatty acid valorization (Scheme 1). Recently, cytochrome P450 monooxygenases exhibiting alpha-, beta-, delta- or omega-selectivity for the hydroxylation of non-activated C-H-bonds in fatty acids have been reported;[2] interestingly fungal peroxygenases also display activity. While peroxygenase-catalysed hydroxylations benefit from the intrinsically much simpler reaction mechanism compared to P450 enzymes, their practical usefulness is hampered by their generally low selectivity towards fatty acids.[3] In our recent protein engineering study on the prototype evolved peroxygenase from Agrocybe aegerita (AaeUPO-PaDa-I) for selective fatty acid hydroxylations, a new mutant (AaeUPO-Fett) was reported for high regioselectivity (almost exclusively located at omega-1).[4] In this contribution, we extended the synthetic potential of non-functionalised fatty acids as starting materials for value-added products and building blocks using AaeUPO-Fett as a promising catalysts to add functionality to the alkyl chain. The primary products (i.e. hydroxyl fatty acids (esters)) are interesting building blocks for lactone and polyester synthesis. If oxidised to the corresponding ketoacids, further synthetic possibilities arise as demonstrated by the Baeyer-Villiger oxidation and the reductive amination reactions in multi-enzyme cascades, thereby paving the way for new fatty acid valorization pathways. |
| References: | [1] a) C. Aouf, E. Durand, J. Lecomte, M.-C. Figueroa-Espinoza, E. Dubreucq, H. Fulcrand, P. Villeneuve, Green Chem. 2014, 16, 1740-1754; b) U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. Meier, J. O. Metzger, Angew. Chem. Int. Ed. 2021, 60, 20144-20165. [2] L. Hammerer, C. K. Winkler, W. Kroutil, Catal. Lett. 2018, 148, 787-812. [3] a) A. Olmedo, C. Aranda, J. C. Del Rio, J. Kiebist, K. Scheibner, A. T. Martinez, A. Gutierrez, Angew. Chem. 2016, 128, 12436-12439; b) C. Aranda, J. Carro, A. Gonzalez-Benjumea, E. D. Babot, A. Olmedo, D. Linde, A. T. Martinez, A. J. B. A. Gutierrez, Biotechnol. Adv. 2021, 51, 107703. [4] P. Gomez de Santos, A. Gonzalez-Benjumea, A. Fernandez Garcia, Y. Wu, A. But, P. Molina-Espeja, D. M. Mate, D. Gonzalez-Parez, W. Zhang, J. Kiebist, Angew. Chem. Int. Ed. 2023, 62, e2022173. |
| Figures: | 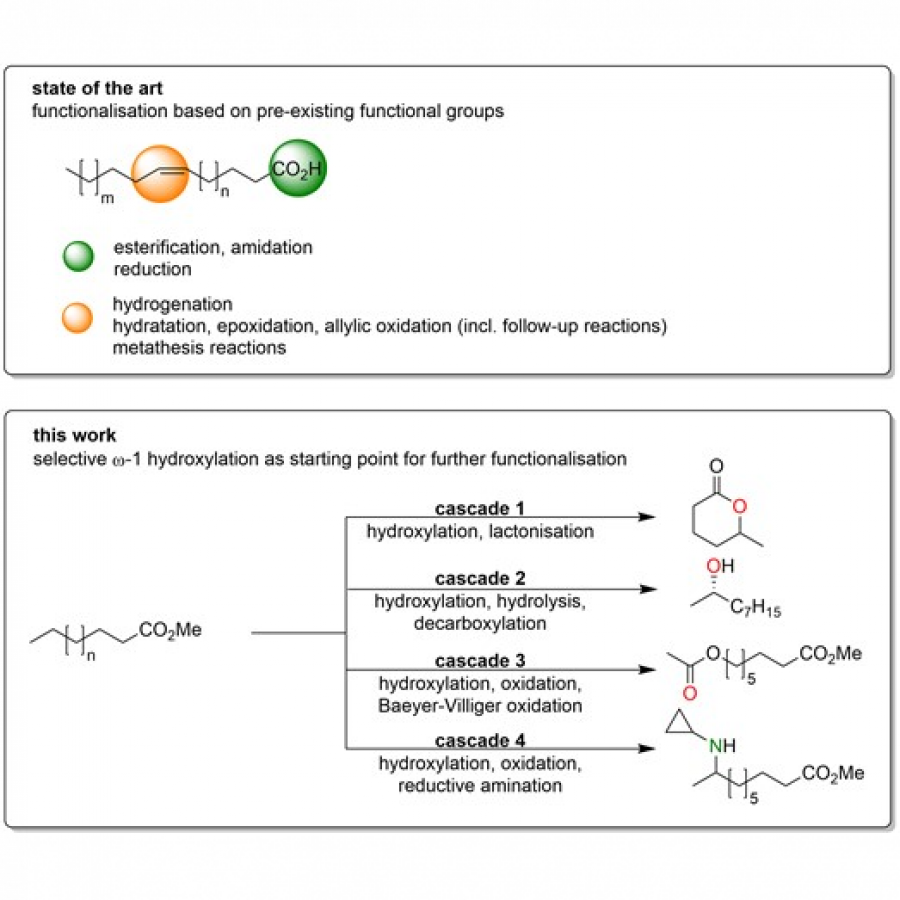 Scheme 1. Established fatty acid valorisation approaches utilise existing functionalities such as the carboxylate group or pre-existing C=C-double bonds. In this contribution, we add functionality via selective, peroxygenase-catalysed hydroxylation enabling further |
#520 | Enzyme-assisted CO2 capture from industrial emissions using mine tailings as co-sequestrating agents |
|
| Presenting author: | Io ANTONOPOULOU of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Other authors: | AYANNE DE OLIVEIRA MACIEL of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY ULRIKA ROVA of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY PAUL CHRISTAKOPOULOS of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | CO2 capture / Carbonic anhydrase / Mining tailings / Climate change |
| Purpose: | Global temperatures are expected to rise by at least 2.6 °C by the end of the century, even if all signatory states effectively carry out the commitments set in the 2015 Paris Agreement, targeting global mean temperatures below 2 °C by 2100 [1]. It is evident that achieving net zero emissions would need significant emission reductions, which will make necessary the investments in novel and technologically advanced solutions as a large share of the reductions will come from technologies that are currently at the demonstration or prototype phase. The mining industry has a high potential for Carbon Capture and Storage (CCS). Enhanced weathering, where the natural reactions between CO2 and silicate minerals that produce dissolved bicarbonate ions are accelerated, has the potential to remove substantial amounts of CO2 from the environment. The global mining industry produces huge volumes of waste that could be utilized as feedstock for enhanced weathering. The annual global enhanced weathering potential of mined metal commodity tailings from silicate-hosted deposits is estimated to 31 and 125% of the industry’s primary emissions. However, current knowledge suggests that dissolution rates of many minerals are relatively slow, such that only a small percentage (∼3–21%) of this potential may be realized on timescales of <50 years. Thus, methodologies for accelerating weathering reactions, must be developed [2]. Carbonic anhydrase (CA) is one of the fastest enzymes in nature, catalyzing the hydration of CO2 to bicarbonate [3]. Integrating carbonic anhydrase in a natural weathering process, will boost bicarbonate formation which is key for further carbonate dissolution to form bicarbonate. The liquid bicarbonate solution, which is a temporary storage form for captured CO2, could subsequently be pumped into old or mined-out mine shafts/drifts to facilitate carbonation of the bedrock and thereby storing atmospheric carbon dioxide. Thus, formation of bicarbonate is strongly urged in order to provide both an efficient CO2 capture step and easy transportation for further permanent storage. Aim of this work is to assess a novel evolved CA from Desulfovibrio vulgaris [4,5] for its potential to enhance the weathering of mine tailing waste. Preliminary results showed that, by adding CA into an aqueous solution of mine tailing waste, the silicate dissolution rate was increased 16.45 times at the first 40 min of reaction, while the CO2 capture yield was improved 2.29 times. The CA was also characterized for its stability towards potential inhibitors found in mine tailings (e.g. sulfates, carbonates). Further optimization will elucidate the potential of enzymatic CO2 hydration towards Carbon Dioxide Removal (CDR) practices to achieve the Paris Agreement ambition and mitigate climate change. This work is supported by the Swedish Energy Agency funded project TAILOR-MADE (Use of mine tailings to carbonate minerals by a symbiotic CC and DAC bio-strategy, Dnr number P2022-01123). |
| References: | 1. IPCC (2022) Climate Change 2022: Impacts, Adaptation and Vulnerability. https://www.ipcc.ch/report/ar6/wg2/. 2. Bullock et al. Global Carbon Dioxide Removal Potential of Waste Materials From Metal and Diamond Mining. Global Carbon Dioxide Removal Potential of Waste Materials From Metal and Diamond Mining. Front. Clim. 3:694175 (2021). 3. De Oliveira Maciel, A., Rova, U., Christakopoulos, P., Antonopoulou I. (2022). Carbonic anhydrase to boost CO2 sequestration: Improving carbon capture utilization and storage (CCUS). Chemosphere, 299, 134419. 4. Alvizo, O., Nguyen, L.J., Savile, C.K. et al. (2014). Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas. PNAS, 111(46), 16436-16441. 5. Sjöblom, M., Antonopoulou, I., Giménez, I.G., de Oliveira Maciel, A., Khokarale, S.G., Mikkola, J.-P., Rova, U., Christakopoulos, P. (2020). Enzyme-assisted CO2 absorption in aqueous amino acid ionic liquid amine blends. ACS Sustainable Chemistry & Engineering, 36(8), 13672-13682. |
#522 | Enantio-complementary synthesis of 2-substituted pyrrolidines and piperidines via transaminase-triggered cyclizations |
|
| Presenting author: | Christian HECKMANN of DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Caroline E. PAUL of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Transaminase / chiral amines / N-heterocycles / Asymmetric synthesis |
| Purpose: | Transaminases are powerful biocatalysts for the synthesis of chiral primary amines. These primary amines may then be further reacted to access secondary and/or tertiary amines, through e.g. spontaneous formation of cyclic imines followed or organo-catalysed Mannich-reaction,[1] enzymatic or phosphate-catalysed Pictet–Spengler reactions,[2] and Pd-catalysed arylation.[3] Less explored is the utilization of the newly formed amine in spontaneous SN2-type reactions.[4] Here, we employ transaminases to form reactive amine intermediates, which then spontaneously undergo cyclizations via SN2-type reactions to give valuable chiral 2-substituted pyrrolidines and piperidines (Figure 1), a common motif in many active pharmaceutical ingredients. To this end, a panel of (S)- and (R)-selective amine transaminases was screened against a variety of substituted chloro-ketones, achieving analytical yields of up to 90% and enantiomeric excesses of up to >99.5%, with access to both enantiomers. This strategy was then applied to synthesize (R)-2-(p-chlorophenyl)pyrrolidine on a 300 mg scale, affording 84% isolated yield, with >99.5% ee. |
| References: | [1] F. Taday, R. Cairns, A. O’Connell, E. O’Reilly, Chem. Commun. 2022, 58, 1697-1700. [2] V. Erdmann, B. R. Lichman, J. Zhao, R. C. Simon, W. Kroutil, J. M. Ward, H. C. Hailes, D. Rother, Angew. Chem. Int. Ed. 2017, 56, 12503-12507. [3] C. M. Heckmann, F. Paradisi, Chem. Eur. J. 2021, 27, 16616-16620. [4] C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman, G. J. Hughes, Science 2010, 329, 305-310. |
| Figures: | 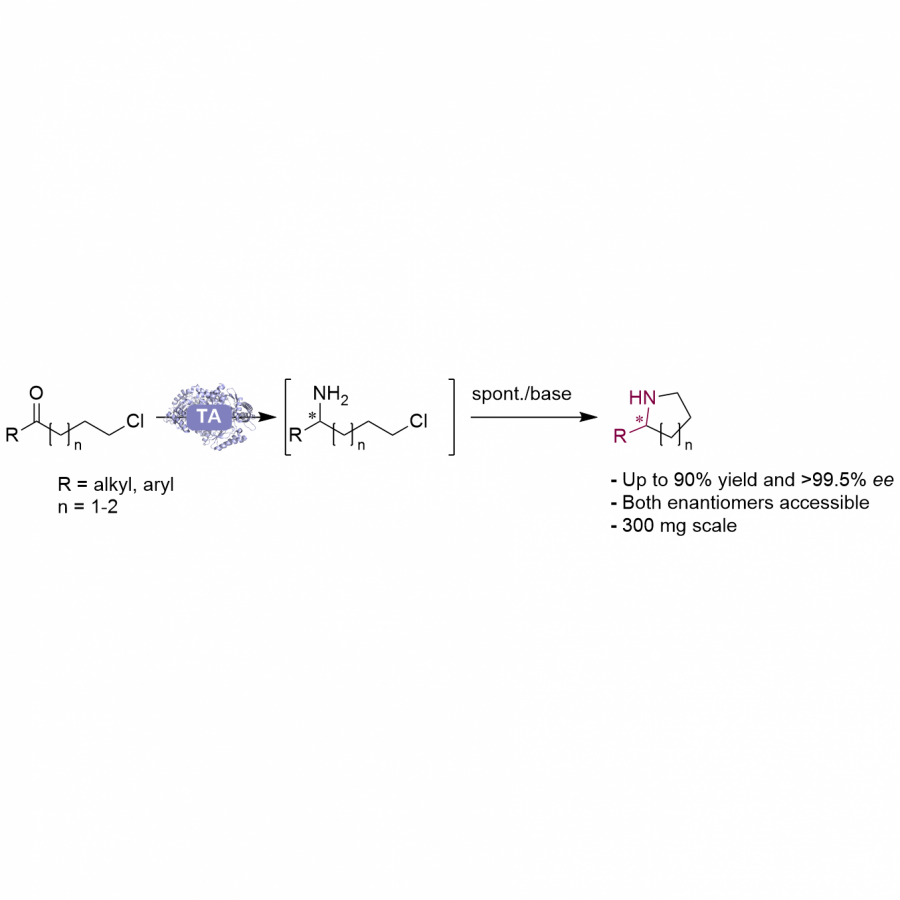 Figure 1 Transaminase-triggered SN2-type cyclization to 2-substituted pyrrolidines and piperidines. |
#526 | Novel Artificial Metalloenzymes for Copper-Catalyzed Enantioselective Michael Addition to Nitroalkenes |
|
| Presenting author: | Ru JIANG of UNIVERSITY OF GRONINGEN |
| Corresponding author: | Gerard ROELFES of UNIVERSITY OF GRONINGEN |
| Other authors: | Fabrizio CASILLI of UNIVERSITY OF GRONINGEN |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzyme / copper catalysis / Michael addition of nitroalkenes / enantioselectivity |
| Purpose: | To combine the attractive features of transition metal catalysis and biocatalysis, artificial enzymes, which consist of abiological catalytic moieties incorporated into protein scaffolds, have emerged as a promising strategy to realize non-natural reactions in biocatalysis. LmrR, a small, homodimeric protein that contains a large hydrophobic pore at its dimer interface, is a privileged scaffold for artificial metalloenzyme (ArM) design. Here, we incorporated the metal binding unnatural amino acid bipyridine alanine (BpyAla) into the protein scaffold LmrR through in vivo stop codon suppression and created a proficient and stereoselective artificial metalloenzyme (LmrR_XBpy) for catalytic asymmetric Michael addition of 2-acetyl azaarenes to nitroalkenes (up to 89±0% yield, 96±1% ee) (Fig 1). In our design, 2-acetyl azaarene 1 is activated by the new created [Cu-Bpy] site to form the enolate, whereas the aryl substituted nitroalkene 2 is the Michael acceptor, which is bound by the tryptophan residues (W96 and W96’) via π-stacking interactions, to give the addition product 3. Furthermore, derivatizing the nitro compounds 3 into potential natural products or drug targets renders this methodology attractive for the green synthesis. |
| References: | [1] (a) Pecoraro, V. L. et al. Chem. Rev. 2014, 114, 3495. (b) Pàmies, O.; Diéguez, M.; Bäckvall, J.-E. Adv. Synth. Catal. 2015, 357, 1567. (c) Schwizer, F. et al. Chem. Rev. 2018, 118, 142. [2] (a) Madoori, P. K.; Agustiandari, H.; Driessen A. J. M.; Thunnissen, A.-M. W. H. EMBO J. 2009, 28, 156. (b) Roelfes, G. Acc. Chem. Res. 2019, 52, 545. [3] (a) Wang, L.; Brock, A.; Herberich B.; Schultz, P. G. Science 2001, 292, 498. (b) Drienovská, I.; Rioz-Martínez, A.; Draksharapu, A.; Roelfes, G. Chem. Sci. 2015, 6, 770. |
| Figures: | 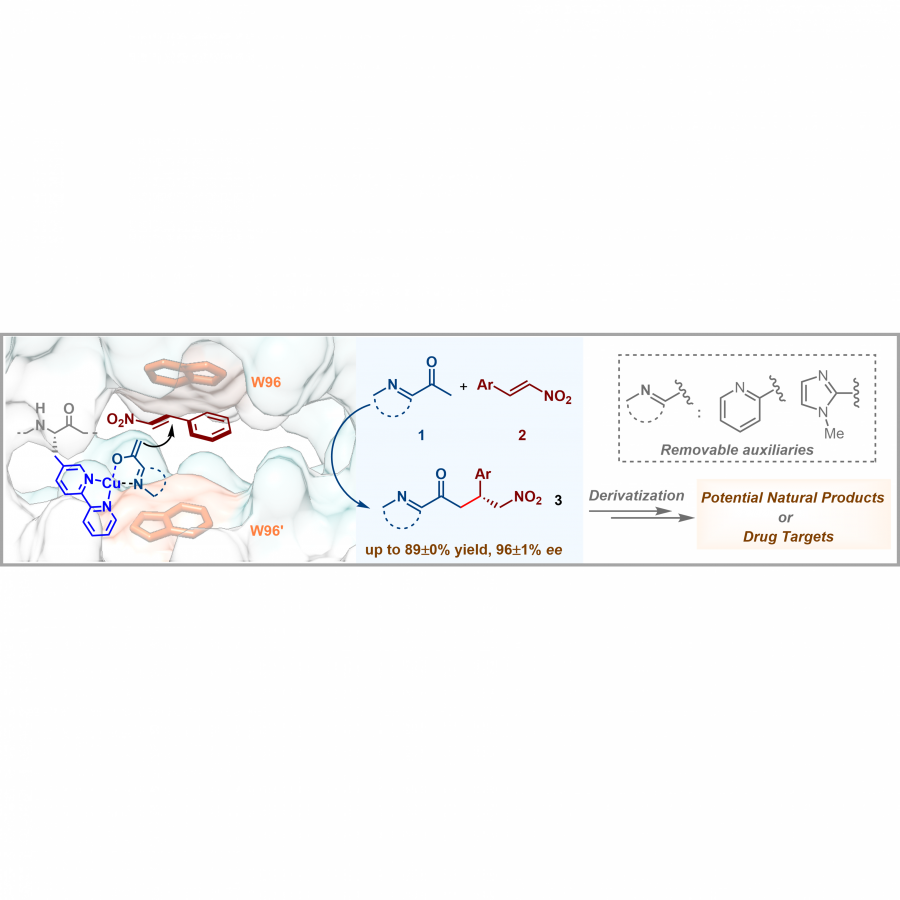 Fig. 1 Cu-catalyzed asymmetric Michael addition of 2-acetyl azaarenes to nitroalkenes in an artificial enzyme This figure shows that our reaction design, inculding reaction mechanism and derivatization of products. |
#527 | Engineering of bacterial terpene synthases for improved terpenoid production in Escherichia coli |
|
| Presenting author: | Nicole LEFERINK of FUTURE BIOMANUFACTURING RESEARCH HUB |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme engineering / Synthetic biology / Terpene synthase / Terpenoids |
| Purpose: | Terpenoids are a highly diverse group of natural products that are of considerable industrial interest. Monoterpenoids (C10) are, for example, used as flavour and fragrance ingredients, as antimicrobials in household products, but also as precursors for anti-cancer drugs and alternative fuels. Increasingly, engineered microbes are used for the production of terpenoids to replace natural extracts and chemical synthesis. All terpenoids are produced from the C5 building blocks isopentenyl diphosphate and dimethylallyl diphosphate, which are combined by prenyl pyrophosphate synthases resulting in substrates of varying lengths. Terpene synthases (TSs), the enzymes responsible for the wide structural diversity of terpenoids, guide the linear carbocationic intermediates along one of many cyclisation paths via exertion of subtle steric and electrostatic control, before the reaction is terminated by deprotonation or nucleophilic water attack. In addition to plants, bacteria are a rich source for TS activity. Bioinformatics searches of available bacterial genomes revealed over 2000 genes with a class I TS domain. In addition to 2-methyl isoborneol and geosmin synthases, TSs long known to be present in soil bacteria, many classical TSs were identified in a wide variety of bacterial species [1]. Most of these enzymes consist of a single class I TS domain only and exhibit sesqui- (C15) or di-terpene (C20) synthase activity. Until recently, only two known bacterial monoterpene synthases (MTSs) had been identified, a bi-functional linalool/nerolidol synthase (bLinS) and a cineole synthase (bCinS). Interestingly, these enzymes show a high-level of product fidelity, where plant MTSs are often highly promiscuous yielding product mixtures. We used a multi-disciplinary approach involving, data-mining, bio-informatics, X-ray crystallography, site-directed mutagenesis and computational chemistry to unravel the mechanism of high-fidelity bacterial TSs. Linalool is a valuable acyclic monoterpenoid product. bLinS from Streptomyces clavuligerus produces linalool from geranyl diphosphate (GPP, C10) and nerolidol from farnesyl diphosphate (FPP, C15). We engineered bLinS for increased linalool production in E. coli by constricting the active site, improving both linalool titre and purity [2]. bCinS from S. clavuligerus is the only known ‘true’ bacterial MTS and produces bi-cyclic 1,8-cineole from GPP at high purity, unlike plant CinS from Greek sage (CinS_Sf). We identified residues involved in key steps in the bCinS cyclisation cascade, including water attack and carbocation stabilisation. We also show that bCinS produces cineole almost exclusively via S-a-terpineol, where CinS_Sf appears to use both S- and R-isomers [3,4]. 10-epi-cubebol synthase from Sorangium cellulosum (ScCubS) forms >93% products with a tri-cyclic cubebane scaffold from FPP. We identified key residues that control the distribution between several early-stage cations which determine whether the final product is derived from the mono-, bi or tri-cyclic germacrane, cadalane, or cubebane hydrocarbon scaffold respectively [5]. Our results show that each terpene product requires a tailored active site, where each residue has a unique and precise function, and unlike high-fidelity enzymes, promiscuous enzymes may supply multiple routes to the end product. However, progress is slow and knowledge often does not translate to other TSs. Due to the absence of strong protein interactions with the carbocation intermediates, there is a remarkable lack of sequence-function relationship within the class I TS family, making product-outcome predictions from sequences alone highly challenging. Future work is therefore directed towards combining sequence diversity data, with product profiles and structural information in data-driven approaches for the predictive engineering of TS activity [6]. |
| References: | [1]. Reddy, G. K.; Leferink, N. G. H.; Umemura, M.; Ahmed, S. T.; Breitling, R.; Scrutton, N. S.; Takano, E., Exploring novel bacterial terpene synthases. PLoS One 2020, 15 (4), e0232220. [2]. Ferraz, C. A.; Leferink, N. G. H.; Kosov, I.; Scrutton, N. S., Isopentenol Utilization Pathway for the Production of Linalool in Escherichia coli Using an improved bacterial linalool/nerolidol synthase. Chembiochem 2021, 22 (13), 2325-2334. [3]. Leferink, N. G. H.; Ranaghan, K. E.; Battye, J.; Johannissen, L. O.; Hay, S.; van der Kamp, M. W.; Mulholland, A. J.; Scrutton, N. S., Taming the reactivity of monoterpene synthases to guide regioselective product hydroxylation. Chembiochem 2020, 21 (7), 985-990. [4]. Leferink, N. G. H.; Escorcia, A. M.; Ouwersloot, B. R.; Johanissen, L. O.; Hay, S.; van der Kamp, M. W.; Scrutton, N. S., Molecular determinants of carbocation cyclisation in bacterial monoterpene synthases. Chembiochem 2022, 23 (5), e202100688. [5]. Whitehead, J. N.; Leferink, N. G. H.; Reddy, G. K.; Levy, C. W.; Hay, S.; Takano, E.; Scrutton, N. S., How a 10-epi-cubebol synthase avoids premature reaction quenching to form a tricyclic product at high purity. ACS Catalysis 2022, 12 (19), 12123-12131. [6]. Leferink, N. G. H.; Scrutton, N. S., Predictive engineering of class I terpene synthases using experimental and computational approaches. Chembiochem 2022, 23 (5), e202100484. |
#528 | Δ1-Piperidine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase from Pseudomonas syringae (DpkAPsyrin) as catalyst for the synthesis 2-hydroxy-4-arylbut-3-enoic acid derivatives |
|
| Presenting author: | CARLOS JOSE MORENO FONTALBA of CATALONIA INSTITUTE FOR ADVANCED CHEMISTRY-(IQAC)- |
| Other authors: | Karel HERNANDEZ of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY Samantha GITTINGS of PROZOMIX LTD Dieter SCHOLLMEYER of JOHANNES GUTENBERG-UNIVERSITAET MAINZ, DEPARTMENT CHEMIE, ZENTRALE ANALYTIK Jesús JOGLAR of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC Jordi BUJONS of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC Teodor PARELLA of SERVEI DE RESSONÀNCIA MAGNÈTICA NUCLEAR. UNIVERSITAT AUTÒNOMA DE BARCELONA Pere CLAPÉS of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ketorreductase / trans-o-hydroxybenzylidene pyruvate hydratase-ald / 2-Hydroxy-4-arylbut-3-enoic acids / biocatalytic cascade |
| Purpose: | NAD(P)H-dependent Δ1-piperideine-2-carboxylate/Δ1-pyrroline-2-carboxylate reductase from Pseudomonas syringae pv. tomato (DpkAPsyrin, EC 1.5.1.21) catalyzes the stereoselective reduction of Δ1-piperideine-2-carboxylate and Δ1-pyrroline-2-carboxylate to L-pipecolic acid and L-proline respectively. Furthermore, DpkAPsyrin is also able to catalyze the enantioselective synthesis of N-methyl-L-amino acids from methylamine and various 2-oxo acids [1]. In our research group, we observed that in addition to the imine reductase activity, DpkAPsyrin have a promiscuous ketoreductase activity and, consequently, we consider interesting to exploit its underdeveloped synthetic capabilities for the synthesis of 2-hydroxy-4-arylbut-3-enoic acid (4). These 2-hydroxyacids are intermediates in the synthesis of 2-hydroxy-4-arylbutanoic acid, important building blocks for the production of a variety of angiotensin converting enzyme (ACE) inhibitors (e.g. enalapril, lisinopril, cilapril, and benazepril) [2]. In this communication we reported the preparation of structurally diverse (S)-2-hydroxy-4-arylbut-3-enoic acid (4) (isolated yield: 57%-78% and ee: 87%->99%) from aromatic aldehydes (1) and pyruvate (2) by enzymatic cascade reaction using trans-o-hydroxybenzylidene pyruvate hydratase-aldolase from Pseudomonas putida HBPAPputida) [3] and DpkAPsyrin (Scheme 1). The cascade process was possible because it was found that the ketoreductase preferentially reduced the aldol condensation adducts (3) rather than the pyruvate (2). This allows for high levels of 2-hydroxy-4-arylbut-3-enoic acids (4) conversions ( >95% after 24 h). |
| References: | [1] Goto, M.; Muramatsu, H.; Mihara, H., et al., J. Biol. Chem. 2005, 280, 40875-40884. [2] (a) Larissegger-Schnell, B.; Kroutil, W.; Faber, K., Synlett 2005, 2005, 1936-1938; (b) Zhu, L.; Meng, Q.; Fan, W., et al., J. Org. Chem. 2010, 75, 6027-6030. [3] Fansher, D. J.; Ngwira, N.; Salehi, A. R., et al., Synthesis 2022, 55, 75-89. |
| Figures: | 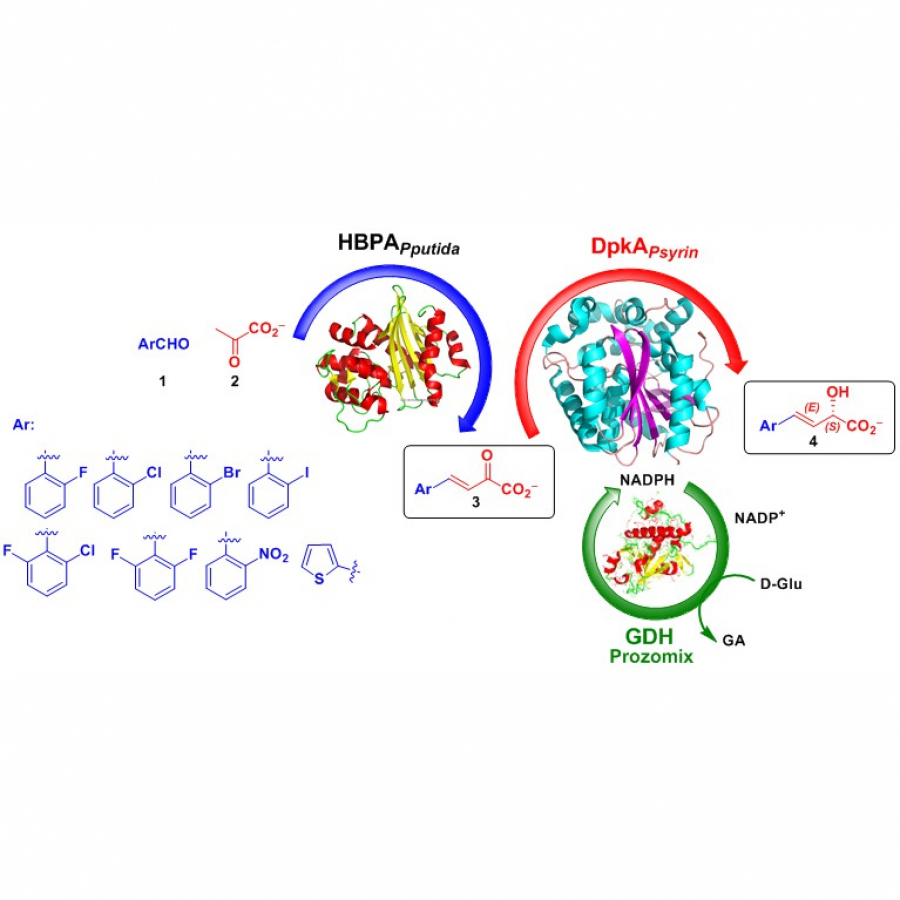 Scheme 1 Synthesis of (S)-2-hydroxy-4-arylbut-3-enoic acid (4) through enzymatic cascade reaction. |
#535 | Unlocking Promiscuous Activity of the Photoenzyme Fatty Acid Photodecarboxylase from C. variabilis |
|
| Presenting author: | Florian WEISSENSTEINER of UNIVERSITY OF GRAZ |
| Other authors: | Emilia IGLESIAS of UNIVERSITY OF GRAZ Moritz RIEGLER of UNIVERSITY OF GRAZ Stefan SIMIC of UNIVERSITY OF GRAZ Isabel OROZ-GUINEA of UNIVERSITY OF GRAZ Christoph WINKLER of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme promiscuity / photobiocatalysis / C-C-bond formations / protein engineering |
| Purpose: | Strategic C-C bond formations are among the most important synthetic reactions in organic chemistry to construct the carbon backbone of complex molecules from small building blocks. Traditional organic approaches often display low atom economy, thus producing higher amounts of waste compared to applying enzymes because of the need of sophisticated reagents. The photoenzyme Fatty Acid Photodecarboxylase (CvFAP) from the microalgae C. variabilis, was recently identified and characterized to decarboxylate fatty acids into their corresponding Cn-1 alkanes or alkenes under a continuous flux of blue light via a radical mechanism [1-3]. Inspired by precedence in chemical photocatalysis utilizing carboxylic acids for radical decarboxylative couplings, [4] we investigated the capability of CvFAP to catalyze the decarboxylation of unsaturated fatty acids, bearing an internal electron acceptor within the chain. The intermediate radical generated upon decarboxylation then undergoes an intramolecular cyclization leading to alkylated ring systems. Herein we demonstrate the proof-of-concept for the desired promiscuity by illuminating suitable fatty acids bearing internal electron acceptors, with variants of CvFAP, in an in-house built photoreactor (Figure 1, A and B). Suitable variants resulted in different ratios of cyclized and decarboxylated product. |
| References: | [1] D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugière, Y. Couté, D. Nurizzo, P. Müller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier, F. Beisson, Science 2017, 357, 903-907. [2] D. Sorigué, K. Hadjidemetriou, S. Blangy, G. Gotthard, A. Bonvalet, N. Coquelle, P. Samire, A. Aleksandrov, L. Antonucci, A. Benachir, S. Boutet, M. Byrdin, M. Cammarata, S. Carbajo, S. Cuiné, R. B. Doak, L. Foucar, A. Gorel, M. Grünbein, E. Hartmann, R. Hienerwadel, M. Hilpert, M. Kloos, T. J. Lane, B. Légeret, P. Legrand, Y. Li-Beisson, S. L. Y. Moulin, D. Nurizzo, G. Peltier, G. Schirò, R. L. Shoeman, M. Sliwa, X. Solinas, B. Zhuang, T. R. M. Barends, J. P. Colletier, M. Joffre, A. Royant, C. Berthomieu, M. Weik, T. Domratcheva, K. Brettel, M. H. Vos, I. Schlichting, P. Arnoux, P. Müller, F. Beisson, Science 2021, 372. [3] D. J. Heyes, B. Lakavath, S. J. O. Hardman, M. Sakuma, T. M. Hedison, N. S. Scrutton, ACS Catal. 2020, 10, 6691-6696. [4] Q.-Q. Zhou, Y. Wei, L.-Q. Lu, W.-J. Xiao, Sci. Synth. 2018, 6, 167-218, DOI: 10.1055/sosSD-229-00103. |
| Figures: | 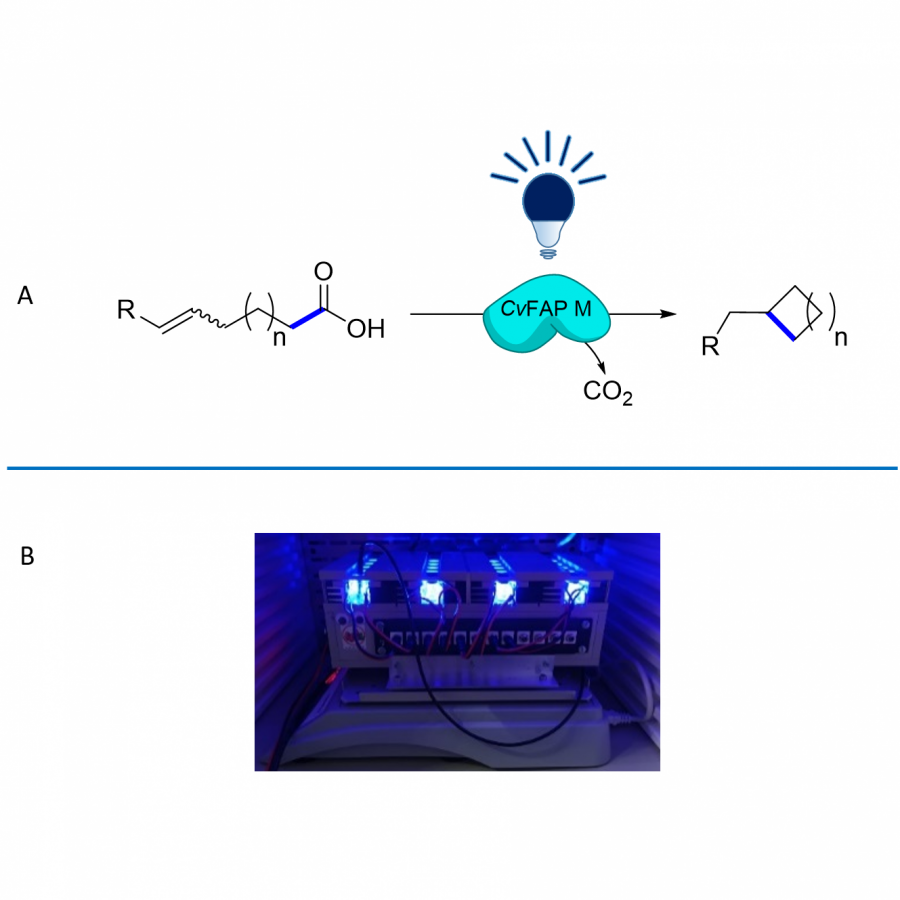 Promiscuous activity of CvFAP towards radical intramolecular cyclizations. (A) Generalized scheme for promiscuous intramolecular C-C bond formations using a suitable CvFAP mutant. (B) Photodecarboxylation in an in-house built photoreactor. |
#536 | Engineering enzymes to enhance lactonase activity towards bacterial quorum sensing control |
|
| Presenting author: | Raphael BILLOT of GENE&GREENTK |
| Corresponding author: | David DAUDE of GENE&GREENTK |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | quorum sensing / quorum quenching / lactonase / enzyme engineering |
| Purpose: | Many bacteria use quorum sensing (QS), a bacterial communication system based on the diffusion and perception of small signaling molecules, to synchronize their behavior in a cell-density dependent manner. This mechanism enables bacteria to adapt their behavior according to their population size and regulate the expression of genes involved in virulence, antimicrobial resistance and biofilm formation. Methods have emerged to inhibit bacterial communication and counteract its noxious traits. Chemical inhibitors, sequestering antibodies and degrading enzymes have been developed and proved efficient to decrease bacterial virulence. Among these, enzymes from various origins having lactonase or acylase activities were particularly studied for their ability to degrade acyl-homoserine lactones (AHL) involved in the communication of numerous Gram-negative bacteria. Our work focuses on the extremophilic lactonase SsoPox able to degrade a wide panel of AHL. This enzyme has a natural preference for the hydrolysis of AHL with long acyl chains (>C8) and a relatively low activity for short-chain AHL while these latter are widely involved in the QS of numerous pathogens. Enzyme engineering was applied to increase the performance of SsoPox towards various types of AHL. Structural analysis led to the identification of a mobile loop surrounding the active site that highly impacts the lactonase activity spectrum. Combining alanine scanning, saturation mutagenesis and semi-rational approaches, variant libraries were generated and improved variants with up to 297-fold activity increase towards short chain AHL were isolated. Tridimensional structures of these variants revealed rearrangement of the active site cavity leading to the reorientation of lactone acyl chain in favor of small AHL recognition. The potential of the best identified variant to disrupt short-AHL based QS was then studied in vitro towards the bacterium Serratia sp. 39006, which mainly uses C4 AHL. A multi-faceted approach combining phenotypic, proteomic and metabolomic analyses allowed to demonstrate the ability of the enzyme to drastically alter strain behavior by disrupting various phenotypes including biofilm formation, production of antibiotics, mobility and floatability. Development of these more effective QS disrupting enzymes will pave the way for developing concrete biotechnological applications to tackle the virulence of bacterial pathogens affecting animal nutrition, agriculture and human health. |
| Figures: | 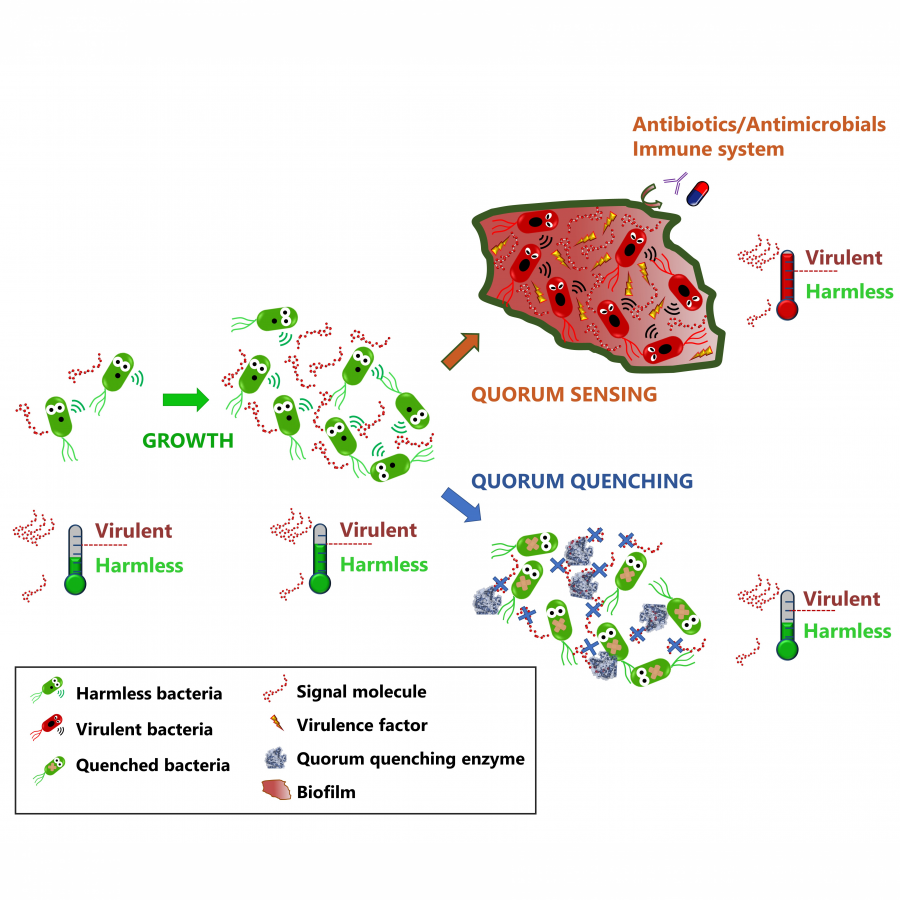 Disrupting bacterial quorum sensing (QS) with enzymes: a promising approach against pathogens. Bacteria secrete diffusible molecules during growth. When the concentration reaches a threshold, bacteria synchronize the expression of genes involved in virulence and biofilm. Enzymes able to degrade signal molecules disrupt QS and keep bacteria harmless 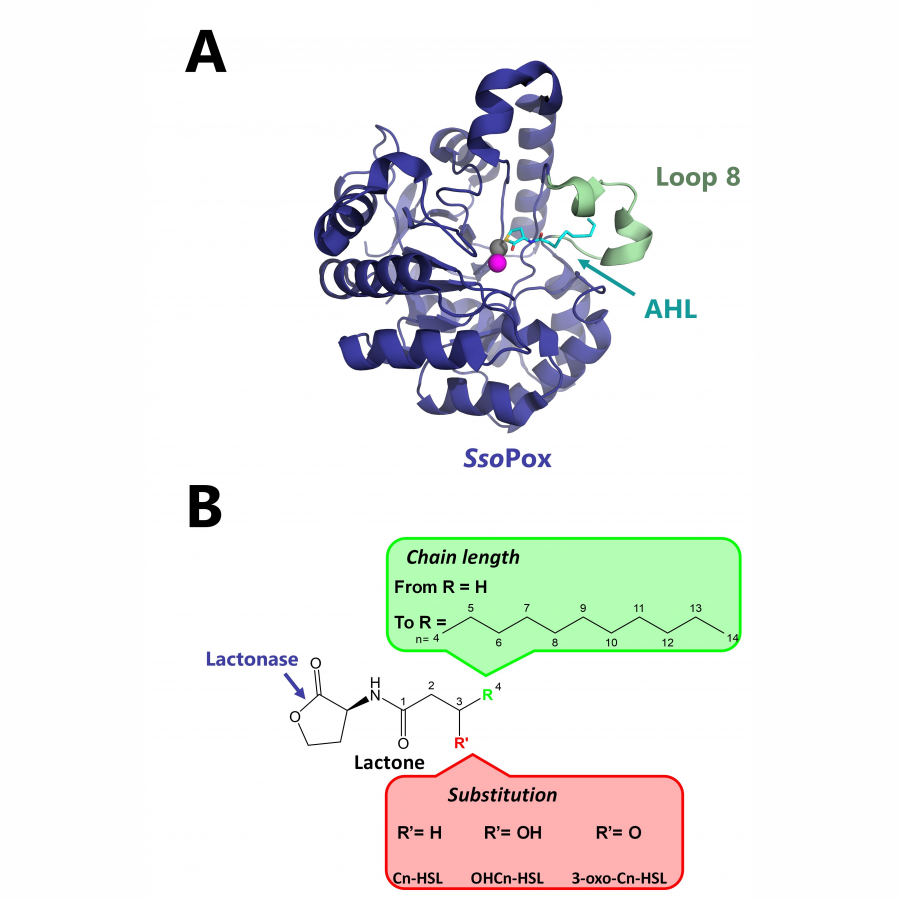 The SsoPox enzyme and its various AHL substrates. (A) Tridimensional structure of SsoPox (dark blue), in complex with an AHL (cyan). The mobile loop (loop 8) is highlighted in green. (B) General structure of QS AHL (HSL, oxo-HSL and OH-HSL). Lactonases degrade AHL by opening lactone rings. |
#544 | Shifting the balance: Soluble ADAM10 as a potential treatment for Alzheimer’s disease |
|
| Presenting author: | Ayelet FISHMAN of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY |
| Other authors: | Ayelet Sarah HERSHKOVITS of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY Sivan GELLEY of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY Rawad HANNA of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY Oded KLEIFELD of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY Avidor SHULMAN of TECHNION-ISRAEL INSTITUTE OF TECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alzheimer’s disease / ADAM10 / Proteomics / Amyloid beta |
| Purpose: | Accumulation of amyloid β in the brain is regarded as a key initiator of Alzheimer’s disease pathology. Processing of the amyloid precursor protein (APP) in the amyloidogenic pathway yields neurotoxic amyloid β species. In the non-amyloidogenic pathway, APP is processed by membrane-bound ADAM10, the main α-secretase in the nervous system. Here we present a new enzymatic approach for potential treatment of Alzheimer’s disease using a soluble form of ADAM10. The ability of the soluble ADAM10 to shed overexpressed and endogenous APP was determined with an ADAM10 knockout cell line and a human neuroblastoma cell line, respectively. The soluble enzyme hydrolyzes APP and releases the neuroprotective soluble APPα when exogenously added to cell cultures. We further demonstrated by thioflavin T fluorescence, HPLC and confocal microscopy its ability to degrade different amyloid β species inhibiting the formation and aggregation of characteristic amyloid β extracellular neuronal plaques. Using N-terminal and C-terminal enrichment proteomic approaches we investigated its substrates, identifying new and verifying others, such as VGF and N-cadherin, respectively. Lastly, a truncated soluble ADAM10, based on the catalytic domain, was expressed in E. coli for the first time and its activity evaluated. The truncated variant also exhibited α-secretase capacity as shown with a specific ADAM10 fluorescent substrate in addition to shedding overexpressed and endogenous APP. Our in vitro work demonstrates that exogenous treatment with a soluble variant of ADAM10 would shift the balance towards the non-amyloidogenic pathway. The potential of such a treatment for Alzheimer’s disease needs to be further evaluated in vivo. |
#548 | Characterization of a novel hyperthermophilic glycosyltransferase GT-2 from the Archaea Pyrococus horikoshii |
|
| Presenting author: | Ghazal KHALED of ECOLE NATIONALE SUPÉRIEURE DE CHIMIE RENNES ENSCR |
| Corresponding author: | Sylvain TRANCHIMAND of ECOLE NATIONALE SUPÉRIEURE DE CHIMIE RENNES ENSCR |
| Other authors: | Hala CHAMIEH of LEBANESE UNIVERSITY FACULTY OF SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Glycosyltransferase GT-2 / Hyperthermophilic Archaea / Truncated protein / Glycosaminoglycan |
| Purpose: | Glycosyltransferases (GTs) are enzymes involved in the synthesis of glycoconjugates and polysaccharides, molecules with a wide range of biological activities, and as such are very attractive for biotechnological applications. GTs catalyze the formation of glycosidic bonds through the transfer of activated sugar donors to a diversity of acceptor molecules such as proteins, lipids or carbohydrate residues, with a very tight control either on the nature of both donor and acceptor substrates and on the stereochemistry of the glycosidic bond itself [1]. GTs are classified in more than 116 families according to the Carbohydrate-Active enzyme (CAZY) database [2].. Enzymes from the GT-2 family are involved in the synthesis of several linear polysaccharides such as cellulose, chitin or hyaluronic acid. The latter is of particular interest due to its numerous clinical and cosmetic applications. A genome-wide survey analysis of GT-2 encoding sequences in the three domains of life was performed and revealed an uncharacterized separate clade, closely related to a clade including hyaluronan synthases [3](Khaled et al., Unpublished; Amin et al., EPO under revision). Members of this family are typically processive enzymes, displaying a GT-A fold with two Rossman fold-like domains. A topology prediction revealed that proteins of this family are transmembrane proteins and show a similar folding as the hyaluronan synthase (HAS), either human or bacterial HAS, and hence were named HAS-like. Sequences corresponding to this new clade were particularly found in hyperthermophilic Archaea genomes. Enzymes from hyperthermophilic microorganisms are of high biotechnological interest as they generally display high robustness and thermostability allowing their use in various industrial processes requiring elevated temperatures [4]. The objective of our study was therefore to biochemically characterize a representative of this clade from the hyperthermophilic archaea Pyrococcus horikoshii, thereafter named Ph-has-like. The recombinant protein Ph-has-like was expressed in E.coli using different expression systems. The full-length transmembrane protein (1-314 aa) was first produced but was difficult to overexpress and purify. Domain analysis of the protein showed that the catalytic domain was located mainly in the N-terminal soluble cytoplasmic region. Site-directed mutagenesis was then performed to express a truncated form (1-232 aa) of Ph-Has-like displaying the catalytic domains but not the transmembrane domain. Several constructions were performed to express the soluble form of the protein with and without tags. After several optimizations using different E.coli strains and induction conditions, the protein was successfully produced as a soluble form from the BL21 (DE3) pLysS E. coli strain and purified by affinity chromatography. The biochemical activity of the protein is currently under investigation, particularly, the activity of the hyperthermophilic enzyme on a wide range of substrates used by processive GT-2 of the same family. For that purpose we have developed an HPLC-based method for the evaluation of such GT activity. Our work represents one of the few characterized GT-2 enzymes from Archaea so far. It will contribute to the elucidation of the enzymatic mechanism of polysaccharide synthesis and enlarge our understanding on the behavior of these enzymes at high temperature. |
| References: | L. L. Lairson, B. Henrissat, G. J. Davies, and S. G. Withers, “Glycosyl transferases: Structures, functions, and mechanisms,” Annu. Rev. Biochem., vol. 77, pp. 521-555, 2008, doi: 10.1146/annurev.biochem.76.061005.092322. [2] S. Fushinobu, V. D. Alves, and P. M. Coutinho, “Multiple rewards from a treasure trove of novel glycoside hydrolase and polysaccharide lyase structures: New folds, mechanistic details, and evolutionary relationships,” Curr. Opin. Struct. Biol., vol. 23, no. 5, pp. 652-659, 2013, doi: 10.1016/j.sbi.2013.06.001. [3] Khaled et al., “Genome-wide analysis of GT-2 families from the three domains of life.” [4] K. Amin, “Metabolic engineering for the production of a hyaluronic Like GAG from hyperthermophilic archaea in E.coli,” Thèse de doctorat Biotechnologie appliquée,ENSCR- Université Libanaise, 2020. |
| Figures: | 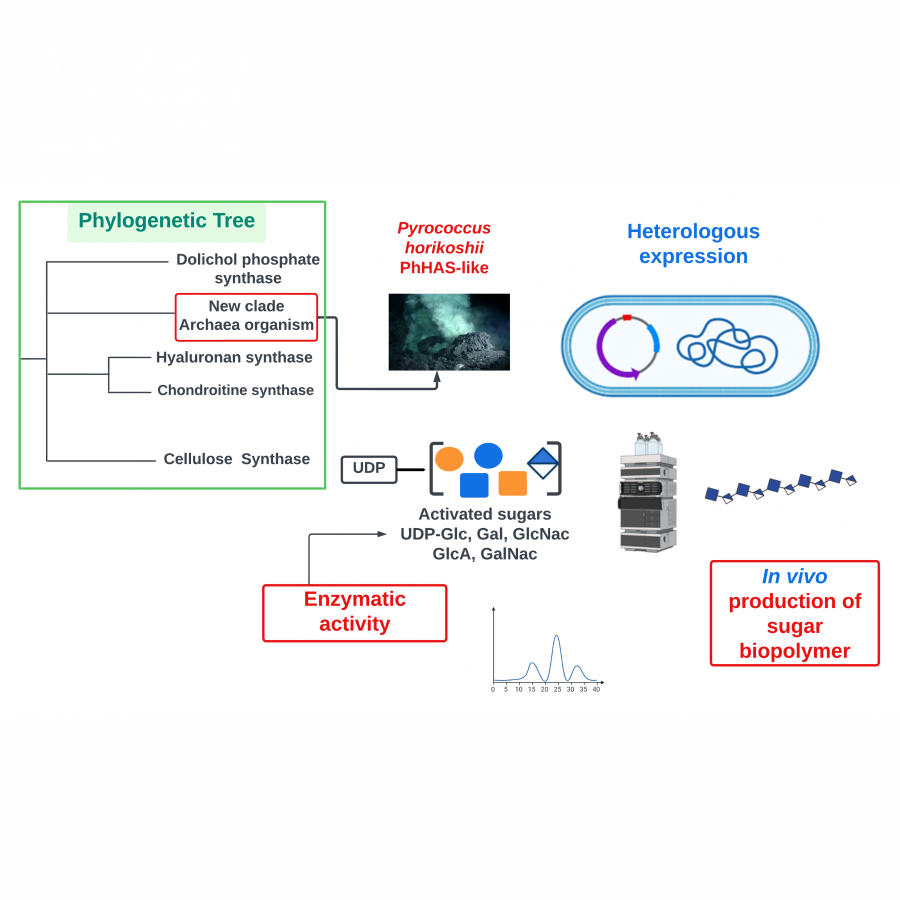 Overview of stategy developed to characterize a novel GT-2 from Pyrococcus Horikoshii Enzymatic activity of recombinant PhHas-like purified (left). In vivo production of the new polysaccharide (right). |
#555 | Identification and characterization of archaeal and bacterial F420-dependent thioredoxin reductases |
|
| Presenting author: | Gaung YANG of UNIVERSITY OF GRONINGEN |
| Corresponding author: | Marco FRAAIJE of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Deazaflavin / F420 / Thioredoxin reductase / Cofactor specificity |
| Purpose: | The thioredoxin pathway is an antioxidant system present in most organisms, in which thioredoxin reductase plays a key role. Recently, it was shown that it can also be used in biocatalysis for cofactor regeneration [1]. Most known thioredoxin reductases are NADP(H)-dependent, enabling regeneration of NADPH. Yet, in 2016, a new type of thioredoxin reductase was discovered in archaea which utilizes instead a deazaflavin cofactor, F420. For this reason, the respective enzyme was named deazaflavin-dependent flavin-containing thioredoxin reductase (DFTR). To explore the potential of DFTRs, we set out to express and study microbial DFTRs. First, we identified and characterized two new archaeal representatives. A detailed kinetic study, which included pre-steady state kinetic analyses, revealed these two DFTRs are highly specific for F420H2 while displaying marginal activity with NADPH. A detailed structural analysis led the identification of two key residues that tune cofactor specificity of DFTRs. This allowed us to define a DFTR-specific sequence motif that enabled for the first time the identification and experimental characterization of a bacterial DFTR. All discovered DFTRs are highly thermostable and efficient in utilizing F420 as cofactor. Thus, they provide an additional tool for the regeneration of this deazaflavin cofactor. |
| References: | [1] Zhang N., Müller B., Kirkeby T. Ø., Kara S., Loderer C., ChemCatChem, 2022, 14, e202101625. [2] Susanti D., Loganathan U., Mukhopadhyay B., J Biol Chem.,2016, 291, 23084-23100. |
#558 | Colorimetric assay for the of Alcohol Dehydrogenase activity assessment. |
|
| Presenting author: | Maria Cristina CANCELLIERI of POLITECNICO DI MILANO |
| Corresponding author: | Francesco Gilberto GATTI of POLITECNICO DI MILANO |
| Other authors: | Augusto BIGANZOLI of DIPARTIMENTO DI SCIENZE DELL’AMBIENTE E DELLA TERRA, UNIVERSITÀ DEGLI STUDI DI MILANO BICOCCA Danilo COLOMBO of POLITECNICO DI MILANO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | colorimetric assay / ADH activity / chemoselective bio-reduction / resorufin dye |
| Purpose: | It is a matter of fact that the importance of enzymatic catalysis has significantly increased during the last decades, indeed, biocatalysts seem to be one of the most promising answers to the urgent need of environmentally friendly chemical processes. Although a plethora of new enzymes have been discovered or bioengineered, their employment at preparative scale always requires the knowledge of activity and stereoselectivity. For example, the Alcohol Dehydrogenases (ADH), which are very likely one of the most popular redox enzymes, can reduce prochiral ketones and aldehydes into alcohols, but the determination of their activity is tedious and a time-consuming practice [1]. Herein we describe a new colorimetric assay suitable to the rapid and efficient screening of ADH activity [2]. Our colorimetric probe is formed by the levulinc carboxylic acid coupled to the resorufin dye, and it should be a suitable substrate for the ADHs. The chromogenic signaling is due to the ADH triggered release of resorufin dye, that when it is free exhibits a brilliant pink color, instead, when it is functionalized has a pale-yellow color, as shown in the Figure. The chemoselective bio-reduction of the ketone group of levulinate affords the corresponding hydroxyester, which in turn ring-closes very rapidly affording the γ-lactone and the resorufin dye. The suggested ADH triggered cyclization was studied by 1H-NMR, UV-vis, and fluorescence measurements. |
| References: | [1] de Miranda A.S.; Milagre C.D.F., Hollmann F., Frontiers in Catalysis, 2022, 2. [2] Choi M.G.; Hwang J., Eor S., Chang S.K., Organic Letters, 2010, 12(24), 5624-5627. |
| Figures: | 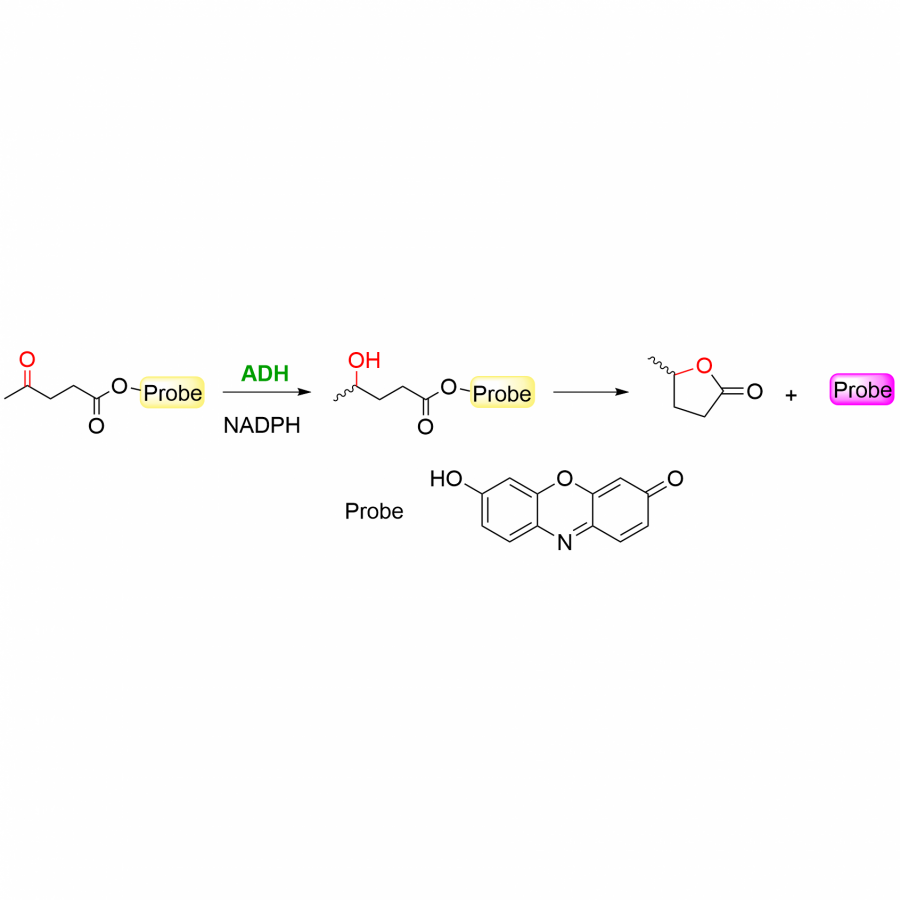 Colorimetric assay for the screening of Alcohol Dehydrogenase activity The formation of γ-lactone and the release of resorufin dye are afforded rapidly after the ADH mediated chemoselective reduction of ketone group of levulinate. |
#564 | Study of a versatile amide bond synthetase and its application in pharmaceutical amide synthesis |
|
| Presenting author: | Qingyun TANG of UNIVERSITY OF YORK |
| Corresponding author: | Gideon GROGAN of UNIVERSITY OF YORK |
| Other authors: | Mark PETCHEY of UNIVERSITY OF YORK Anibal CUETOS of UNIVERSITY OF YORK Benjamin ROWLINSON of UNIVERSITY OF YORK Richard LLOYD of GSK MEDICINES RESEARCH CENTRE Ian FAIRLAMB of UNIVERSITY OF YORK |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Amide ligase / Enantioselectivity / Protein engineering / Pharmaceutical amides |
| Purpose: | The synthesis of amide bonds is one of the most frequently performed reactions in pharmaceutical industry. 74 % of the top 50 selling small molecule drugs in 2021 contain an amide bond linkage [1]. The requirement of stoichiometric quantities of coupling agents to activate acyl groups in chemical synthesis methods has prompted interest in biocatalytic alternatives [2]. Amide bond synthetases (ABSs) activate carboxylic acid substrates through ATP-dependent adenylation, followed by acylation of amine nucleophiles, both within the active site of the enzyme. Following our earlier studies on the ABS from Marinactinospora thermotolerans [3-4], we further investigated a versatile ABS from Streptomyces hindustanus (ShABS). It is highly active towards a wide range of carboxylic acids and amines and displayed superior enantioselectivity to different chiral substrates. Alanine scanning of both substrate pockets revealed key residues influencing the enzyme activity and enantioselectivity. Crystal structures of ShABS showed a dramatic conformational change of the enzyme which might associate with the two half-reactions catalyzed. Furthermore, pharmaceutical amides such as the anticonvulsant drug ilepcimide and the antiemetic drug trimethobenzamide were synthesised on preparative milligram scales using ShABS as a catalyst. |
| References: | [1] https://njardarson.lab.arizona.edu/content/top-pharmaceuticals-poster; [2] M. Petchey, et al. Adv. Synth. Catal. 2019, 361, 3895. [3] M. Petchey, et al. Angew. Chem. Int. Ed. 2018, 57, 11584. [4] M. Petchey, et al. ACS Catal. 2020, 10, 4659. |
| Figures: | 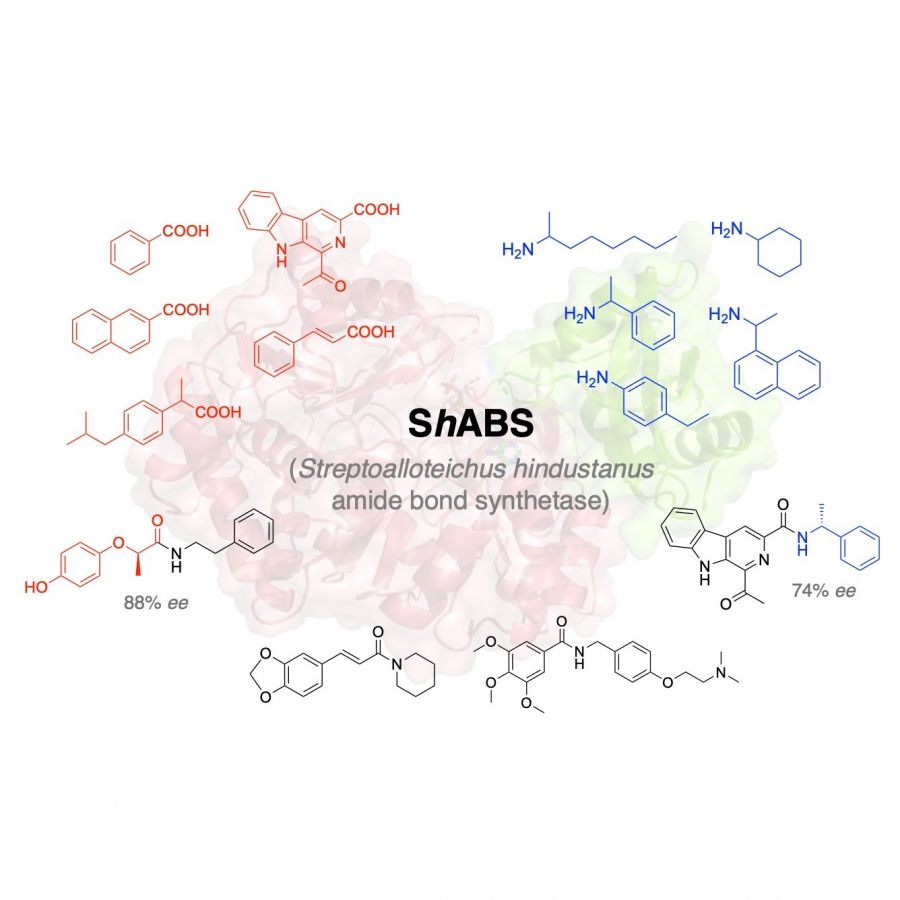 Substrate scope and enantioselectivity of the Streptomyces hindustanus amide bond synthetase (ShABS). ShABS accepts a wide range of carboxylic acids and amines and displayed superior enantioselectivity to different chiral substrates. It also catalyses the synthesis of the pharmaceutical amides ilepcimide and trimethobenzamide. 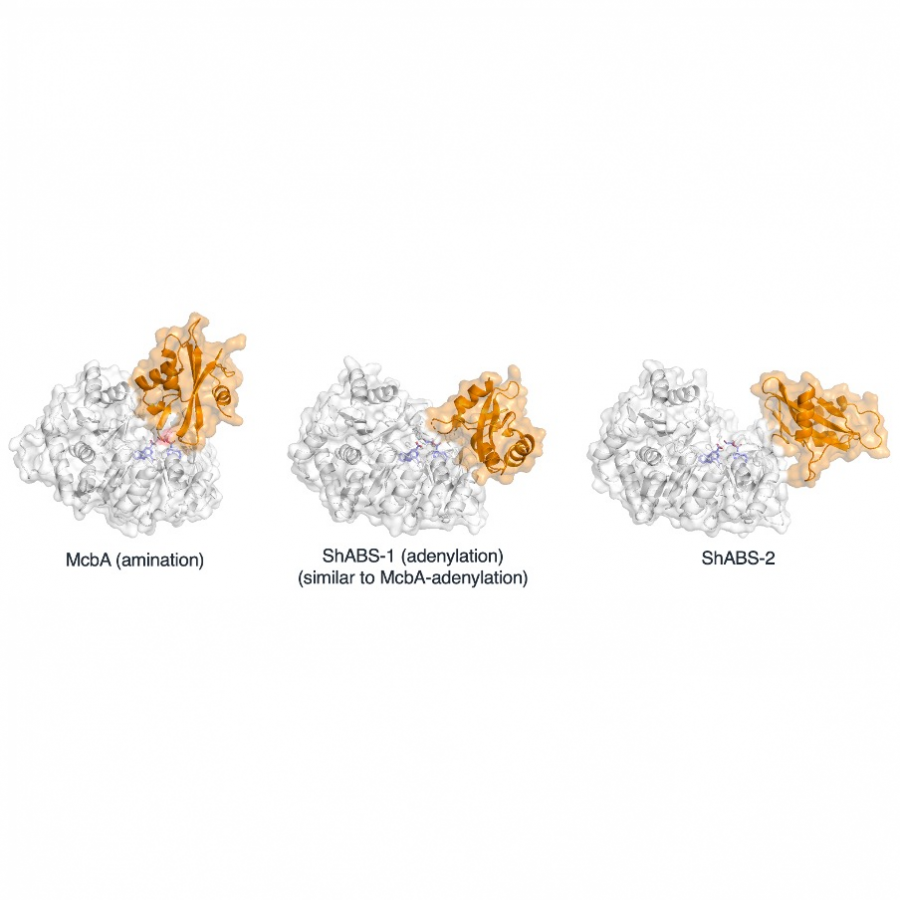 Conformational change of ShABS associated with the different reaction stages. Crystal structures of ShABS showed a dramatic conformational change of the enzyme which might associate with the different reaction stages. Acid and amine substrate pockets are coloured white and orange, respectively. |
#569 | Chemoenzymatic cascades involving gold and enzyme catalysis |
|
| Presenting author: | VICENTE GOTOR-FERNANDEZ of UNIVERSITY OF OVIEDO |
| Other authors: | Lorena ESCOT of UNIVERSITY OF OVIEDO Sergio GONZALEZ-GRANDA of UNIVERSITY OF OVIEDO Ivan LAVANDERA of UNIVERSITY OF OVIEDO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alcohol dehydrogenases / Amine transaminases / Chemoenzymatic cascades / Gold catalysis |
| Purpose: | The development of catalytic methods is a useful solution for synthetic challenges towards the preparation of valuable (enantioenriched) organic molecules. The combination of metal and enzyme catalysis has emerged a few years ago, allowing the design of more efficient and novel selective catalytic routes.[1] Moreover, these multicatalytic approaches have become more popular when they are carried out concurrently or sequentially in the same reaction vessel. However, they are usually hampered by (partial) incompatibility of the different catalyst types. The design of metalloenzymatic cascades is particularly attractive, by means of one-pot sequential or cascade approaches towards chiral products.[2] In this context, the use of gold species allows to activate multiple C–C bonds under mild conditions, while enzymes provide multiple solutions for stereoselective synthesis (Figure 1). Herein, the combined use of gold(I) species, particularly N-heterocyclic carbene complexes, and biocatalysts such as alcohol dehydrogenases or amine transaminases, have permitted the straightforward transformation of alkynes into chiral alcohols and amines with exquisite selectivity. The development of gold-catalyzed Meyer-Schuster rearrangements[3,4] and hydration reactions[5,6] will be disclosed, to subsequently perform on the carbonyl intermediates asymmetric bioreduction or biotransamination transformations in a concurrent or sequential manner. |
| References: | [1]. S. Gonzalez-Granda, J. Albarran-Velo, I. Lavandera, V. Gotor-Fernandez, Chem. Rev. 2023, 123, https://doi.org/10.1021/acs.chemrev.2c00454. [2]. S. Gonzalez-Granda, L. Escot, I. Lavandera, V. Gotor-Fernandez, Angew. Chem. Int. Ed. 2023, https://doi.org/10.1002/anie.202217713. [3]. S. Gonzalez-Granda, I. Lavandera, V. Gotor-Fernandez, Angew. Chem. Int. Ed. 2021, 60, 13945-13951. [4]. S. Gonzalez-Granda, N. V. Tzouras, S. Nolan, I. Lavandera, V. Gotor-Fernandez, Adv. Synth. Catal. 2022, 364, 3856-3866. [5]. S. Gonzalez-Granda, L. Escot, I. Lavandera, V. Gotor-Fernandez, ACS Catal. 2022, 12, 2552-2560. [6]. L. Escot, S. Gonzalez-Granda, V. Gotor-Fernández, I. Lavandera, Org. Biomol. Chem. 2022, 20, 9650-9658. |
| Figures: |  Figure 1 Figure 1. Combination of gold species and enzyme for the development of chemoenzymatic cascades |
#572 | Selective peroxygenase-catalysed oxidation of toluene derivates to benzaldehydes |
|
| Presenting author: | Yutong WANG of DELFT UNIVERSITY OF TECHNOLOGY |
| Corresponding author: | Frank HOLLMANN of DELFT UNVERSITY OF TECHNOLOGY |
| Other authors: | Wuyuan ZHANG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY, CHINESE ACADEMY OF SCIENCES Niklas TEETZ of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN Dirk HOLTMANN of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN Miguel ALCALDE of INSTITUTE OF CATALYSIS, ICP-CSIC Jacob HENGST of DELFT UNVERSITY OF TECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalytic oxidation / Selective oxyfunctionalisation / Peroxygenase / Solvent-free biocatalysis |
| Purpose: | In recent years, unspecific peroxygenases (UPOs) have emerged as promising biocatalysts in the selective activation of C-H bonds,[1] which is still challenging for organic chemistry. However, for the conversion of toluene, the selectivity of the archetypal UPO from Agrocybe aegerita (AaeUPO) is rather poor comprising both ring-hydroxylation and oxidation of the alkyl substituent.[2] In contrast, the AaeUPO-catalyzed hydroxylations of the toluene homologue ethyl benzene proceed highly selectively with (R)-1-phenyl ethanol as the sole product.[3] Inspired by the smart-substrate strategy, we hypothesize that substitution of aromatic ring might influence the selectivity of AaeUPO-catalyzed oxyfunctionalization of toluene derivates. In silico docking studies suggest that indeed, further substituents at the aromatic ring should improve the substrate binding selectivity, leading to a preferable position for benzylic oxyfunctionalization. In fact, a range of ring-substituted toluene derivates were highly selectively converted to corresponding benzaldehydes with a selectivity above 92%, while the overoxidation to benzoic acids were hardly detected. Due to the poor aqueous solubility of the starting materials, a neat system was employed with the toluene derivates as sole solvent and AaeUPO immobilized on a solid carrier. The immobilized AaeUPO was stable in the organic phase under high peroxide dosing, giving on average 100 mM of the desired benzaldehyde products, corresponding to more than 10 g L-1. A highest product concentration of 185 mM was achieved in the formation of p-nitro-benzaldehyde. |
| References: | [1] Hobisch, M.; Holtmann, D.; Gomez de Santos, P.; Alcalde, M.; Hollmann, F.; Kara, S., Biotechnol. Adv. 2021, 51, 107615. [2] Ullrich, R.; Hofrichter, M., FEBS Lett. 2005, 579 (27), 6247-6250. [3] Kluge, M.; Ullrich, R.; Scheibner, K.; Hofrichter, M., Green Chem. 2012, 14 (2), 440-446. |
| Figures: | 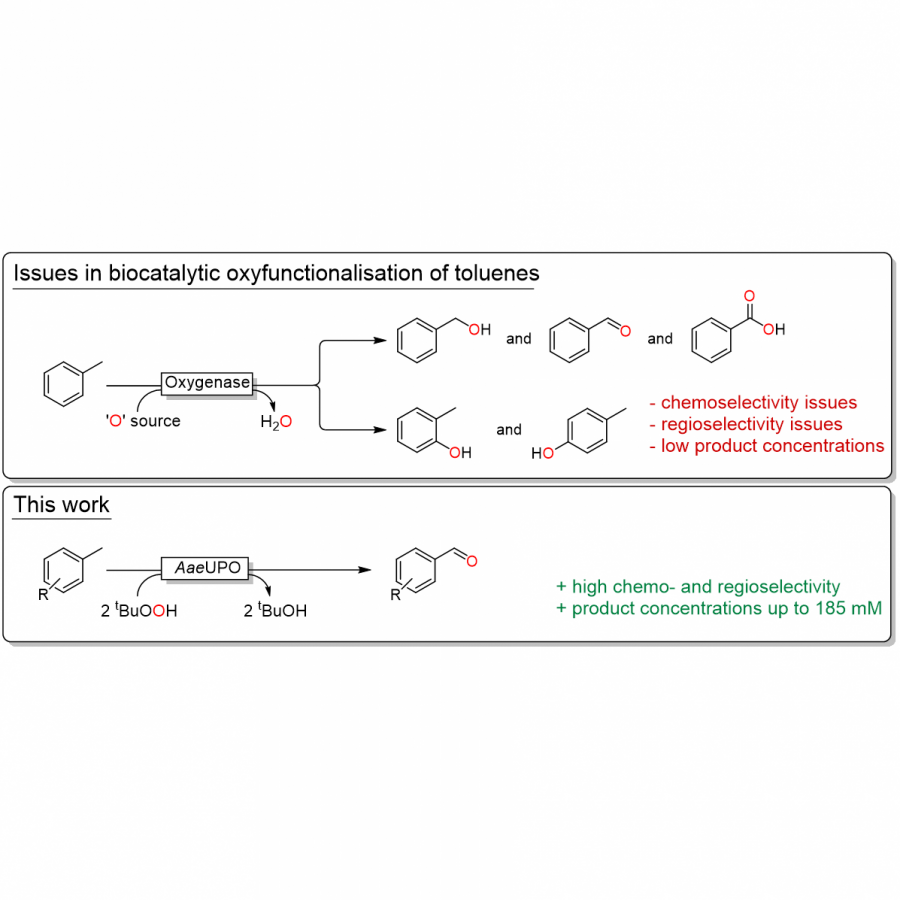 Scheme Selective issues and low product concentration in the biocatalytic oxyfunctionalisation of toluene (derivates) addressed by the combined substrate- and reaction engineering approach. 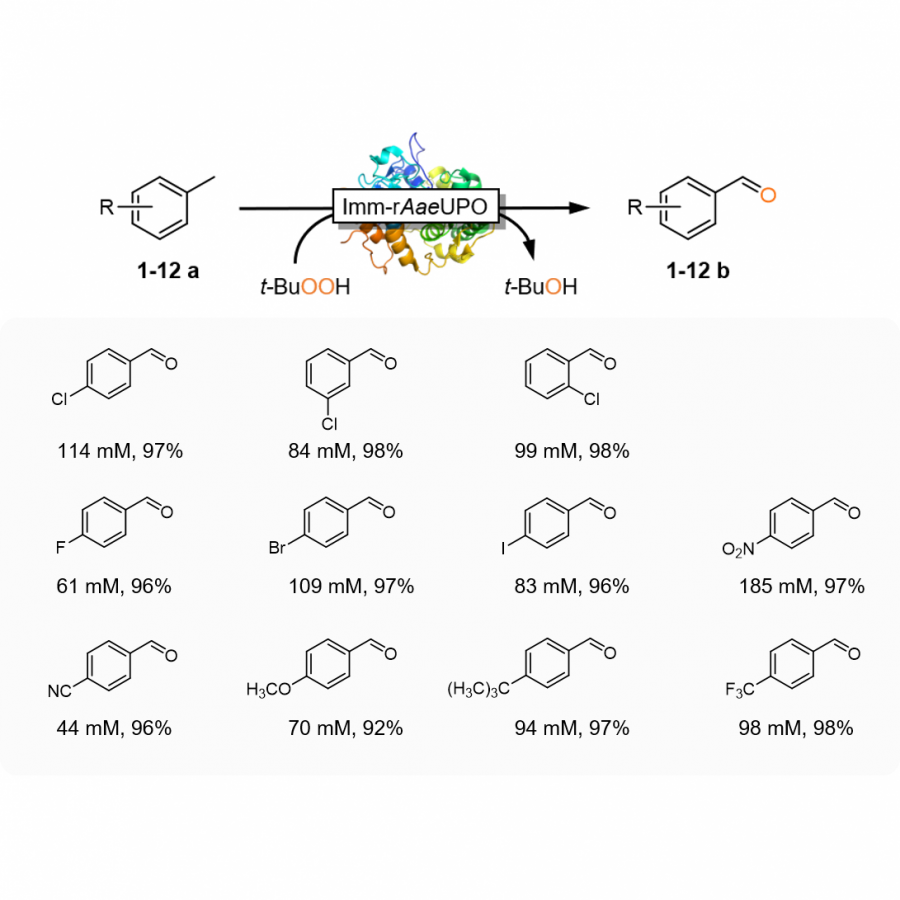 Substrate scope Substrate scope is shown along with product concentration and selectivity. All reactions were conducted under neat condition using immobilised peroxygenase (imm-UPO). |
#574 | Developing inhibitors for human tyrosinase |
|
| Presenting author: | Inbal RUSH of TECHNION |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | tyrosinase / yeast surface display / inhibition / peptides |
| Purpose: | Tyrosinases are ubiquitous enzymes mainly responsible for the formation of melanin in skin pigmentation and in fruit and vegetable browning. Disorder in melanin formation has been linked to a variety of skin diseases in humans such as melasma, solar lentigo, and even melanoma. Therefore, tyrosinase inhibitors are highly warranted by the pharmaceutical, cosmetics, and food industries. Developing inhibitors for human tyrosinase is especially difficult due to the low expression yields in mammalian cells. The present research focuses on designing new peptide-based tyrosinase inhibitors using yeast surface display peptide libraries. Yeast surface display is an in vitro screening method utilizing flow-cytometry. It is a novel approach for this purpose, which will be used to screen and identify strong inhibitors for both human and bacterial tyrosinases. Initially, recombinant human and bacterial tyrosinases were expressed and purified in their active form, and utilized for the screening of several soluble rationally-designed peptides with potential inhibitory effect. Results showed a 4-fold lower IC50 value with human tyrosinase for peptide P8 (HSWMDWVPTPWAA), the most inhibiting peptide screened, compared to kojic acid, a commercially available tyrosinase inhibitor. Further characterization of soluble P8 with bacterial tyrosinase gave an IC50 value of 98 µM and a KD value of 4 µM, when measured using microscale thermophoresis analysis. Therefore, P8 serves as the peptide-library template in the yeast display system. To generate combinatorial libraries, four sets of sub-libraries, consisting of NNK codons in 3 or 4 adjacent positions in the P8 amino acid sequence were designed. In parallel, the conditions for the yeast display and flow-cytometry assay were calibrated, comprising an incubation temperature of 40°C and concentration of 0.5 µM of bacterial tyrosinase for binding. Optimization resulted in 60% expression levels of the displayed construct in flow cytometry analysis. Subsequent work will focus on optimizing binding of the libraries to human tyrosinase. Development of small molecule inhibitors was also pursued. Several resorcinol-based hemiindigoid derivatives, were designed, synthesized and evaluated on human tyrosinase. Two compounds showed superior inhibition in both human melanoma cell lysates and purified human tyrosinase in comparison to kojic acid. In kinetic studies, a partial competitive inhibition mode was obtained from the Lineweaver-Burk correlation for both compounds, and Ki values were four orders of magnitude lower for the new compounds than for kojic acid, 0.55 and 0.25 µM versus 1100 µM, respectively. When the two compounds were tested as well with bacterial tyrosinase, IC50 values of 15 and 30 µM were defined, versus IC50 of 52 for kojic acid [1]. |
| References: | [1] Roulier B#, Rush I#, Lazinski LM, Pérès B, Olleik H, Royal G, Fishman A, Maresca M, Haudecoeur R. 2023. Eur J. Med. Chem. 246:114972. # equal contribution. |
#578 | A semi-automated discovery pipeline for compostable plastic degrading enzymes |
|
| Presenting author: | Gorjan STOJANOVSKI of UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Helen HAILES of UNIVERSITY COLLEGE LONDON |
| Other authors: | Maria BAWN of JOHNSON MATTHEY Jack JEFFRIES of UNIVERSITY COLLEGE LONDON John WARD of UNIVERSITY COLLEGE LONDON |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme discovery and screening / Automation / Microbial Plastic degradation / Bioinformatics |
| Purpose: | Annual global plastic production has reached a staggering 360 – 400 million metric tonnes in recent years, of which 60% is estimated to be lost into the environment [1]. This growing plastic waste problem has triggered greater interest in compostable plastic alternatives such as polylactic acid (PLA) and polybutylene adiptate terephthalate (PBAT). Despite their renewable nature, compostable plastics such as PLA and PBAT can be relatively resistant to microbial degradation in soil, and do not degrade optimally in existing waste management solutions such industrial composting and anaerobic digestors [2]. Given these issues, we aim to identify novel plastic degrading microbes and enzymes which could be used as innocula or microbial consortia to improve compostable plastic degradation in industrial composting and anaerobic digestion systems. In pursuit of this goal, we performed soil enrichment cultures in the presence of PLA and PBAT plastic films. From these we isolated 23 PLA/PBAT degrading microbes all of which were Bacilli bacteria, not previously shown to degrade plastics. To identify and characterise the enzymes involved, we developed an in silico and wet lab semi-automated pipeline for PLA and PBAT degrading enzyme discovery. This approach utilizes and integrates existing bioinformatic search tools such SignalP and HMMER to filter candidate genes by sequence-based searches and comparison with literature-characterised enzymes. Through this, we narrowed our search to 105 candidate genes from an initial pool of 108,000 genes across the 23 strains. These genes were then cloned and characterized in a high-throughput approach using emulsified agar plate screens. We envision that this pipeline will help further our understanding of the PLA/PBAT degradation activity observed in our strain isolates and inform us of which strains could be useful to generate synthetic microbial consortia for enhanced PLA/PBAT degradation. |
| References: | [1] Chow, J. et al. Microbial enzymes will offer limited solutions to the global plastic pollution crisis, Microbial Biotechnology, 2023, 16, 195-217. [2] Kim, M. et al. Polymer film-based screening and isolation of polylactic acid (PLA)-degrading microorganisms, Journal of Microbiology and Biotechnology, 2017, 27, 342-349. |
| Figures: | 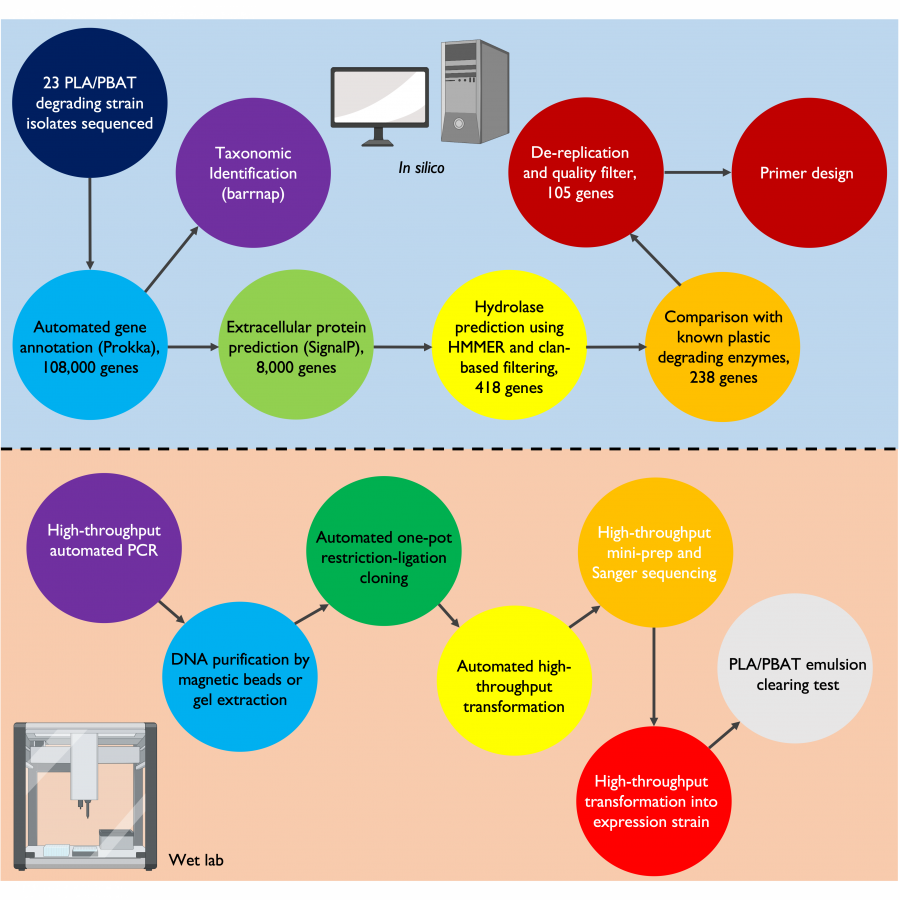 Figure 1 Schematic overview of the semi-automated discovery pipeline developed |
#582 | Solar Biomanufacturing: Engineering photosynthetic electron transfer for P450 biocatalysis in Chlamydomonas reinhardtii |
|
| Presenting author: | Bernadius AGUSTINUS of UNIVERSITY OF QUEENSLAND |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cytochrome P450 / photosynthesis / light-driven / biocatalysis |
| Purpose: | Biocatalysis is increasingly of interest in the pharmaceutical and fine chemical industries for sustainable production of high-value and structurally complex chemicals. Cytochrome P450 monooxygenases (P450s) are attractive biocatalysts for industrial applications due to their ability to transform a wide range of substrates in a stereo- and regiospecific manner. However, the industrial application of P450s is limited by their dependence on costly NADPH and an auxiliary redox partner protein, cytochrome P450 reductase (CPR). The aim of the present study was to couple P450s with the photosynthetic machinery of Chlamydomonas reinhardtii, to take advantage of photosynthetically generated electrons for driving catalysis and eliminate the dependence on a reducing cofactor. Highly thermostable ancestral P450s have been shown previously to be active towards a wide range of drug substrates. To explore the potential of these P450s for light-driven biocatalysis, an in vitro system was developed initially using the P450 ancestor, CYP2ABGSFTCEH_N13, thylakoid membranes from C. reinhardtii, and a soluble C. reinhardtii ferredoxin (PetF) as a redox partner. This system was assessed for the ability to catalyse the hydroxylation of the small molecule substrates, diclofenac, in the presence and absence of light and PetF. The reaction was found to be light and PetF-dependent with an apparent reaction rate 2.6 times higher than catalysed by the same P450 supported by NADPH and human CPR. The ability of the ancestral P450s to couple to the photosynthetic machinery was then assessed in vivo, using C. reinhardtii mutants expressing CYP2ABGSFTCEH_N13 in the chloroplast. Western blot analysis demonstrated that the P450 was expressed and bound to the chloroplast of C. reinhardtii. Furthermore, using diclofenac as a substrate, C. reinhardtii mutants expressing CYP2ABGSFTCEH_N13 showed approximately three-fold higher diclofenac 5-hydroxylation compared to the wildtype C. reinhardtii. These results show that the photosynthetic machinery of C. reinhardtii can act as an alternative electron delivery system to CYP2ABGSFTCEH_N13 via ferredoxin in the presence of light. These findings highlight the potential of C. reinhardtii as a chassis for light-driven P450 biocatalysis to produce fine chemicals and pharmaceuticals. |
| Figures: | 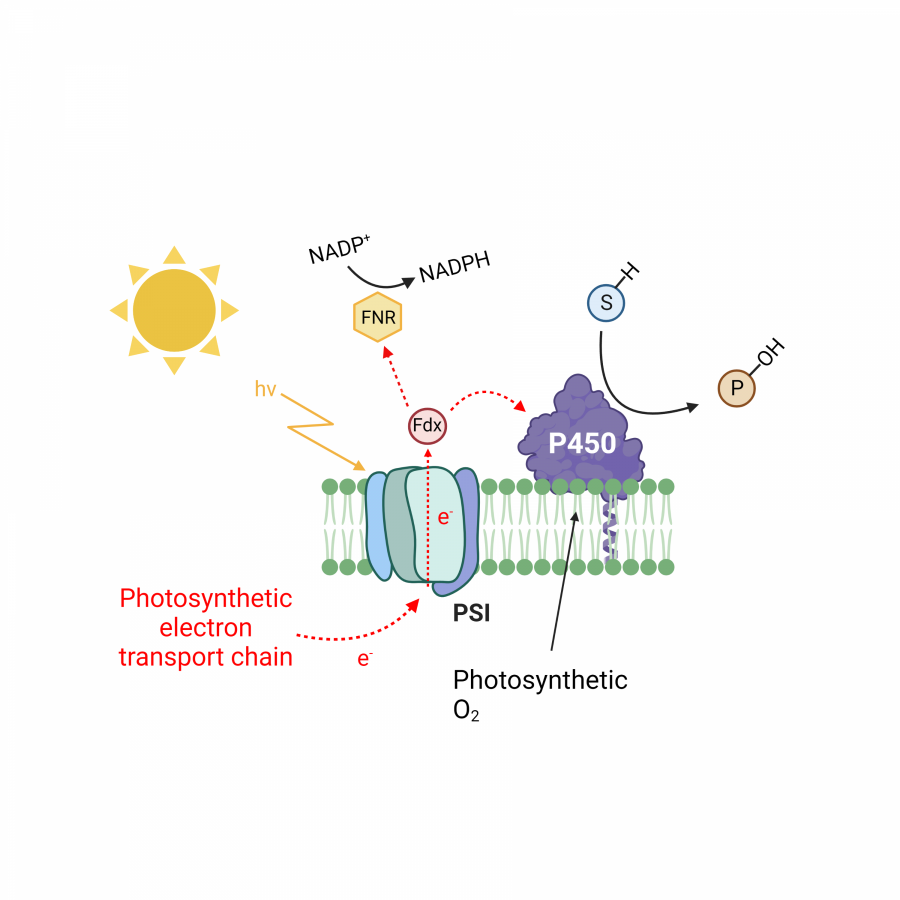 Fig. 1 P450 biocatalysis powered using photosynthetic reducing power. The red dashed arrows indicate photosynthetic electron flow. Fd, ferredoxin; P450, cytochrome P450 monooxygenase, S-H, substrate; P-OH, product 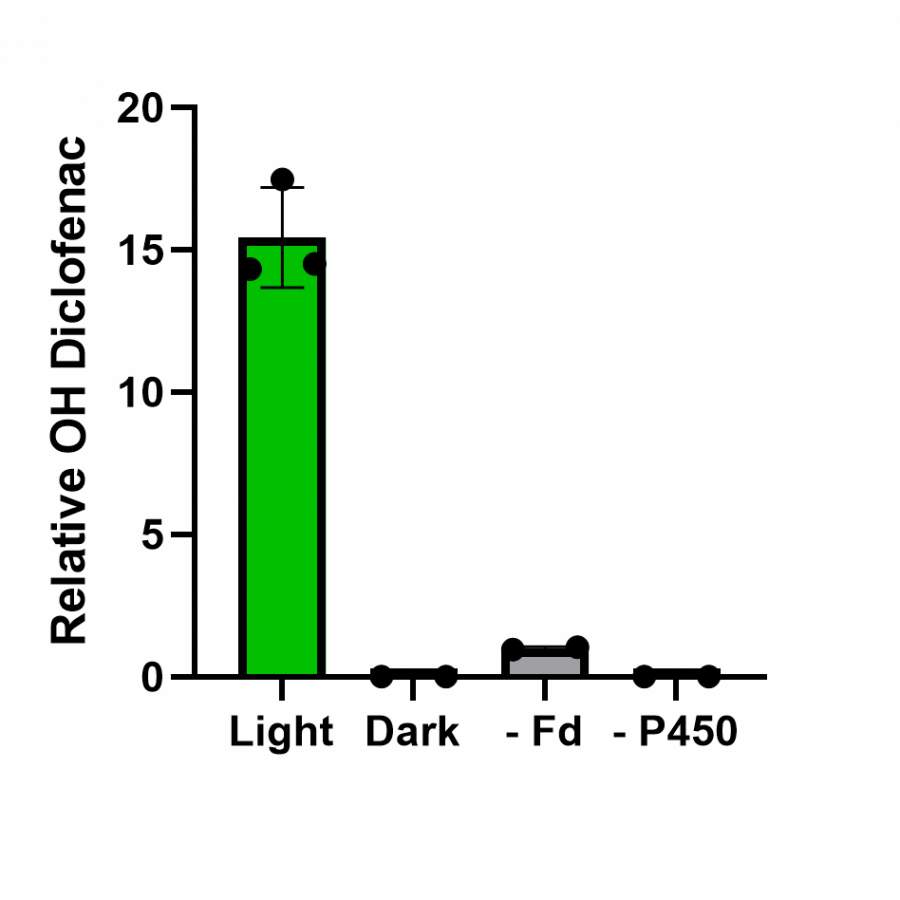 Fig. 2 relative hydroxy (OH) metabolite of diclofenac in in vitro light-driven system using P450 ancestor, CYP2ABGSFTCEH_N13, C reinhardtii thylakoid membrane and ferredoxin Light and dark, reactions in the presence and absence of light, respectively. -Fd and -P450 control reactions without ferredoxin or P450, respectively |
#586 | Seed meal of Fagopyrum tataricum as catalyst for transrutinosylation of tyrosol and hydroxytyrosol |
|
| Presenting author: | Elena KARNIŠOVÁ POTOCKÁ of INSTITUTE OF CHEMISTRY, SLOVAK ACADEMY OF SCIENCES |
| Other authors: | Mária MASTIHUBOVÁ of INSTITUTE OF CHEMISTRY, SLOVAK ACADEMY OF SCIENCES Vladimír MASTIHUBA of INSTITUTE OF CHEMISTRY, SLOVAK ACADEMY OF SCIENCES |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | rutinosidase / Fagopyrum tataricum / tyrosol / hydroxytyrosol |
| Purpose: | Enzymatic synthesis using diglycosidases offer effective, environmentally friendly procedures and more convenient strategies in preparation of structured oligoglycosides. Seeds of Fagopyrum tataricum possess high rutinosidase activity, therefore are able to transfer the whole rutinosyl moiety from the donor to an acceptor in one step, while containing up to 2.4 % of rutin. Tyrosol rutinoside and hydroxytyrosol rutinoside were synthesised for the first time with rutinosidase of this origin. First one was previously synthetised with rutinosidase from Sophora japonica [1] and as a mixture of hardly separated isomers using rutinosidase from Aspergillus niger [2]. Synthesis was preoptimized in terms of pH, added rutin, tyrosol and amount of catalyst in the form of defatted seed meal using HPLC analysis. The preparative reaction was then carried out under following conditions: pH 6.5, 33 mM rutin, 72 mM tyrosol and catalyst in amount 3% (w/vol.). The maximal conversion of tyrosol rutinoside achieved more than 61 % (respective to rutin) and the isolated yield 35 % with purity ca. 97 %. Isolation was achieved with combination of chromatographic separation on Al2O3, Diaion HP 20 and silica gel. The rutinosylation proceeds regioselectively and results in formation of only one product glycosylated on the primary hydroxyl of tyrosol (as confirmed by NMR). Hydroxytyrosol rutinoside was prepared under the same conditions. This work was supported by the Slovak Research and Development Agency under the contract No. APVV 18-0188 and by the Slovak Grant Agency for Science VEGA (grant number 2/0111/22). The work was inspired by scientific interactions that evolved within the COST Action CA20127 - Waste biorefinery technologies for accelerating sustainable energy processes (WIRE). |
| References: | [1] Karnišová Potocká, E., Mastihubová, M., Mastihuba, V. Food Chem. 2021, 336, 127674 [2] Bassanini, I., Krejzová, J., Panzeri, W., Monti, D., Křen, V., Riva, S. ChemSusChem. 2017, 10, 2040-2045. |
#593 | Glycosynthase-based synthesis of peptidoglycan oligosaccharides to decipher bacterial cell-wall elongation processes |
|
| Presenting author: | Sebastien FORT of UNIV. GRENOBLE ALPES, CNRS, CERMAV |
| Other authors: | Antoine ROUSSEAU of UNIV. GRENOBLE ALPES, CNRS, CERMAV Dindet Steve-Evanes KOFFI TEKI of UNIV. GRENOBLE ALPES, CNRS, CERMAV Louis BRIGANDAT of UNIV. GRENOBLE ALPES, CNRS, CEA, IBS Emeline RICHARD of UNIV. GRENOBLE ALPES, CNRS, CERMAV Isabel AYALA of UNIV. GRENOBLE ALPES, CNRS, CEA, IBS Jean-Pierre SIMORRE of UNIV. GRENOBLE ALPES, CNRS, CEA, IBS Sylvie ARMAND of UNIV. GRENOBLE ALPES, CNRS, CERMAV Sylvain COTTAZ of UNIV. GRENOBLE ALPES, CNRS, CERMAV Catherine BOUGAULT of UNIV. GRENOBLE ALPES, CNRS, CEA, IBS |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Glycosynthase / Oligosaccharides / Peptidoglycane / |
| Purpose: | Most antibiotics used in human medicine inhibit the biosynthesis of peptidoglycan, an essential component of the bacterial wall. However, antibiotic resistance is rising to dangerously high levels, becoming one of the biggest threats to global health.[1] Combating this burden therefore requires the detailed study of peptidoglycan metabolism and implies the need to produce well-defined molecular probes.[2,3] In this context we devised a glycosynthase-based chemo-enzymatic synthesis of peptidoglycan oligosaccharides using the D52S mutant of hen egg-white lysozyme (D52S HEWL).[4] Size-control of the oligosaccharides during glycosylation was achieved using a non-polymerizable donor. A galactosyl group, whose introduction and deprotection can be done enzymatically was chosen as temporary protecting group. Trisaccharide Gal-GlcNAc-1,6-anhMurNAc was produced by metabolically engineering E. coli cells and chemically fluorinated at the reducing end to provide the target donor. Successive rounds of glycosylation using D52S HEWL followed by degalactosylation using a commercial -galactosidase afforded the expected tetra-, hexa- and octasaccharides in 60-70% yields. These compounds were used to decipher the impact of the charge on the lactoyl group of MurNAc, of the anhydro at the reducing end of the oligosaccharide, and of the oligosaccharide chain length on the interaction with E. coli DedD, a SPOR-domain-containing protein involved in bacterial cell-division.[5] The information retrieved using NMR spectroscopy in this study sheds a new light on the role of this protein. Acknowledgments: This work was supported by the French National Research Agency (ANR) through Glyco_SWIM (ANR-20-CE07-0012-01) |
| References: | [1] O'Neill, J. Nat. Rev. Drug Discov. 2016, 15(8), 526-526 [2] Lee, M., Hesek, D., Lastochkin, E., Dik, D. A., Boggess, B., Mobashery, S. ChemBioChem 2017, 18(17), 1696-1702 [3] Wang, N., Hasegawa, H., Huang, C. Y., Fukase, K., Fujimoto, Y. Chem. Asian J. 2017, 12(1), 27-30 [4] Rousseau, A., Armand, S., Cottaz, S., Chem. Eur. J. 2021, 27(70), 17637-17646 [5] Pazos, M., Peters, K., Boes, A., Safaei, Y., Kenward, C., Caveney, N. A., Laguri, C., Breukink, E., Strynadka, N. C. J., Simorre, J. P., Terrak, M., Vollmer, W. 2021, mBio, e02796-20 |
| Figures: | 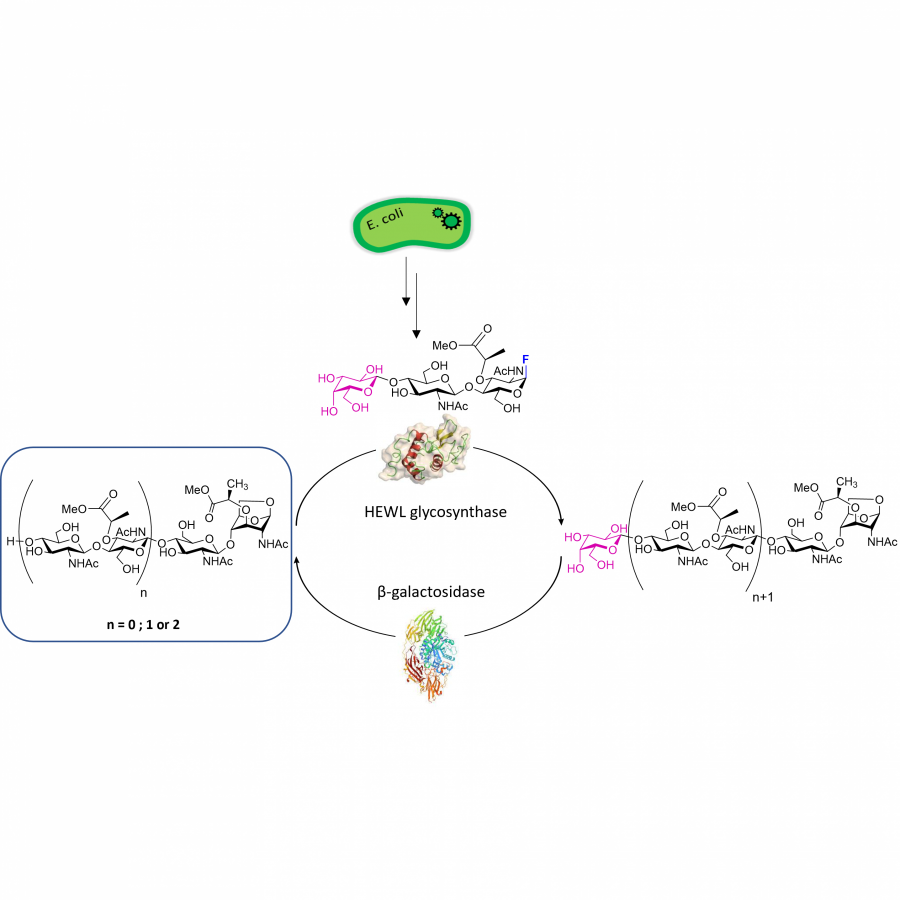 Glycosynthase-based synthesis of peptidoglycan oligosaccharides Iterative synthesis of peptidoglycan oligosaccharides using a HEWL-glycosynthase/galactosidase tandem from precursors obtained by metabolically engineered bacteria 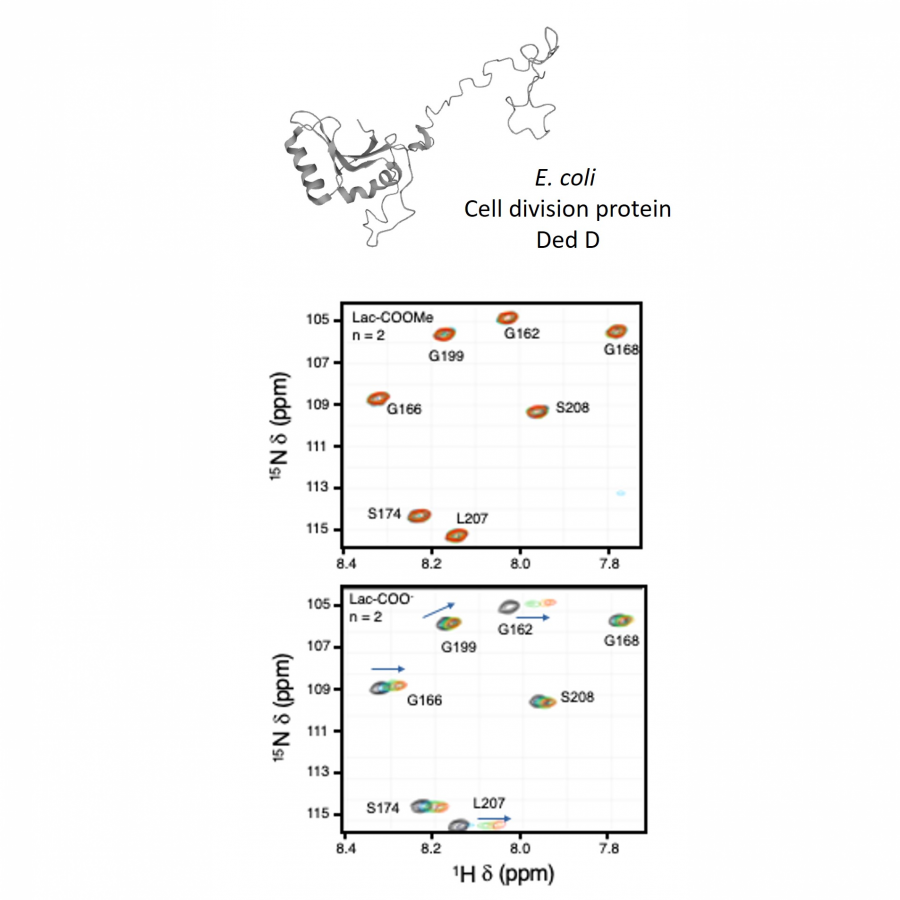 Characterization of E. coli DedD oligosaccharides interaction by NMR Proton NMR spectra of DedD protein in presence of peptidoglycan oligosaccharides containing a free lactoyl group or its methyl ester form |
#597 | ANALYSIS OF DIFFERENT SET-UPS FOR SCALING-UP THE BIOCATALYTIC SYNTHESIS OF PANTHENYL MONOESTERS |
|
| Presenting author: | Susana NIETO CERON of UNIVERSITY OF MURCIA |
| Other authors: | Juana M. BERNAL of UNIVERSITY OF MURCIA Rocio VILLA of UNIVERSITY OF MURCIA Eduardo GARCIA-VERDUGO of UNIVERSIDAD JAUME I Antonio DONAIRE of UNIVERSIDAD DE MURCIA Pedro LOZANO of UNIVERSITY OF MURCIA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sustainable / biocatalysis / scaling-up / green metrics |
| Purpose: | The incorporation of bioactive molecules to commodities is under increasing demand because of their benefits to health. Nonetheless, the use of solely active ingredients has some hints derived from their structure, which determines a previous step of modification to improve their miscibility with different formulas, their performance, stability, or duration effects. In the field of cosmeceuticals the acylation of bioactive molecules is the predominant modification, which improves the dermal absorption of hydrophilic biomolecules and stabilizes the antioxidant activity of the aromatic ones. However, the concerns about the impact of chemical modifications on health and the environmental burden demand the design of more sustainable approaches (1 ). At this point, solvent-free reactions catalyzed by enzymes stand out as the more promising strategies from the sustainable point of view. When working in the absence of solvent is not always possible due to substrates’ non-miscibility, the formation of a eutectic mixture between substrates is a smart strategy to achieve their coexistence in the same phase (2 ). In addition, the enzymes show outstanding biodegradability, efficiency, and selectivity which permits on-demand customization of the substrates leading to innovative compounds. For example, polyalcohol panthenol is the pro-vitamin B5, which acylated derivative is highly demanded in the cosmetic and pharmacological fields because of its bioactive properties (humectant, wound-healing, etc.) (3 ,4 ). However, it has been reported how the selective single esterification of panthenol, achieves the same result but also provides an additional surfactant activity which is also very convenient for the aforementioned sectors (2). This work shows the optimization and validation of a gradual scaling-up process for the solvent-free biocatalytic synthesis of panthenol monoesters, based on eutectic mixtures with different free fatty acids (FFAs), and different size sept-ups (ultrasounds, orbital shaking, rotary evaporator, and mechanically stirred reactor) to reach a final 500 g reaction size (Figure 1) ( 5). The intensification of the process at higher scales has been assessed by kinetic studies and the productivity (monoester (g) / reaction time (h) x biocatalyst (g)) achieved. The results point to 3 key parameters to shift the equilibria towards the synthesis: (1) the longer alkyl chain in the FFA (i.e. the yield increases from 63 % yield for panthenyl butyrate to 83 % yield for panthenyl myristate), (2) an efficient agitation and (3) high-performing water removal (i.e. systems coupled to vacuum). The last two parameters, dependent on the set-up, are improved at higher scale systems (rotary evaporator, and mechanically stirred reactor) affording not only the best results (87 %-95 % monoester yield), but also a significant five-fold reduction of the amount of biocatalyst which in addition can be recovered and reuse for 5 cycles. In addition, different Green Metrics Parameters (ε , AE, 1/S, MRP, RME, PMI, TCR, E-factor, EcoScale) have guided the scaling process from the benchmark scale to the 500 g size, highlighting the higher sustainability of this strategy respect to others previously reported. These results demonstrate the process of scaling-up this solvent-free reaction, based on eutectic mixtures and biocatalysis, from the benchmark to a relevant environment, is feasible and provides an intensification of the performance with economical and sustainable up-gradings. Acknowledgments This work was partially supported by 21640/PDC/21 and 21884/PI/22 (Fundación SENECA-CARM), and by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021- 124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN –European Union Next Generation EU-PRTR. |
| References: | 1. Alvarez, E.; Villa, R.; Nieto, S.; Donaire, A.; García-Verdugo, E.; Luis, S. V.; Lozano. P. The suitability of lipases for the synthesis of bioactive compounds with cosmeceutical applications. Mini-Rev. Org. Chem. 2021, 18 (4), 515-528, DOI: 10.2174/1570193X17999200805215623 2 . Lozano, P.; Alvarez, E.; Nieto, S.; Villa, R.; Bernal, J.M.; Donaire, A. Biocatalytic synthesis of panthenyl monoacyl esters in ionic liquids and deep eutectic solvents. Green Chem. 2019, 21, 3353-3361, DOI: 10.1039/C9GC01076A 3 . Gorski, J.; Proksch, E.; Baron, J.M.; Schmid, D.; Zhang, L. Dexpanthenol in wound healing after medical and cosmetic interventions (postprocedure wound healing). Pharmaceuticals, 2020, 13, Art Nº 138. DOI: 10.3390/ph13070138 4 . Ferreira, M.S.; Sousa Lobo, J.M.; Almeida, I.F. Sensitive skin: Active ingredients on the spotlight. Int. J. Cosmet. Sci. 2022, 44, 56-73, DOI: 10.1111/ics.12754 5. Nieto, S., Bernal, J.M., Villa, R., Garcia-Verdugo, E., Donaire, A., Lozano, P. Sustainable set-ups for the biocatalytic production and scale-up of panthenyl monoacyl esters under solvent-free conditions. ACS Sustainable Chem. Eng. 2023. In press. DOI 10.1021/acssuschemeng.3c00266 |
| Figures: | 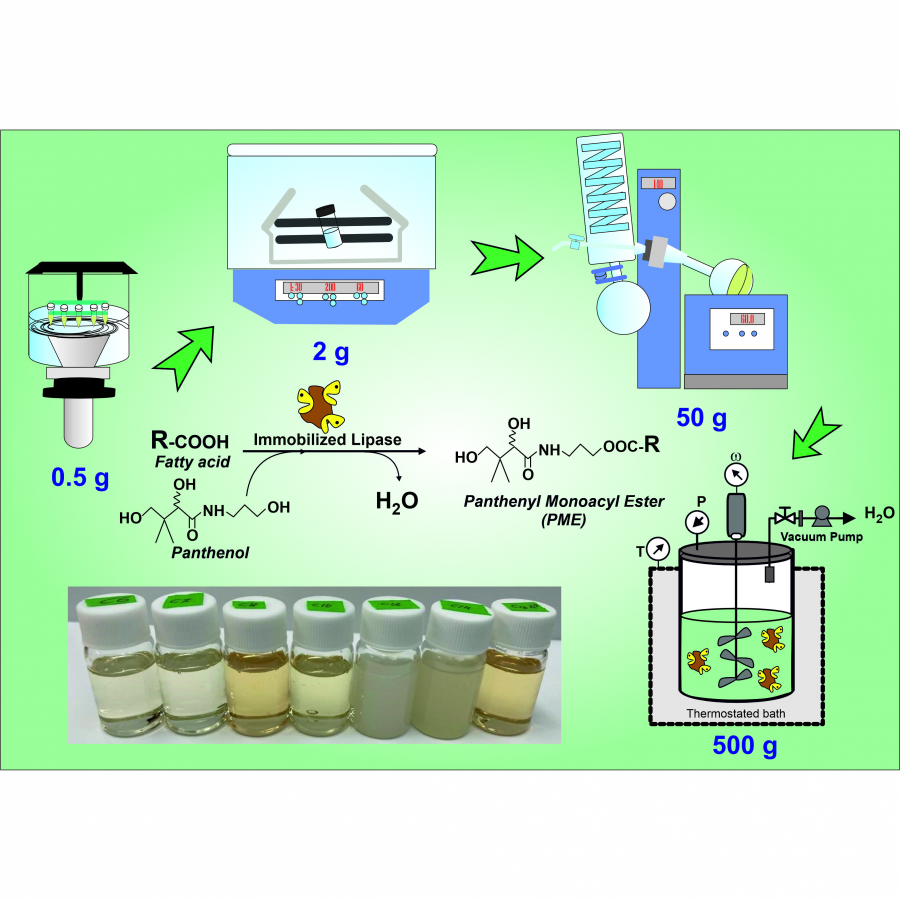 Figure 1. Scheme of biocatalytic esterification of FFAs with panthenol and set-ups for scaling-up. Bottom left: picture of panthenyl monoesters with different alkyl-chain lengths |
#600 | Green biocatalysis: chemo-enzymatic valorisation of industrial hemp waste from essential oil production |
|
| Presenting author: | Fabio PARMEGGIANI of POLITECNICO DI MILANO |
| Other authors: | Davide TESSARO of POLITECNICO DI MILANO Fabio SANGALLI of POLITECNICO DI MILANO Celeste NOBBIO of POLITECNICO DI MILANO Daniele FIORITO of POLITECNICO DI MILANO Elisabetta BRENNA of POLITECNICO DI MILANO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | agri-food waste / lipase / cannabinoids / chemo-enzymatic synthesis |
| Purpose: | Waste from the agri-food industry is gaining increasing value in the modern economy, and exploiting the potential of these resources requires a systemic change: the use of renewable organic residues as starting material for a sustainable value creation. In this work, the valorization of the organic waste connected with the production of Cannabis sativa essential oil has been considered as a potential source of valuable chemicals.[1] The essential oil is produced by steam distillation of the apical part of industrial hemp plant, which removes all the volatile components. The spent distillation residue, in the form of wet biomass, constitutes more than 90% of the original material loaded and is normally disposed of as agricultural waste or returned to the fields. However, many potentially useful natural products are still present in the matrix and would be discarded. The broad family of cannabinoids, in particular, are not sufficiently volatile to be removed by distillation, and remain in the solid wet residue. The naturally occurring cannabinoids (>100 molecules), as well as their synthetic or functionalised analogues, have been studied for many years due to their extremely interesting combination of bioactivities and pharmacological properties.[2] Although some of the cannabinoids, most notably tetrahydrocannabinol (THC), have psychoactive properties and are considered illegal substances in most countries, industrial hemp crops have been specifically selected for their lower content of psychotropic cannabinoids. Industrial hemp can be cultivated without special authorisations for multiple industrial applications, including the production of fiber, seeds and essential oil, and still contains non-psychoactive cannabinoids which are very valuable for the consumer market and the pharmaceutical industry. The possibility to recover cannabinoids has been investigated in this work, analysing selective extraction techniques from the distillation waste of the industrial hemp C. sativa Futura 75, leading to an optimised protocol for the recovery of pure cannabidiol (CBD) from the spent waste with a yield of approximately 10-20 g/kg, depending on the variety and quality of the crop (Figure 1). The CBD recovered can be used in a range of consumer products for its antioxidant, anti-inflammatory and neuroprotective properties, as well as a natural food supplement. In the perspective of maximising the value of the material recovered, a procedure for the chemo-enzymatic functionalisation has been studied to convert the recovered CBD into other valuable cannabinoid derivatives. Exploiting an optimised lipase-mediated perhydrolysis in the presence of ethyl acetate, CBD has been converted into epoxy-cannabidiol (CBO) and further into cannabielsoin (CBE), cannabinoids with reported activity for pain management and as neuroprotective agents. [3,4] Overall, the method (Figure 1) is sustainable and environmentally friendly, involving only an immobilised enzyme, a benign solvent and hydrogen peroxide as the oxidant, to afford a high-value product starting from agricultural waste, complying with the highly prized principles of green chemistry and circular economy. |
| References: | [1] A. Sainz Martinez, et al., Nat. Prod. Rep. 2023,40, 676-717 [2] A. Bilbao et al., BMC Medicine 2022, 20, 259. [3] Y. Nalli, et al., Bioorg. Med. Chem. Lett. 2019, 29, 1043-1046. [4] A. Z. Monroe, et al., Chem. Commun. 2021, 57, 5658-5661. |
| Figures: | 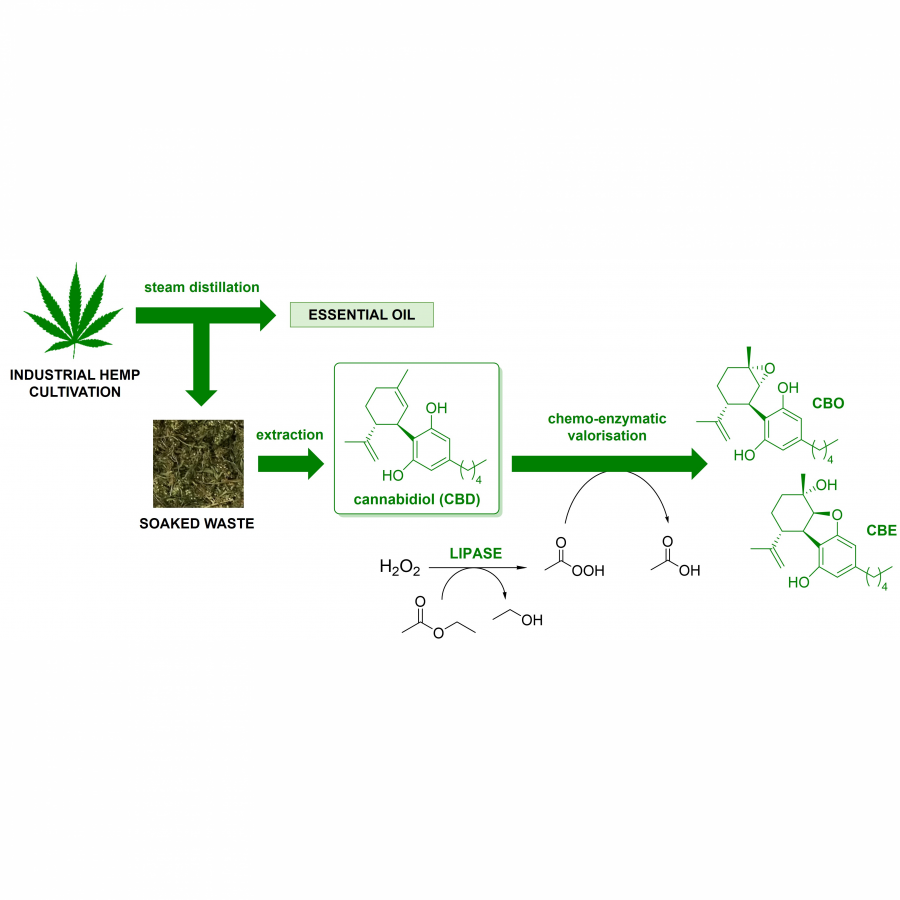 Figure 1. Recovery and chemo-enzymatic transformations of CBD from hemp distilation waste |
#602 | The power of phylogeny: Rational redesign of bacterial eugenol oxidases towards non-natural substrates |
|
| Presenting author: | Daniel EGGERICHS of RUHR-UNIVERSITY BOCHUM |
| Other authors: | Laura ALVIGINI of UNIVERSITY OF PAVIA Nils WEINDORF of RUHR-UNIVERSITY BOCHUM Heiner Gerald WEDDELING of RUHR-UNIVERSITY BOCHUM Andrea MATTEVI of UNIVERSITY OF PAVIA Dirk TISCHLER of RUHR-UNIVERSITY BOCHUM |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme mining / enzyme engeering / structure-function relations / side-saturation mutagenesis |
| Purpose: | Bacterial eugenol oxidases (EUGOs) and their fungal homologues, vanillyl alcohol oxidases (VAOs), form an enzyme family of versatile biocatalysts.[1,2] They catalyze a variety of different reaction types on a broad substrate range.[3] The oxidation of alkanes or alcohols in benzylic position is the characteristic reaction of both subfamilies including the oxidation of eugenol and vanillyl alcohol.[4,5] As phenolic compounds are important building blocks in the chemical industry for polymers, fragrances or flavors, we intended to widen the substrate scope towards none-natural substrates. Therefore, we created an R-based GUI-driven software to streamline enzyme mining and identify crucial residues in the enzyme structure. These positions were explored by mutagenesis, and the crystal structure of the most promising candi-date was obtained. The use of computer-guided approaches in the field of enzyme design increases in popularity and recently great success was achieved by tailoring the eugenol oxidase from Rhodococcus jostii RHA1 (RjEUGO) through an in silico strategy to produce iso-eugenol.[6] Despite this recent advancement, the overall sequences space of EUGOs is rather poorly investigated with the mentioned RjEUGO representing the only member so far. Thus, we explored homologous enzymes in order to find naturally improved variants. Among the enzyme family, we compared key residues and selected a set of seven candidates for subsequent characterization. The substrate profile of each enzyme was correlated with a respective structural model. As example, the sterically size of three residues was found to effect the conversion of 3-bromo-4-hydroxybenzyl alcohol (Figure 1), a motive commonly found in bromo dyes like pH indicators. Subsequently, we performed site-saturation mutagenesis screening ~100 clones for every position individually (>95% library coverage) using a hydrogen peroxide sensitive assay in the cell-free crude extract. Characterization of the best enzyme variant highlighted a nine-fold improved vmax (14.7 ± 0.5 U/mg) compared to the wild type and even higher activities compared to other EUGOs (Figure 2). In our effort to explore non-natural substrates for EUGOs, we obtained sixteen active enzyme variants and achieved improved reaction rates compared to the respective most active natural enzyme for the dehydrogenation of 4-cyclopentylphenol (2.5-fold, ScEUGO V427I L282M), the deamination of vanillyl amine (2.2-fold, ScEUGO E378T), the ether cleavage of butyl vanillyl ether (2.5-fold, ScEUGO V427I) and, as a side effect, for the hydroxylation of eugenol (1.4-fold, ScEUGO V427I). As the enzyme from Streptomyces cavernae (ScEUGO) appeared as the overall most interesting enzyme, we obtained the crystal structure up to 1.8 Å resolution to validate our models and explain the observed effects on a molecular level. Thus, we managed to construct robust relationships between structure and function of EUGOs. Crucial residues for selectivity were identified and can be used to design a tailored oxidase. Further, the presented computational tools allow a transfer of this methodology to other enzyme families and will significantly improve the quality and pace of enzyme mining. |
| References: | [1] M. Garcia-Bofill, P.W. Sutton, H. Straatman, J. Brummund, M. Schuermann, M. Guillen, G. Alvaro, Appl. Catal. A, 2021, 610, 117934. [2] M. Habib, M. Trajkovic, M.W. Fraaije, ACS Catal., 2018, 8, 5549-5552. [3] T.A. Ewing, A. van Noord, C.E. Paul, W.J.H. van Berkel, Molecules, 2018, 23, 164. [4] R.H.H. van den Heuvel, M.W. Fraaije, A. Mattevi, C. Laane, W.J.H. van Berkel, J. of Mol. Catal. B, 2001, 11, 185-188. [5] Q.-T. Nguyen, G. de Gonzalo, C. Binda, A. Rioz-Martinez, A. Mattevi, M.W. Fraaije, Chembiochem, 2016, 17, 1359-1366. [6] Y. Guo, L. Alvigina, M. Trajkovic, L. Alonso-Cotchico, E. Monza, S. Savino, I. Maric, A. Mattevi, M.W. Fraaije, Nat. Commun., 2022, 13, 7195. |
| Figures: | 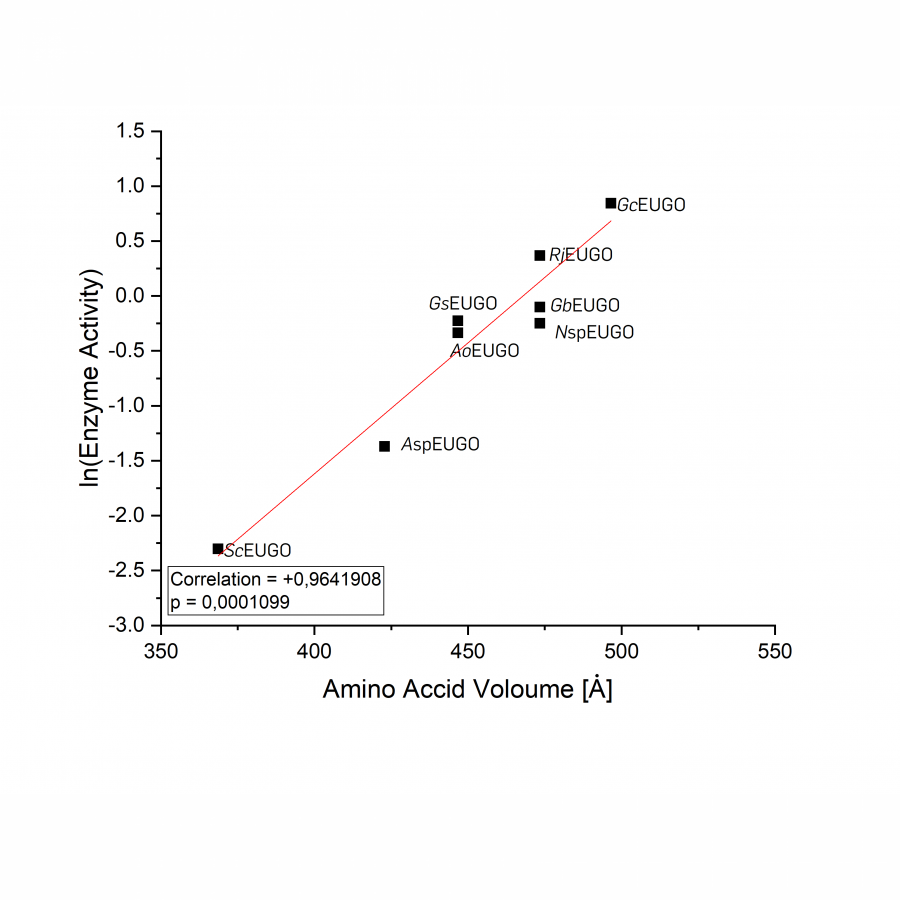 Activity correlation Semi-logarithmic plot of the enzyme activity on 3-bromo-4-hydroxybenzyl alcohol of indicated EUGOs against the added volume of amino acids in position 166, 391 and 427 (RjEUGO numbering). Enzyme activity increases with the sterical demand of these residue 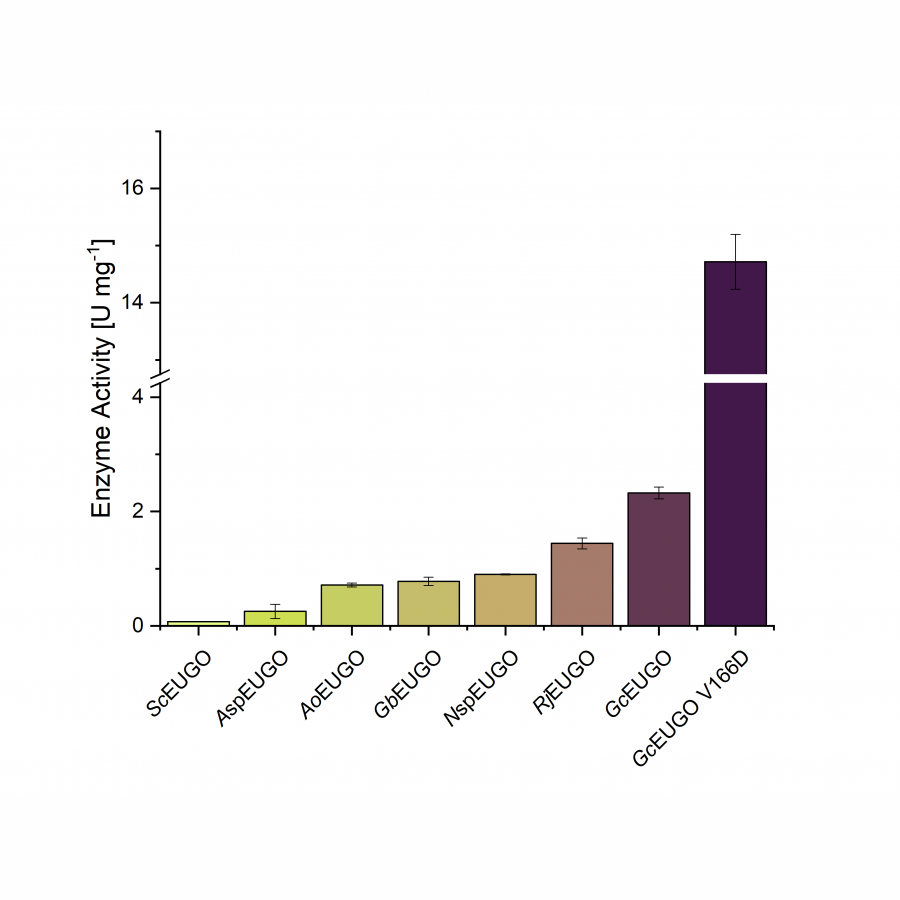 Mutagenesis Enzyme activity on 3-bromo-4-hydroxybenzyl alcohol by natural enzymes in comparison with GcEUGO V166D. The variant was found to be most active in the mutagenesis studies. |
#603 | Enzymatic Degradation of PET Plastic by Mechanical Agitation |
|
| Presenting author: | Esther AMBROSE-DEMPSTER of UNIVERSITY COLLEGE LONDON |
| Other authors: | Helen HAILES of UNIVERSITY COLLEGE LONDON |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | polyethylene terephthalate (PET) / PETase enzymes / mechanical agitation / natural environment |
| Purpose: | Due to the rising problem of plastic pollution, it is becoming necessary to develop new methods of removing plastic waste from the natural environment. As a plastic generally used for single use purposes, polyethylene terephthalate (PET) is produced on an annual scale of over 50 million tonnes.[1] A large proportion of the PET produced ends up as plastic waste in the environment, despite it being fully recyclable. In 2016, an enzyme able to degrade PET by using it as its sole carbon source was discovered, providing a potential solution to part of the plastic waste problem.[2] In this work, PETase enzymes have been used in whole cell form to degrade PET substrates by both traditional biocatalytic methods and mechanical agitation. By HPLC analysis and SEM imaging, it has been found that optimal time lengths of mechanical agitation and aging periods improved the degradation of PET substrates, achieving higher yields of breakdown products than seen previously. This sustainable method of PET breakdown offers potential for molecular recycling, using reduced solvent volumes in comparison with traditional approaches and utilises enzymatic methods as a sustainable approach to breaking down the plastic waste polluting our natural environment. |
| References: | 1. U. T Bornscheuer, Science, 2016, 351, 1154-1155. 2. S. Yoshida, K. Hiraga, T. Takehana, I. Taniguchi, H. Yamaji, Y. Maeda, K. Toyohara, K. Miyamoto, Y. Kimura and K. Oda, Science, 2016, 351, 1196-1199. |
| Figures: | 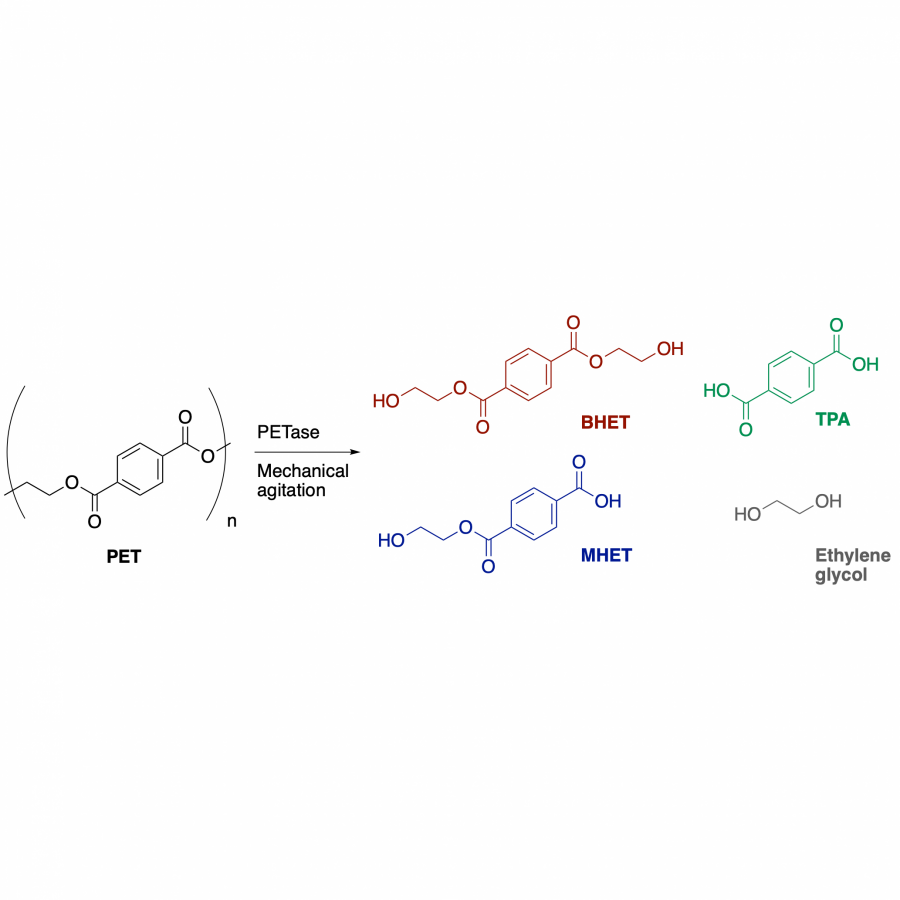 PET breakdown Scheme showing PET breakdown into monomers: bishydroxyethyl terephthalate (BHET), mono(2-hydroxyethyl) terephthalic acid (MHET), terephthalic acid (TPA) and ethylene glycol (EG).  SEM image SEM imagery showing PET powder prior to enzymatic breakdown. |
#605 | APPLICATION OF GREEN METRIC PARAMETERS TO THE EVALUATION OF THE SUSTAINABILITY OF A BIOCATALYTIC SOLVENT-FREE SCALE-UP PROCESS |
|
| Presenting author: | Susana NIETO CERON of UNIVERSIDAD DE MURCIA |
| Other authors: | Francisco J. RUIZ of UNIVERSIDAD DE MURCIA Francisco MARTINEZ-MORA of UNIVERSIDAD DE MURCIA Inmaculada LOZANO of UNIVERSIDAD DE MURCIA Rocio VILLA of UNIVERSIDAD DE MURCIA Eduardo GARCIA-VERDUGO of UNIVERSIDAD JAUME I Pedro LOZANO of UNIVERSIDAD DE MURCIA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / green metrics / optimization / scaling-up |
| Purpose: | Chemistry has contributed to society’s evolution and well-being by means of providing different commodities. However, nowadays the environmental burden of chemical transformations is in the spotlight of government agencies and society itself, because of the impact on the biosphere and the depletion of resources that mortgages future generations. The current compromise of chemists is the development of more sustainable strategies based on the more efficient use of renewable resources and energy and with a lower environmental impact as advocated by the Twelve Principles of Green Chemistry. Nonetheless, merely the application of some of those principles is not enough, being necessary to quantitatively certify the sustainability of the processes. For this, different Green Metrics Parameters have been developed as tools to measure different aspects of the upstream and downstream stages of a process. This work describes how the application of some Green Metrics Parameters can be very useful in the optimization of a biocatalytic process for synthesizing new non-ionic surfactants based on the direct selective mono-esterification of a poly-alcohol (panthenol), as well as to assess the sustainability of the intensified strategy after a scaling-up process (1 ). As can be seen in Table 1, while the Atom Economy (AE), Stoichiometric Factor (S), and Yield are parameters that exclusively refer to the reactivity of the process, the Material Recovery Parameter (MRP), Reaction Mass Efficiency (RME), Process Mass Intensification (PMI), Total Carbon Release (TCR) and E-factor, also consider auxiliary reagents and downstream processes of purification and wastes treatments. In addition, the EcoScale is a tool that integrates ecological and economical aspects to perform a preliminary study of the Life Cycle Assessment (LCA) of a process ( 2). An initial study performed with AE, S, Yield, MRP, and RME permitted to identify the optimal molar ratio panthenol : FFA in order to achieve the higher yield in the synthesis of specific panthenyl monoesters. Figure 1.A. shows a radial diagram, as proposed by Andraos, where it is assumed that as the values of the Green Metrics plot a balanced pentagon with a radius close to 1, the greener is the process (3 ). These results highlight that the maximum yield is achieved with an equimolar ratio of substrates and that an excess of either substrate only leads to the accumulation of wastes or non-desired products. In addition, these values point to the high economy of the strategy based on the direct esterification in solvent-free system since the negligible mass weight water, as the only by-product, improves the economy of the process, to which must be added the recovery and reuse of the biocatalysts. To compare the impact of the synthesis with different set-ups, the PMI, TCR, E-factor and EcoScale parameters were used (Figure 1B). Again, the values of these parameters support the sustainability of the process due to its´ high efficiency, the lack of solvent, and that no purification step is needed, as non-reacted substrates are also cosmetic ingredients. However, best productivity and selectivity because of the intensification at higher reaction scales, points to the reactor system at 500 g size as the most sustainable and economical set-up. This intensification seems to be related to a more efficient agitation (that improves mass transfer) and water removal (because of the coupled vacuum system) along the reaction. This study evidence that the use of Green Metric Parameters is highly recommended whenever a (bio)chemical process is being developed for industry implementation. Acknowledgments This work was partially supported by 21640/PDC/21 and 21884/PI/22 (Fundación SENECA-CARM), and by MICINN-FEDER-AEI 10.13039/501100011033 (PID2021- 124695OB-C21/C22 and PDC2022-133313-C21/C22), MICINN –European Union Next Generation EU-PRTR. |
| References: | 1. Nieto, S., Bernal, J.M., Villa, R., Garcia-Verdugo, E., Donaire, A., Lozano, P. Sustainable set-ups for the biocatalytic production and scale-up of panthenyl monoacyl esters under solvent-free conditions. ACS Sustainable Chem. Eng. 2023. In press.DOI 10.1021/acssuschemeng.3c00266 2 . Van Aken, K.; Strekowski, L.; Patiny, L. EcoScale, a semi-quantitative tool to select an organic preparation based on economical and ecological parameters. Beilstein J. Org. Chem. 2006, 2, 3, DOI:10.1186/1860-5397-2-3 3 . Andraos, J.; Sayed, M. On the use of “green” metrics in the undergraduate organic chemistry lecture and lab to assess the mass efficiency of organic reactions. J. Chem. Educ. 2007, 84 (6), 1004-1010, DOI: 10.1021/ed084p1004. 4 . Sheldon, R. A. Metrics of Green Chemistry and Sustainability: Past, Present, and Future. ACS Sustainable Chem. eng. Green Chem., 2018, 6, 32-48, DOI: 10.1021/acssuschemeng.7b03505 5 . https://www.acs.org/content/acs/en/greenchemistry/research-innovation/tools-for-green-chemistry.html |
| Figures: |  Table 1. Green Metric Parameters, equations, and definitions. (1,4,5) 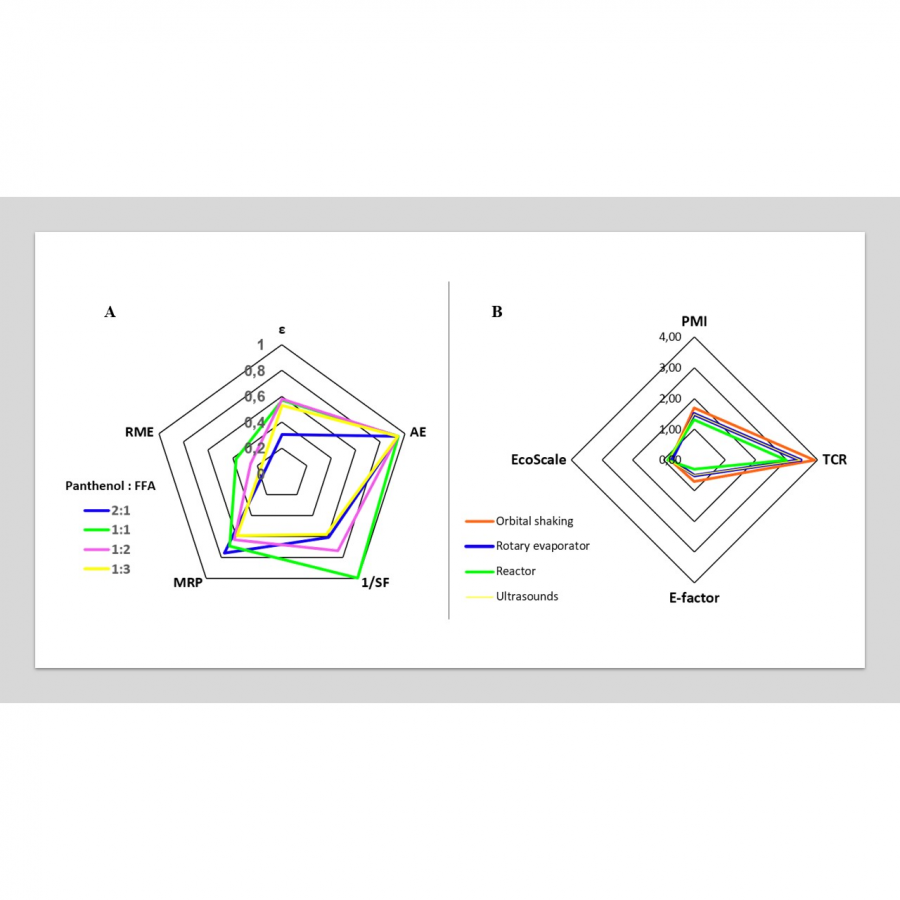 Figure 1. Diagram representations of the Green Metrics analyses. A. Radial pentagon in the optimization analysis of substrates molar ratios. B. Assessment of the environmental impact of different set-ups. |
#612 | Substrate Profiling of Anion Methyltransferases for Promiscuous Synthesis of S-Adenosylmethionine Analogs from Haloalkanes |
|
| Presenting author: | Kai SCHÜLKE of UNIVERSITÄT BIELEFELD |
| Other authors: | Stephan HAMMER of UNIVERSITÄT BIELEFELD |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Methyltransferases / Biocatalysis / Cascade / Cosubstrate Analogs |
| Purpose: | Biocatalytic alkylation reactions can be performed with high chemo-, regio- and stereoselectivity using S-adenosyl-L-methionine (SAM)-dependent methyltransferases (MTs) and SAM analogs.[1–3] Currently, however, this methodology is limited in application due to the rather laborious protocols to access SAM analogs. It has recently been shown that halide methyltransferases (HMTs) enable synthesis and recycling of SAM and its analogs from S-adenosyl-L-homocysteine (SAH) and readily available haloalkanes as starting material.[4–6] We further expanded this work by using substrate profiling of the anion MT enzyme family to explore promiscuous SAM analog synthesis.[7] Our study shows that anion MTs are in general very promiscuous with respect to the alkyl chain as well as the halide leaving group. Substrate profiling further suggests that promiscuous anion MTs cluster in sequence space. Next to iodoalkanes, cheaper, less toxic, and more available bromoalkanes have been converted and several haloalkanes bearing short alkyl groups, alkyl rings, and functional groups such as alkene, alkyne and aromatic moieties are accepted as substrates. Further, we applied these SAM analogs as electrophiles in highly selective C-N and C-C bond formations with N-heteroarens and coumarins.[8,9] |
| References: | 1. I. J. W. McKean, P. A. Hoskisson, G. A. Burley, ChemBioChem 2020, 21, 2890-2897. 2. T. D. Huber, B. R. Johnson, J. Zhang, J. S. Thorson, Curr. Opin. Biotechnol. 2016, 42, 189-197. 3. F. Ospina, K. H. Schülke, S. C. Hammer, ChemPlusChem 2022, 87, e202100454. 4. C. Liao, F. P. Seebeck, Nat. Catal. 2019, 2, 696-701. 5. L. L. Bengel, B. Aberle, A. Egler‐Kemmerer, S. Kienzle, B. Hauer, S. C. Hammer, Angew. Chem. Int. Ed. 2021, 60, 5554-5560. 6. Q. Tang, C. W. Grathwol, A. S. Aslan‐Üzel, S. Wu, A. Link, I. V. Pavlidis, C. P. S. Badenhorst, U. T. Bornscheuer, Angew. Chem. Int. Ed. 2021, 60, 1524-1527. 7. K. H. Schülke, F. Ospina, K. Hörnschemeyer, S. Gergel, S. C. Hammer, ChemBioChem 2022, 23, e202100632. 8. F. Ospina, K. H. Schülke, J. Soler, A. Klein, B. Prosenc, M. Garcia-Borràs, S. C. Hammer, Angew. Chem. Int. Ed. 2022, 61, e202213056. 9. A. Hoffmann, K. H. Schulke, S. Hammer, A. Rentmeister, N. Cornelissen, Chem. Commun. 2023, DOI 10.1039/D3CC01036H. |
| Figures: | 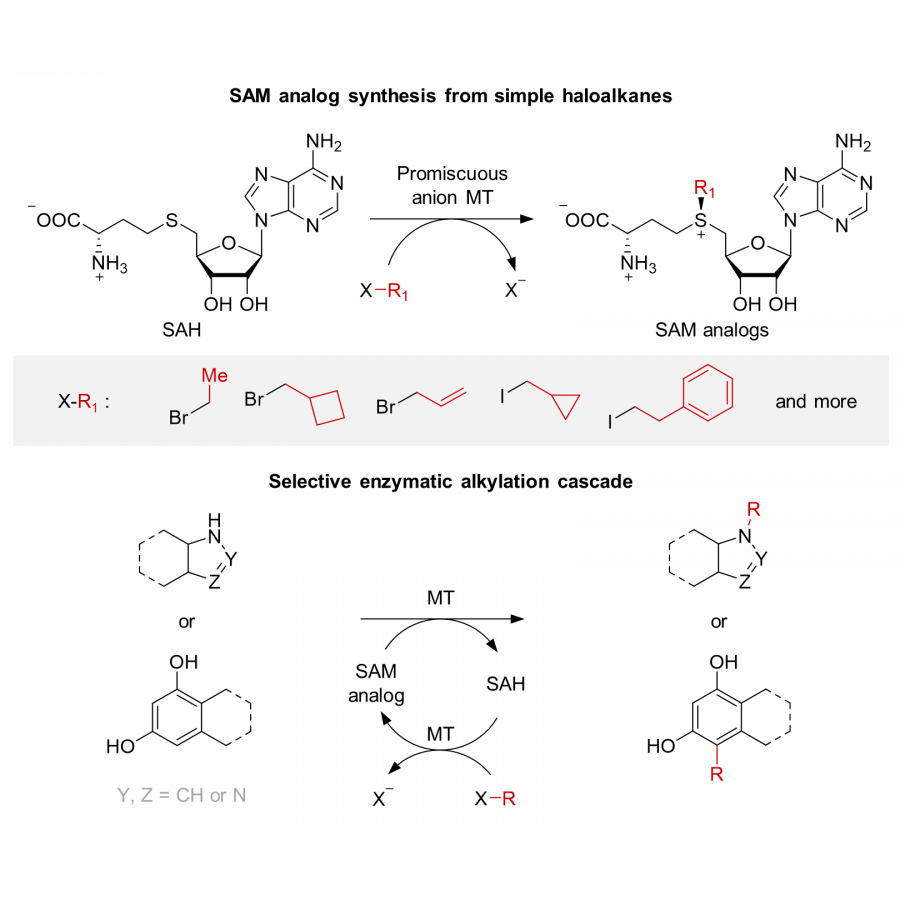 Enzymatic regeneration of SAM analogs via anion MT mediated alkylation of SAH with haloalkanes. SAM analogs used as electrophiles in MTs catalyzed alkylation cascades. |
#613 | Enzymatic synthesis of benzylisoquinoline alkaloids using a parallel cascade strategy and tyrosinase variants |
|
| Presenting author: | Yu WANG of DEPART OF CHEMISTRY |
| Corresponding author: | Helen HAILES of DEPART OF CHEMISTRY |
| Other authors: | Fabiana SUBRIZI of DEPART OF CHEMISTRY Eve CARTER of DEPART OF CHEMISTRY Tom SHEPPARD of DEPART OF CHEMISTRY John WARD of DEPART OF BIOCHEMICAL ENGINEERING Helen HAILES of DEPARTMENT OF CHEMISTRY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / benzylisoquinoline alkaloid / Enzyme cascade / |
| Purpose: | Benzylisoquinoline alkaloid derived pharmaceuticals are widely applied in modern medicines. Recent studies on the microbial production of benzylisoquinolines have highlighted key biological syntheses towards these natural products.1-3 Routes to non-natural benzylisoquinolines have been less explored, particularly halogenated compounds which are more challenging. In our previous work, up to seven enzymes were combined into one-pot cascades, yielding natural BIAs in good yields and enantiomeric excesses.4-5 Here, we show the use of a parallel cascade design incorporating a tyrosinase, tyrosine decarboxylase, transaminase, and norcoclaurine synthase, in order to generate halogenated benzylisoquinoline alkaloids in high enantiomeric excesses. Notably, mutagenesis studies were applied to generate tyrosinase variants, which enhanced the acceptance of halogenated tyrosines for use in the biocatalytic cascades developed.6 |
| References: | [1] H. Minami, J. Kim, N. Ikezawa, T. Takemura, T. Katayama, H. Kumagai, F. Sato. Proc. Natl Acad. Sci. 2008, 105, 7393-7398. [2] A. Nakagawa, H. Minami, JS. Kim, T. Koyanagi, T. Katayama, F. Sato, H. Kumagai. Nat. Commun. 2011, 2, 326. [3] A. Nakagawa, E. Matsumura, T. Koyanagi, T. Katayama, N. Kawano, K. Yoshimatsu, K. Yamamoto, H. Kumagai, F. Sato, H. Minami. Nat. Commun. 2016, 7, 10390. [4] Y. Wang, N. Tappertzhofen, D. Méndez-Sánchez, M. Bawn, B. Lyu, JM. Ward, HC. Hailes. Angew. Chem. Int. Ed. 2019, 58, 10120-10125. [5] F. Subrizi, Y. Wang, B. Thair, D. Méndez-Sánchez, R. Roddan, M. Cárdenas-Fernández, J. Siegrist, M. Richter, JN. Andexer, JM. Ward, HC. Hailes. Angew. Chem. Int. Ed. 2021, 60,18673-18679. [6] Y. Wang, F. Subrizi, EM. Carter, TD. Sheppard, JM. Ward, HC. Hailes. Nat. Commun. 2022, 13, 5436. |
| Figures: | 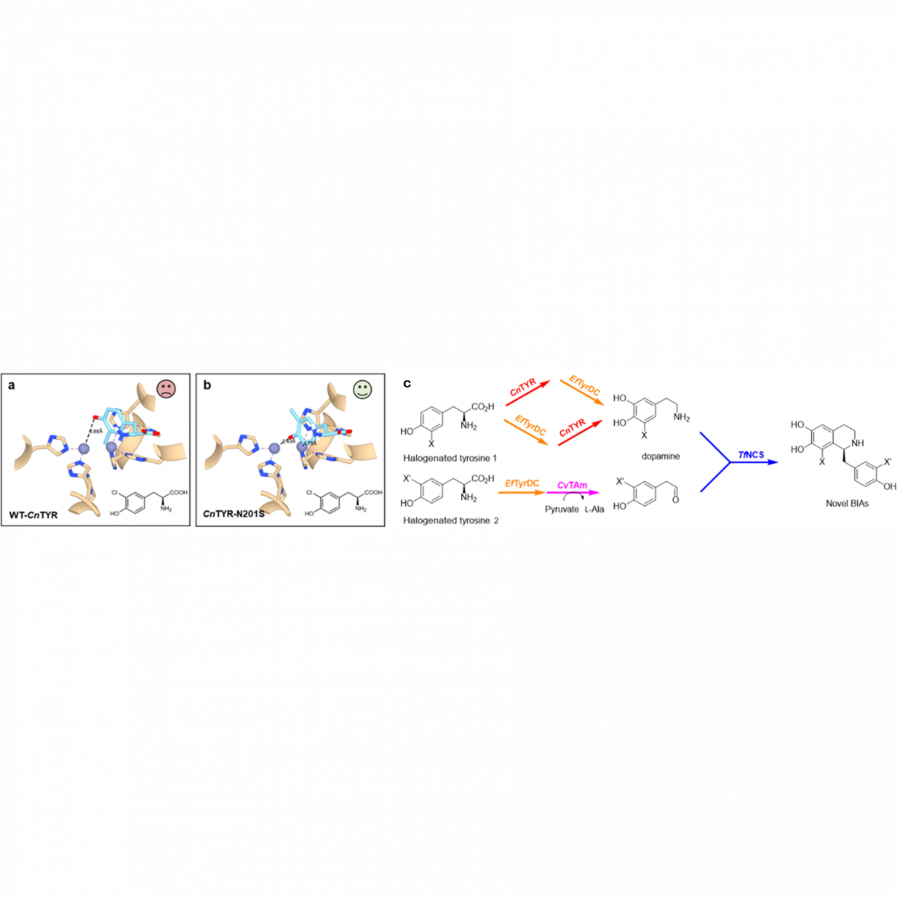 Figure 1. Scheme for parallel cascade design and molecular docking studies with WT-CnTYR and the variant. a. Docking of 3-Cl-L-tyrosine with WT-CnTYR: the substrate can fit into the active sites of WT-CnTYR but not in a productive orientation. b. Docking of 3-Cl-L-tyrosine with CnTYR-N201S: the substrate fits well into the active sites of CnTYR-N201S. The fun |
#619 | Diversity-oriented synthesis of cyclohexenes by combining enzymatic intermolecular Diels- Alder reactions and decarboxylative functionalizations |
|
| Presenting author: | Jin WANG of PEKING UNIVERSITY |
| Corresponding author: | Xiaoguang LEI of PEKING UNIVERSITY |
| Other authors: | Lei GAO of PEKING UNIVERSITY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | intermolecular Diels-Alderase / decarboxylative functionalization / diversity-oriented synthesis / chemoenzymatic synthesis |
| Purpose: | Substituted cyclohexanes are common scaffolds found in both natural products and drug molecules. Diels-Alderases that can efficiently catalyze intermolecular Diels-Alder reactions to generate cyclohexene ring systems have received considerable interest. However, the synthetic power of Diels-Alderases is incomparable with chemo-catalysts due to their limited substrate scopes. Here, we report a new chemo-enzymatic strategy for the diversity-oriented syntheses of functionalized cyclohexenes. A natural Diels-Alderase variant M3 were generated with focused rational iterative site-specific mutagenesis, which shows a 34-fold increase in catalytic efficiency, broad substrate scope, and good to perfect stereoselectivity. Diverse transition-metal-catalyzed decarboxylative coupling reactions were sequentially used to functionalize the enzymatic Diels-Alder products. This work offers an efficient synthetic route to structurally diverse cyclohexenes that are not accessible by solely biocatalysis or chemo-catalysis and illustrates how chemo-catalysis can cooperate with biocatalysis to expand the synthetic application of biocatalysts. |
| References: | [1] Wang, J., Ke, H., Yang, J., Guo, N., Hu, K., Tang, R., Ding, Q., Gao, L., Lei, X. Chem. Catal. 2023, 3, 100451 [2] Gao, L., Su, C., Du, X., Wang, R., Chen, S., Zhou, Y., Liu, C., Liu, X., Tian, R., Zhang, L., et al. Nat. Chem. 2020, 12, 620-628. [3] Gao, L., Zou, Y., Liu, X., Yang, J., Du, X., Wang, J., Yu, X., Fan, J., Jiang, M., Li, Y., Houk, K.N., Lei, X. Nat. Catal. 2021, 4, 1059-1069. |
| Figures: | 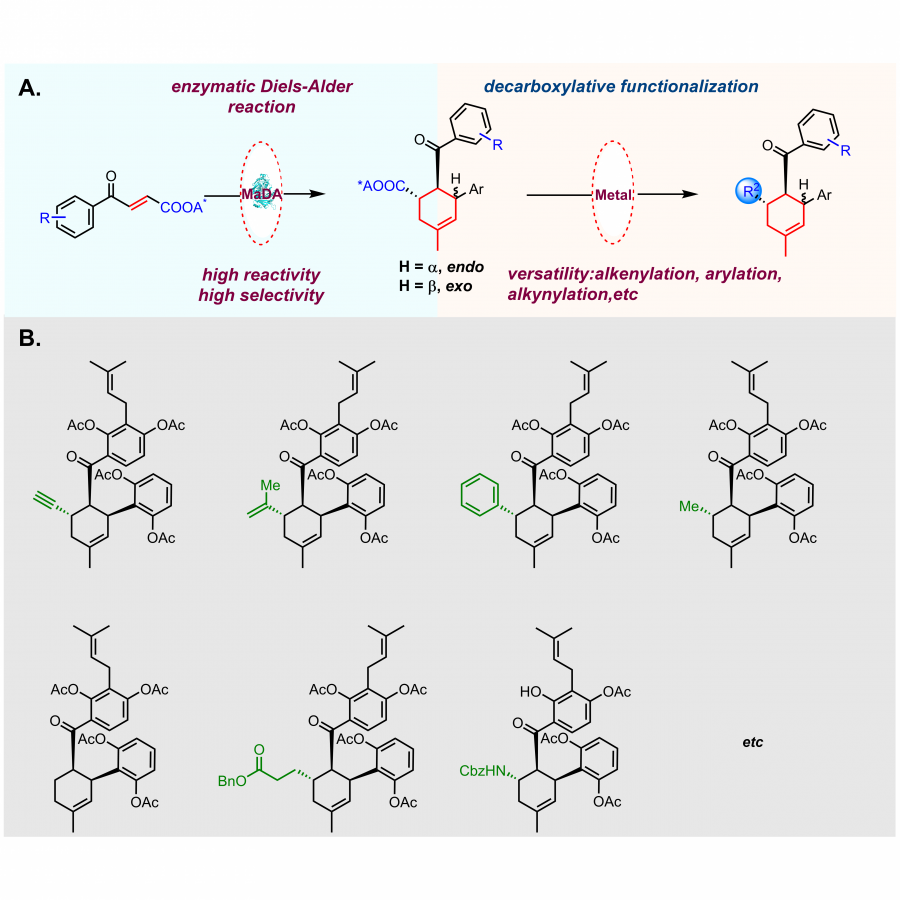 Chemoenzymatic strategy toward the diversity-oriented synthesis of enantiopure cyclohexenes A. General workflow for
the chemoenzymatic strategy
B. Synthesized structurally diverse enantiopure cyclohexanes 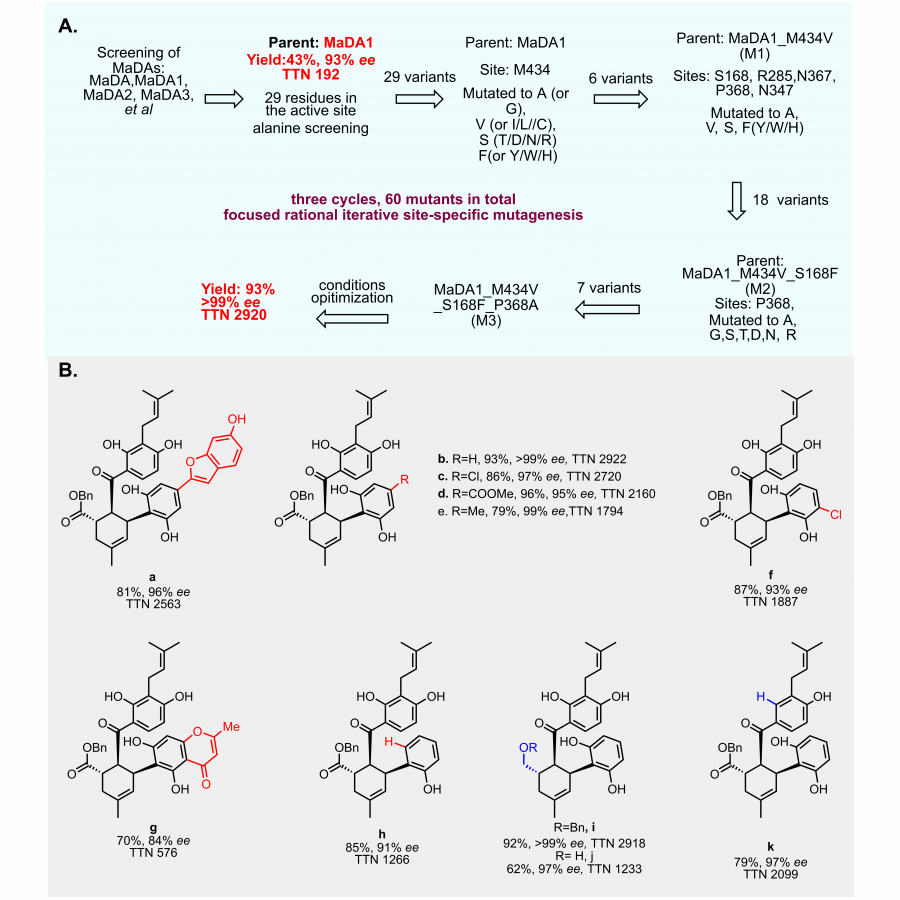 Protein engineerig of MaDA1 and substrate scope of MaDA1_M3 A. General workflow for protein engineering
B. Substrate scope of MaDA1_M3-catalyzed intermolecular Diels-Alder reactions |
#626 | Rational Design of Transketolase with Non-Natural Amino Acids to Accelerate a Novel Reaction for Pharmaceutical Synthesis |
|
| Presenting author: | Yiwen LI of BIOCHEMICAL ENGINEERING DEPARTMENT, UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Paul DALBY of BIOCHEMICAL ENGINEERING DEPARTMENT, UNIVERSITY COLLEGE LONDON |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Rational design / non-natural amino acid / smart library / industrial biocatalysis |
| Purpose: | Transketolase (TK) catalyses the reversible transfer of a C2-ketol unit from a donor substrate to an aldehyde acceptor, but its narrow substrate scope limits its potential for industrial applications. It was initially engineered by our research group to accept both the unnatural donor substrate pyruvate and acceptor substrate 3-formylbenzoic acid (3-FBA) to produce an analogue of phenylacetylcarbinol (PAC) [1], a crucial pharmaceutical intermediate. To accelerate this reaction, we first established a "push and pull" rational design strategy that incorporated non-natural amino acids to reshape the enzyme's binding pocket and guide substrates into its active sites. In-silico approaches, including the Molecular Mechanics Poisson-Boltzmann Surface Area (MMPBSA) method, were employed to evaluate the binding affinities of the transketolase-cofactor complex and substrates. Subsequently, a smart library of transketolase mutants was designed and screened, resulting in the identification of a lead mutant exhibiting a 25.6% improvement in substrate conversion in comparison to the best-performing historical variant [2]. Additionally, this mutant displayed a significant increase in stability, reflected by a 6.7°C enhancement in its melting temperature [2]. Furthermore, our investigations revealed a strong correlation between the root-mean-square deviation (RMSD), obtained through molecular dynamic simulations, and the thermal stability of transketolase within diverse scaffolds, providing important insights for designing thermally stable proteins. Overall, the significant improvements identified demonstrate the potential of this strategy in computer-aided protein design. These findings are not only valuable for the development of transketolase-based biocatalysts but also hold great promise for the engineering of other proteins with enhanced properties for a wide range of industrial and biotechnological applications. [1] Yu, H., Hernández López, R. I., Steadman, D., Méndez-Sánchez, D., Higson, Cázares-Körner, A., Sheppard, T. D., Ward, J. M., Hailes, H. C., & Dalby, P. A. (2020). Engineering transketolase to accept both unnatural donor and acceptor substrates and produce α-hydroxyketones. The FEBS journal, 287(9), 1758-1776. [2] Li, Y., Dalby, P. A. Rational Design of Transketolase with Non-Natural Amino Acids to Accelerate a Novel Reaction for Pharmaceutical Synthesis. unpublished results |
| References: | [1] Yu, H., Hernández López, R. I., Steadman, D., Méndez-Sánchez, D., Higson, Cázares-Körner, A., Sheppard, T. D., Ward, J. M., Hailes, H. C., & Dalby, P. A. (2020). Engineering transketolase to accept both unnatural donor and acceptor substrates and produce α-hydroxyketones. The FEBS journal, 287(9), 1758-1776. [2] Li, Y., Dalby, P. A. Rational Design of Transketolase with Non-Natural Amino Acids to Accelerate a Novel Reaction for Pharmaceutical Synthesis. unpublished results |
| Figures: | 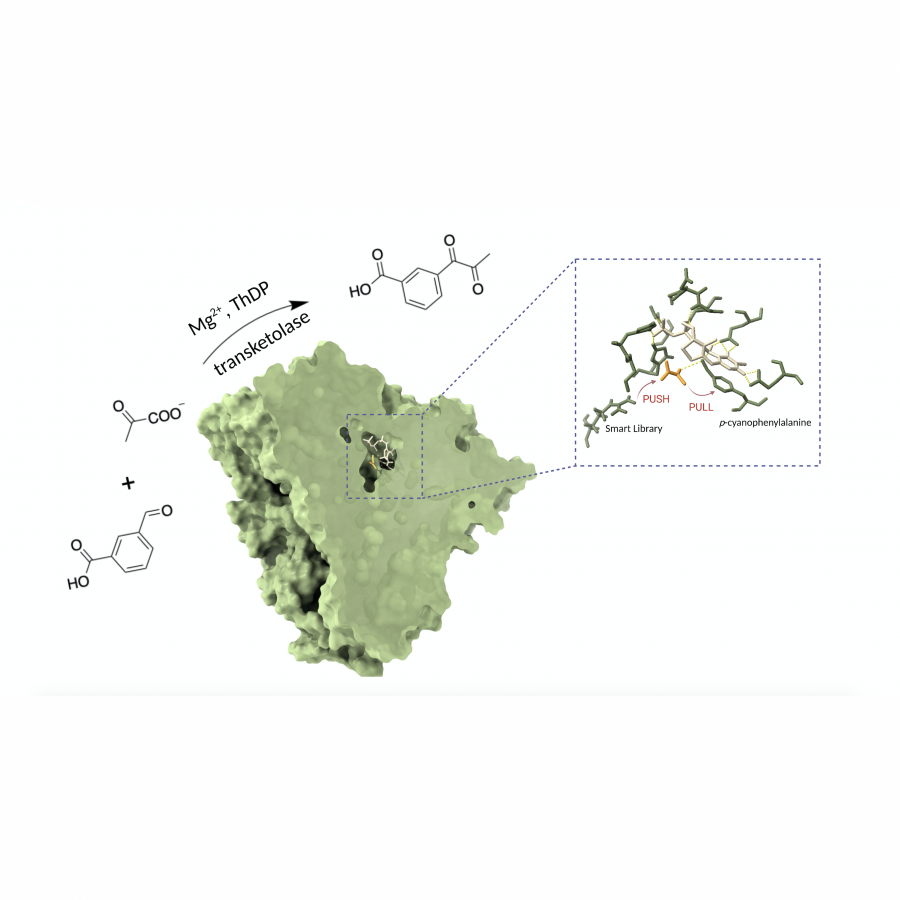 The push and pull mechanism of transketolase catalysing a Novel Reaction for Pharmaceutical The push and pull rational design strategy mechanism of transketolase catalysing a Novel Reaction for Pharmaceutical |
#627 | Retracing the evolutionary emergence of stereoselectivity in borneol-type dehydrogenases |
|
| Presenting author: | Jasmin ZUSON of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, TU GRAZ |
| Other authors: | Andrea M. CHÁNIQUE of INSTITUTE FOR BIOLOGICAL AND MEDICAL ENGINEERING, PONTIFICAL CATHOLIC UNIVERSITY OF CHILE Katarína KAVČIAKOVÁ of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, TU GRAZ Bernhard LOLL of FREIE UNIVERSITÄT BERLIN, INSTITUTE FOR CHEMISTRY AND BIOCHEMISTRY, LABORATORY OF STRUCTURAL BIOCHEMISTRY Daniel KRACHER of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, TU GRAZ Robert KOURIST of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, TU GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ancestral Sequence Reconstruction / borneol-type dehydrogenases / camphor / Enantioselectivity |
| Purpose: | The exceptional stereoselectivity of enzymes is a key feature for industrial applications, and elucidating the structural basis thereof is a critical step toward their optimization. Even though directed evolution and rational design are successful methods for the improvement of stereoselectivity [1], it remains unclear how Nature evolved stereoselectivity within enzyme classes. Enzymes within the family of plant borneol dehydrogenases (BDHs), although closely related, display striking differences in enantiospecificity towards borneol enantiomers [2, 3]. In this context, ancestral sequence reconstruction (ASR) has emerged as a powerful approach to infer the evolutionary history of BDH enzymes and identify key mutations contributing to their stepwise diversification. Following Nature’s footsteps, we conducted a comprehensive analysis of the effect of the active site and peripheral mutations on BDH selectivity, toward rationalizing natural enzyme evolution. To this end, ASR was employed to create common ancestors of selective and non-selective dehydrogenases in order to retrace the emergence of selectivity in BDHs via a plausible natural evolutionary pathway to the enantiospecific SrBDH1 [2] (Figure 1). Ancestral enzymes were recombinantly produced, biochemically characterized, and their structures predicted. Consecutively, guided by ASR and de-novo protein structure prediction, selected residues were substituted to further deepen the insights into the structure-function relation in BDHs. This work elucidates the importance of the structural and evolutionary changes in enzymes and exemplifies how the application of computationally-guided enzyme engineering tools and structural analysis leads to a deeper understanding of natural evolutionary principles in less investigated enzyme classes. |
| References: | [1] u.t. bornscheuer, g.w. huisman, r.j. kazlauskas, s. lutz, j.c. moore, k. robins, nature. 2012, 485, 185-194. [2] a.m. chánique, n. dimos, i. drienovská, e. calderini, m.p. pantín, c.p.o. helmer, m. hofer, v. sieber, l.p. parra, b. loll, r. kourist, chemcatchem. 2021, 13, 2262-2277. [3] r. croteau, c.l. hooper, m. felton, arch. biochem. 2012, 188, 182-193. |
| Figures: | 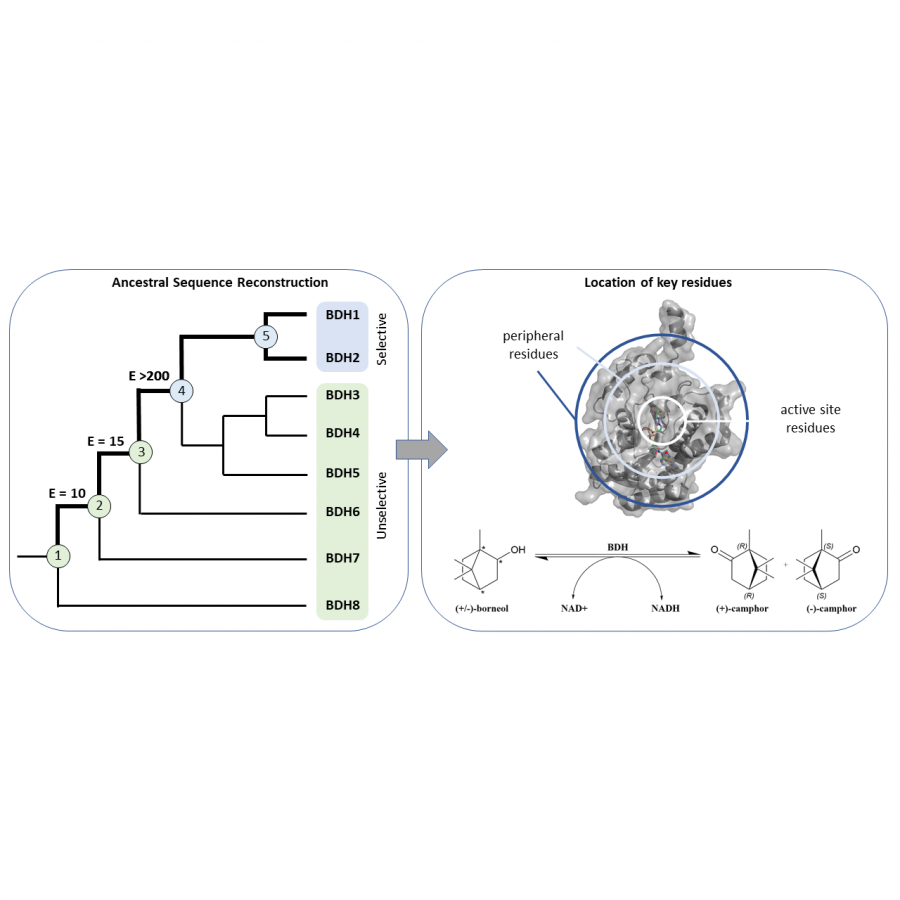 Ancestral sequence reconstruction as guide for enzyme engineering. Ancestral sequence reconstruction is thereby utilized to identify peripheral and active site mutations in BDH ancestors leading to changes in enantioselectivity. |
#628 | Green Chemo-Enzymatic Synthesis of Pure Enantiomers of β-Antagonists Bisoprolol and Betaxolol |
|
| Presenting author: | Elisabeth JACOBSEN of NORWEGIAN UNIVERSITY OF SCIENCE & TECHNOLOGY |
| Other authors: | Susanne TROØYEN of NORWEGIAN UNIVERSITY OF SCIENCE & TECHNOLOGY Lucas BOCQUIN of UNIVERSITY OF BIELEFELD |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | β-Blockers are one of the classes of drugs which plays a relevant role in the treatment of various human diseases such as hypertension, angina pectoris, migraines and tremors. β-Blockers are β-adrenergic antagonists, which affects the β-adrenergic receptors found in a variety of places in the human body, with most efficiency in the heart and vascular system [1]. Only a few β-blockers are manufactured with the single enantiomer Active pharmaceutical ingredient (API), even though for most β-blockers the (S)-enantiomers are the more potent enantiomer [2]. Several negative side effects from the “wrong” enantiomer are also known. Due to this there is a need for effective and green methods for production of single enantiomers of these widely used drugs. We have for some time studied the synthesis mechanisms for production of single enantiomers of several β-blockers [3-5]. Here we present our results of producing the pure enantiomers of (S)-bisoprolol and (S)-betaxolol (Figure 1)[6-8]. |
| References: | [1] Khan, M. Cardiac Drug Therapy, 8th ed., Humana Press: 2015, p 7. [2] Agustian, J. et al. Process Biochem. 2010, 45, 1597-1604. [3] Lund, I.T. et al. Tetrahedron 2016, 72, 7288-7292. [4] Blindheim, F.H. et al. Catalysts 2018, 8, 516-524 [5] Gundersen, M.A et al. Catalysts 2021, 11, 503-518. [6] Troøyen S. et al Catalysts 2022, 12, 980-992 [7] Troøyen S. et al. Catalysts 2022, 12, 1645-1657 [8] Bocquin L. et al Catalysts 2023, 13, 54-63 |
| Figures: |  Figure 1 Figure 1. (S)-Bisoprolol (left) and (S)-betaxolol (right). |
#642 | ENZYMES FOR GREENER CONSUMER PRODUCTS AND FOR A HEALTHY ZERO-PLASTIC POLLUTED ENVIRONMENT |
|
| Presenting author: | Paula VIDAL RAMÓN of ICP-CSIC |
| Other authors: | Manuel FERRER MARTÍNEZ of ICP-CSIC Laura FERNÁNDEZ LÓPEZ of ICP-CSIC Cristina COSCOLÍN GALÁN of ICP-CSIC David ALMENDRAL NIETO of ICP-CSIC Patricia MOLINA ESPEJA of ICP-CSIC |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | natural resources / hydrolases / climate change / engineer |
| Purpose: | Today, the chemosphere’s and biosphere’s compositions of the planet are changing faster than experienced during the past thousand year. This is a consequence of, first, the rising of CO2 emissions from fossil fuel combustion, including those from processing, manufacturing and consuming everyday products [1]. Second, the increase of the pollution level; indeed, about 140000 artificial chemicals and mixtures of chemicals, including plastics and micro-plastics, are currently produced, 220 billion-tons of which are produced and disposed each year [1]. How do we deal with the green transition to minimize global warming like CO2 and pollution? One of the solutions is to use renewable natural resources, namely, the working parts of its living systems, the enzymes. We review the consequences of climate change and chemical pollution at multiple scales and how enzymes can counteract or mitigate them. We then focus on how they mobilize sustainable and greener innovations in consumer products that have a high contribution to global carbon emissions, and how they can help reaching a healthy zero-pollution environment. Lessons learnt from past and present effort to retrieve enzymes through naïve- and bioinformatics-based metagenomics, and to engineer variants by machine learning and meta-data integration, are discussed. Recent findings in a number of target enzymes, particularly, hydrolases relevant to lipid’ and plastic’ hydrolysis, are finally discussed. Acknowledgements. This research was funded by the FuturEnzyme Project, funded by the European Union’s Horizon 2020 Research and Innovation Program under Grant Agreements No. 101000327 and 101060625. We also acknowledge financial support under Grants PID2020-112758RB-I00, PDC2021-121534-I00, and TED2021-130544B-I00 from the Ministerio de Economía, Industria y Competitividad, Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación (AEI) (Digital Object Identifier 10.13039/501100011033) and the European Union (“NextGenerationEU/PRTR”). |
| References: | [1] Intasian P., Prakinee K., et al. Enzymes, in vivo biocatalysis, and metabolic engineering for enabling a circular economy and sustainability (2021). Chem Rev 121:10367-10451. |
#646 | Hydroxysteroid dehydrogenases: promiscuous enzymes for diverse synthetic applications |
|
| Presenting author: | Ivan BASSANINI of NATIONAL RESEARCH COUNCIL OF ITALY |
| Other authors: | Erica Elisa FERRANDI of NATIONAL RESEARCH COUNCIL OF ITALY Chiara TOGNOLI of NATIONAL RESEARCH COUNCIL OF ITALY Daniela MONTI of NATIONAL RESEARCH COUNCIL OF ITALY Sergio RIVA of NATIONAL RESEARCH COUNCIL OF ITALY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Hydroxysteroid dehydrogenases / Enzyme promiscuity / Oxidoreductases / Chiral Synthons |
| Purpose: | Hydroxysteroid dehydrogenases (HSDHs) are NAD(P)H-dependent enzymes belonging to the superfamily of short-chain dehydrogenases/reductases (SDRs). In Nature, these enzymes catalyze the reversible oxidoreduction of the hydroxyl/keto groups of neutral steroids, bile acids and other steroid derivatives with exquisite selectivity. Specifically, HSDHs have been shown to display very high regioselectivity for the hydroxyl groups at different positions, e.g., at C-3, C-7, and C-12 of bile acids. For each one of these positions, HSDHs usually show high stereoselectivity for either the hydroxyl group above (β configuration) or below (α configuration) the plane of the steroid molecule. [1,2] The use of HSDHs on substrates not showing the core structure of steroid molecules, i.e. the investigation of their potential substrate promiscuity, represent an enabling tool for synthetic organic chemists seeking for novel, biocatalyzed and selective processes. For instance, some years ago a study reported the stereoselective reductions of α-ketoesters catalyzed by HSDHs, [3] but, after a careful literature check, very few reports can be found on this topic. Therefore, the presented work aims at describing the efforts made by us to fill this gap by exploring the substrate promiscuity on different non-steroid molecules of a library of microbial HSDHs, showing either 7α, 7β, or 12α activity, and originated by already reported sources, as well as from newly identified (meta)genomic sequences. Accordingly, the selected enzymes were tested as biocatalysts for the stereoselective reduction of a panel of different substrates (Figure 1). Figure 1. Examples of the substrate promiscuity of HSDHs. At first, simple ketones and aldehydes were tested, then α-keto esters were studied as synthons of pharmaceutical interest along with selected cyclic ketones. [4,5] After this, the HSDHs-mediated biotransformation of racemic “Wieland-Miescher ketone”, an important intermediate widely used in the total synthesis of complex natural terpenoid products, [6] was implemented in multi-enzymatic entry to isolate its enantiomers and alcohol derivatives. [7] Moreover, HSDHs performances were tested on complex natural, bioactive products, like gingerol and gingerdiols, as well as towards “bulky-bulky” ketones like aryl decorated γ-keto esters, synthetic precursors of valuable bioactive metabolites. [8] |
| References: | [1] E. E. Ferrandi, S. Bertuletti, D. Monti, S. Riva, European Journal of Organic Chemistry 2020, 2020, 4463-4473. [2] S. Bertuletti, E. E. Ferrandi, D. Monti, G. Fronza, I. Bassanini, S. Riva, ChemCatChem 2021, 13, 4948-4953. [3] D. Zhu, J. E. Stearns, M. Ramirez, L. Hua, Tetrahedron 2006, 62, 4535-4539. [4] E. E. Ferrandi, I. Bassanini, S. Bertuletti, S. Riva, C. Tognoli, M. Vanoni, D. Monti, International Journal of Molecular Sciences 2022, 23, 12153. [5] S. Bertuletti, E. E. Ferrandi, S. Marzorati, M. Vanoni, S. Riva, D. Monti, Advanced Synthesis and Catalysis 2020, 362, 2474-2485. [6] H. Hagiwara, Natural Product Communications 2020, 15, 1934578X2092534. [7] S. Bertuletti, I. Bayout, I. Bassanini, E. E. Ferrandi, N. Bouzemi, D. Monti, S. Riva, European Journal of Organic Chemistry 2021, 2021, 3992-3998. [8] R. Nasti, I. Bassanini, E. E. Ferrandi, F. Linguardo, S. Bertuletti, M. Vanoni, S. Riva, L. Verotta, D. Monti, ChemBioChem 2022, 23, e202200105. |
| Figures: |  Figure 1 Examples of the substrate promiscuity of HSDHs. |
#648 | Engineering Cytochrome P450 Monooxygenase Biocatalysts for Regioselective Fatty Acid Hydroxylation |
|
| Presenting author: | Bethan JONES of UNIVERSITY OF MANCHESTER |
| Other authors: | Joseph SHARRATT of UNIVERSITY OF MANCHESTER Jordi SOLER of INSTITUT DE QUÍMICA COMPUTACIONAL I CATÀLISI AND DEPARTAMENT DE QUÍMICA, UNIVERSITATDE GIRONA Marc GARCIA-BORRÀS of INSTITUT DE QUÍMICA COMPUTACIONAL I CATÀLISI AND DEPARTAMENT DE QUÍMICA, UNIVERSITATDE GIRONA Sílvia OSUNA of INSTITUT DE QUÍMICA COMPUTACIONAL I CATÀLISI AND DEPARTAMENT DE QUÍMICA, UNIVERSITATDE GIRONA Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | P450 / Hydroxylation / Regioselectivity / Fatty acids |
| Purpose: | Hydroxylation of fatty acids to chiral hydroxy acids yields a diverse array of high value compounds that have useful applications in the fragrances and flavoring industries. The synthesis of these compounds is challenging due to activation of unreactive C-H bonds, especially with regards to the appropriate stereo- and regioselective introduction of the new hydroxyl group. The chemical oxy-functionalization typically requires harsh reaction conditions including transition metal catalysts, peracids and organic solvents. The regioselective targeting of a methylene group can be achieved under milder reaction conditions using cytochrome P450 biocatalysts. Whilst regioselective terminal fatty acid hydroxylation catalyzed by P450s has been reported, the oxygenation at highly subterminal and mid-chain positions remains challenging. In this study we describe engineering and molecular dynamic simulations to provide insights into the unique regioselective mid-chain hydroxylation of decanoic acid catalyzed by a wild type, class VII P450. All variants showed a varying array of expression, activity, and selectivity. A range of different regioselectivity from C5 to C10 hydroxylated products was observed, where key mutations shifted the mid-chain selectivity. We have demonstrated that engineering wild-type class VII P450s yielded useful biocatalysts for mid-chain C-H activation for selective hydroxylation of fatty acids. |
| References: | 1. Manning, J., Tavanti, M., Porter, J. L., Kress, N., De Visser, S. P., Turner, N. J. & Flitsch, S. L. (2019). Regio - and enantio-selective chemo-enzymatic C-H-lactonization of decanoic acid to (S)-δ-decalactone, Angew. Chemie Int. Ed., 58, 5668-5671. |
#651 | Methyltransferase Cascade Reactions for the Benzylation of mRNA at the 5´-Cap |
|
| Presenting author: | Nicolas CORNELISSEN of UNIVERSITY OF MÜNSTER |
| Corresponding author: | Andrea RENTMEISTER of UNIVERSITY OF MÜNSTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Methyltransferase / S-adenosyl-L-methionine / SAM analogues / Methionine adenosyltransferase |
| Purpose: | Methyltransferases (MTases) provide excellent specificity in late-stage alkylation of biomolecules. Their dependence on S-adenosyl-L-methionine (SAM) mandates efficient access to SAM analogues for biocatalytic applications. We recently engineered a methionine adenosyltransferase (MAT) from Methanocaldococcus jannaschii to access SAM analogues with benzylic moieties. Here we explore their utility in MAT/MTase cascade reactions with NovO for regioselective benzylation of a coumarin substrate. Further, we apply the cascade for post-synthetic modification of mRNA at the 5´ cap. In the case of the 4-chlorobenzyl group, protein production increased by 28 % in HeLa and 39-50 % in HEK cell lines compared to mRNA with a regular 5´ cap, underlining the potential to improve mRNA based therapeutics. |
| References: | [1] F. Michailidou*, N. Kloecker*, N. V. Cornelissen*, R. K. Singh, A. Peters, A. Ovcharenko, D. Kuemmel and A. Rentmeister, Angew. Chem. Int. Ed. 2021, 60, 480-485. [2] A. Peters, E. Herrmann, N. V. Cornelissen, N. Kloecker, D. Kuemmel and A. Rentmeister, Chembiochem 2022, 23, e202100437. [3] S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris and J. S. Thorson, Angew. Chem. Int. Ed. 2014, 53, 3965-3969. [4] H. Stecher, M. Tengg, B. J. Ueberbacher, P. Remler, H. Schwab, H. Griengl and M. Gruber-Khadjawi, Angew. Chem. Int. Ed. 2009, 48, 9546-9548. *Authors contributed equally. |
| Figures: |  MjMAT L147A/I351A (PC-MjMAT) Crystal structure of PC-MjMAT. A) The homodimeric structure (blue, dark blue) forms two active sites buried behind a "gating loop" (green) (PDB: 7P84). B) The active site with AdoONB. C) The active site with AdoDMNB (PDB: 7P8M). 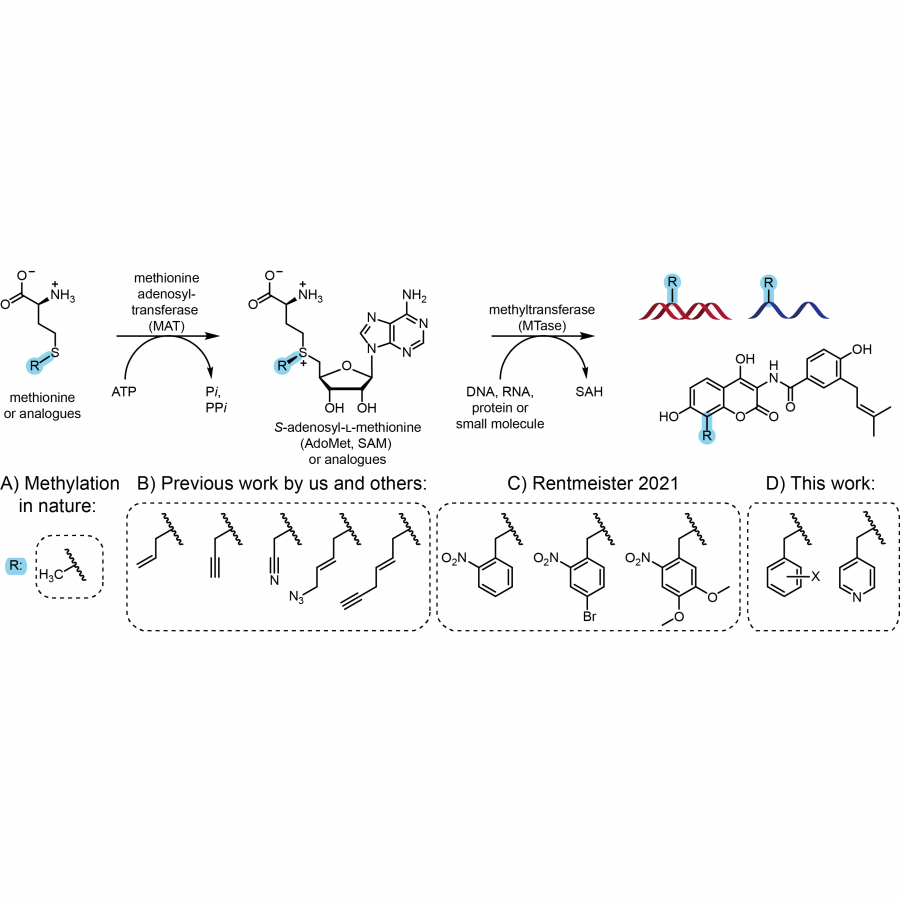 MAT/MTase cascade reaction A) methylation in nature and application for site-specific B) alkylation, C) photocaging and D) (hetero-) benzylation. |
#663 | Oxidation of indole derivatives by engineered fungal peroxygenases |
|
| Presenting author: | Alejandro BELTRAN-NOGAL of INSTITUTE OF CATALYSIS, CSIC |
| Corresponding author: | Miguel ALCALDE of INSTITUTE OF CATALYSIS, CSIC |
| Other authors: | Ivan MATELJAK of EVOENZYME Patricia GOMEZ DE SANTOS of EVOENZYME David GONZALEZ-PEREZ of INSTITUTE OF CATALYSIS, CSIC |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | directed evolution / UPO / indigo / combinatorial saturation mutagenesis |
| Purpose: | Indigo blue is one of the oldest dyes used throughout Human History since early times (as far as 6,000 years ago) [1, 2]. A precursor of indigo is indole, an aromatic heterocycle constituted by a benzene ring bonded to an imidazole moiety, that is widely present in biomolecules. Indole can be oxidized to indoxyl (3-oxindole) and then undergo spontaneous oxidation towards the formation of indigo (Figure 1) [3]. Traditionally, this reaction has been achieved by fermentation of indican-producing plants – a protected O-glycosylated version of indoxyl – and later by fossil-based processes relying on aniline and hazardous chemicals [1]. The biotechnological synthesis of indigo is an alternative to the chemical processes that depart from unrenewable resources. These processes constitute microbial fermentations and enzymatic reactions [1]. Among the enzymes that produce indigo are naphthalene dioxygenases, multicompetent phenol hydroxylases, P450s, and flavin-dependent monooxygenases. However, most of these depend on expensive cofactors and/or electron transport chains that can hamper their industrial implementation. On the contrary, fungal unspecific peroxygenases (UPOs) are stable biocatalysts that simply require hydrogen peroxide to bring about selective oxygen-insertion reactions in unactivated C-H bonds, including indole and indole-derivative compounds [3, 4]. Here we present a directed evolution campaign focused on obtaining UPO variants with high activity to produce indigo and halogenated indigo-derivatives. The UPO from Daldinia sp. EC12 (DspUPO) was subjected to iterative rounds of mutagenesis and selection in order to increase its secretion levels in the heterologous host Saccharomyces cerevisiae. Upon increasing DspUPO functional expression, a study of the amino acids lining the active site channel was carried out. Five residues were selected to be targets of combinatorial saturation mutagenesis and codon degeneracy with NDTs codons in independent libraries that were evaluated by their ability to produce indigo, 5,5’-dicloroindigo and 6,6’-dibromoindigo (also known as Tyrian purple or Royal purple). The best mutant hits were isolated and biochemically characterized aiming to assess the potential of these enzyme candidates for the synthesis of different indigo derivatives. |
| References: | [1] N. Fabara, A., W. Fraaije, M., Appl. Microbiol. Biotechnol. 2020, 104, 925-933. [2] C. Splitstoser, J., D. Dillehay, T., Wouters, J., Claro, A., Sci. Adv. 2016, 2, e1501623. [3] Ullrich, R., Poraj-Kobielska, M., Herold-Majumdar, O., Vind, J., Hofrichter, M., Catalysts 2021, 11, 1495. [4] Beltran-Nogal, A., Sanchez-Moreno, I., Mendez-Sanchez, D., Gomez de Santos, P., Hollmann, F., Alcalde, M., Curr. Opin. Struct. Biol. 2022, 73, 102342. |
| Figures: | 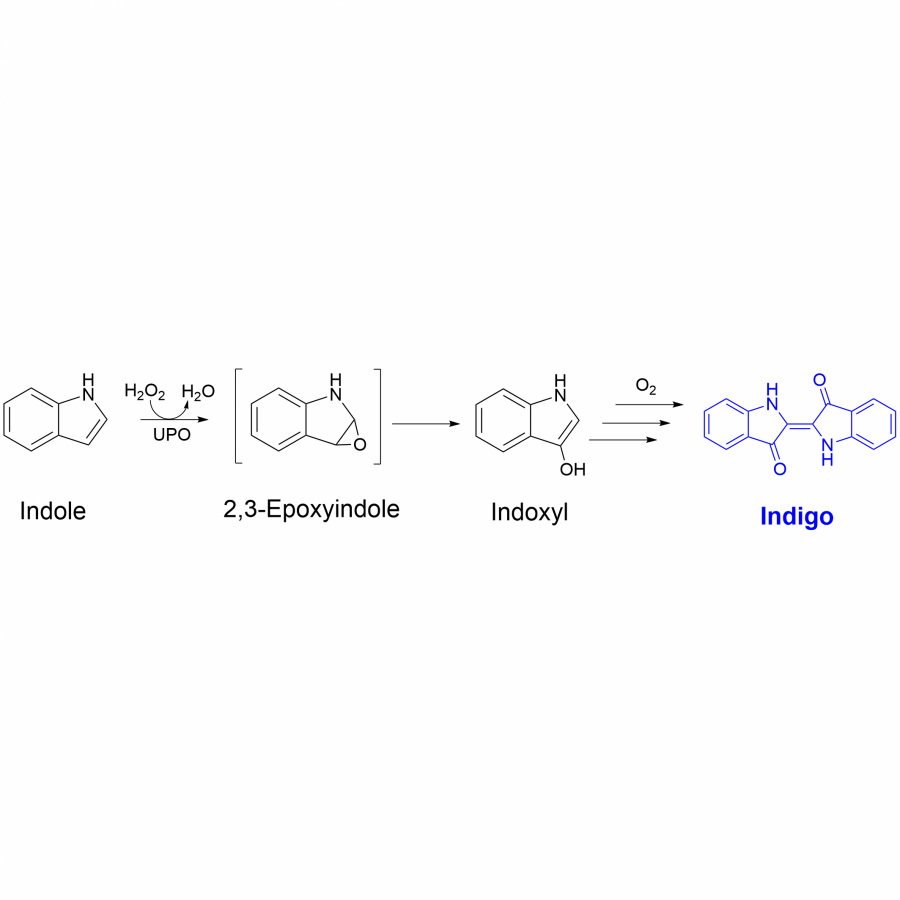 Oxidation of indole. Proposed reaction of the oxidation of indole to indigo by UPOs [3]. |
#667 | Microbial transformation of bromo and chloro flavonoids in the culture of entomopathogenic filamentous fungi |
|
| Presenting author: | Martyna PERZ of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Monika DYMARSKA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Tomasz JANECZKO of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Edyta KOSTRZEWA-SUSŁOW of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | microbial transformation / entomopathogenic filamentous fungi / halogenated flavonoids / antibiotic resistance |
| Purpose: | Bacterial resistance to antibiotics is a significant global problem nowadays. The pharmaceutical industry is constantly looking for new compounds that prevent antibiotic resistance. Flavonoid compounds with bromine and chlorine atoms may be the solution, as some of them show bacteriostatic/bactericidal properties [1] [2]. Many naturally bioactive chemical compounds, including flavonoids, occur in nature in the form of glycosides, which improves their solubility [3]. Obtaining flavonoid glycosides using a chemical method is extremely difficult and requires toxic reagents. Contrary, biotransformation is a simple method that allows obtaining flavonoid derivatives, including glycosides, under mild reaction conditions [4] [5]. The presented research aimed to combine chemical synthesis with microbial transformation to obtain flavonoid glycosides with bromine and chlorine atoms. As a result of the Claisen-Schmidt condensation reaction and its further transformations, chalcone, flavanone, and flavone containing -Br and -Cl in the A ring were obtained. Then, the biotransformations of these compounds were carried out in cultures of entomopathogenic strains of the filamentous fungi Beauveria bassiana KCH J1.5, Isaria fumosorosea KCH J2, and Isaria farinosa KCH J2.6. For the compound 3'-bromo-5'-chloro-2'-hydroxychalcone, the biotransformation was the most effective in the culture of the B. bassiana KCH J1.5 strain. As a result of the reaction, 8-bromo-6-chloroflavanone 3'-O-β-D-(4"-O-methyl)-glucopyranoside was obtained. Another compound, 8-bromo-6-chloroflavanone, was transformed by I. fumosorosea KCH J2 to give 8-bromo-6-chloroflavan-4-ol 4'-O-β-D-(4”-O-methyl)-glucopyranoside. 8-bromo-6-chloroflavone was most efficiently transformed by I. farinosa KCH J2.6 into 8-bromo-6-chloroflavone 4'-O-β-D-(4”-O-methyl)-glucopyranoside. Because the properties of the obtained glycosides have never been described in the literature, activity prediction of the above products was carried out. Pass online and Swiss ADME platforms have shown that all obtained glycoside products are potential CDP-glycerol glycerophosphotransferase inhibitors with a probability of over 90%. CDP-glycerol glycerophosphotransferase polymerizes the main chain of wall teichoic acid (WTA) associated with the cell wall of gram-positive bacteria, which in turn is responsible, among others, for antibiotic resistance. In the absence of WTA, bacteria are sensitive to external factors (temperature, salt content), which can contribute to the fight against pathogenic gram-positive bacteria in the human body [6]. Moreover, activity predictions of the products of the biotransformation process showed many other properties, such as anti-carcinogenic, anti-inflammatory, or hepatoprotective. However, further in vitro studies are needed to confirm them. The research is co-financed by the Wrocław University of Environmental and Life Sciences (Poland) as the Ph.D. research program ,,Innowacyjny Doktorat”, no. N070/0004/22. |
| References: | [1] m. a. rahim, m. m. h. bhuiyan, m. m. matin, m. r. alam., j. sci. res. 2018, 10, 67-76. [2] y. xie, w. yang, f. tang, x. chen, l. ren., curr. med. chem. 2014, 22, 132-149. [3] j. a. salas, c. méndez., trends microbiol. 2007, 15, 219-232. [4] a. krawczyk-łebek, m. dymarska, t. janeczko, e. kostrzewa-susłow., int. j. mol. sci. 2022, 23, 5558-5580. [5] a. krawczyk-łebek, m. dymarska, t. janeczko, e. kostrzewa-susłow., catalysts. 2020, 10, 1148-1166. [6] s. brown, j. r. santa maria, s. walker., annu. rev. microbiol. 2013, 67, 313-336. |
#668 | Tungsten aldehyde oxidoreductase/ hydrogenase forms an enzymatic decorated protein nanowire |
|
| Presenting author: | Maciej SZALENIEC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY POLISH ACADEMY OF SCIENCE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | tungsten enzyme / hydrogenase / aldehyde oxidoreductase / cryo-EM |
| Purpose: | Tungsten aldehyde oxidoreductase/ hydrogenase forms an enzymatic decorated protein nanowire Agnieszka Winiarska1, Fidel Ramírez-Amador2, Gabriela Pacek1, Ivana Aleksic1, Dominik Hege3, Yvonne Gemmecker3, Simone Prinz4, Georg Hochberg5, Johann Heider*2,3, Jan Michael Schuller*2, Maciej Szaleniec*1 1 - Jerzy Haber Institute of Catalysis and Surface Chemistry Polish Academy of Sciences 2 - SYNMIKRO Research Center and Department of Chemistry, University of Marburg, Marburg, Germany. 3 - Faculty of Biology, Philipps-Universität Marburg, Marburg, Germany 4 - Max Planck Institute of Biophysics, Frankfurt am Main, Germany 5 - Max Planck Institute for Terrestrial Microbiology, Marburg, Germany The multi-subunit aldehyde oxidoreductase from the denitrifying betaproteobacterium Aromatoleum aromaticum (AORAa) catalyzes the reversible oxidation of aldehydes to carboxylic acids with electron acceptors like NAD+ or viologens (1). Recently we showed the ability of AORAa to use hydrogen as an electron donor for acid or NAD+ reduction, which was not shown before for any tungsten enzyme and qualifies AOR as a new type of hydrogenase (2). Although the catalytic W-containing subunit (AorB) is highly similar to AOR from Pyrococcus furiosus (pdb code 1AOR), AORAa also contains an additional FeS cluster-rich polyferredoxin-like subunit (AorA) and a FAD-containing subunit (AorC). Recently, we resolved the first molecular structure of AORAa, with a global resolution of 3.22 Å using cryo-electron microscopy (3). The study resulted in the molecular structure of an Aor(AB)2C complex, although the subunit stoichiometry of AORAa varies and additional electron density depicting an Aor(AB)3C complex was obtained. These results, supported by mass spectrophotometry, revealed that the polyferredoxin-like subunit AorA oligomerizes from a single AorC subunit to form an electron-conducting nanowire that is decorated with enzymatically active and W-cofactor (W-co) containing AorB subunits (Fig. 1). This supermolecular structure model of AORAa was additionally supported by disrupting the interface between AorAB protomers via mutagenesis, which led to a simple AorABC complex. The structure of the AorB subunit revealed the binding mode of the substrate benzoate in the active site. This, together with EXAFS spectrometry and QM:MM modelling of the W-co, enabled the proposal of a catalytic mechanism describing the oxidation of aldehydes and the H2-dependent reduction of carboxylic acids. We characterized the catalytic activity of AORAa in H2-dependent reductions. A series of reactors proved that an enzymatic cascade from carboxylic acids to alcohols can be catalyzed at both acidic and neutral pH, ruling out an electron bifurcation mechanism. We have also demonstrated that whole-cells with recombinant AOR can be applied for substrate conversion. Acknowledgement: This project has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (Two-CO2-One, grant agreement no. 101075992) (JMS); European Molecular Biology Organization Scientific Exchange Grant 9048 (AW); EU Project POWR.03.02.00-00-I004/16 (AW); National Science Centre, Poland grant PRELUDIUM 2017/27/N/ST4/02676 (AW); German Research Foundation grant He2190/15-1 (JH); Emmy Noether grant SCHU 3364/1-1 (JMS); We gratefully acknowledge Poland’s high-performance computing infrastructure PLGrid (HPC Centers: ACK Cyfronet AGH) for providing computer facilities and support within computational grant no. PLG/2022/015584 |
| References: | [1] F. Arndt, et al. , Characterization of an Aldehyde Oxidoreductase From the Mesophilic Bacterium Aromatoleum aromaticum EbN1, a Member of a New Subfamily of Tungsten-Containing Enzymes. Front Microbiol. 10 (2019), doi:10.3389/fmicb.2019.00071. [2] A. Winiarska, et al., Tungsten Enzyme Using Hydrogen as an Electron Donor to Reduce Carboxylic Acids and NAD +. ACS Catal. 12, (2022) 8707-8717. [3] A. Winiarska, et al., A bacterial tungsten-containing aldehyde oxidoreductase forms an enzymatic decorated protein nanowire, BioRxiv (2023) doi: 10.1101/2023.01.23.525143 |
| Figures: | 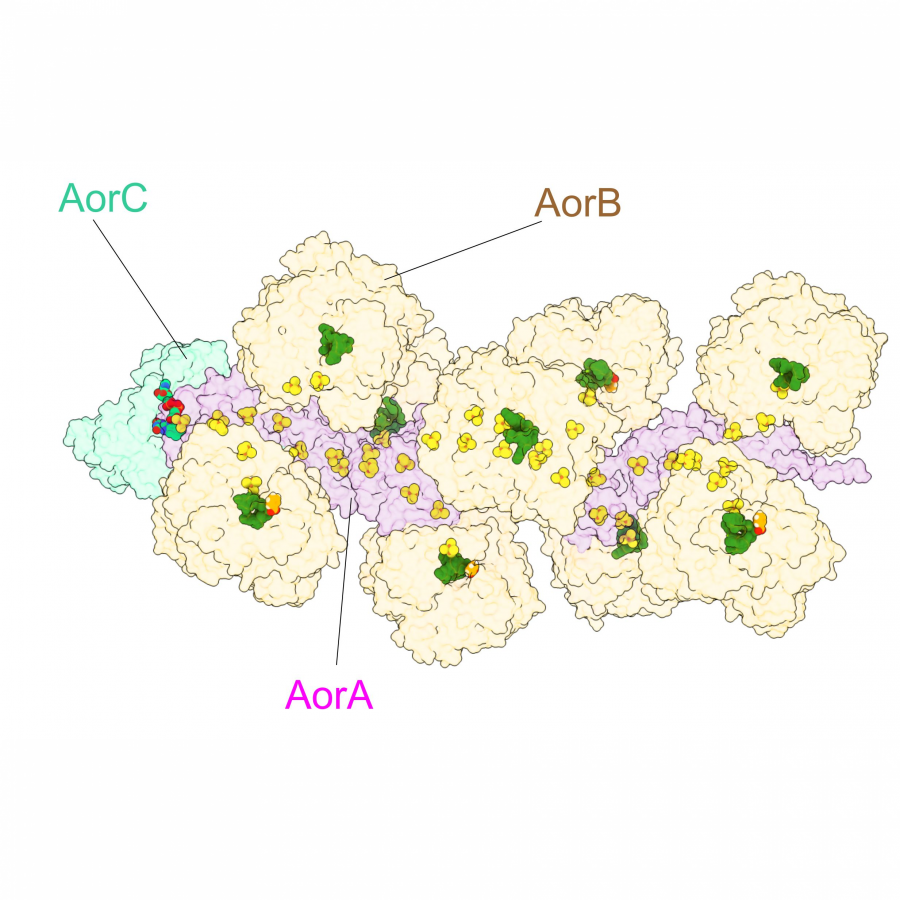 Fig. 1. Filament formation in the AORAa complex - modelled nanowire complex of composition Aor(AB)5C; |
#674 | Impact of different light sources on CvFAP photodecarboxylation |
|
| Presenting author: | Gabriela BREDA of UFRJ |
| Corresponding author: | Rodrigo SOUZA of UFRJ |
| Other authors: | Alexandre FRANÇA of UFRJ Luiza BENINCA of SENAI Raquel LEÃO of UFRJ Kleber OLIVEIRA of UFSCAR Rodrigo ALMEIDA of UFRJ Frank HOLLMAN of TUDELFT |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | photobiocatalysis / CvFAP / green chemistry / sunlight |
| Purpose: | Chlorella variabilis fatty acid photodecarboxylase (CvFAP) catalyzes the elimination of CO2 from fatty acids (Cn) producing the corresponding hydrocarbon (Cn-1) from photoexcitation of the FAD prosthetic group [1]. The production of alkanes by biological route is of great biotechnological interest in the high-performance biofuel sector. However, despite the large number of publications in recent years, photodecarboxylation using CvFAP needs optimizations to increase yields and alternatives to reduce costs associated with this process. Therefore, the present project aims to intensify the photodecarboxylation reactions of palmitic acid using batch reactions with different light sources such as high-power LEDs and sunlight [2]. First, batch reactions with enzymatic extract containing CvFAP and 13 mM of palmitic acid as substrate were performed using illumination of blue LED lamps of different powers (50, 100, 200 and 300 W), which demonstrated that reductions in reaction time are possible by increasing the power of the light source (84% conversion using 300 W against 48% conversion using 100 W in 20 minutes reactions). A white LED lamp (300 W) connected to a solar panel was also used, which presented a slightly slower kinetic profile compared to the blue lamp of the same power (73% conversion using white LED against 94% conversion with blue LED in 30 minutes reactions). This alternative light source is beneficial as the energy consumption is lower and a solar panel is used. The further investigation studied the reaction application in flow reactors, as the scale-up remains a challenge for photodecarboxylation reactions using batch-type process. Reagents were added to a light-protected flask and pumped through a tubular reactor (FEP tubing) with the illumination of high-power LED (300 W white or blue LED). The product was collected in the end of the tubing and different residence times (flow rates) were tested. Full conversion (>99%) were obtained with 15 minutes of residence time for the blue LED and 60 minutes for the white LED. The results showed that continuous-flow approach is a potential alternative for scaling-up photodecarboxylation processes. Finally, batch reactions were also carried out using sunlight, which presented a higher yield when compared to the results with LED lamps reported in this work and in the literature so far, although using a lower light intensity (13.11 μmol.s-1.m-2 photon flux density for sunlight on a sunny day against 317.77 μmol.s-1.m-2 for 300 W blue LED lamp). After 12 minutes reaction using the sunlight, a full conversion (>99%) was obtained on a sunny day. A reduction in the efficiency of our process was also observed on cloudy day with >99% conversion after 60 minutes, which can be attributed to the lower sunlight intensity (4.21 μmol.s-1.m-2). The results of the present work correspond to the first reports of photodecarboxylation using CvFAP with white LED lamp and sunlight as alternative light sources, demonstrating its feasibility. The contributions of different wavelength ranges of the electromagnetic spectrum of sunlight, such as the <400 nm range together with the blue range (≈ 430-490 nm), could justify the higher yields using sunlight when compared to the blue LED lamps, that presents a shorter spectrum (439 nm). |
| References: | [1] sorigue, d., et al., plant physiol. 2016, 171, 2393-405. [2] beninca, l., et al., molecular catalysis. 2022, 528, 112469. |
#677 | Lecithin biotransformation into various lysolecithin-fatty acid liposomes by Thermostable Phospholipase A2 from Sciscionella marina as a key enzyme |
|
| Presenting author: | Hyo-Ran LEE of EWHA WOMANS UNIVERSITY |
| Corresponding author: | Jin-Byung PARK of EWHA WOMANS UNIVERSITY |
| Other authors: | Seung-Yeon KWON of EWHA WOMANS UNIVERSITY Jeong-Hoo LEE of DR.S MEDI CO.,LTD Ye-Eun PARK of EWHA WOMANS UNIVERSITY Kyungmoon PARK of HONGIK UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | phospholipase A2 / Sciscionella marina / crystal structure / biotransformation |
| Purpose: | The phospholipase A2 (PLA2) hydrolyzes the sn-2 acyl bond of lecithin and phospholipids in a Ca2+-dependent manner [1]. The PLA2 from porcine pancreas, which is the most widely used in industry, is unstable at high temperatures. Herein, we investigated molecular characterization of PLA2 originated from marine bacterium, Sciscionella marina (SmPLA2). The X-ray crystallography study revealed that SmPLA2 is composed of five helixes containing two disulfide bonds and one Ca2+-binding site, which is similar to that of the PLA2 from Streptomyces violaceoruber (SvPLA2) [2]. Notably, the SmPLA2 was more thermo-stable than SvPLA2 and porcine PLA2, probably due to Ca2+-binding loop's anchoring by Trp41 into the rest of the protein body. The lecithin biotransformation into lysolecithin-fatty acid liposomes by SvPLA2, SmPLA2, and porcine PLA2 was investigated. The SmPLA2 led to the production of the lysolecithin-fatty acids to the highest concentration due to greater thermal stability. The 200 g/L lecithin were transformed into lysolecithin and fatty acids to a conversion of over 80%. The resulting fatty acids in lysolecithin liposomes could be further transformed into various fatty acids derivatives such as hydroxy fatty acids, hydrocarbons, and secondary fatty alcohols in lysolecithin liposomes by combinatorial reactions of a fatty acid double-bond hydratase from Stenotrophomonas maltophilia (SmOhyA2) [3] and a photoactivated decarboxylase from Chlorella variabilis NC64A (CvFAP) [4] (Fig. 1 and 2). The resulting lysolecithin-fatty acid liposomes were approximately 100 nm in diameter, globular in shape, and monodispersed. Comparing lysolecithin-fatty acids liposomes to lecithin liposomes, they had equal levels of emulsifying activity and encapsulation efficiency. The antibacterial effects against Porphyromonas gingivalis were also examined by measuring minimal inhibition concentration (MIC). Lysolecithin-fatty acids liposomes showed significantly greater antibacterial activity against P. gingivalis, compared to the free form of fatty acids and lecithin liposome. This study will contribute to lecithin and phospholipid biotransformations into various lysolecithin-fatty acid liposomes for food and cosmetic industries. |
| References: | [1] Cerminati, S., Paoletti, L., Aguirre, A., Peirú, S., Menzella, H. G., & Castelli, M. E., Appl. Microbiol. Biotechnol. 2019, 103, 2571-2582. [2] Sugiyama, M., Ohtani, K., Izuhara, M., Koike, T., Suzuki, K., Imamura, S., & Misaki, H., J. Biol. Chem. 2002, 277, 20051-20058. [3] Jeon, E. Y., Seo, J. H., Kang, W. R., Kim, M. J., Lee, J. H., Oh, D. K., & Park, J. B., ACS Catal. 2016, 6, 7547-7553. [4] Huijbers, M. M., Zhang, W., Tonin, F., & Hollmann, F., Angew. Chem. Int. Ed. 2018, 57, 13648-13651. |
| Figures: | 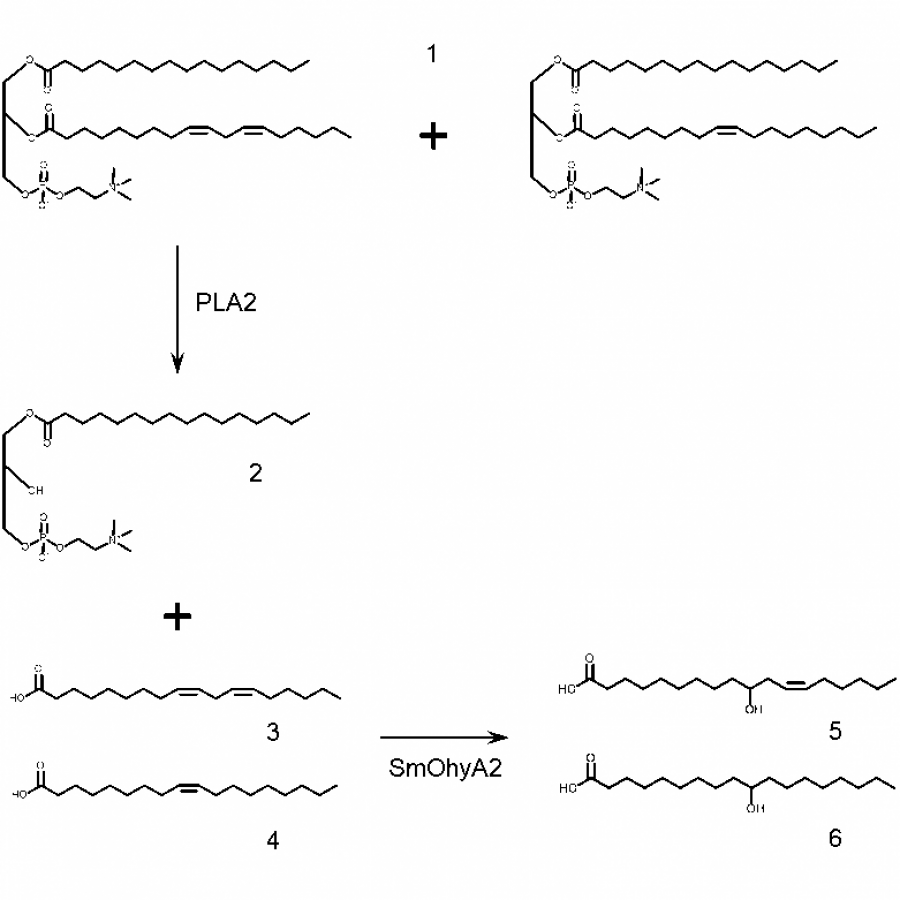 Biotransformation of lecithin (1) into lysolecithin (2) and free fatty acids (3 and 4) and into hydroxy fatty acids (5 and 6). The cascade reactions were catalyzed by a phospholipase A2 (PLA2) and a fatty acid double bond hydratase from S. maltophilia (SmOhyA2). 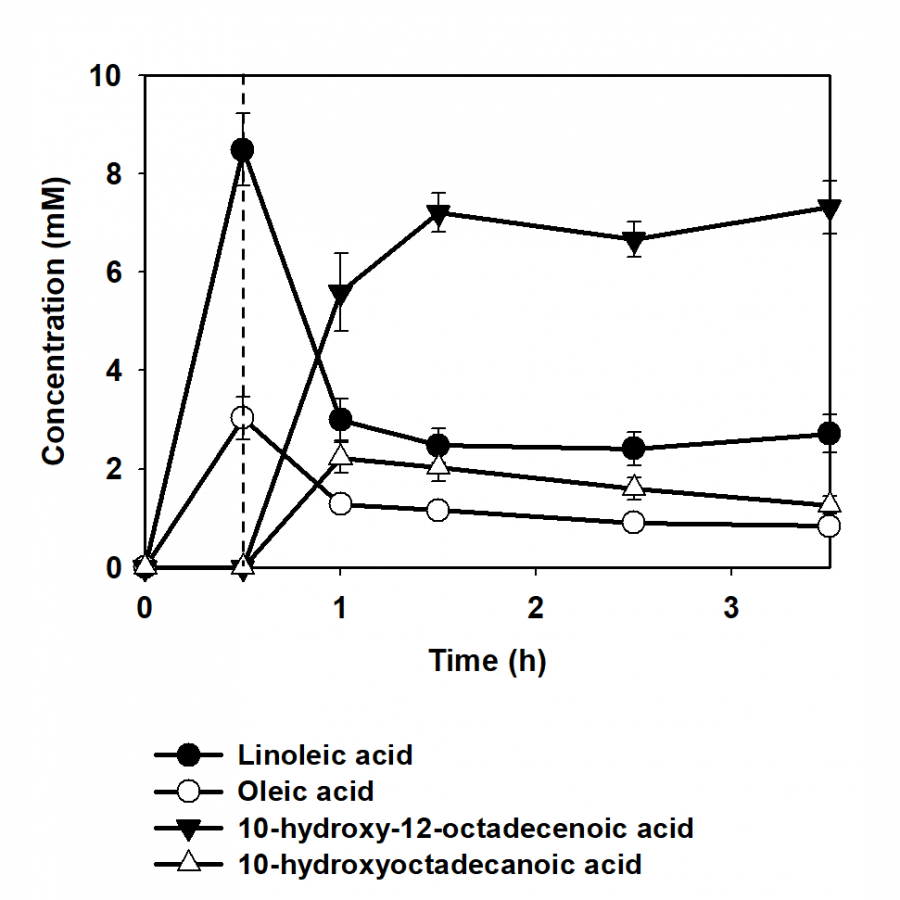 Time course of soy lecithin (1) biotransformation into lysolecithin (2) and hydroxy fatty acids (5 and 6) via lysolecithin (2) and free fatty acids (3 and 4). The biotransformation of soy lecithin (1) into lysolecithin (2) and hydroxy fatty acids (5 and 6) was carried out by the cascade reactions by PLA2 and SmOhyA2. |
#680 | Biological reaction engineering for the preparation of C9 chemicals from oleic acid: n-nonanoic acid, 9-hydroxynonanoic acid, 1,9-nonanedioic acid, 9-aminononanoic acid, 1,9-nonanediol, 9-amino-1-nonanol, and 1,9-diaminononane |
|
| Presenting author: | Se-Yeun HWANG of EHWA WOMANS UNIVERSITY |
| Corresponding author: | Jin-Byung PARK of EHWA WOMANS UNIVERSITY |
| Other authors: | Ji-Min WOO of EHWA WOMANS UNIVERSITY Go Eun CHOI of EHWA WOMANS UNIVERSITY Joo-Hyun SEO of KOOKMIN UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme Catalysis / Oleic acid / C9 Chemicals / 1,9-Diaminononane |
| Purpose: | Engineering of native and recombinant enzyme reactions in whole-cell biocatalysis may allow to produce a variety of chemicals. In particular, fine-tuning of the reactions may enable to prepare a desired product to a high conversion. Here, we present that various C9 chemicals such as n-nonanoic acid, 9-hydroxynonanoic acid, 1,9-nonanedioic acid, 9-aminononanoic acid, 1,9-nonanediol, 9-amino-1-nonanol, and 1,9-diaminononane could be produced to a high conversion from C18 oleic acid (Fig. 1). The C9 chemicals could be prepared as follows; 1) 9-Hydroxynonanoic acid and n-nonanoic acid were produced from oleic acid by using the recombinant Escherichia coli, expressing an oleate double bond hydratase, a secondary alcohol dehydrogenase, and a Baeyer-Villiger monooxygenase, and a lipase from Thermomyces lanuginosus. [1] 2) 9-Hydroxynonanoic acid was converted into 1,9-nonanedioic acid by the recombinant E. coli expressing the primary alcohol dehydrogenase and aldehyde dehydrogenase (ChnDE). [2, 3] 3) 9-Aminononanoic acid was prepared from 9-hydroxynonanoic acid by the combination of the primary alcohol dehydrogenase (ChnD) and ω-aminotransferase from Agrobacterium fabrum (ω-AT_Afab) expressed in recombinant E. coli. Moreover, 9-hydroxynonanic acid was converted into 1,9-nonanediol by carboxylic acid reductase (CAR) and aldehyde reductase expressed in recombinant E. coli. [4] 4) 1,9-Diaminononane and/or 9-amino-1-nonanol were produced from 1,9-nonanediol by the recombinant E. coli expressing ChnD and ω-AT_Afab. As a representative example, activation of 7 recombinant enzyme reactions and 1 native enzyme reaction with repression of 1 native enzyme reaction in E. coli led to formation of 1,9-diaminononane with the conversion of ca. 70% from oleic acid (Fig. 2). These results suggest that engineering biological reactions allow to produce various industrially relevant chemicals from renewable fatty acids to a high conversion. |
| References: | [1] H. J. Cha, E. J. Seo, et al., Adv. Synth. Catal. 2018, 360, 696-703. [2] H. J. Lee, Y. S. Kang, et al., Polymers 2019, 11, 1690. [3] J. W. Song, J. H. Lee, et al., Adv. Synth. Catal. 2014, 356, 1782-1788. [4] H. W. Yoo, H. S. Jung, et al., Green Chem. 2022, 24, 2222-2231. |
| Figures: | 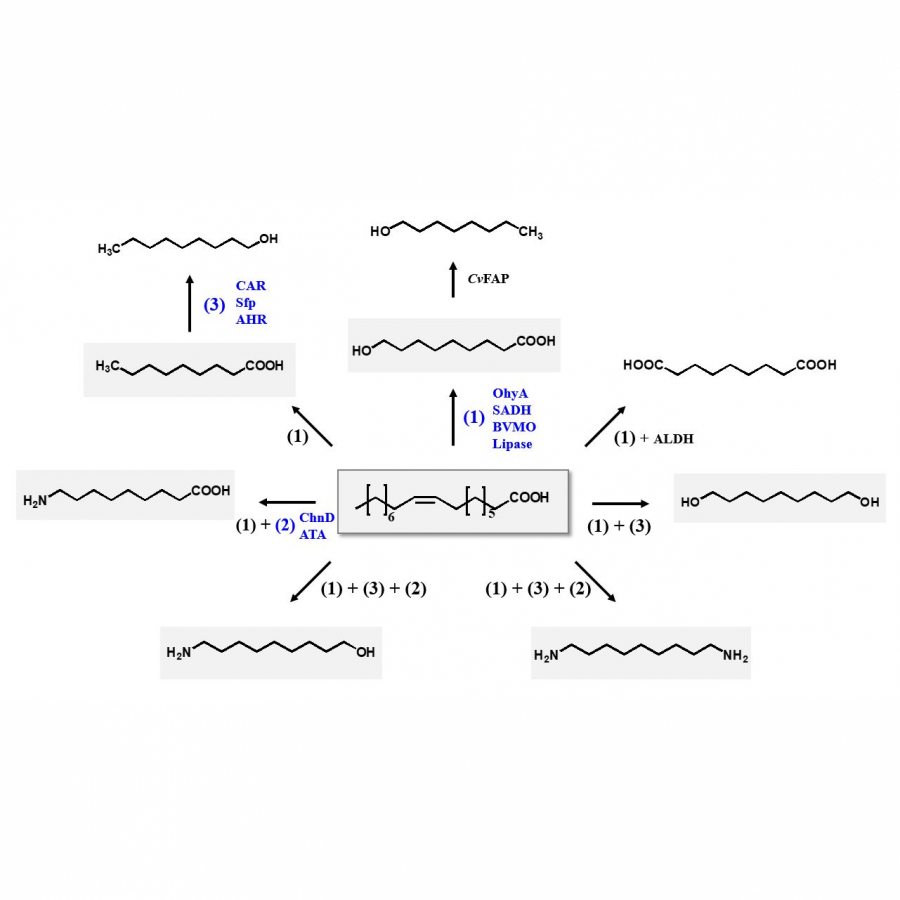 Enzymatic preparation of various C9 chemicals from oleic acid . 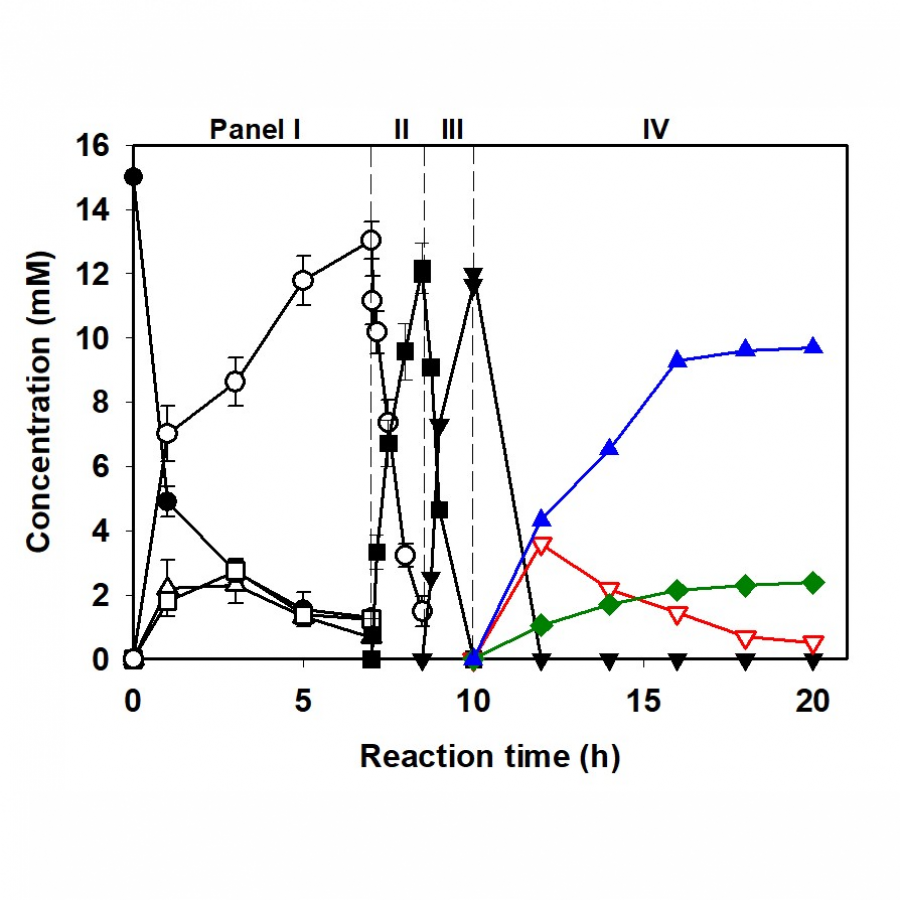 Cascade preparation of 1,9-diaminononane from oleic acid by E. coli-based whole-cell biocatalysts. The symbols indicate the concentration of oleic acid (closed circle), 1,9-nonanediol (closed down triangle), 9-ANA (closed diamond), 9-amino-1-nonanol (opened down triangle), and 1,9-diaminononane (closed triangle). |
#682 | Biotransformation of formaldehyde (C1) into erythrulose (C4) by ThDP-dependent Carbon-Carbon Ligases without by-product formation |
|
| Presenting author: | HUIJIN CHEON of EWHA WOMANS UNIVERSITY |
| Other authors: | HYEJIN JO of EWHA WOMANS UNIVERSITY JunHong KIM of CHONNAM NATIONAL UNIVERSITY JeongSun KIM of CHONNAM NATIONAL UNIVERSITY JinByung PARK of EWHA WOMANS UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Erythrulose / Formaldehyde / Carboligase / Enzyme cascade |
| Purpose: | Erythrulose (C4) is used as an active ingredient for tanning in the cosmetic industry [1]. Thereby, we examined an enzyme cascade reaction system to selectively produce C4 compounds (e.g., erythrulose) from C1 carbon source (e.g., formaldehyde) without by-product formation (e.g., dihydroxyacetone) (Fig. 1). The enzyme cascade consisted of two thiamine-diphosphate dependent carbon-carbon ligases. The thermostable glyoxylate carboligase from Escherichia coli K-12 (EcGCL), which is able to condense two molecules of formaldehyde into one molecule of glycolaldehyde [2,3], was selected as the first enzyme. The EcGCL was engineered to improve catalytic efficiency based on the crystal structure with glycolaldehyde. The SucA, a decarboxylating E1 subunit of the α-ketoglutarate dehydrogenase complex of Vibrio vulnificus (VvSucA) was chosen as the second enzyme (i.e., C2 carboligase), because it catalyzed the condensation of two molecules of acetaldehyde into one molecule of acetoin [4]. The coupling of EcGCL mutant (i.e., EcGCL_R484M/N283Q/L478M/M488L /R284K) and VvSucA_K228L allowed to produce 0.9 g/L erythrulose from 3 g/L formaldehyde via glycolaldehyde (Fig. 2). By-products (e.g., dihydroxyacetone) were not observed. This study will contribute to valorization of C1 gas into industrially relevant multi-carbon products in an environment-friendly way. |
| References: | 1. Samed, G., Vanessa, W., André, P., & Volker, S., Green Chem., 2021, 23, 6583-6590. 2. Jo, H. J., Kim, J. H., Kim, Y. N., Seo, P. W., Kim, C. Y., Kim, J. W., Yu, H. N., Cheon, H. J., Lee, E. Y., Kim, J. S., & Park, J. B., Green Chem., 2021, 24(1), 218-226. 3. Jo, H. J., Yu, H. N., Cheon, H. J., Kim, J. H., Kim, G. Y., Kim, B. S., Kim, J. S., & Park, J. B., ACS Sustainable Chem. Eng., 2023, 11(3), 1078-1086. 4. Seo, P. W., Jo. H. J., Hwang, I. Y., Jeong, H. Y., Kim, J. H., Kim, J. W., Lee, E. Y., Park., J. B., & Kim, J. S, Catal Sci Technol., 2020, 10, 79-85. |
| Figures: | 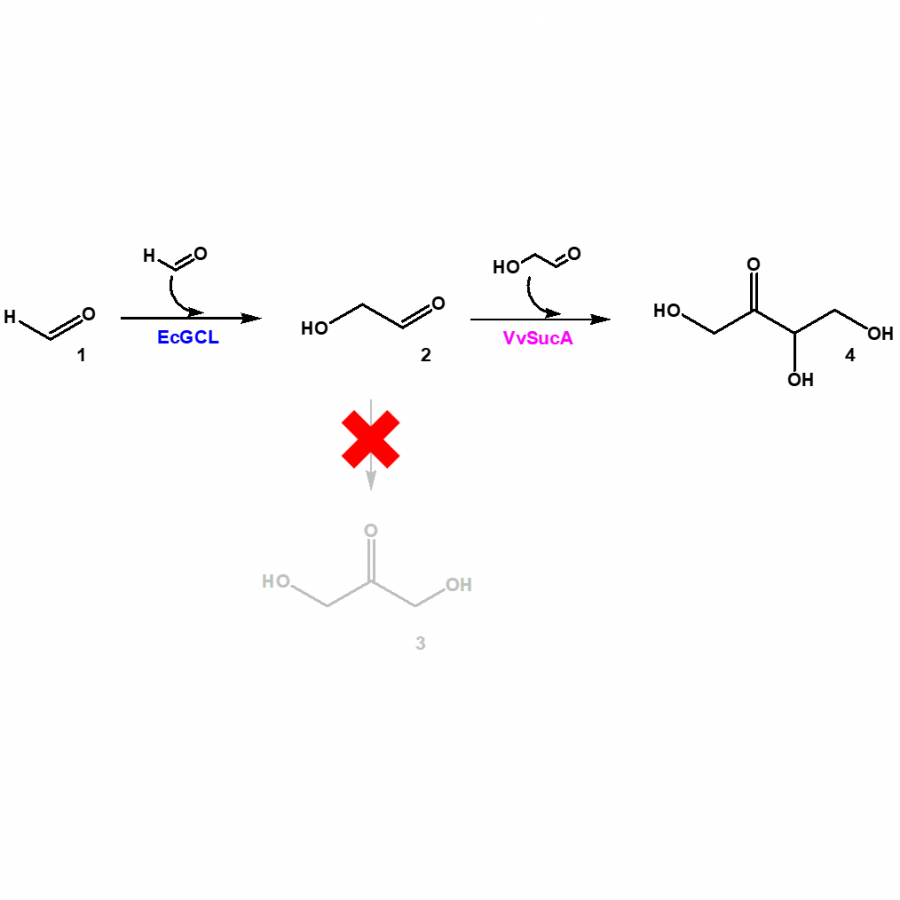 Biocatalytic pathway for producing erythrulose (3) from formaldehyde (1) via glycolaldehyde (2). Erythrulose (4) can be made from formaldehyde (1) without dihydroxyacetone (3) by combination of EcGCL and VvSucA. 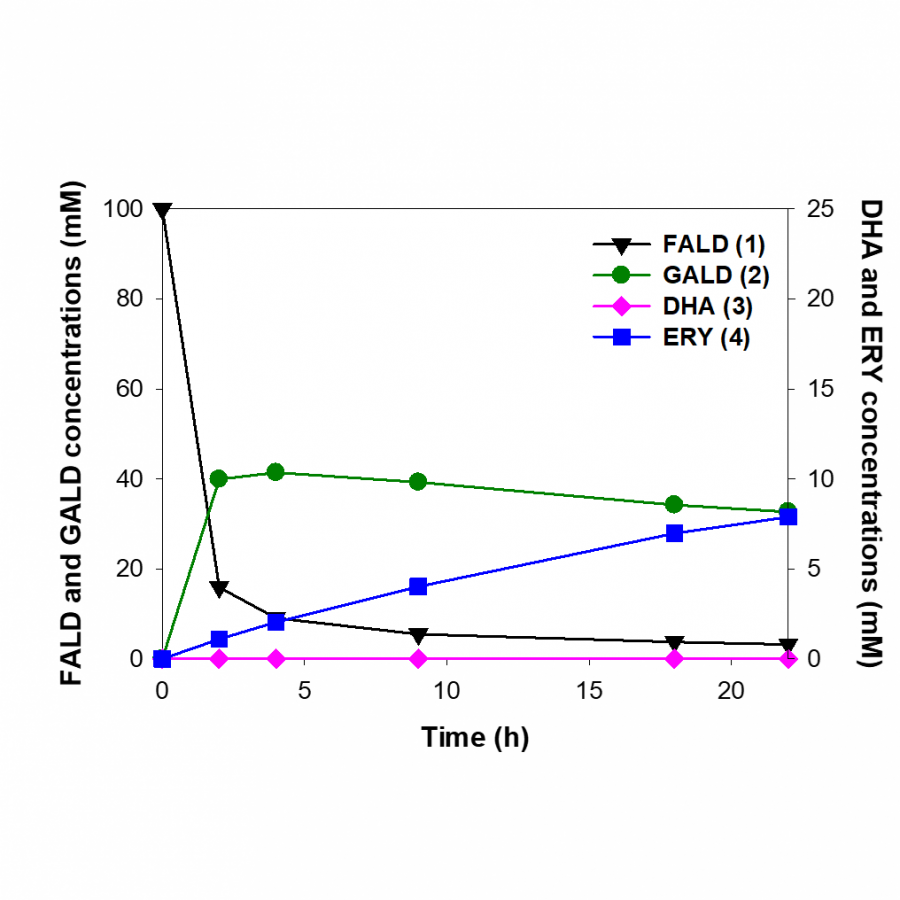 Biotransformation of formaldehyde (1) to erythrulose (4) via glycolaldehyde (2) by coupling EcGCL R484MN283QL478MM488LR284K and VvSucAK228L The symbols reflect the change in concentration of formaldehyde (▼), glycolaldehyde (●), dihydroxyacetone (◆) and erythrulose (■) over time. |
#685 | Precise Regulation of the substrate selectivity of BVMO to Minimize Overoxidation of sulfoxide |
|
| Presenting author: | Huilei YU of EAST CHINA UNIVERSITY OF SCIENCE AND TECHONOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Baeyer-Villiger monooxygenase / chiral sulfoxide / protein engineering / substrate selelctivity |
| Purpose: | Oxidases-catalyzed molecular functionalization is undoubtedly one of the most attractive research areas. Highly selective oxidation reaction is critical for chiral molecules construction. A large number of studies have focused on modification of the stereoselectivity and regioselectivity of oxidases, but the problem of overoxidation caused by the poor substrate selectivity is still unsolvable. Baeyer–Villiger monooxygenases can catalyze the asymmetric oxidation of sulfides to chiral sulfoxides. However, BVMOs can also catalyze the undesired overoxidation of sulfoxides to sulfones, which limits their synthetic application. To minimize this overoxidation, the present study aimed to establish a precise method for regulating the substrate selectivity of BVMOs in sulfide oxidation. The sulfone content of variant F277L was less than 1% (mol/mol), compared with 65% for the wild-type AcPSMO, in the pyrmetazole oxidation reaction after 24 h. The methodology for precise control of substrate selectivity developed in this work will also help to enhance the substrate specificity of other oxygenases and solve the long-standing overoxidation problem in other biooxidation reactions. Before this work, understanding of the structural basis of the substrate selectivity in sulfide oxidation is elusive. In this work, crystal structure determination, molecular dynamics simulations, and QM/MM computations were conducted to elucidate the mechanism of sulfide/sulfoxide substrate selectivity in BVMOs. Furthermore, the redesigned mutants of AcPSMO according to the mechanism we elucidated were also successfully applied for the controllable synthesis of other three chiral prazole sulfoxides. These insights will hopefully lead to a general approach to further mitigate the overoxidation of sulfoxide to sulfone by other BVMOs in a predictable manner. The sulfur atom oxidation mechanism elucidated in this work also sheds light on the electrophilic oxygenation paradigm of BVMOs. |
#699 | Exploring the mechanism of metalloenzymes using using microsecond timescale rapid mixing techniques |
|
| Presenting author: | Peter-Leon HAGEDOORN of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | rapid kinetics / heme enzyme / reaction mechanism / spectroscopy |
| Purpose: | Roughly one third of all proteins contain one or more metal cofactor, and the identity, oxidation state and coordination environment of the metal cofactor provides essential information about the function of the protein. Furthermore, spectroscopy can be used to probe the electronic structure of metal cofactors while the enzyme is converting its substrate, which helps to identify catalytic intermediates and ultimately resolve a catalytic mechanisms. Enzymes are superior catalysts and can achieve turnover rates of more than a thousand per second. This makes it technically challenging to study the mechanism of such enzymes. Special ultrafast mixing techniques are necessary to solve this. I will present the use of ultrafast mixing techniques to establish kinetic mechanisms in the case of metalloprotein folding, metalloenzyme catalysis, and metal to protein binding. Two different in-house developed ultrafast kinetic techniques were used: Nanospec, ultrafast continuous flow UV-vis spectrophotometry; MHQ, microsecond timescale rapid freeze hyperquenching. The dead times of these instruments are 100X shorter than for commercially available devices. Cytochrome c refolding kinetics during a pH jump from 2 (unfolded) to 4.7 (near native state) was studied in the submillisecond timescale.1 The combination of the transient UV-vis and EPR data show that within 1 millisecond cytochrome c folding proceeds through three different steps: 1) His18 binding to the heme cofactor, 2) a conformational change that affects the heme environment, 3) Met80 binding to the heme cofactor in concert with a significant increase of the alpha helical content of the protein. There is evidence for further slower conformational steps towards the stable folded state at pH 4.7. Novel intermediate states were identified in the catalytic mechanism of the fast heme enzyme chlorite dismutase.2 Chlorite dismutase is a unique heme b dependent enzyme that catalyzes the conversion of chlorite (ClO2-) to molecular oxygen (O2) and chloride (Cl-). This reaction involves O-O bond formation, which is rare in nature. The enzyme catalyzes a single turnover in less than a millisecond. The catalytic mechanism of chlorite dismutase was investigated using microsecond timescale mixing techniques and the natural substrate chlorite. The UV-visible, EPR and Resonance Raman spectra of catalytic intermediates were obtained. Distinct intermediates were found that are not observed with the artificial substrate peracetic acid. Most notably a triplet state EPR signal that we attribute to two weakly coupled amino acid based cation radicals, ‘compound T’, was transiently formed. The formation of compound T is direct evidence of a two electron transfer process which means that the Cl-O bond break is heterolytic, unlike the most recent proposed mechanism for this enzyme. To our knowledge such a triplet state has never been identified in any heme enzyme. The kinetics of Cu2+ binding to the metal binding motifs of human high affinity copper uptake protein 1 (CTR1) showed the formation of transient binding intermediates.3 The N-terminal domain of this protein has a well-known high affinity Cu2+ binding site, but only the properties of the final complex have been studied previously. Using MHQ discrete Cu2+ binding intermediates were identified in the submillisecond to second scale. These intermediates represent the subsequent coordination to one, two and three protein ligands, clearly suggesting a binding mechanism. Furthermore some of the short lived intermediates have lifetimes and redox properties that have important physiological consequences for Cu transport and the involvement of Cu in human diseases. |
| References: | 1 Srour, B., Strampraad, M.J.F., Hagen, W.R., Hagedoorn, P.L., Refolding kinetics of cytochrome c studied with microsecond timescale continuous-flow UV-vis spectroscopy and rapid freeze-quench EPR, J. Inorg. Biochem. 184 (2018) 42-49. https://doi.org/10.1016/j.jinorgbio.2018.04.011 2 Püschmann, J., Mahor, D., De Geus, D.C., Strampraad, M.J.F., Srour, B., Hagen, W.R., Todorovic, S., Hagedoorn, P.L., A unique biradical intermediate in the mechanism of the heme enzyme chlorite dismutase, ACS Catal. 11 (2021) 14533-14544. https://doi.org/10.1021/acscatal.1c03432 3 Kotuniak, R., Strampraad, M.J.F., Bossak-Ahmad, K., Wawrzyniak, U.E., Ufnalska, I., Hagedoorn, P.L., Bal, W., Key intermediate species reveal the Cu(II) exchange pathway in biorelevant ATCUN/NTS complexes, Angew. Chemie. Int. Ed. 59 (2020)11234-11239. https://doi.org/10.1002/ange.202004264 |
| Figures: | 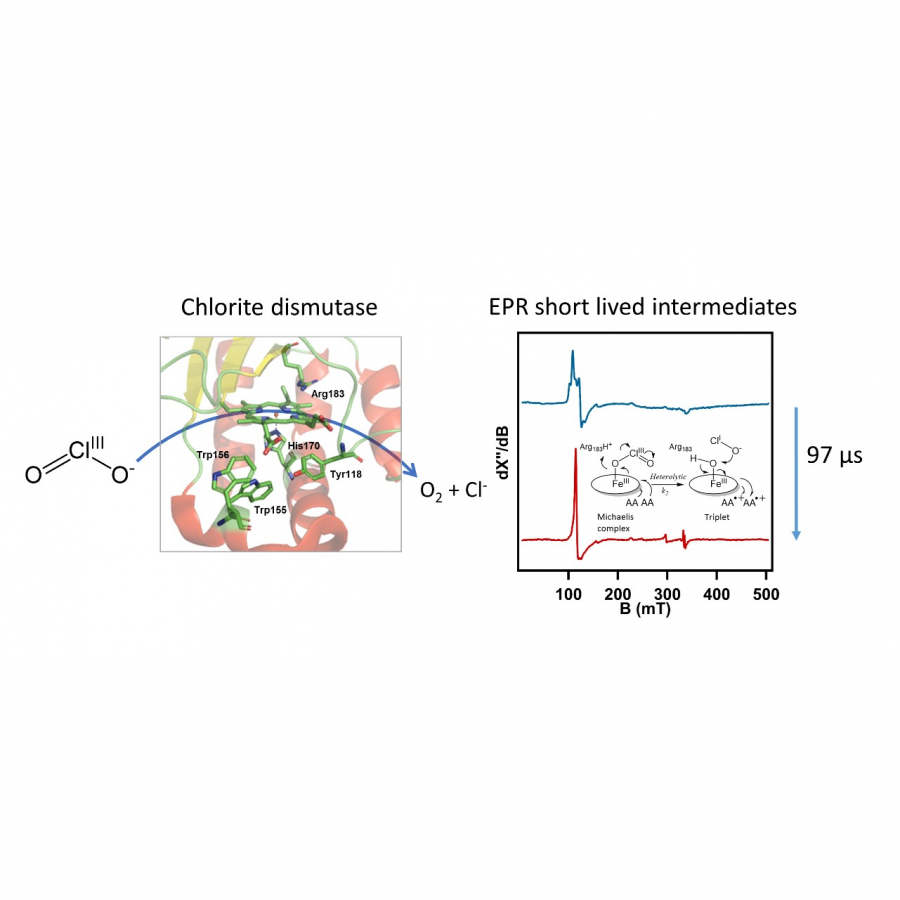 Chlorite dismutase rapid kinetics Chlorite dismutase converts chlorite to molecular oxygen and chloride. Novel reaction intermediates were identified using microsecond timescale rapid kinetics techniques. |
#704 | IN SILICO ENZYME DISCOVERY AND OPTIMIZATION FOR UNNATURAL REACTIONS |
|
| Presenting author: | Lur ALONSO of ZYMVOL BIOMODELING SL |
| Corresponding author: | Maria Fátima LUCAS of ZYMVOL BIOMODELING SL |
| Other authors: | Jesús SECO of ZYMVOL BIOMODELING SL Ryoji TAKAHASHI of ZYMVOL BIOMODELING SL Marina CAÑELLAS of ZYMVOL BIOMODELING SL Ferrán SANCHO of ZYMVOL BIOMODELING SL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme engineering / Enzyme discovery / Computational tools / |
| Purpose: | Enzymes have the potential of increasing the sustainability of industry by substituting polluting chemical processes with environmentally friendly reactions. This is possible thanks to the wide spectrum of reactions natural enzymes can perform as well as the inherent catalytic features many of them own, like specificity and enatioselectivity. However, these capabilities are often not sufficient to satisfy industrial needs, and natural enzymes need to be tuned, which is usually a slow and costly process addressed by directed evolution campaigns. These strategies have revealed over the last decades how distal mutations can be the key to boosting enzymatic properties [1]. In the last years, and with the advance of computation, the scientific community has tried to understand the fundamentals for predicting distal mutations [2], so that directed evolution studies, which are tedious, expensive and depend on efficient screening approaches, can be outperformed. Still, engineering approaches are often insufficient, in particular, when natural catalysts for specific abiological reactions do not exist or have not yet been discovered. Genomic and metagenomic-based techniques are the strategies of choice for identifying enzymes with novel biocatalytic activities [3]. However, such genome-mining techniques are typically time-consuming and laborious, highlighting the need to develop novel high-throughput enzyme discovery technologies capable of matching the pace of business. In Zymvol, we have focused our efforts in covering these two hurdles, aiming to speed up the application of biocatalysts to industry and therefore its transition towards greener practices. In particular, we have developed ZYMEVOLVER (ZYV), able to boost the enzymatic properties through targeting both active site and distal mutations; and BIOMATCHMAKER (BMM), that combines bioinformatics with reliable physics-based simulations at distinct theoretical levels to perform enzyme discovery for abiological reactions. Here, the capabilities of both ZYV and BMM pipelines will be discussed, highlighting various case studies that have led to the identification and optimization of novel enzymes for non-biological reactions that are of high interest for the chemical and pharmaceutical industries. |
| References: | [1] K. Chen, F. Arnold, Nat. Catal., 2020, 3, 203-213 [2] S. Osuna, Wires. Comput. Mol. Sci., 2020, 11 [3] S. L. Robinson, Nat. Prod. Rep., 2021, 38, 1994-2023 |
#710 | Bienzymatic ATP-dependent Bacterial System Fueled by Metaphosphate as Efficient Tool to Break the Chemical Stability of δ-Lactams |
|
| Presenting author: | Mélanie HALL of UNIVERSITY OF GRAZ |
| Other authors: | Sebastian ROTH of UNIVERSITY OF GRAZ Somayyeh GANDOMKAR of UNIVERSITY OF GRAZ Federico ROSSI of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | valerolactam / ATP recycling / cascade / oxoprolinase |
| Purpose: | Medium-sized 5- and 6-membered ring lactams are – in contrast to smaller 4-ring β-lactams – molecules with remarkable stability, a feature which appears connected to the increased resonance stabilization of the amide bond and the higher partial C-N double bond character in these lactams [1,2]. Chemical hydrolysis to the corresponding amino acids requires harsh reaction conditions (reflux and strong acid) and up to now, no enzyme active on monocyclic γ- and δ-lactams has been reported. Our goal is to establish a biocatalytic platform for the ring opening of γ- and δ-lactams under mild reaction conditions. In this context, we explored the biocatalytic potential of bacterial ATP-dependent oxoprolinases, which function in pair (OplA and OplB) [3]. Strong activity in the presence of excess of ATP could be detected on δ-valerolactam and a range of derivatives. An ATP recycling system (Figure 1) based on cheap Graham’s salt (sodium hexametaphosphate) and a polyphosphate kinase [4] allowed the use of catalytic amounts of ATP (1 mol%). Further improvements were obtained by co-expressing OplA and OplB using the pETDUET1 vector, a strategy which enhanced the soluble expression yield and the protein stability. Finally, a range of alternate phosphodonors were investigated in place of ATP. In some cases, activity was detected, providing hints on a possible mechanism, which was further studied by 31P-NMR. Acknowledgements Funding by the Austrian Science Fund (FWF P32815-N) is acknowledged. We thank Prof. Yakunin for the kind gift of the kinase plasmid. |
| References: | [1] M. Szostak, J. Aube, Chem. Rev. 2013, 113, 5701. [2] Z. Assaf, K. Faber, M. Hall, J. Biotechnol. 2016, 235, 11; Z. Assaf, E. Eger, Z. Vitnik, W. M. F. Fabian, D. Ribitsch, G. M. Guebitz, K. Faber, M. Hall, ChemCatChem 2014, 6, 2517. [3] A. Marjanovic, H. J. Rozeboom, M. S. de Vries, C. Mayer, M. Otzen, H. J. Wijma, D. B. Janssen, Proteins 2021, 89, 1079. [4] B. Nocek, S. Kochinyan, M. Proudfoot, G. Brown, E. Evdokimova, J. Osipiuk, A. M. Edwards, A. Savchenko, A. Joachimiak, A. F. Yakunin, Proc. Natl. Acad. Sci. USA 2008, 105, 17730. |
| Figures: | 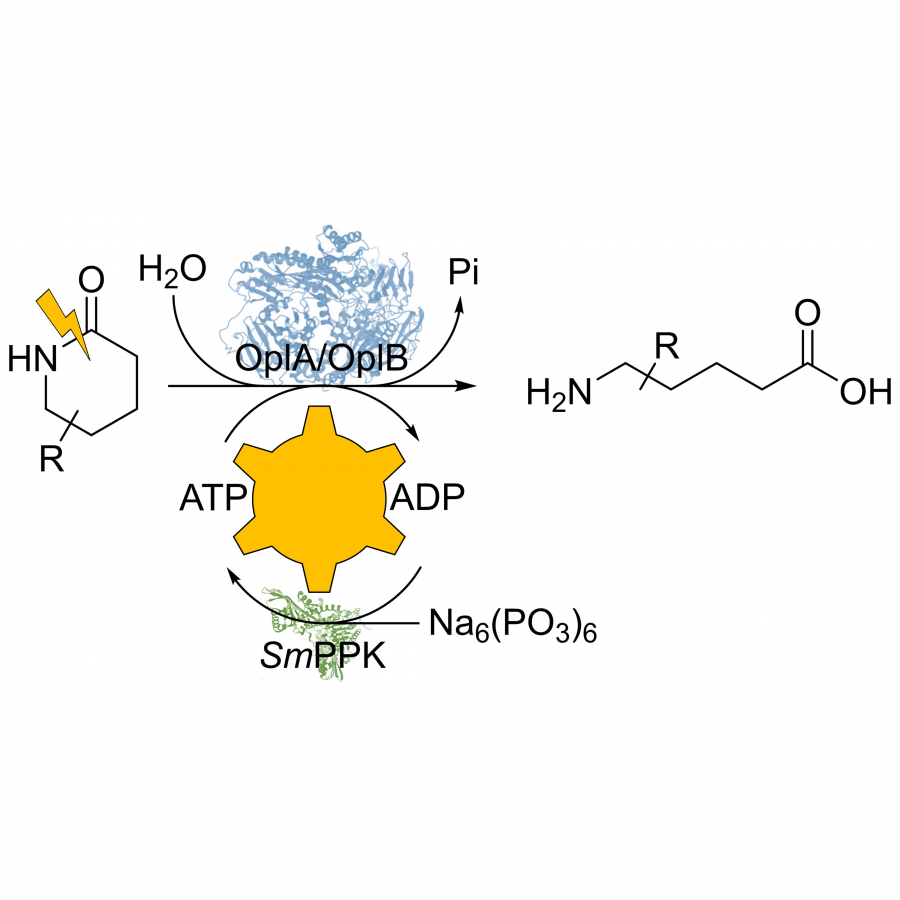 Biocatalytic metaphosphate-fueled ring-opening of delta-valerolactam derivatives by ATP-dependent-enzymes |
#715 | SELECTIVE SYNTHESIS OF INDUTRY RELEVANT PRODUCTS BASED ON 5-HYDROXYMETHYLFURFURAL (HMF) CATALYZED BY SELF-SUFFICIENT IMMOBILIZED MULTI-ENZYME SYSTEMS |
|
| Presenting author: | Jakub Franciszek KORNECKI of CIC BIOMAGUNE / CASCAT GMBH |
| Corresponding author: | Fernando LÓPEZ GALLEGO of CIC BIOMAGUNE |
| Other authors: | André PICK of CASCAT GMBH |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme immobilization / Alcohol dehydrogenase / Glucose dehydrogenase / HMF reduction |
| Purpose: | Production of 2,5-dihydroxymethyl furan (BHMF) via the reduction of 5-hydroxymethyl furan (HMF) through an enzyme based biocatalytic process is a promising and more sustainable alternative to the manufacturing of building blocks for plastic production, which is normally based on fossil raw materials [1]. The replacement of traditional processes to produce plastics with the polymerization of BHMF obtained via enzymatic reaction is a clear path to a sustainable and environmentally friendly system without having to deny the use of plastics as a cheap and easily available material. The biosynthesis of BHMF starting from bio-based HMF is challenging when using enzymes as biocatalysts due to the need of a high input of cofactor, which is depleted extremely fast. In this work, we have tackled this limitation via the co-immobilization of a redox cofactor (NADH) and two dehydrogenases capable of orthogonally reducing the HMF and recycling NADH in situ and inside the porous structure of the solid supports. This approach has allowed recycling both enzymes and cofactor for consecutive batch cycles. YjgB Ec LND [2], an alcohol dehydrogenase from Escherichia coli is the enzyme in charge of the reduction of HMF into BHMF, while the thermostable glucose dehydrogenase developed by Figueroa [3] was chosen as the counterpart capable of delivering a cofactor recycling reaction. Both dehydrogenases were sequentially immobilized on a survey of supports to find the most optimal carrier and immobilization conditions that allowed reusing the co-immobilized enzymatic system over sequential HMF reduction/oxidation reactions. Purolite ECR8204F chemically modified with E-Co2+ and glyoxyl [4] groups turned out to be the best choice from such intense immobilization screening. The two enzymes achieved an immobilization yield of over 99% during the co-immobilization over E-Co2+ and glyoxyl carriers, and recovering 1.2 U/grsupport and 1.4 U/grsupport in the case of YjgB Ec LND, respectively for each carrier and 2.14 U/grsupport and 0.8 U/grsupport respectively in the case of the GDH. This system is able to quantitatively produce BHMF by the reduction of HMF, regenerating the NADH in situ. The solid biocatalyst was reused up to fifteen times, in consecutive reaction courses of 1 h each, substituting the reaction media every cycle, and no decrease in conversion capabilities were detected along these cycles. The resulting bi-functional heterogeneous biocatalysts was further coated with cationic polymers capable of binding cofactor into their matrix, bringing the system one step closer to the true meaning of a self-sufficient catalyst, where exogeneous supply of cofactor is no longer needed and both enzymes and cofactors integrated into the solid carrier can be recycled simultaneously. |
| References: | [1] A. A. Rosatella, S. P. Simeonov, R. F. M. Frade, C. A. M. Afonso, Green Chem. 2011, 13, 754. [2] A. Pick, B. Rühmann, J. Schmid, V. Sieber, Appl. Microbiol. Biotechnol. 2013, 97, 5815-5824. [3] E. Vázquez-Figueroa, J. Chapparro-Riggers, A. S. Bommarius, ChemBioChem 2007, 8, 2295-2301 [4] R. Fernandez-Lafuente, Enzyme Microb. Technol. 2009, 45, 405-418. |
| Figures: | 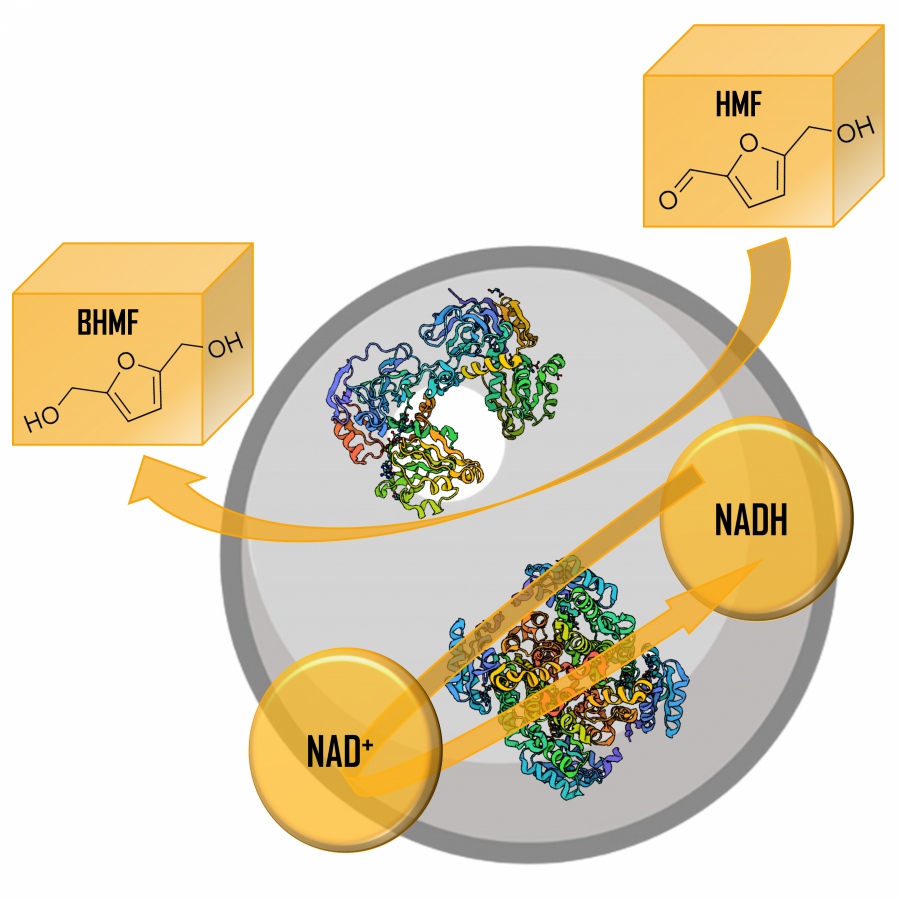 Scheme of the solid biocatalyst Graphical representation of how the solid biocatalytic system that includes both alcohol and glucose dehydrogenases, is able to reduce HMF into BHMF at the same time as it is recycling the available cofactor (NADH). |
#718 | Bioengineering of metabolic pathways of pyrrocidines for the generation of chemical diversity in this family of compounds. |
|
| Presenting author: | Steffi SEWSURN of MUSEUM NATIONAL D'HISTOIRE NATURELLE DE PARIS |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Pyrrodicidines / Metabolic engineering / PKS-NRPS / Secondary metabolites |
| Purpose: | Pyrrocidines are secondary metabolites isolated from diverse fungi and characterized by antimicrobial1 and apoptosis-inducing activities2, as well as potential inhibitory effects of SARS-CoV2 RNA-dependent RNA polymerase3. These molecules belong to a growing family of fungal natural products sharing a decahydrofluorene core connected to a highly strained paracyclophane moiety. Each member possesses different combinations of configuration at the chiral centers present in the paracyclophane-decahydrofluorene motif such as in hirsutellone B and xenoacremone C. In order to understand the steps leading to this unique polycyclic structure, we investigated pyrrocidine biosynthesis in the fungal producing strain Sarocladium zeae by gene knock-out and thorough metabolic analysis. Thus, a biosynthetic gene cluster encoding a hybrid polyketide synthase – non-ribosomal peptide synthetase (PKS-NRPS) and its auxiliary enzymes was identified. This work reveals an intrinsic plasticity of the pyrrocidine pathway and leads to the generation of complex metabolites with new cyclic backbones in the mutants. In this process, the megaenzyme PKS-NRPS forms a linear precursor which then undergoes diverse cyclisation and redox steps catalyzed by the auxiliary enzymes. Among them, several enzymes are attractive tools for biocatalytical approaches like the α,β-hydrolase PrcH catalyzing the formation of pyrrolidinone ring by a Knoevenagel reaction and PrcI performing the reduction of this five-member ring, or the lipocalin-like protein PrcX controlling a Diels-Alder cycloaddition. Our work also focuses on the reprogramming of the PKS-NRPS by reassembling this multi-domain protein to generate a chemical diversity of linear precursors and integrates it in synthetic biology approaches. The latest results will be presented. |
| References: | 1 D. T. Wicklow and S. M. Poling, Phytopathology, 2009, 99, 109-115. 2 S. Uesugi, N. Fujisawa, J. Yoshida, M. Watanabe, S. Dan, T. Yamori, Y. Shiono and K. Kimura, J. Antibiot. (Tokyo), 2016, 69, 133-140. 3 K. S. Ebrahimi, M. Ansari, M. S. Hosseyni Moghaddam, Z. Ebrahimi, Z. salehi, M. Shahlaei and S. Moradi, Comput. Biol. Med., 2021, 135, 104613. |
| Figures: | 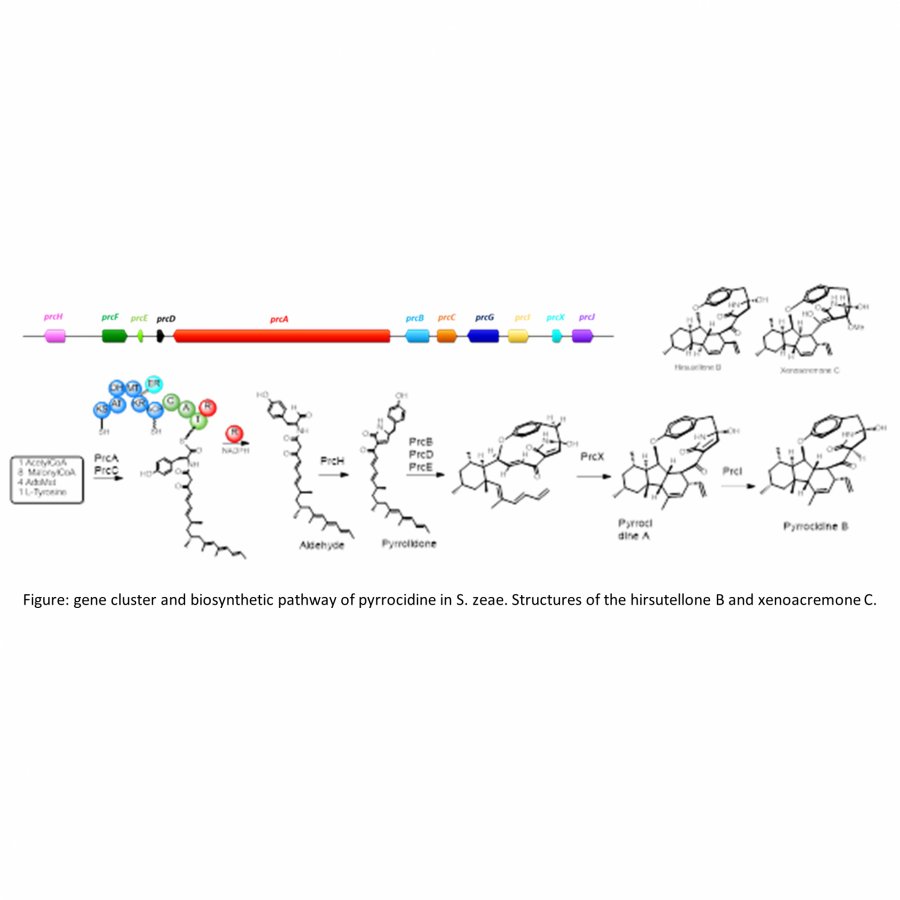 Figure gene cluster and biosynthetic pathway of pyrrocidine in S. zeae. Structures of the hirsutellone B and xenoacremone C. |
#723 | Use of the bacterial two-hybrid system for the development of quasi-homodimeric papain-like cysteine proteases |
|
| Presenting author: | Ana OBAHA of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIV |
| Other authors: | Marko NOVINEC of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIV |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Protein engineering / Xylellain / Oligomerization / |
| Purpose: | Oligomeric proteins account for 30-50 % of all proteins in nature [1]. The abundance of oligomers is explained by their functional and structural advantages over monomeric variants. These include increased stability, resistance to denaturation and degradation, additional ability to regulate enzyme activity, and cooperativity [2]. Oligomeric proteins are formed in nature in several ways. The most common is the interaction of polypeptide chains via complementary surfaces with non-covalent interactions, but it is also possible to form oligomers via the domain swapping mechanism, formation of common metal ions and disulfide bonds. The subject of our research is the family of papain-like cysteine proteases, which are mostly monomeric. Our goal is to produce these enzymes in the form of stable dimers. Due to the advantages of the oligomeric state of enzymes compared to the monomeric state, we expect that the produced recombinant dimeric enzymes will have better regulatory mechanisms and kinetic properties compared to natural enzymes, making them interesting for biotechnological applications. Our main target is the prokaryotic cysteine protease xylellain from the bacterium Xylella Fastidiosa, which is a suitable model protein due to its ease of production in the bacterial expression system. In addition, we aim to produce dimeric forms of ficin, stem bromelain, and fruit bromelain, three representatives of the plant papain-like proteases that are indispensable in biotechnological processes in the production of meat, dairy products, and juices. Our approach is based on the production of dimeric forms of the target by optimizing the existing surfaces of the protein. We have identified the back and bottom surfaces of the protein with respect to the standard orientation as suitable surfaces for engineering oligomerization, as these surfaces are planar and large enough to allow the formation of stable interfaces. We have prepared several site-directed saturated libraries to be studied using a commercially available bacterial two-hybrid system originally developed for the discovery of interaction partners in-vivo [3]. The two-hybrid system is based on co-transformation of bacteria with plasmids pUT18 and pKT25, which express the fusion of the target protein with one of the two fragments of adenylate cyclase. Screening is performed on plates using the blue-white screening protocol. Because the system uses two differently mutated forms of the target protein that otherwise contain the mutation at the same position, we have termed the resulting dimers quasi-homodimers. So far, we have identified 15 pairs of potential quasi-homodimers of xylellain using the bacterial two-hybrid system. The identified mutants had predominantly hydrophobic residues on the interaction surface, but cysteine, arginine, glutamic acid, threonine, and histidine also occurred. Mutants expected to contribute to quasi-homodimerization were mutated in-silico, and the potential structures of the symmetric dimers were calculated using the program M-ZDOCK. Experimental verification using purified recombinant proteins is underway. |
| References: | [1] e. d. levy, s. teichmann, prog. mol. biol transl. sci., 2013, 117, 25-51. [2] d. s. goodsell, a. j. olson, annu. rev. biophys. biomol. struct., 2000, 29, 105-153. [3] m. g. olson, m. goldammer, e. gauliard, d. ladant, s. p. ouellette, methods mol. biol., 2018, 1794, 75-96. |
#725 | Styrene oxide isomerase performing both epoxidation and isomerization of styrene in presence of hydrogen peroxide |
|
| Presenting author: | Selvapravin KUMARAN of RUHR UNIVERSITY BOCHUM |
| Corresponding author: | Dirk TISCHLER of RUHR UNIVERSITY BOCHUM |
| Other authors: | Shanice OLANIPEKUN of RUHR UNIVERSITY BOCHUM Peter-Leon HAGEDOORN of TECHNISCHE UNIVERSITEIT DELFT |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | SOI / heme / epoxidation and isomerization / biotransformation |
| Purpose: | Styrene oxide isomerase (SOI), a membrane bound enzyme in the side chain oxygenation of styrene degradation pathway, believed to be co-factor independent. SOI catalyses the isomerization of styrene oxide (SO) into phenylacetaldehyde (PA). SOIs are approximately 20kDa in size as a monomer. During protein enrichment the membrane fraction comprising SOI was always reddish1, but this had never been explained. The mechanism of SOI was recently proved to be 1,2 hydride/methyl shift, but the active site remained as an enigma2. It was also known that SOI are highly stable at high temperature as well as wide range of pH1 potentially making this enzyme a candidate for various reaction in biotechnological application. We discovered the presence of styrene degrading gene cluster from an alphaproteobacteria Zavarzinia compransoris Z-1155 by in silico genome mining. The ZcSOI-gene from Z. compransoris Z-1155 was cloned into pET vector and heterologously expressed in E. coli (yield: 6.1 mg/mL). A purification protocol for the membrane protein was established. Hence, this provided the chance to characterize ZcSOI in more detail which we did in comparison to related enzymes from Rhodococcus and Pseudomonas. SOIs are functional as trimer possessing an iron containing heme B at the interface of two protomers, comprising three active sites. This explains the previously observed red colour of the enriched protein. We confirmed the presence of the heme B cofactor by several methods (ICP-MS, UV/vis spectroscopy, EPR) and believe it is key for epoxide conversion. The direct kinetics of SOI for its natural substrate showed a turnover of around 500s-1 and the catalytic efficiency with just one order of magnitude short of catalase. The stopped-flow measurement of heme reduction with sodium dithionite showed the presence of reducible heme. Biochemically the enzyme behaved similar to that of other SOIs from previous studies1,3. Owing to the fact that SOI has reducible heme, performs the isomerization of SO, we aimed at exploring the biochemical potential of ZcSOI for complete conversion of styrene to phenylacetaldehyde (Fig. 1). Preliminary test of enzyme with styrene and hydrogen peroxide showed the production of both SO and PA. The extended incubation resulted in only PA as the final product. In order to optimize, the kinetic of H2O2 for the enzyme ZcSOI was tested spectrophotometrically using ABTS as substrate. The ZcSOI showed a Vmax of about 1568.9 µmoles/min with Km 1.6 mM. This assay proves that the enzyme acts like a peroxidase while the earlier assay with H2O2 mediated combined conversion of styrene to phenylacetaldehyde provides some hint that the enzyme could possibly play the role of peroxygenase in epoxidation while the produced epoxides being the natural substrate for ZcSOI instantly converted into phenylacetaldehyde. Both the products having significance in industrial applications, a single enzyme performing the combined reaction, possessing redox potential heme which accepts H2O2 makes this enzyme remarkably a trend setter in various peroxygenase, peroxidase and many more combined biotechnological reactions. |
| References: | 1. m. Oelschlägel, j.a. gröning, d. tischler, s.r. kaschabek, m. schlömann, appl. environ. microbiol. 2012, 78(12), 4330-7. 2. r. xin, w.w.l. see, h. yun, z. li, angew. Chem. Int. ed engl. 2022, 61(28), e202204889. 3. j.p.s. choo, r.a. kammerer, x. li, z. li, adv. synth. catal. 2021, 363(6), 1714-1721. |
| Figures: | 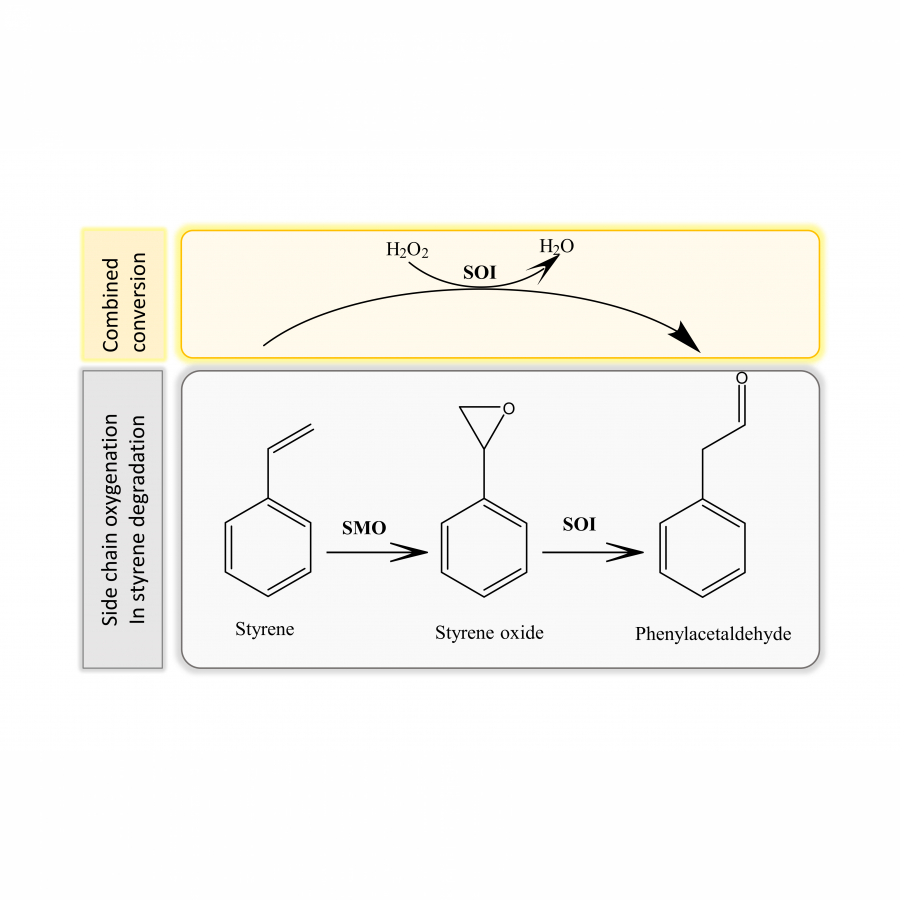 Combined conversion of styrene to phenylacetaldehyde by SOI Schematic diagram showing the classical side chain oxygenation of styrene biodegradation in gray and the combined conversion of Styrene to PA by SOI in yellow (this study) |
#729 | Methylformamide Conversion by Formate Dehydrogenase to Fuel Reductive Aminase with NADPH and Amine in a Cascade Setup |
|
| Presenting author: | Artur MAIER of RUHR UNIVERSITY BOCHUM |
| Other authors: | Tanja KNAUS of UNIVERSITY OF AMSTERDAM Francesco MUTTI of UNIVERSITY OF AMSTERDAM Dirk TISCHLER of RUHR UNIVERSITY BOCHUM |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | NADPH regeneration / cascade / FDH / reductive amination |
| Purpose: | Formate dehydrogenase (FDH) from Candida biodinii is well studied and applied in various setups as a NADH-regeneration system. Attempts were made to increase stability and due to its clear preference for NAD+, to shift the substrate specificity towards NADP+ through mutagenesis. Here, the impact of various known stability mutations [1,2] on the NADP+ accepting FDH (D195Q/Y196R/Q197N) [3], in the following FDH-QRN, and the bioinformatically predicted T130I mutation on wild type and mutants are elucidated. FDH (C23S/T130I) showed a 6-fold lower Km value (8.5 ± 0.32 µM vs 51.0 ± 1.0 µM) and an approx. 3.5-fold increase in catalytic efficiency (235.7 s-1¬ mM-1¬) compared to the wild type (66.4 s-1 mM-1). FDH-QRN (C23S/T130I) showed a 2-fold lower Km and increase of the catalytic efficiency to 18.37 ± 1.4 s-1 mM-1 compared to the FDH-QRN (16.74 ± 1.55 s-1 mM-1). FDHs are known to accept formate derivatives such as methyl-, ethyl- and phenylformate, among others [4]. In silico docking and molecular dynamics simulations suggested that formamides might be suitable substrates for FDHs as well. All variants were screened for activity towards formamide (F), N-methylformamide (MF) and N,N-dimethylformamide (DMF), showing that all variants have the ability to accept formamides, with highest activities, with NAD+ as cofactor, achieved by FDH (T130I) for F with 83.1 mU/mg and for MF and DMF by FDH (C23S) with 68.3 mU/mg and 7.4 mU/mg, respectively. With NADP+ as cofactor, the FDH-QRN reached activities with F, MF and DMF of 6.2, 14.6 and 3.5 mU/mg. The NADP+-accepting variants were employed for NADPH regeneration in a cascade reaction for the reductive amination of cyclohexanone by reductive aminase from Aspergillus oryzae [5] with MF as the sole electron and amine donor, reaching conversion rates up to 63% in a whole cell approach, broadening the applicability of FDHs in biocatalysis. |
| References: | [1] Slusarczyk, H. et al., Eur. J. Biochem. (2000) 267, 1280-1289 [2] Zheng, J. et al., Appl. Environ. Microbiol. (2016) 83(2):e02624-16 [3] Wu, W. et al., J. Mol. Cat. (2009) 61, 157-161 [4] Froehlich, P. et al., Org. Biomol. Chem. (2011) 26;9(22):7941-50 [5] Aleku, G. et al. Nature Chem. (2017) 9, 961-969 |
| Figures: | 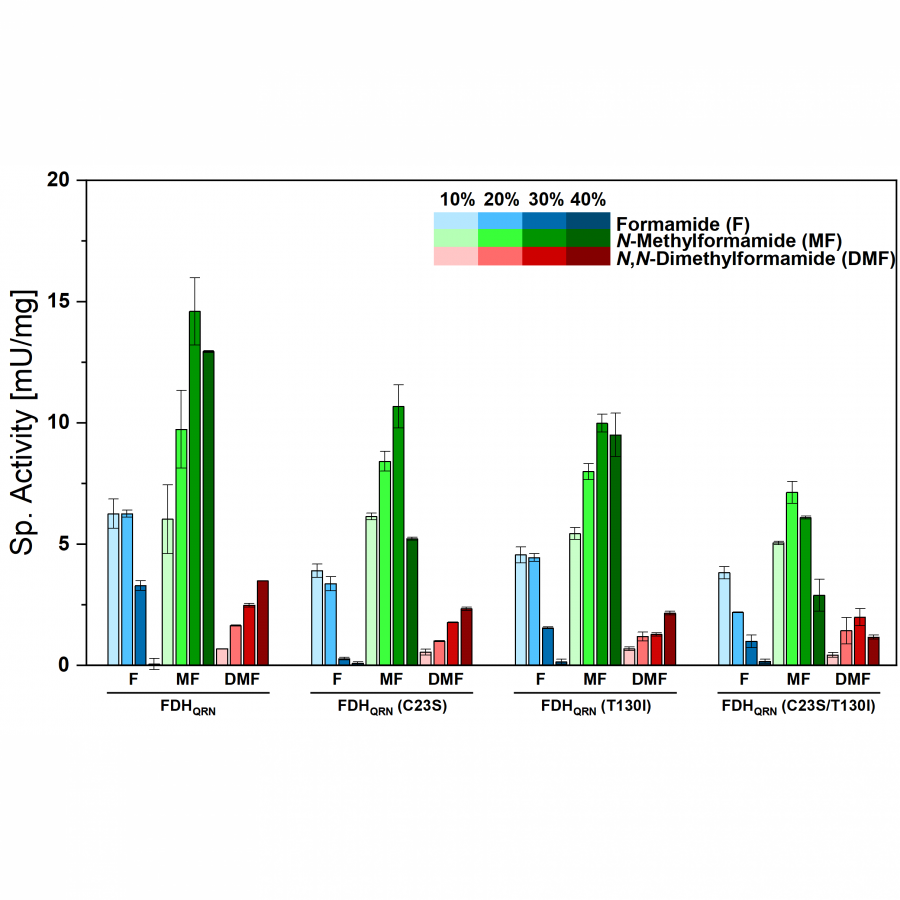 FDH Activity with formamide derivatives Specific activity of the respective NADP+ accepting FDH variants with formamide (F), N-methylformamide (MF) and N,N-dimethylformamide (DMF) as the sole electrone donor at concentrations ranging from 10-40% (v/v). 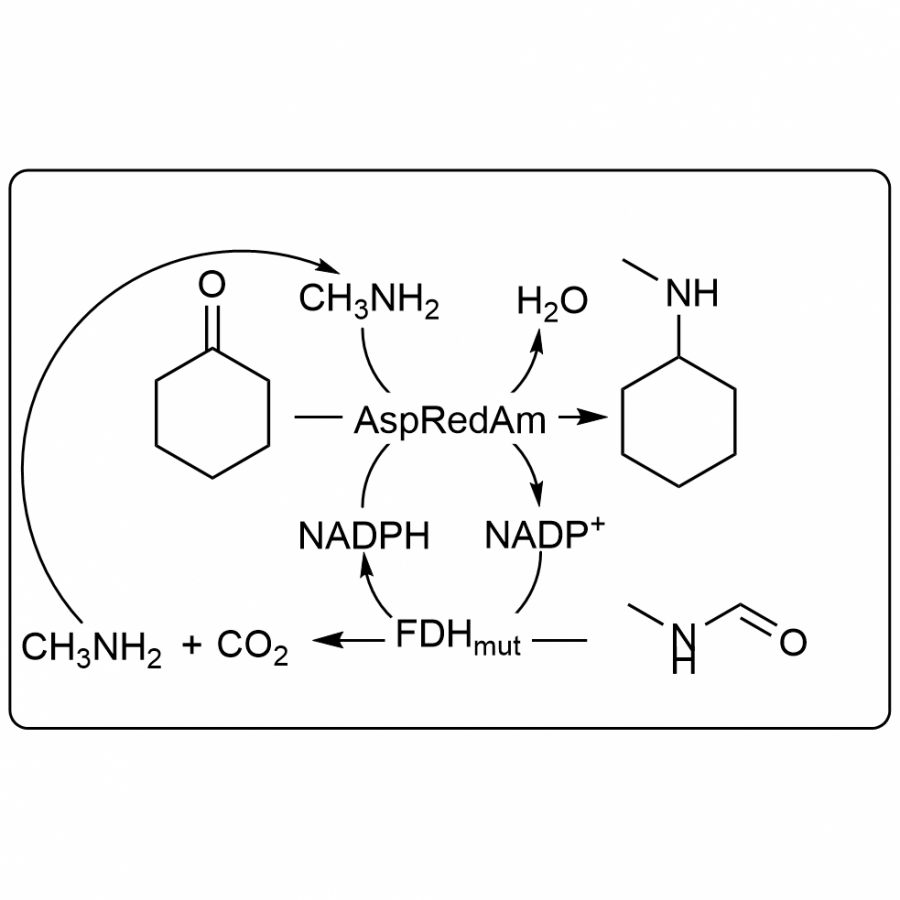 Reductive amination cascade Scheme of the reductive amination of cyclohexanone catalyzed by AspRedAm coupled with a FDH mutant utilizing N-methylformamide for NADPH regeneration. N-methylformamide serves as the sole electrone and methylamine donor. |
#732 | Repurposing EREDs for Regioselective Radical Cross-Coupling |
|
| Presenting author: | Mark PETCHEY of ASTRAZENECA |
| Other authors: | Yuxuan YE of CORNELL UNIVERSITY Victor SPELLING of ASTRAZENECA Magnus JOHANSSON of ASTRAZENECA Martin HAYES of ASTRAZENECA Todd HYSTER of CORNELL UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Radical C-C bond formation / Enzyme engineering / FMN dependent EREDs / New-to-nature chemistry |
| Purpose: | Single-electron radical cross coupling reactions display unique reactivity, expediting the requirement for protecting group chemistry, functional group interconversions and unproductive redox cycling.(1) However, using traditional small-molecule catalysts, regio- and stereoselective radical C-C bond formation is challenging due to the inherent short-lived nature of highly reactive radical species.(2,3) Nature has evolved biocatalysts, such as radical S-adenosyl (SAM) enzymes and cobalamin, to catalyze these transformations in the synthesis of structurally complex natural products. However, the high specificity of these enzymes often militates against their application for small molecule synthesis, due to their limited substrate scope. The introduction of abiological transformations to natural proteins can serve as a powerful tool to advance enzymatic catalysis to reaction landscapes unexplored by natural evolution. Pioneering work by the Hyster group showed that flavoenzymes could be reprogrammed for non-natural single electron transfer mechanisms, coupling α-halo ketones and amides with styrenyl derivatves. In these examples the chiral centre is set via asymmetric hydrogen atom transfer (HAT) from the flavin.(4, 5) By investigating a diverse range of radical precursors and coupling partners, we have discovered that FMN-dependent ‘ene’-reductases (EREDs) are also able to catalyse regioselective radical cross-coupling additions. Rational enzyme engineering was used to probe the enantio- and regioselectivity, revealing how radical termination is intricately controlled by hydride transfer from the flavin cofactor. |
| References: | (1) Yan, M; Lo, J. C.; Edwards, J. T.; Baran, P. S. J. Am Chem.l Soc. 2016, 138, 12692-12714. (2) Studer, A.; Curran, D. P. Angew. Chem., Int. Ed. 2016, 55, 58-102. (3) Beckwith, A. L. J. Chem. Soc. Rev. 1993, 22, 143- 151. (4) Biegasiewicz, K. F.; Cooper, S. J.; Gao, X.; Oblinsky, D. G.; Kim, J. H.; Garfinkle, S. E.; Joyce, L. A.; Sandoval, B. A.; Scholes, G. D.; Hyster, T. K. Science 2019, 364, 1166- 1169 (5) Fu, H.; Lam, H.; Emmanuel, M. A.; Kim, J. H.; Sandoval, B. A.; Hyster, T. K. J. Am. Chem. Soc. 2021, 143, 9622- 9629. |
#742 | Engineering Cyclodextrin Glycosyltransferase to favour higher Degree of polymerization products |
|
| Presenting author: | Matthew TREADELL of UNIVERSITY OF MANCHESTER |
| Other authors: | Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Directed evolution / Glycoscience / Carbohydrate active enzymes / |
| Purpose: | Cyclodextrin Glycosyltransferase (CGTase) is a member of GH13 family of enzyme used in the production of Cyclodextrin (CD). More recently CGTase is being used to for its ability to glycosylate substrates using CD as a donor molecule in a reaction called coupling. Coupling transfers the amount of glucose units found in a CD to the acceptor, however due to the multiple activities of CGTase a product distribution that favours low degree of polymerization (DP) products than the coupling products is attained. In this study, a mutant of Paenibacillus macerans CGTase was engineered in the acceptor binding region to see the effect on product distribution. It was found that the mutation resulted in a relatively greater production of higher DP products. The results suggest that it is possible to engineer the CGTase enzyme to favour higher DP products, and presents a first step towards efficient enzymatic synthesis of higher DP products. |
| References: | [1 ] J. Zhou, Z. Feng, S. Liu, F. Wei, Y. Shi, L. Zhao, W. Huang, Y. Zhou, H. Feng and H. Zhu, Mol. Plant Pathol., 2021, 22, 130-144. [2] M. Y. Rather, K. Z. G. Ara, E. N. Karlsson and P. Adlercreutz, Process Biochem., 2015, 50, 722-728. [3] R. K. Gudiminchi, A. Towns, S. Varalwar and B. Nidetzky, ACS Catal., 2016, 6, 1606-1615. [4] H. Hamada, K. Nishida, T. Furuya, K. Ishihara and N. Nakajima, J. Mol. Catal. B Enzym., 2001, 16, 115-119. |
#746 | Elucidation and optimisation of the glycosyl-polymerisation activity of the RlpE variovoracin enzyme for synthesis of novel sugar-based biosurfactants |
|
| Presenting author: | David EVANS of UNIVERSITY OF MANCHESTER |
| Other authors: | Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Glycosyltransferase / Biosurfactant / / |
| Purpose: | Surfactants are required in formulations of many products, enabling oil and aqueous mediums to mix and be stable. There has been increased interest in biosurfactants made by microorganisms, as traditional surfactants are derived from petrochemical sources. One such biosurfactant, variovoracin, is produced by the bacteria V. paradoxus and is classed as a glycolipopeptide. The structure consists of a hydrophobic lipid oligomer covalently linked to a peptide chain, a carbohydrate moiety, and a serine- leucinol dipeptide link, which possesses excellent surfactant properties. Within the biosynthetic pathway the gene RlpE, coding for a rhamnosyltransferase, is responsible for adding a second rhamnose moiety to the variovoracin structure, however, structures with 3 and 4 additional rhamnose moieties have been detected in trace amounts which improves hydrophilicity. The aim of this project is to optimise reaction conditions and characterise the enzyme. With the end goal being to generate a panel of RlpE mutants that access these longer chain biosurfactants for industrial application. |
| References: | EP. Pat., EP3440179B1, 2020. |
#747 | Multi-enzymatic cascade for Sustainable Biosynthesis of Tryptamine and Tryptamine Analogues and Future Scalability Towards a Closed-Loop Semi-Automated Process |
|
| Presenting author: | Lucia ROBUSTINI of UNIVERSITY OF BERN |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | flow-biocatalyses / tryptamine / sustainability / enzyme immobilisation |
| Purpose: | Tryptamine is a biogenic amine that is formed from the catabolism of tryptophan. Despite its well-known hallucinogenic properties, recent research has demonstrated potential applications of tryptamine and its analogues, 5-methoxytryptamine and 5-hydroxytryptamine, in the treatment of depression and Post Traumatic Stress Disorder (PTSD). [1] Moreover, these analogues serve as precursors for two other widely used neurotransmitters, 5-hydroxytryptamine, commonly known as Serotonin, and N-acetyl-5-methoxytryptamine, better known as melatonin, which are utilized as food supplements and nutraceuticals.[2] This study focuses on the continuous production of acetylated tryptamine products at a scale of 50 mM using cost-effective starting materials such as Serine and Indole or its 5-substituted analogues. The initial building blocks are modified through a three-step cascade reaction involving the enzymes tryptophanase (EcTnaA)[3], decarboxylase (RgTDC) [4] and acyltransferase (MsAcT) [5], with yields up to a 100% demonstrating the feasibility of this approach for the production of acetylated tryptamine compounds. |
| References: | [1] Elsouri, et al , Cureus 2022, 14 (5), e25235. [2] Arnao B.; Hernández-Ruiz, Josefa, Molecules 2018, 23 (1) [3] Ku, Shao Yang; et al, Acta crystallographica. Section D, Biological crystallography 62 (Pt 7), 2006, pp. 814-823 [4] Williams, B et al. ,Cell host & microbe, 2014, 16 (4), pp. 495-503. [5] Contente, M. L ; Pinto, A. ; Molinari, F; Paradisi, F., Adv. Synth. Catal. 2018, 360 (24), pp. 4814-4819. |
| Figures: | 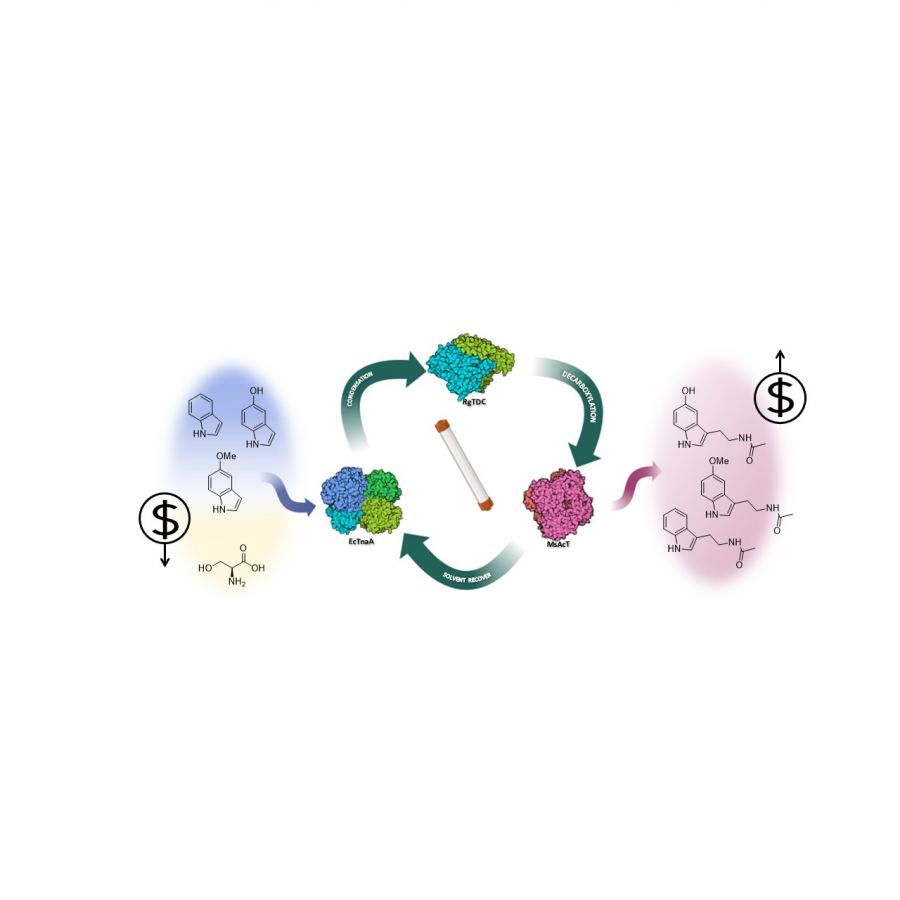 three steps closed-loop process for the synthesis of tryptamine analogues The biosynthesis of tryptamine analogues from serine and indole analogues goes through 3 enzymatic steps: an E. coli tryptophanase (EctnaA) condenses the first two building blocks. The obtained tryptophan (or its derivatives) is the substrate for a decarb |
#748 | Novel artificial metalloenzymes for olefin metathesis based on modified Grubbs-Hoveyda complexes |
|
| Presenting author: | Aaron A. INGRAM of INSTITUTE OF INORGANIC CHEMISTRY, RWTH AACHEN UNIVERSITY |
| Corresponding author: | Jun OKUDA of INSTITUTE OF INORGANIC CHEMISTRY, RWTH AACHEN UNIVERSITYRWTH AACHEN UNIVERSITY |
| Other authors: | Ulrich SCHWANEBERG of INSTITUTE OF BIOTECHNOLOGY, RWTH AACHEN UNIVERSITY |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzymes / olefin metathesis / β-barrel protein / |
| Purpose: | Artificial metalloenzymes represent an attractive approach for the design of novel biocatalysts by combining homogeneous catalysis and enzyme catalysis.[1,2] A successful example for artificial metalloenzymes are ruthenium-based Grubbs-Hoveyda complexes incorporated into protein scaffolds which catalyse olefin metathesis, a powerful C-C bond formation reaction with numerous applications in organic chemistry which no natural enzyme is reported to catalyse. In previous studies, we reported the covalent conjugation of Grubbs-Hoveyda type complexes with maleimide functionalised NHC backbone to the β-barrel protein nitrobindin from Arabidopsis thaliana via thiol-ene reaction with a free cysteine residue. The engineered cavity of the protein provides a defined hydrophobic second coordination sphere around the central ruthenium atom, which improves the turnover number of the catalyst compared to the free metal complex.[3,4] This approach also enables cascade reactions with an artificial hydrogenase[5] and a fatty acid decarboxylase[6] which do not proceed with the free metal complex due to inactivation issues. Artificial metalloenzymes often suffer from comparably low activities and total turnover numbers in comparison to natural enzymes. Consequently, there is a high interest in strategoes for their optimisation. On one hand engineering of these artificial metalloenzymes by directed evolution of the protein scaffold gained increased attention.[7] On the other hand, a deep understanding about the behaviour of the metal catalyst in aqueous solutions, which is often different compared to dry organic solvents, is crucial for the effective design of these de novo biocatalysts as well. In this work, we present novel artificial metalloenzymes for olefin metathesis based on Grubbs Hoveyda complexes with substituted halide ligands cojugated to nitrobindin variants which we characterised by ESI-MS, CD-spectroscopy and UV-vis spectroscopy. We studied the effect of common parameters in aqueous biotransformations such as buffer, pH-value, salt additives and organic co-solvents on the catalyst activity and stability. In order to elucidate the effect of the substituted halide ligands in interaction with the protein scaffold, we compared the catalytic performance to free catalysts with the same substitution pattern. |
| References: | [1] B. J. Bloomer, D. S. Clark, J. F. Hartwig, Biochemistry, 2023, 62, 221-228. [2] F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis, T. R. Ward, Chem. Rev. 2018, 118, 142-231. [3] D. F. Sauer, T. Himiyama, K. Tachikawa, K. Fukumoto, A. Onoda, E. Mizohata, T. Inoue, M. Bocola, U. Schwaneberg, T. Hayashi, J. Okuda, ACS Catal. 2015, 5, 7519-7522. [4] A. R. Grimm, D. F. Sauer, M. D. Davari, L. Zhu, M. Bocola, S. Kato, A. Onoda, T. Hayashi, J. Okuda, U. Schwaneberg, ACS Catal. 2018, 8, 3358-3364. [5] D. F. Sauer, Y. Qu, M. A. S. Mertens, J. Schiffels, T. Polen, U. Schwaneberg, J. Okuda, Catal. Sci. Technol. 2019, 9, 942-946. [6] M. A. S. Mertens, D. F. Sauer, U. Markel, J. Schiffels, J. Okuda, U. Schwaneberg, Catal. Sci. Technol. 2019, 9, 5572-5576. [7] M. Jeschek, R. Reuter, T. Heinisch, C. Trindler, J. Klehr, S. Panke, T. R. Ward, Nature 2016, 537, 19114. |
| Figures: | 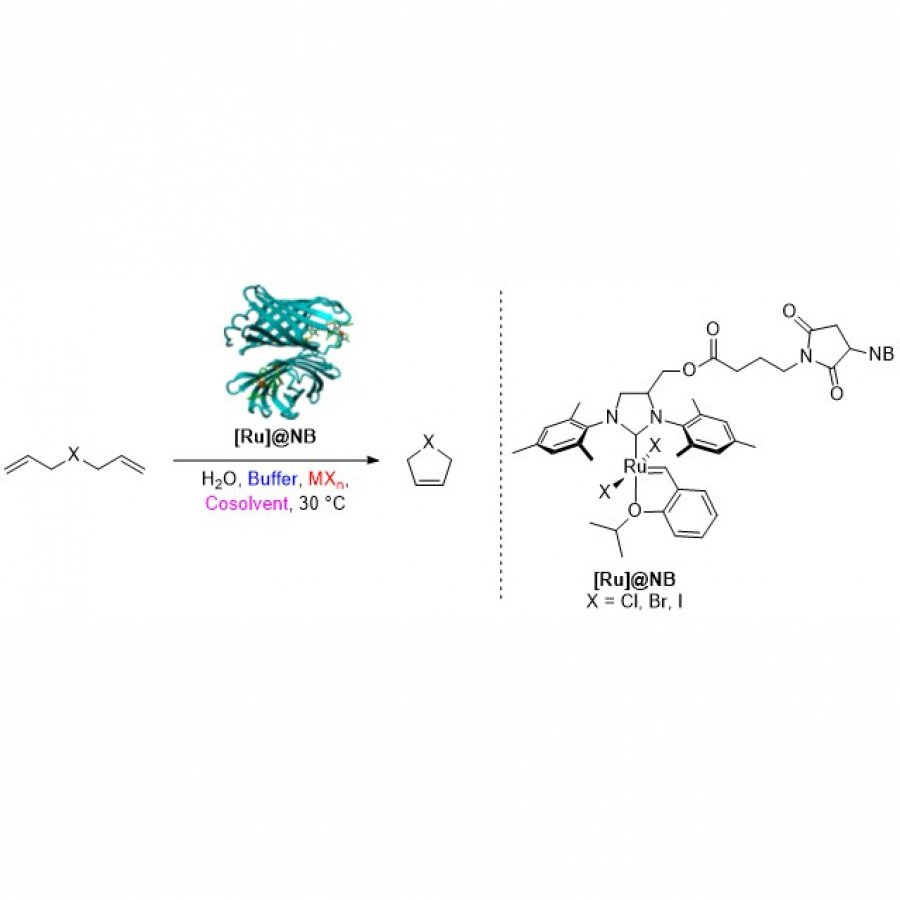 Figure 1 Ring-closing metathesis with artificial metalloenzymes based on halide-substituted Grubbs-Hoveyda catalysts. |
#757 | The road to large-scale production of enantiopure epoxides: kinetic resolution with the glutathione S-transferase StyI |
|
| Presenting author: | Melody HAARMANN of RUHR UNIVERSITY BOCHUM |
| Other authors: | Daniel EGGERICHS of RUHR UNIVERSITY BOCHUM Artur MAIER of RUHR UNIVERSITY BOCHUM Dirk TISCHLER of RUHR UNIVERSITY BOCHUM |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Kinetic resolution / Enantiopure epoxides / Glutathione s-transferase / Upscaling |
| Purpose: | Epoxides are important platform chemicals but are also a common motive in nature. Especially chiral epoxides are of high interest in industry as they have a wide range of application like in drug or agrochemical synthesis [1]. One of the major obstacles in modern organic chemistry is to achieve a high yield production while maintaining both: enantio- and regioselectivities. Enzymatic epoxidation allows for both characteristics but is limited by the selectivity of the respective enzymes and thereby not universal applicable. Styrene monooxygenases (SMOs) for example, produce the (S)-epoxide with over 99% ee but no easy access to the (R)-epoxide was identified so far. Thus, kinetic resolution emerges as alternative solution. A few enzymes were described to convert epoxides with a certain selectivity like epoxide hydrolases (EHs), glutathione S-transferases (GSTs), CoM-transferases (CoMTs) or haloalcohol dehalogenases (HADHs) [2]. So far, a certain selectivity has only been reported for GSTs which brought these enzymes to our focus [2]. Until now, investigations on these enzymes have only been made for isoenzymes from rat liver and a restricted substrate amount. The overall function of GSTs in epoxide metabolism and their potential as enantioselective catalyst remain ambiguous [1]. Recently, a bacterial glutathione S-transferase (StyI) was found in the styrene degradation pathway of the actinobacterium Gordonia rubripertincta CWB2 [3]. Unlike other styrene degraders, strain CWB2 demonstrated its ability to metabolize a variety of styrene-related compounds, which was exploited biotechnologically to produce ibuprofen in a co-metabolic process [4]. The unusual presence of GSTs within bacterial styrene degradation appears to broaden the substrate spectrum of this pathway. We therefore produced StyI heterologously in E. coli for subsequent characterization of the substrate selectivity and enantiomeric preference. Our studies revealed a three times higher activity with (S)-styrene oxide compared to its (R)-enantiomer (Figure 1). This was also supported by a 16-times higher affinity and a 1.8 fold higher vmax for (S)-styrene oxide, giving a clear evidence of StyI's stereoselectivity. This observation is in agreement with the proposed natural role of the enzyme as StyI is the subsequent enzyme of the (S)-enantioselective styrene monooxygenase [5]. To investigate the potential of the GST StyI for enantioselective epoxide opening, chiral GC-FID analysis was performed. More than 10 substrates comprising different structural motives (aromatic and aliphatic) were tested within the substrate screening. For styrene oxide derivatives, like para-fluoro- or para-chlorostyrene oxide, also a high selectivity for the (S)-enantiomer was found. In contrast, the enantiomeric preference of more diverse structures such as phenylpropylene oxide, indene oxide, and aliphatic epoxides revealed a reduction in enantioselectivity. Despite this, the overall substrate scope for StyI was remarkably large. Since StyI showed high enantioselectivity towards (S)-styrene oxide and therefore indicating its promising application in production of enantiopure (R)-epoxide, enzymatic kinetic resolution was performed. Subsequently, the reaction volume was increased to 150 mL scale to proof the feasibility of large-scale kinetic resolution. The approach yielded about 18 mg pure (R)-styrene oxide which was validated by NMR spectroscopy. In conclusion, we found the glutathione S-transferase StyI to be highly enantioselective for the native substrate (S)-styrene oxide and structural analogous. Further, a certain selectivity was observed for non-aromatic substrates which highlights the broad application range for this enzyme. This led to the subsequent upscaling of the process enriching pure (R)-styrene oxide. Hence, this study reveals StyI’s potential within the road to large-scale production of enantiopure epoxides like styrene oxide. |
| References: | [1] Moschona et al. (2020). Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts. 2020; 10(10):1117. [2] de Vries & Janssen (2003). Biocatalytic conversion of epoxides. Current opinion in biotechnology, 14(4), 414-420. [3] Heine et al. (2018). On the Enigma of Glutathione-Dependent Styrene Degradation in Gordonia rubripertincta CWB2. Applied and environmental microbiology, 84(9), e00154-18. [4] Oelschlägel et al. (2015). Co-metabolic formation of substituted phenylacetic acids by styrene-degrading bacteria. Biotechnology reports (Amsterdam, Netherlands), 6, 20-26. [5] Heine et al. (2020). Accessing Enantiopure Epoxides and Sulfoxides: Related Flavin-Dependent Monooxygenases Provide Reversed Enantioselectivity. ChemCatChem 12, 199-209. |
| Figures: | 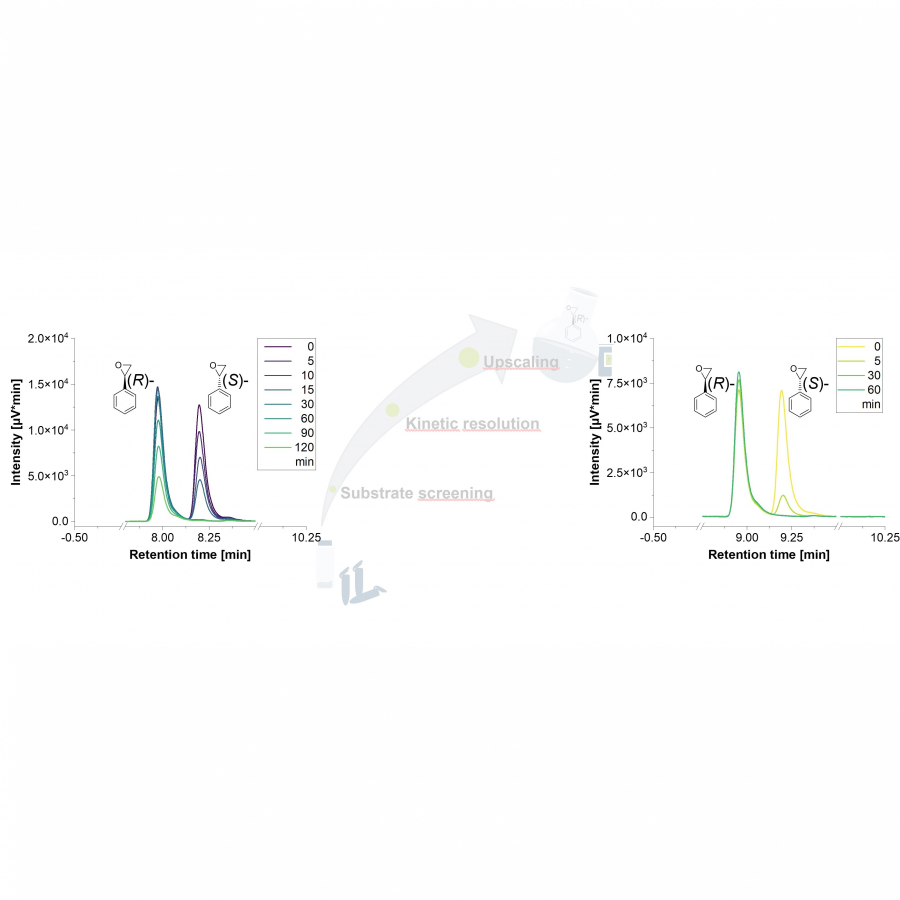 Workflow from enantioselective substrate consumption to the upscaling of kinetic resolution with StyI. Left: Conversion of the chiral substrate styrene oxide by StyI determined by GC-FID. StyI shows a strong enantiomeric preference for (S)-styrene oxide, whereas the (R)-epoxide is consumed afterwards. Right: Kinetic resolution of racemic styrene oxide. |
#760 | SAM regeneration goes radical |
|
| Presenting author: | Lukas GERICKE of UNIVERSITY OF FREIBURG |
| Corresponding author: | Jennifer N. ANDEXER of UNIVERSITY OF FREIBURG |
| Other authors: | Dipali MHAINDARKAR of UNIVERSITY OF FREIBURG Lukas C. KARST of UNIVERSITY OF FREIBURG Sören JAHN of UNIVERSITY OF FREIBURG Marco KUGE of UNIVERSITY OF FREIBURG Jana GAGSTEIGER of UNIVERSITY OF FREIBURG Michael K. F. MOHR of UNIVERSITY OF FREIBURG Silja MORDHORST of UNIVERSITY OF TÜBINGEN Florian P. SEEBECK of UNIVERSITY OF BASEL Gunhild LAYER of UNIVERSITY OF FREIBRUG Christoph LOENARZ of UNIVERSITY OF FREIBURG |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alkylation / Cofactor regeneration / SAM Analogues / Radical SAM Enzyme |
| Purpose: | S-adenoslymethionine (SAM) is a highly versatile cofactor used by methyltransferases, amino(carboxy)propyltransferases and radical SAM enzymes. Hence, it is involved in a wide range of reactions with polar or radical mechanisms.[1] However, the instability and high cost of SAM and byproduct inhibition impedes the usage of SAM-dependent enzymes.[2] Therefore, in situ SAM regeneration systems have been developed. These systems are highly effective and are able to support up to 500 turnovers with catalytic amounts of cofactor.[3–5] Nevertheless, in all systems SAM regeneration relies on S-adenosylhomocysteine (SAH), the byproduct of methyltransferase reactions. Amino(carboxy)propyltransferases and radical SAM enzymes are not supported as they produce 5’-methylthioadenosine (MTA) and 5’-deoxyadenosine as byproducts, respectively. The goal of the work presented was to develop a versatile SAM regeneration system that supports the vast catalytic spectrum of methyltransferases, amino(carboxy)propyltransferases and radical SAM enzymes. The system relies on byproduct cleavage by MTA/SAH nucleosidase and a polyphosphate-fuelled biomimetic production of phopshoribose pyrophosphate. We could show that methyltransferases, aminopropyltransferases and radical SAM enzymes are functional in this system, supporting up to 80, 57 and 29 turnovers, respectively. Additionally, the system’s flexibility was underlined by the transfer of an ethyl group using a cobalamin-dependent radical SAM methyltransferase. This novel reaction further demonstrates the potential of SAM analogue supply and regeneration for the application of diverse chemistry.[6] |
| References: | [1] M. Fontecave, M. Atta, E. Mulliez, Trends in Biochemical Sciences 2004, 29, 243-249. [2] S. Mordhorst, J. N. Andexer, Nat. Prod. Rep. 2020, 37, 1316-1333. [3] S. Mordhorst, J. Siegrist, M. Müller, M. Richter, J. N. Andexer, Angew. Chem. Int. Ed. 2017, 56, 4037-4041. [4] C. Liao, F. P. Seebeck, Nature Catalysis 2019, 2, 696-701. [5] D. Popadić, D. Mhaindarkar, M. H. N. Dang Thai, H. C. Hailes, S. Mordhorst, J. N. Andexer, RSC Chem. Biol. 2021, 10.1039.D1CB00033K. [6] L. Gericke, D. Mhaindarkar, L. C. Karst, S. Jahn, M. Kuge, M. K. F. Mohr, J. Gagsteiger, N. V. Cornelissen, X. Wen, S. Mordhorst, H. J. Jessen, A. Rentmeister, F. P. Seebeck, G. Layer, C. Loenarz, J. N. Andexer, ChemBioChem 2023, e202300133. |
#763 | New Horizons in Biocatalysis – Exploring the potential of Unspecific Peroxygenases and Iron- and α-Ketoglutarate-dependent Oxygenases |
|
| Presenting author: | Hannah BRASS of AMINOVERSE |
| Other authors: | David SCHÖNAUER of AMINOVERSE |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | The application of biocatalysis in chemical synthesis enables new synthesis routes and offers, amongst others, the great advantage of highest selectivity.[1][2][3] With the selective activation of inert or poorly activated C-H bonds being one of the most important and at the same time most difficult reactions in organic chemistry, harnessing the power of biocatalysis in synthetic chemistry became an essential approach. Out of the group of oxidoreductases, P450 monooxygenases have been extensively studied over the years e.g., in terms of oxyfunctionalization reactions of C-H bonds. To this day, their use comes with several disadvantages, including reliance on expensive cofactor NAD(P)H and loss of it due to uncoupling reactions.[4][5] Since their discovery in 2004, unspecific peroxygenases (UPOs) have increasingly come into focus and have been the subject of intensive research, as they stand out as a very promising alternative to P450 monoxygenases.[6] UPOs catalyze a wide range of reactions such as hydroxylations, epoxidations, alcohol oxidation, hetereoatom oxygenation, and dealkylations. Unlike other enzymes, UPOs require only hydrogen peroxide, serving as both electron acceptor and oxygen donor, and do not rely on complex electron transport chains or expensive cofactors, making them easy to apply and attractive for scaling up. UPOs convert a variety of substrates with different regio- and enantioselectivities, making them ideal biocatalysts for any oxyfunctionalization reaction.[5][7][8] The latest results obtained with the world's largest collection of commercially available UPOs will be shared. Another enzyme class capable of oxyfunctionalization are Iron- and α-ketoglutarate dependent oxygenases (KGOs), which are mainly known for their ability to hydroxylate e.g. amino acids or natural products under the expense of α-ketoglutarate and oxygen.[9][10][11] Despite their great potential for application in biocatalysis and chemical synthesis, the power of KGOs has not yet been fully exploited and commercial availability is still limited. To open up new biocatalytic routes and pave the way to simplified and sustainable chemical synthesis of APIs and intermediates, Aminoverse B.V. offers the world's largest collection of UPOs and KGOs (coming soon) for oxyfunctionalisation reactions. |
| References: | [1] S. Wu, R. Snajdrova, J. C. Moore, K. Baldenius, U. T. Bornscheuer, Angew. Chemie - Int. Ed. 2021, 60, 88-119. [2] E. L. Bell, W. Finnigan, S. P. France, A. P. Green, M. A. Hayes, L. J. Hepworth, S. L. Lovelock, H. Niikura, S. Osuna, E. Romero, K. S. Ryan, N. J. Turner, S. L. Flitsch, Nat. Rev. 2021, 1, 1-21. [3] S. P. France, R. D. Lewis, C. A. Martinez, JACS Au 2022, DOI 10.1021/jacsau.2c00712. [4] D. Holtmann, F. Hollmann, ChemBioChem 2016, 17, 1391-1398. [5] A. Beltran-Nogal, I. Sanchez-Moreno, D. Mendez-Sanchez, P. Gomez de Santos, F. Hollmann, M. Alcalde, Curr. Opin. Struct. Biol. 2022, 73, 102342. [6] R. Ullrich, J. Nueske, K. Scheibner, J. Spantzel, M. Hofrichter, Appl. Environ. Microbiol. 2004, 70, 4575-4581. [7] B. Pogranyi, T. Mielke, A. Diaz-Rodriguez, J. Cartwright, W. P. Unsworth, G. Grogan, Angew. Chemie - Int. Ed. 2022, DOI 10.1002/anie.202214759. [8] D. T. Monterrey, A. Menes-Rubio, M. Keser, D. Gonzalez-Perez, M. Alcalde, Curr. Opin. Green Sustain. Chem. 2023, https://doi.org/10.1016/j.cogsc.2023.100786. [9] S. N. Charlton, M. A. Hayes, ChemMedChem 2022, 17, DOI 10.1002/cmdc.202200115. [10] C. R. Zwick, H. Renata, Nat. Prod. Rep. 2020, 37, 1065-1079. [11] C. Peters, R. M. Buller, Catalysts 2019, 9, DOI 10.3390/catal9030221. |
#768 | Engineered enzymes for the synthesis of key intermediates in valuable drug targets |
|
| Presenting author: | Grayson FORD of UNIVERSITY OF MANCHESTER |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Protein Engineering / Chemoenzymatic Synthesis / Cytidine Deaminase |
| Purpose: | The Covid-19 pandemic has highlighted the need for cost-effective and environmentally friendly drug manufacturing processes. Biocatalysis can provide low-cost routes to access important chiral drug intermediates and reduce synthetic steps. Enzyme candidates can be quickly screened and engineered for the target molecule using biocatalytic retrosynthetic analysis and database searching. Our group has developed concise biocatalytic processes towards Covid-19 and HIV antiviral intermediates and targets. |
| References: | [1] Ashleigh J. Burke, William R. Birmingham, Ying Zhuo, Thomas W. Thorpe, Bruna Zucoloto da Costa, Rebecca Crawshaw, Ian Rowles, James D. Finnigan, Carl Young, Gregory M. Holgate, Mark P. Muldowney, Simon J. Charnock, Sarah L. Lovelock, Nicholas J. Turner, and Anthony P. Green Journal of the American Chemical Society 2022 144 (9), 3761-3765. |
| Figures: | 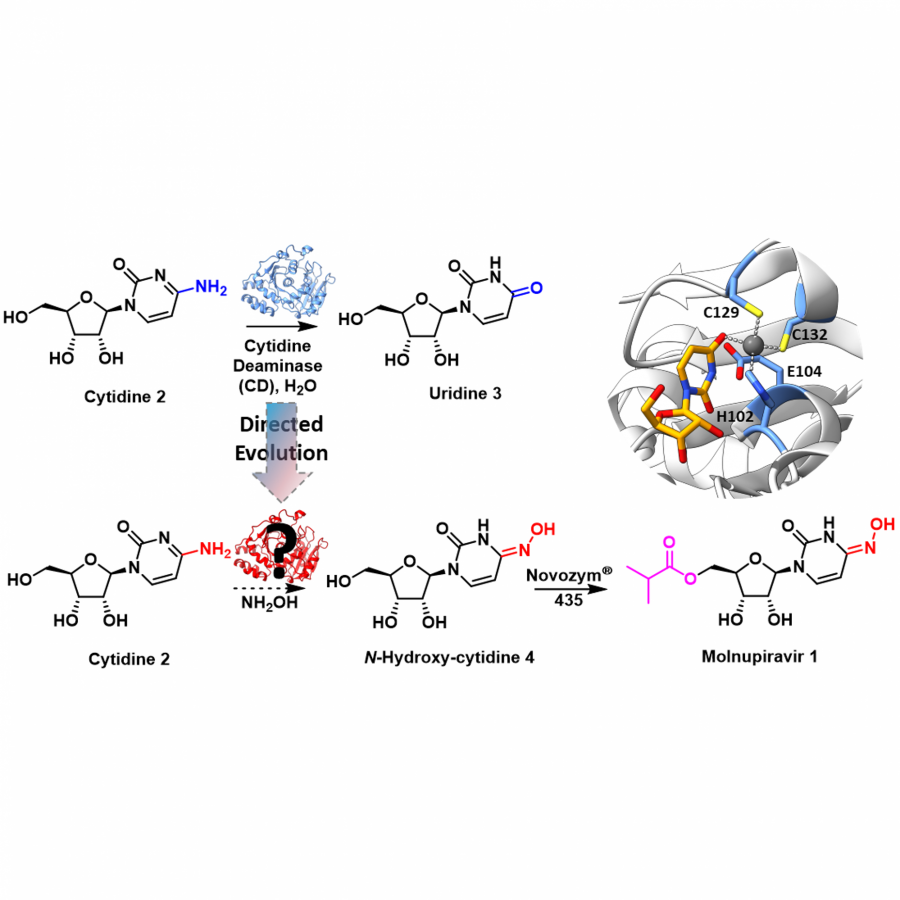 An Engineered Cytidine Deaminase for Biocatalytic Production of a Key Intermediate of the Covid-19 Antiviral Molnupiravir Development of an efficient biocatalyst for the production of N-hydroxy-cytidine through evolutionary adaption of the hydrolytic enzyme cytidine deaminase. This engineered biocatalyst performs >85 000 turnovers in less than 3 h.
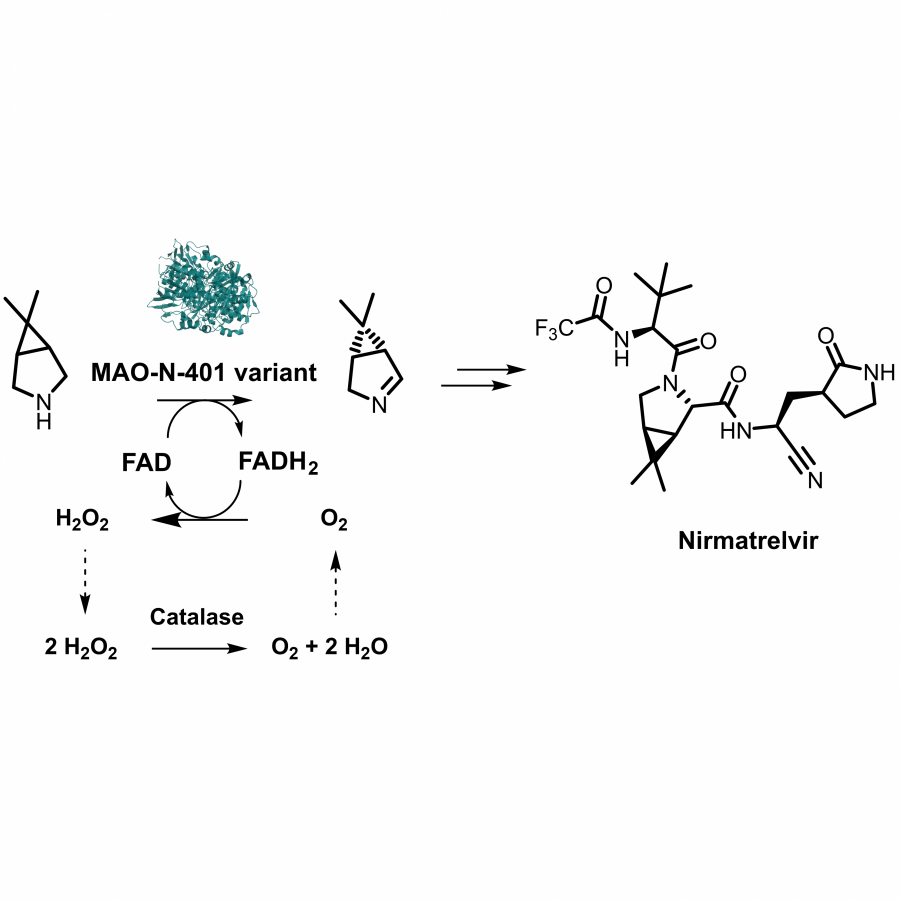 Chemoenzymatic Multicomponent synthesis of Nirmatrelvir Development of a synthetic route to Nirmatrelvir, featuring a highly enantio selective biocatalytic de-symmetrization and diastereoselective multicomponent reaction as key steps. The route avoids use of transitions metals and peptide coupling reagents and |
#769 | Nature goes natural: A novel chemo-enzymatic one-pot cascade for the synthesis of fragrance & flavor aldehydes |
|
| Presenting author: | Stefan GIPARAKIS of TU WIEN |
| Corresponding author: | Florian RUDROFF of TU WIEN |
| Other authors: | Margit WINKLER of TU GRAZ |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | The effective utilization of naturally occurring compounds is of the most importance when the sustainability of the chemical industry needs to be increased. However, due to the multitude of different compound classes with varying usability for chemical applications combined with difficult and inefficient transformation strategies, most natural resources lie fallow for the chemical industry. To tackle this problem a proof of concept for two novel methods was developed that allows the conversion of naturally occurring phenylpropene derivatives (e.g., eugenol and safrole, Figure 1) to valuable aromatic aldehydes (e.g., vanillin and piperonal). Both methods utilize a common approach where a chemical transformation is coupled to whole-cell biocatalysis in a one-pot fashion (Figure 2). The first method (Strategy A) - the “shortcut” approach - involves a palladium-catalyzed isomerization of the terminal olefinic bond of a phenylpropene derivative followed by an enzymatic oxidative cleavage performed by a coenzyme-free aromatic dioxygenase (ADO)[1]. On the other hand, the second strategy (Strategy B), couples a Wacker-oxidation[2] to a three-step enzymatic cascade involving the combination of a Baeyer-Villiger monooxygenase (TmCHMO or PAMO)[3], an esterase (pfe I)[4], and an alcohol dehydrogenase (AlkJ)[5] To demonstrate the scope of these transformations eight phenylpropene derivatives were chosen. Strategy A allowed for the synthesis of one of the desired aldehydes (vanillin) due to limitations in the substrate profile of ADO. Strategy B on the other hand allowed for the transformation of all of the tested phenylpropene derivatives resulting in the formation of seven of the desired aldehydes with yields of up to 42 % after 4 reaction steps (80% for each step). |
| References: | [1] Ni, J.; Wu, Y.-T.; Tao, F.; Peng, Y.; Xu, P., J. Am. Chem. Soc. 2018, 140 (47), 16001-16005. [2] González-Martínez, D.; Gotor, V.; Gotor-Fernández, V., Adv. Synth. Catal. 2019, 361 (11), 2582-2593. [3] Rodríguez, C.; de Gonzalo, G.; Gotor, V., J. Mol. Catal. B: Enzym. 2012, 74 (1), 138-143. [4] Krebsfänger, N.; Zocher, F.; Altenbuchner, J.; Bornscheuer, U. T., Enzyme Microb. Technol. 1998, 22 (7), 641-646. [5] Kirmair, L.; Skerra, A., Appl. Environ. Microbiol. 2014, 80 (8), 2468-2477. [6] Nazrul, M.; Bhuiyan, M.; Begum, J.; Nandi, N.; Akter, F., Afr. J. Plant Sci. 2010, 4, 451-454. [7] Gupta, M. P.; Arias, T. D.; Williams, N. H.; Bos, R.; Tattje, D. H. E., J. Nat. Prod. 1985, 48 (2), 330-330. [8] Prasse, A.; Zeller, K.-P.; Siehl, H.-U.; Berger, S.; Sicker, D., Chem. unserer Zeit 2018, 52 (1), 56-64. |
| Figures: | 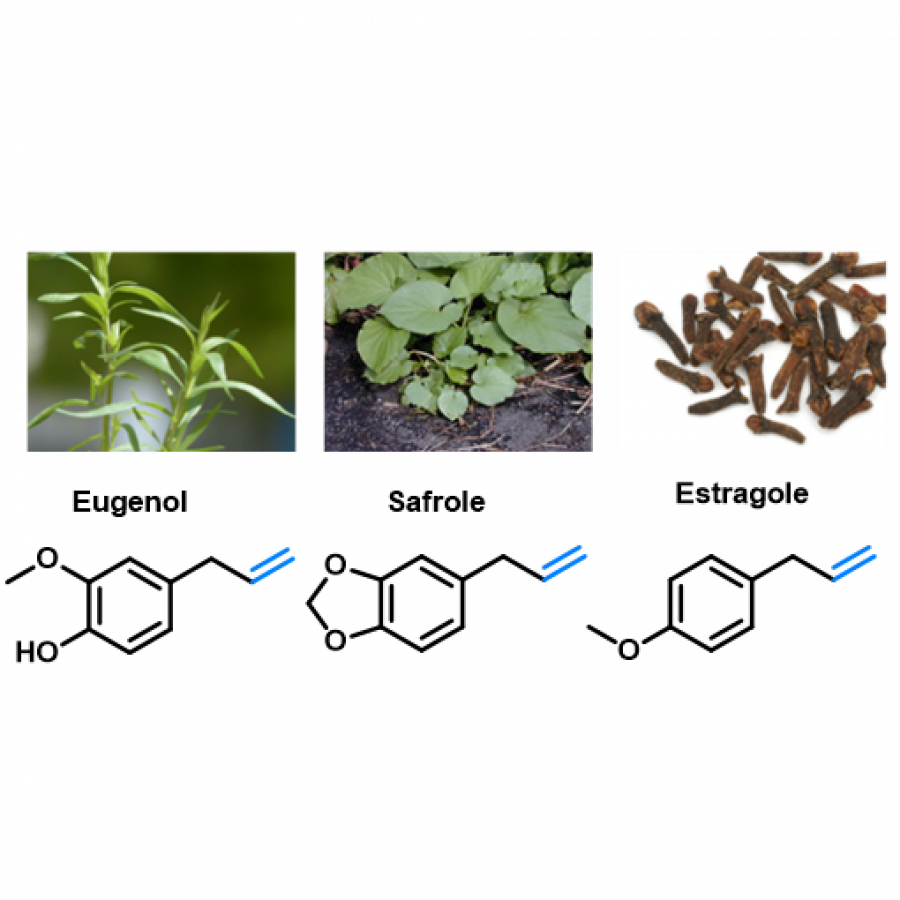 Natural occurring phenylpropenes and their natural source Eugenol from Clove[6].(Image adapted from Brian Arthur (CC BY-SA 4.0)) Safrole from Makulan[7]. (Image: Forest & Kim Starr (CC BY 3.0)) Estragole from Estragon[8]. 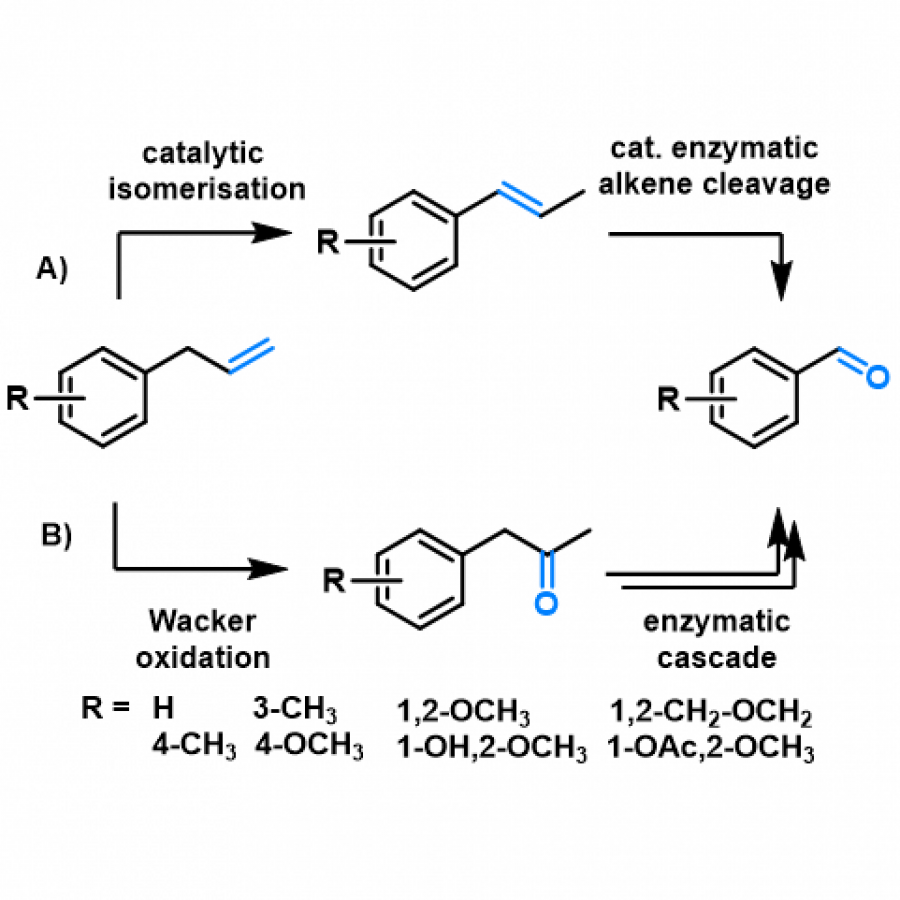 Reaction sequence overview Two different concepts, exploiting oxidation bio/chemistry for the synthesis of fragrance and flavor aldehydes. |
#770 | Enhancement of enzymatic kinetic resolution by a new chemoenzymatic cascade strategy |
|
| Presenting author: | Ryszard OSTASZEWSKI of INSTITUTE OF ORGANIC CHEMISTRY PAS |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Kinetic resolution / chemoenzymatic cascade / byproduct utilization / enzyme activation |
| Purpose: | Aldehydes are common byproducts in enzymatic kinetic resolution (EKR) and enzymatic dynamic kinetic resolution reactions. Due to their high reactivity, physical properties and explosive character they are hardly tolerated in organic laboratories especially in large scale experiments. The most volatile acetaldehyde 4 is formed as byproducts in enzymatic esterification of alcohols, amines or thiols when ethyl acetate is used as an acyl group donor. Hydrolysis of epoxyesters also led to formation of aryl and alkyl aldehyde which are very often very effective enzyme inhibitors. To overcome this common issue in EKR processes we propose a new strategy based on combination of enzyme catalyzed reaction with subsequent aldol reaction into a one-step chemoenzymatic cascade process. In order to eliminate aldehyde's presence in reaction mixture and push the equilibrium towards product formation addition of dimedone 5 into reaction mixture is crucial. This lead to cascade of chemical reaction in which aldehyde 4 the byproduct from enzyme catalyzed reaction immediately reacts with dimedone 5 to form respective aldol product 6 in almost quantitative yield. Elimination of aldehydes presence in reaction mixture makes the process environmentally friendly, and greatly eliminates the enzyme inhibition what is crucial for successful experiments. The validity of the concept on selected examples will be presented and discussed. ACKNOWLEDGMENT The project was supported by the Polish National Science Center project No. OPUS No. 2019/33/B/ST4/01118 and COST action COZYME CA21162. |
| References: | [1] R. A. Sheldon, D. Brady, M. L. Bode, Chem. Sci., 2020, 11, 2587; |
| Figures: | 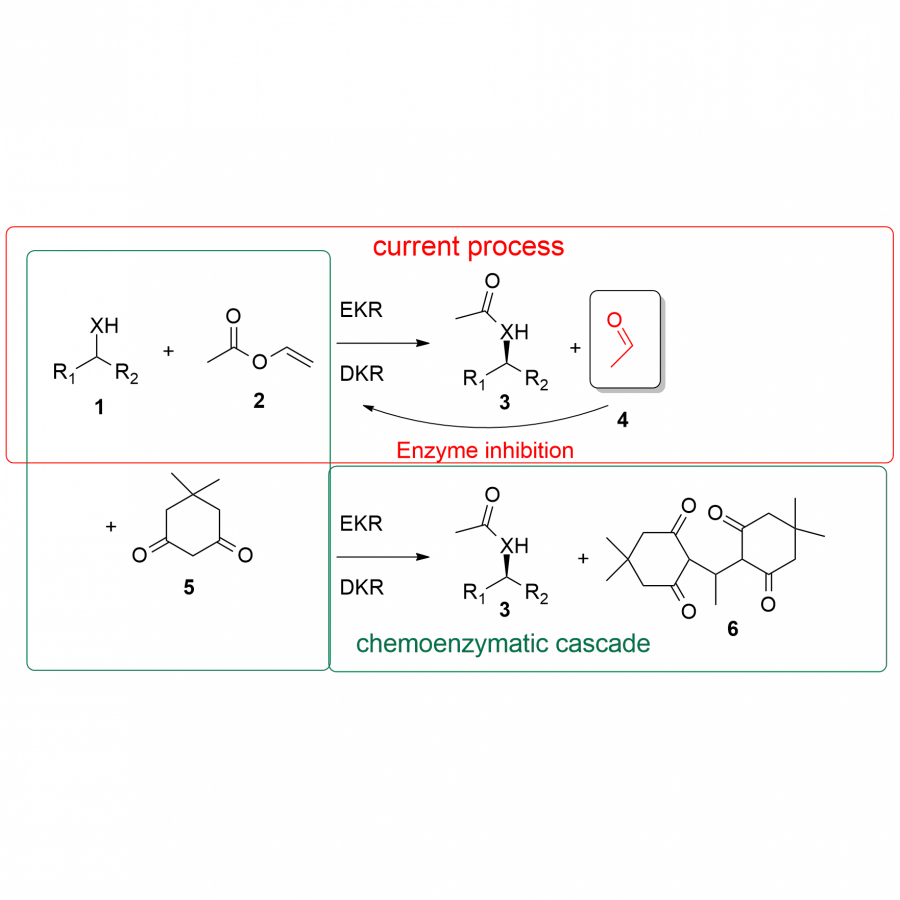 Figure 1 General synthetic concept |
#772 | Exploring the substrate spectrum of two Adenosylcobalamine dependent thermostable ribonucleotide reductases |
|
| Presenting author: | Lobna ELTOUKHY of TU DRESDEN |
| Corresponding author: | Christoph LODERER of TU DREDSDEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ribonucleotide reductases / non-canonical ribonucleotides / substrate specificity / deoxyribonucleotides |
| Purpose: | Deoxyribonucleotides are relevant molecules for medical and biotechnological applications. The synthesis of deoxyribonucleotides for biotechnological purposes requires an enzymatic cascade ending with reduction by ribonucleotide reductase enzymes. In vivo, ribonucleotide reductases are indispensable for all living cells as they provide the elementary segments for DNA biosynthesis and repair (1, 2). The project aims to investigate the substrate specificity of two Adenosylcobalamin-dependent ribonucleotide reductases to discover their potential for biotechnological applications. T..maritima NrdJ (3) and TV74 NrdJm (4) were tested for their substrate specificity and capacity to reduce three categories of ribonucleotides: natural canonical, natural non-canonical, and non-natural non-canonical. The capacity of Thermotoga maritima NrdJ for reducing nucleotides diphosphates and TV74 NrdJm for reducing nucleotides triphosphate was different and mainly dependent on the type of nucleotide and the effector. The broad substrate specificity of both enzymes implies that we can incorporate them in further biological cascades to produce various deoxyribonucleotides di-and triphosphates relevant in medicine or other scientific fields. |
| References: | 1. Lundin, D.; Berggren, G.; Logan, D. T.; SjÖberg, B.-M. The origin and evolution of ribonucleotide reduction. Life (Basel, Switzerland) [Online] 2015, 5 (1), 604-636. 2. Nordlund, P.; Peter Reichard. Ribonucleotide Reductases [Online] 2006. 3. Larsson, K.-M.; Logan, D. T.; Nordlund, P. Structural basis for adenosylcobalamin activation in AdoCbl-dependent ribonucleotide reductases. ACS chemical biology [Online] 2010, 5 (10), 933-942. 4. Loderer, C.; Holmfeldt, K.; Lundin, D. Non-host class II ribonucleotide reductase in Thermus viruses: sequence adaptation and host interaction. PeerJ [Online] 2019, 7, e6700. |
| Figures: | 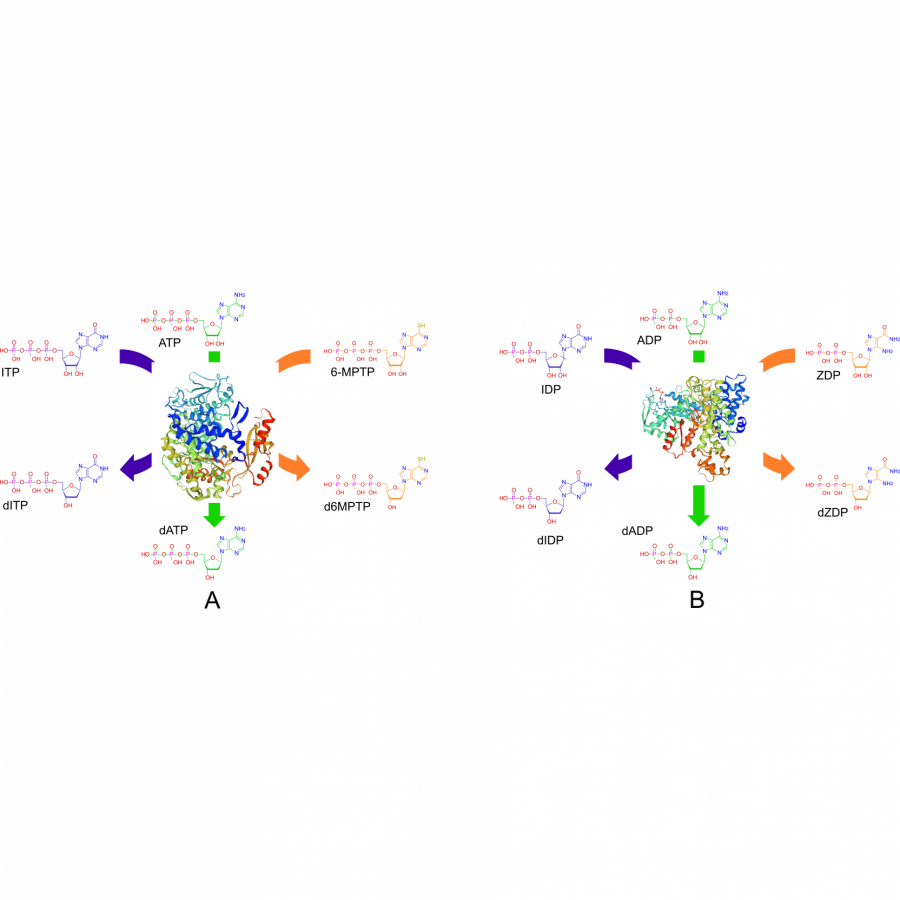 Structures of enzymes and used ribonucleotides di-and triphosphates Figure 1: A. TV74 Nrdjm, B. T.maritima Nrdj reducing the three categories of the nucleotides natural canonical(ATP, ADP), natural non-canonical (ITP/IDP), non-natural non-canonical (6-MPTP, ZDP).
|
#774 | Aromatic hydroxylation catalyzed by an unspecific peroxygenase from Aspergillus brasiliensis |
|
| Presenting author: | Fabian SCHMITZ of INSTITUTE OF BIOCHEMISTRY HEINRICH-HEINE-UNIVER |
| Other authors: | Katja KOSCHORRECK of INSTITUTE OF BIOCHEMISTRY HEINRICH-HEINE-UNIVER Frank HOLLMANN of DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY Vlada B URLACHER of INSTITUTE OF BIOCHEMISTRY HEINRICH-HEINE-UNIVER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | unspecific peroxygenase / Pichia pastoris / aromatic hydroxylation / heterologous expression |
| Purpose: | Unspecific peroxygenases (UPOs, EC 1.11.2.1) are heme-thiolate enzymes secreted by fungi [1]. These enzymes use hydrogen peroxide to form a highly reactive iron-oxo species, Compound I, to perform various oxidation reactions like hydroxylation, epoxidation, and O-dealkylation [2]. Nevertheless, despite of an increasing number of identified UPOs and more than hundreds of identified substrates, aromatic hydroxylation catalyzed by these enzymes is rarely reported while aliphatic hydroxylation reactions have been often described in literature [2, 3]. Aromatic hydroxylation of substituted benzenes gives access to versatile phenolic synthons in the synthesis of dyes, pharmaceuticals, and agrochemicals. In attempt to identify a UPO enabling aromatic hydroxylation we cloned in Pichia pastoris a number putative UPOs. Out of seven UPOs successfully expressed in P. pastoris we focused on an UPO from Aspergillus brasiliensis (AbrUPO). With expression yields of up to 0.75 g/l culture medium, this UPO fulfils a requirement for the production of valuable chemicals at a high scale. Unsimilar to most reported UPOs, AbrUPO was found to catalyze aromatic hydroxylation of substituted benzenes. The chemoselectivity of AbrUPO and its preference for aromatic or benzylic hydroxylation was found to depend on chemical properties and length of the alkyl chain. A comparative analysis of the active site of AbrUPO revealed that it is very different from the active sites of the UPOs unable of aromatic hydroxylation. These observations render AbrUPO not only an interesting biocatalyst for synthetic chemistry but also a good candidate for protein engineering studies to achieve a better insight on the molecular factors governing the chemoselectivity of heme-thiolate enzymes. |
| References: | [1] R, Ullrich et al. Appl. Environ. Microbiol., 2004, 70, pp. 4575 4581. [2] M. Hofrichter et al. in Grand Challenges in Fungal Biotechnology, ed. H. Nevalainen, 2020, ch. 14, pp. 369-397. [3] R. Ullrich et al. Cell. Mol. Life Sci., 2007, 64, pp. 271-293 |
| Figures: | 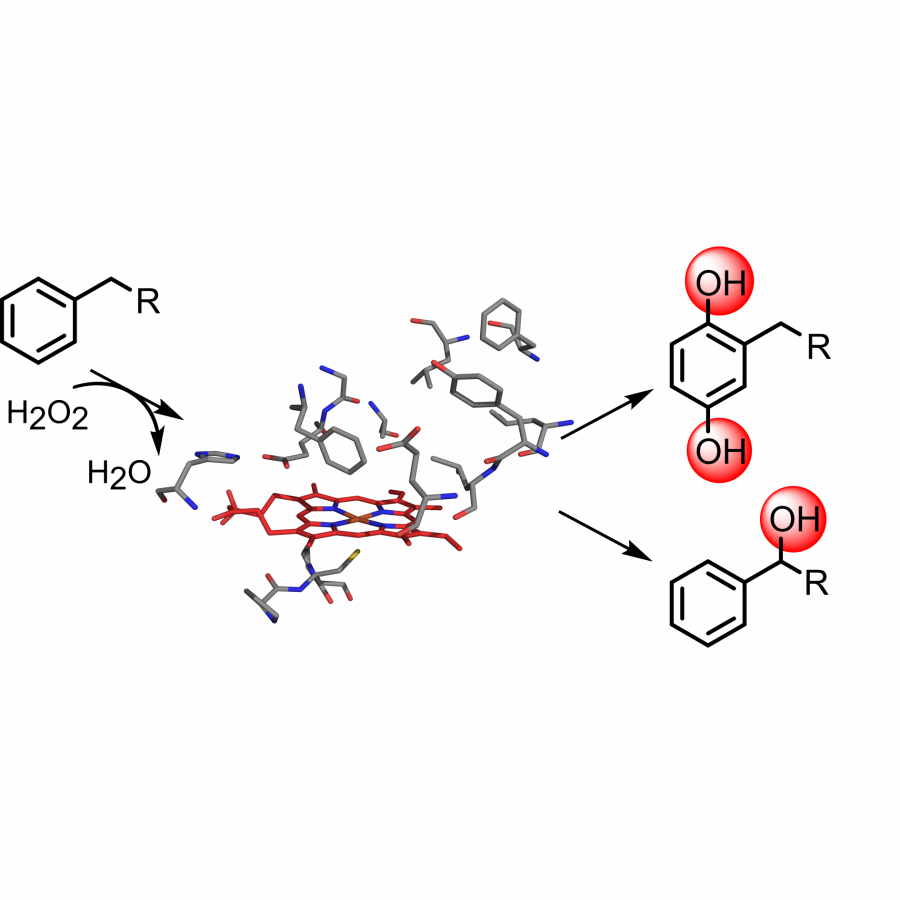 Reaction scheme for AbrUPO An unspecific peroxygenase from Aspergillus brasiliensis highly expressed in Pichia pastoris catalyses aromatic hydroxylation of benzylic compounds. |
#777 | Applications of oxidases in continuous flow |
|
| Presenting author: | Sebastian COSGROVE of KEELE UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Oxidases / Continuous flow / Biocatalysis / |
| Purpose: | Continuous flow offers an alternative way to conduct enzymatic transformations.1 The fine control and abnormal operating windows accessible with flow reactors presents several opportunities for enhanced biocatalyst performance.2,3 I will discuss several approaches using continuous flow to improve the metrics of biocatalytic reactions. It will focus on a range of oxidase enzymes,4–6 and how changing from typical ‘batch’ conditions has improved the synthetic performance of these enzymes, even giving access to new operating windows not obtainable in batch.7 1 S. C. Cosgrove and A. P. Mattey, Chem. Eur. J., 2022, 28, e202103607. 2 J. Britton, S. Majumdar and G. A. Weiss, Chem. Soc. Rev., 2018, 47, 5891–5918. 3 M. P. Thompson, I. Peñafiel, S. C. Cosgrove and N. J. Turner, Org. Process Res. Dev., 2019, 23, 9–18. 4 M. R. Chapman, S. C. Cosgrove, N. J. Turner, N. Kapur and A. J. Blacker, Angew. Chem. Int. Ed., 2018, 57, 10535–10539. 5 S. C. Cosgrove, A. P. Mattey, M. Riese, M. R. Chapman, W. R. Birmingham, A. J. Blacker, N. Kapur, N. J. Turner and S. L. Flitsch, ACS Catal., 2019, 9, 11658–11662. 6 A. P. Mattey, J. J. Sangster, J. I. Ramsden, C. Baldwin, W. R. Birmingham, R. S. Heath, A. Angelastro, N. J. Turner, S. C. Cosgrove and S. L. Flitsch, RSC Advances, 2020, 10, 19501–19505. 7 A. P. Mattey, G. J. Ford, J. Citoler, C. Baldwin, J. R. Marshall, R. B. Palmer, M. Thompson, N. J. Turner, S. C. Cosgrove and S. L. Flitsch, Angew. Chem. Int. Ed., 2021, 60, 18660–18665. |
#817 | Biotransformations of flavonoids with a methyl group or a chlorine atom in cultures of entomopathogenic filamentous fungi Isaria fumosorosea KCH J2 |
|
| Presenting author: | Agnieszka KRAWCZYK-ŁEBEK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Edyta KOSTRZEWA-SUSŁOW of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Monika DYMARSKA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Tomasz JANECZKO of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | fungal biotransformation / flavonoids / glycosylation / O-methylglucosides |
| Purpose: | Flavonoids are widespread in nature secondary metabolites of plants with numerous biological activities, including antimicrobial, anti-inflammatory, antiplatelet, and anti-cancer. Among the various structures of flavonoids, we can distinguish such subclasses as chalcones, flavanones, and flavones. Methylation at the ring position of flavonoid compounds leads to derivatives with increased metabolic stability due to insusceptibility to conjugate with glucuronic acid or sulfate [1]. On the other hand, the introduction of a chlorine atom into the structure of a flavonoid compound enhances its anti-inflammatory activity [2]. In addition, the introduction of a glucose unit into flavonoids leads to the formation of derivatives with improved water solubility and bioavailability [3]. To obtain such flavonoids with a methyl group or a chlorine atom and glucose unit with satisfactory yields, we combined chemical and biotechnological methods. In research of microbial glycosylation of flavonoids, our team obtained compounds with a methyl group at the same position of the flavonoid core, belonging to three subgroups of flavonoids (2’-hydroxy-5’-methylchalcone, 6-methylflavanone, and 6-methylflavone) and biotransformed them using the strain of entomopathogenic filamentous fungi Isaria fumosorosea KCH J2 [3,4]. In further research on the glycosylation of flavonoids, we obtained their counterparts with a chlorine atom (5’-chloro-2’-hydroxychalcone, 6-chloroflavanone, and 6-chloroflavone) and biotransformed them in cultures of the same strain. The resulting compounds were glycosylated at different positions of the flavonoid core. In the case of chalcones, we observed glycosylation of 5’-chloro-2’-hydroxychalcone at A ring (at C-2’), while glycosylation of 2’-hydroxy-5’-methylchalcone occurred at B ring (at C-3). Biotransformation of 6-chloroflavanone led to the formation of one main product glycosylated at B ring (at C-4’) but microbial glycosylation of 6-methylflavanone led to the formation of 4 products and none of them was glycosylated at C-4’. Another biotransformation substrate – 6-chloroflavone was glycosylated at C-4’ (similar to 6-chloroflavanone) and 6-methylflavone was also mainly glycosylated at the same position but with another product glycosylated at C-8. The obtained results show that the regioselectivity of microbial glycosylation conducted by the strain of entomopathogenic filamentous fungi Isaria fumosorosea KCH J2 depends on both: type of substituent (methyl group or chlorine atom) and structure of the flavonoid core (chalcone, flavanone, and flavone), and leads to the formation of biotransformation products glycosylated at different positions. However, some similarities can be observed, such as glycosylation at C-4’ in the case of 6-chloroflavanone, 6-chloroflavone, and 6-methylflavone. The used strain Isaria fumosorosea KCH J2 is able to perform glycosylation of all of the tested flavonoid aglycones and can be used as a valuable tool for microbial glycosylation of various flavonoids. Acknowledgements The work was created as a result of the research project No 2021/41/N/NZ9/01195 financed by the National Science Center. |
| References: | [1] koirala, n., thuan, n.h., ghimire, g.p., thang, d.v., sohng, j.k. Enzym. Microb. Technol. 2016, 86, 103-116. [2] freitas, m., ribeiro, d., tomé, s.m., silva, a.m.s., fernandes, e. Eur. J. Med. Chem. 2014, 86, 153-164. [3] dymarska, m., grzeszczuk, j., urbaniak, m., janeczko, t., pląskowska, e., stępień, ł., kostrzewa-susłow, e. PLoS ONE 2017, 12, e0184885. [4] krawczyk-łebek, a., dymarska, m., janeczko, t., kostrzewa-susłow, e. Catalysts 2020, 10, 1148. |
#818 | Artificial metalloenzyme catalyzed enantiodivergent synthesis of isoindolones |
|
| Presenting author: | Anjali SAIRAMAN of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY |
| Corresponding author: | Prasenjit BHAUMIK of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY |
| Other authors: | Prasun MUKHERJEE of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY Shubhanshu JAIN of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY Sayan ROY of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY Debabrata MAITI of INDIAN INSTITUTE OF TECHNOLOGY BOMBAY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial metalloenzyme / Rational engineering / Enantiodivergence / |
| Purpose: | Isoindolone scaffold is an important pharmacophore present in several medicinal and bioactive compounds. Transition metal catalyzed proximal C−H activation is one of the major routes to construct this N-heterocycle. The Rovis and the Cramer groups have demonstrated efficient synthesis of racemic and enantiopure isoindolones from hydroxamate derivatives and diazo esters using Rh(III) catalysis respectively [1,2]. However, no enzymatic method to assemble the isoindolone scaffold is available till now. Biocatalysis has rightfully been in the spotlight due to the tremendous potential in bringing together greener reagents and methods delivering products with excellent chemo, stereo and regioselectivities. Artificial metalloenzymes complement biocatalysis in substrate scope and protein stability. Streptavidin, a homotetrameric protein, along with a biotinylated metal complex is one of the promising artificial metalloenzymes for its application in diverse non-natural reactions [3]. In this work, we have utilized a streptavidin-biotin-Rh(III) platform to synthesize chiral isoindolones with up to 94:6 e.r. involving a directed inner-sphere C−H activation followed by diazo insertion. A high-resolution crystal structure of streptavidin with unique conformations of the biotinylated Rh(III) cofactor provided interesting insights and inclined us to rationally engineer mutants for enantiodivergence. This is the first report of enzyme catalyzed enantiodivergent route for the synthesis of complex isoindolone derivatives [4]. Our work encompasses a vast substrate scope, crystal structure insights and rational engineering of streptavidin towards enantiodivergence. |
| References: | ex: [1] Hyster, T. K., Ruhl, K. E. & Rovis, T. A. J. Am. Chem. Soc. 2013, 135, 5364-5367 ex: [2] Ye, B. & Cramer, N. Angew. Chem. Int. Ed. 2014, 53, 7896-7899 ex: [3] Hyster, T. K., Knorr, L., Ward, T. R. & Rovis, T. Science 2012, 338, 500-503 ex: [4] Mukherjee, P., Sairaman, A., Jain, S., Roy, S., Maiti, D. & Bhaumik, P. Under revision 2023 |
| Figures: | 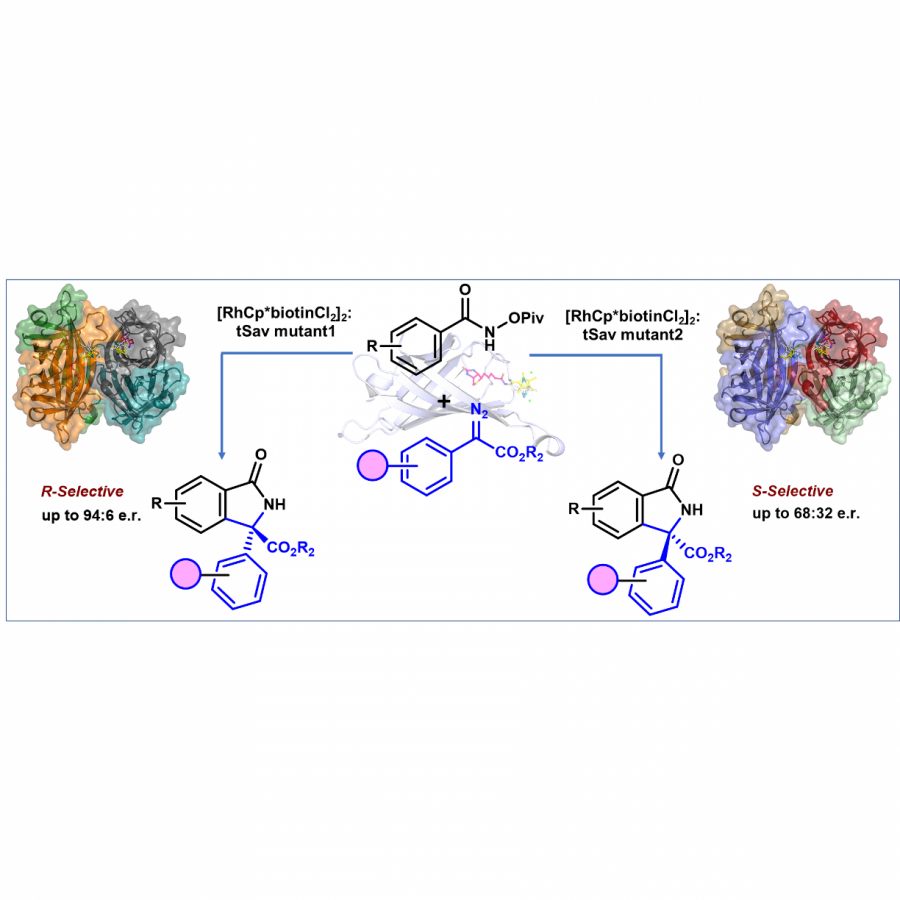 Graphical abstract Enantiodivergent synthesis of isoindolone derivatives |
#820 | Multicopper polyphenol oxidoreductase (laccase) – a novel laccase-mimicking protein family |
|
| Presenting author: | PLEISS Juergen of UNIVERSITY OF STUTTGART |
| Corresponding author: | Marilize LE ROES-HILL of APPLIED MICROBIAL AND HEALTH BIOTECHNOLOGY INSTITUTE |
| Other authors: | Jürgen PLEISS of INSTITUTE OF BIOCHEMISTRY AND TECHNICAL BIOCHEMISTRY Maxim SCHWARZBERG of INSTITUTE OF BIOCHEMISTRY AND TECHNICAL BIOCHEMISTRY Alaric PRINS of APPLIED MICROBIAL AND HEALTH BIOTECHNOLOGY INSTITUTE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Multicopper oxidase / Laccase / Multicopper polyphenol oxidoreductase / STRENDA |
| Purpose: | Enzymes that require copper essential for their catalytic action are collectively referred to as copper oxidases and include the well-known group of multicopper oxidases (MCOs) [1]. Over the past 12 years, an increasing number of bacterial enzymes have been described as ‘laccases’ or MCOs. However, a closer analysis of the sequence data reported in literature clearly reveals a lack of conserved sequence information that is typically associated with the MCO family. In this study, we endeavoured to gain a greater understanding of this group of enzymes that mimic MCO (laccase) activity. The sequences of 20 enzymes published as bacterial laccases and 25 structures (linked to those publications) were analysed and compared to MCOs [2]. The analysis revealed that this group of enzymes belongs to a family of proteins annotated as multicopper polyphenol oxidoreductase (MPO) or MPO laccase (MPOL). MPOLs are found in both prokaryotes and eukaryotes and exhibit biochemical properties similar to that of bacterial laccase-like MCOs, including the oxidation of the ‘typical’ laccase substrates, ABTS, 2,6-dimethoxyphenol, and syringaldazine – characteristics typically used to differentiate MCOs from other copper oxidases. Structural analysis also revealed that there is no similarity between MPOLs and MCOs. The systematic comparison of sequences and structures of large protein families provides an efficient approach for the identification of the structural basis of substrate recognition and catalytic activity and the prediction of activity-specific sequence fingerprints. It further highlighted the need for the incorporation of STRENDA guidelines in the description of novel enzymes, especially in this case where an enzyme of an unrelated protein family was shown to mimic the activities of laccases (and other MCOs) and subsequently erroneously described as a laccase instead of an MPOL. |
| References: | [1]. Gräff, M., Buchholz, P.C.F., Le Roes-Hill, M., Pleiss, J. Proteins 2020, 88, 1329 -1339 [2]. Laccase and Multicopper Oxidase Engineering Database, https://lcced.biocatnet.de/ |
#825 | Synthesis of Pharmaceutically Relevant Arylamines Enabled by a Novel Nitroreductase from Bacillus tequilensis |
|
| Presenting author: | Sara RUSSO of RIJKSUNIVERSITEIT OF GRONINGEN |
| Corresponding author: | Gerrit J POELARENDS of RIJKSUNIVERSITEIT OF GRONINGEN |
| Other authors: | Alejandro PRATS LUJÁN of RIJKSUNIVERSITEIT OF GRONINGEN Henriette J ROZEBOOM of RIJKSUNIVERSITEIT OF GRONINGEN Hein J WIJMA of RIJKSUNIVERSITEIT OF GRONINGEN Marco W FRAAIJE of RIJKSUNIVERSITEIT OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | flavoprotein / nitroreductase / biocatalysis / arylamine |
| Purpose: | Arylamines are essential building blocks for the production of high-value pharmaceuticals. Therefore, strategies for their efficient and cost-effective synthesis are of great interest[1]. The current industrial approach for producing arylamines is based on the catalytic hydrogenation of nitroaromatic precursors, which requires huge catalyst loadings and produces hazardous waste. Besides, these procedures typically lack chemo- and regioselectivity. Therefore, biocatalysis has gained ground as an interesting approach for arylamine synthesis. For example, flavin-dependent nitroreductases are attractive biocatalysts for this, as they perform the reduction of nitroaromatic compounds into aromatic amines with excellent specificity using mild reaction conditions[2–4]. Herein, we assess and characterize a novel nitroreductase from Bacillus tequilensis (BtNTR) that enables the synthesis of pharmaceutically relevant and structurally diverse arylamines. BtNTR was screened towards a panel of nitroarenes, including four intermediates used in the manufacture of blockbuster drugs (Figure 1). BtNTR showed a surprisingly high rate of amine formation (up to 98%) towards a variety of functionalized nitro heterocyclic molecules such as pyridines, benzenes, and pyrazoles. The enzyme has a wide pH optimum for activity and a melting temperature of 58°C, which make it a good candidate for industrial applications. In addition, we elucidated the enzyme crystal structure at 1.15 Å resolution. Guided by the structure, we conducted docking and molecular dynamics studies to better understand the catalytic mechanism. With this aim, we also performed pre-steady-state and steady-state kinetic analyses. Overall, our results suggest that NRs have huge potential as biocatalysts for arylamine production. They constitute a green and selective tool for the synthesis of functionalized anilines that are not easily accessible using standard methods. |
| References: | [1]M. Hoogenraad, J. B. van der Linden, A. A. Smith, B. Hughes, A. M. Derrick, L. J. Harris, P. D. Higginson, A. J. Pettman, Org. Process Res. Dev. 2004, 8, 469-476. [2]E. Akiva, J. N. Copp, N. Tokuriki, P. C. Babbitt, Proc. Natl. Acad. Sci. 2017, 114, E9549. [3]A.-F. Miller, J. T. Park, K. L. Ferguson, W. Pitsawong, A. S. Bommarius, Mol. Basel Switz. 2018, 23, 211. [4]W. Pitsawong, J. P. Hoben, A.-F. Miller, J. Biol. Chem. 2014, 289, 15203-15214. |
| Figures: | 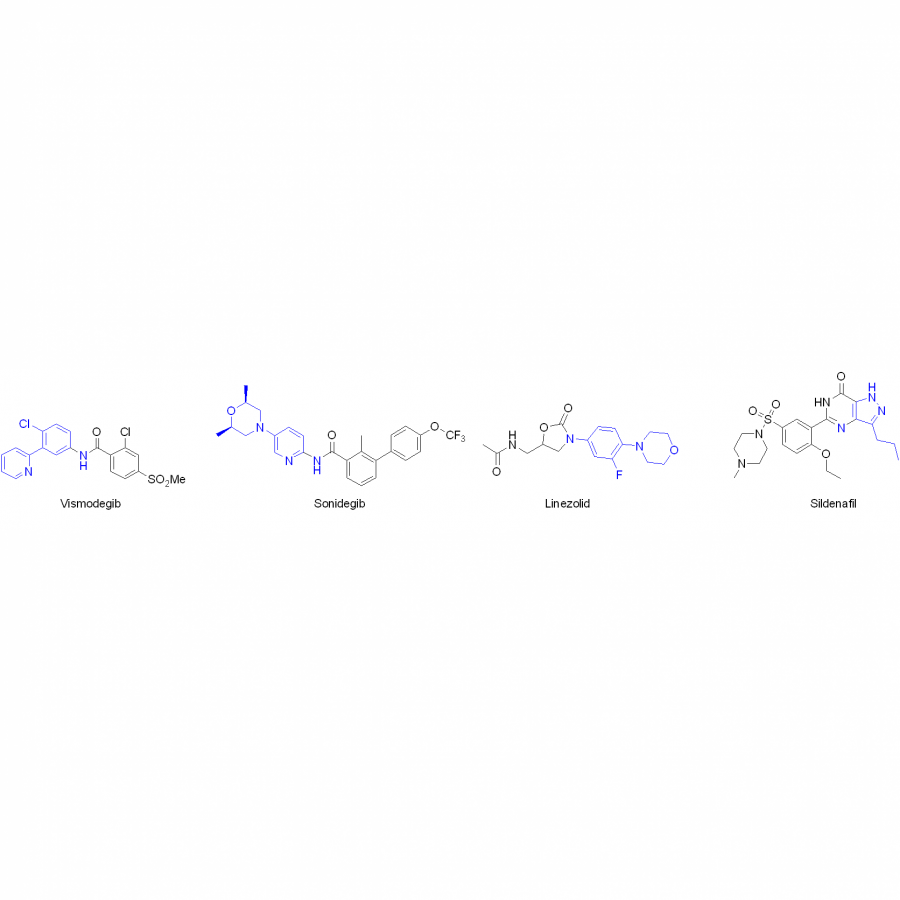 Example of pharmaceutically relevant arylamines . |
#827 | Peroxygenase-Catalysed Selective Oxidation of Silanes to Silanols |
|
| Presenting author: | Jacob VAN HENGST of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | peroxygenase / silanol / / |
| Purpose: | Silanols represent an important product class in organic chemistry as precursors for silicones, as catalyst components or in medicinal chemistry. Syntheses of the state-of-the-art typically start from already functionalised silanes such as chloro- or alkoxysilanes via hydrolysis and producing significant amounts of salt waste-products. Less waste-intensive methods involving dehydrogenative, O2 or H2O2 dependent oxidation of non-functionalised silanols are rare. Even less common are biocatalytic methods for the conversion of silanes. Recently, Arnold and coworkers succeeded in evolving a cytochrome P450-BM3 variant which dramatically increased catalytic activity towards a range of organosilanes Inspired by these pioneering works, we asked ourselves whether so-called unspecific peroxygenases (UPOs) may also be suitable catalysts for this type of transformation. Particularly, we investigated the UPO from Agrocybe aegerita (AaeUPO) as silane oxyfunctionalisation catalyst. UPOs are attractive alternatives to established P450 monooxygenases as they enable drastically simplified, NAD(P)H-independent reaction schemes using simple H2O2 as stoichiometric oxidant (figure 1) |
| Figures: | 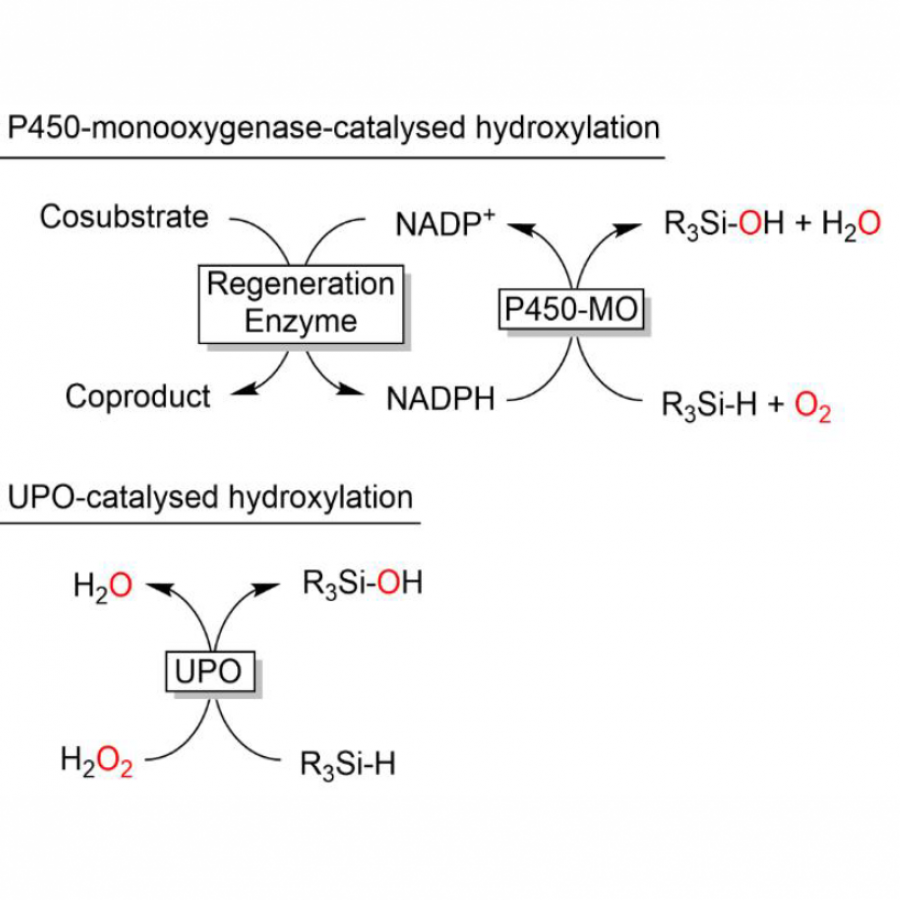 figure 1 Comparison of P450 monooxygenase and peroxygenase catalysed hydroxylation of silanes to silanols |
#829 | Development and Application of Amine and Imine Biocatalysts for the Synthesis of privileged Motifs |
|
| Presenting author: | Tabea GERLACH of THE UNIVERSITY OF MANCHESTER |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | amine oxidase / imine reductase / biocatalytic cascade / stereoselective reduction |
| Purpose: | Development and Application of Amine and Imine Biocatalysts for the Synthesis of privileged Motifs Tabea Gerlach, Lucian Pirvu, Sam Butterworth, Nicholas Turner The synthesis of important amine-containing privileged motifs via a chemo-enzymatic route offers high chemo-, regio- and stereoselectivity with fewer synthetic steps and higher conversions. As opposed to traditional resolution techniques of racemic mixtures that are used in industry, the enzymatic reaction cascade of amine oxidase (AO) and imine reductase (IRED) uses less hazardous reaction conditions and reduces waste production. The chemo-enzymatic route combines the chemical synthesis of the enzymatic substrate, a substituted 1,2,5,6-tetrahydropyridine (THP) with a biocatalytic cascade. The cascade comprises of an AO which oxidises the THP to a conjugated iminium ion. Subsequent double reduction of the iminium by an IRED via an enamine leads to a desired substituted piperidine enantiomer. The chemo-enzymatic route is applied to the synthesis of Niraparib which exemplifies the opportunity this cascade provides for new synthetic strategies of known active pharmaceutical ingredients. |
#831 | Engineering next-generation biocatalysts by in vivo selections |
|
| Presenting author: | Ana Rita OLIVEIRA of UNIVERSITY OF GRONINGEN |
| Corresponding author: | Clemens MAYER of UNIVERSITY OF GRONINGEN |
| Other authors: | Rudy RUBINI of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Direct evolution / In vivo selections / Biocatalysis / |
| Purpose: | Enzymes are impressive catalysts that accelerate reactions with unmatched rates and selectivity. While biocatalysts found in nature are rarely suitable for industrial applications, directed evolution can tailor enzyme properties to a user’s need. Although powerful, enzyme engineering campaigns are notoriously labor- and time-consuming, which derives from the need to assess the activities of enzyme variants one-by-one. To overcome this bottleneck, we have recently provided proof-of-principle for a directed evolution strategy, in which the activity of a model enzyme could be linked to bacterial survival.[1] Critically, this strategy allows us to assess millions of enzyme variants all at once, removing much of the time, cost and technical challenges associated with enzyme evolution. Here, we apply such in vivo selections to the directed evolution of mechanistically diverse enzymes, which are underexploited, yet promising candidates for the sustainable synthesis of valuable compounds. As target enzymes, we selected tyrosine phenol lyases, TPLs, that promote enantioselective C-C bond formations (Fig. 1), and flavin-dependent halogenases, FDHs, that install halogens at aromatic C-H bonds (Fig. 2). To engineer these biocatalysts, we leverage recoded organisms that are addicted to non-canonical amino acids and select improved TPLs and FDHs based on their ability to provide these ncAAs from externally-supplied, synthetic precursors. The resulting next-generation biocatalysts and the designer microbes that produce them will be employed for the synthesis of valuable halogenated building blocks, as well as tyrosine analogs and blockbuster drugs, such as L-DOPA and levothyroxine. |
| References: | 1 - Rubini, R., Jansen, S. C., Beekhuis, H., Rozeboom, H. J. & Mayer, C. Selecting Better Biocatalysts by Complementing Recoded Bacteria. Angewandte Chemie International Edition 62, 1-22 (2023). |
| Figures: | 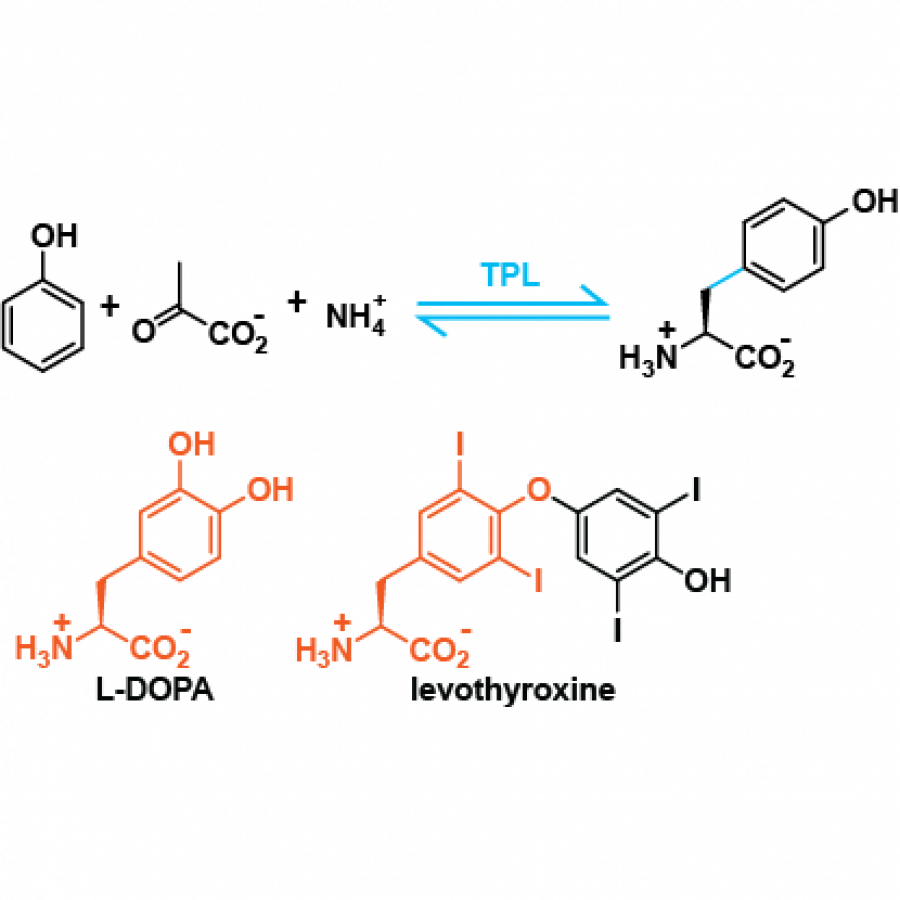 Tyrosine phenol lyase General reactivity of TPLs, and their potential industrial targets. 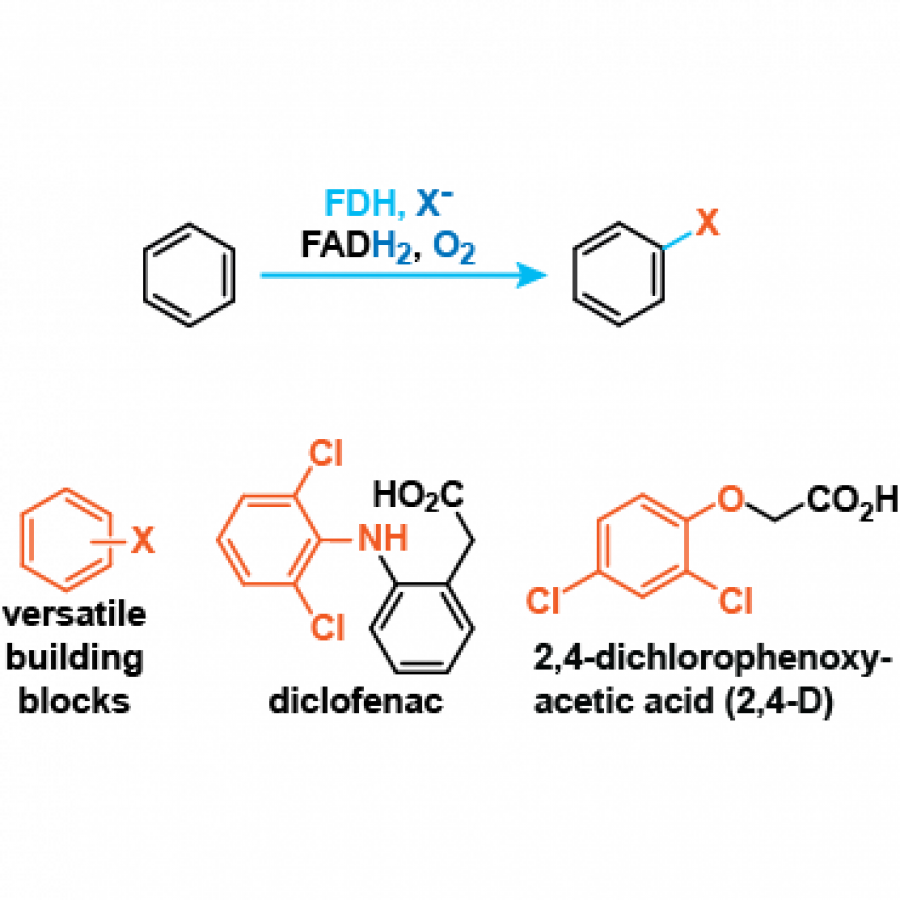 Flavin-dependent halogenases General reactivity of FDHs, and their potential industrial targets. |
#832 | COMPUTATIONAL INSIGHTS INTO THE MUTATIONS OF AN OMEGA-TRANSAMINASE USING EVOLUTIONARY FOOTPRINTS |
|
| Presenting author: | HANDE ABES of YEDITEPE UNIVERSITY |
| Other authors: | NIHAN CELEBI of YEDITEPE UNIVERSITY TURKAN HALILOGLU of BOGAZICI UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | evolution / transaminase / mutation / enzyme activity |
| Purpose: | Transaminases are attractive enzymes to produce enantiopure amines. In the last decade, they are increasingly used to synthesize optically pure chiral amines, which are important intermediates in the synthesis of pharmaceuticals and other fine chemicals.[1] However, the limited activity of these enzymes often causes a bottleneck of their application in biocatalysis. For this reason, the design of new enzymes with improved activity and also operability under industrial process conditions is a significant issue. [2] Although a variety of studies focus on the development of new variants by mutations in the active site of the enzymes, it remains inadequately understood how beneficial the mutations far from the active site confer improved catalytic properties. Getting an insight into this issue would aid the design of new enzymes with higher catalytic activities. Moreover, for natural evolution, it is also shrouded in mystery how mutations far from the active site affect the active sites' functions and also stability. Thus, the current challenge is to figure out how nature alters the function and biophysical properties of the enzymes by amino acid substitutions in both allosteric sites and active sites during evolution. In this study, the objective is to develop an understanding of the mutations that occurred during the natural evolution of a member of the omega-transaminases family, using computational tools and developing a new design conception. With this study, it is also possible to understand the role of cooperativity between allosteric sites and active sites of the enzymes in functionality. To achieve the stated purposes, a modern-day 4-aminobutyrate transaminase, and its 4 ancestral variants having improved catalytic activity and higher promiscuity in substrate selectivity [3] were chosen to study. All mutations that occurred during their evolution were outside the active sites of the enzymes and active sites were totally conserved. Functional collective motions within the variants were evaluated by using computational methods, such as Gaussian Network Model (GNM) and Anisotropic Network Model (ANM). Effects of the mutations that occurred at the dynamic domain interfaces were put under the scope so that mutations could be classified and the important ones for the catalytic activity in addition to substrate selectivity could be identified. Molecular dynamic simulations were performed to develop an understanding of the interactions within and nearby the active sites, as well as the changes in the global behavior of the variants. With this work, the computational results obtained from the stated methods will be presented and discussed. |
| References: | [1] Slabu, I., Galman, J. L., Lloyd, R. C., Turner, N. J., ACS Catalysis, 2017, 7(12), 8263-8284. [2] Kim, G. H., Jeon, H., Khobragade, T. P., Patil, M. D., Sung, S., Yoon, S., Won, Y., Choi, I. S., Yun, H., Enzyme and Microbial Technology, 2019, 120, 52-60. [3] Wilding, M., Peat, T. S., Kalyaanamoorthy, S., Newman, J., Scott, C., Jermiin, L. S., Green Chemistry, 2017, 19(22), 5375-5380. |
#835 | Biocatalytic Synthesis of Selectively Isotopically Labelled Molecules for Specialised Protein NMR Spectroscopy |
|
| Presenting author: | Alison TAM of UNIVERSITY OF OXFORD |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Isotopic labelling / Cofactor recycling / Amino acid synthesis / Sugar synthesis |
| Purpose: | Enantioselective synthesis of isotopically labelled biomolecules is not trivial, with most chemical methods requiring precious-metal catalysts and expensive starting materials. Issues such as limited selectivity and low isotopic purities can also create downstream purification steps and waste. Biocatalytic synthesis offers improved selectivity, but typically relies upon costly precursors, such as labelled formate or glucose. In this project, we explore cost-effective routes towards synthesising isotopically labelled biomolecules (such as amino acids and sugars) using chemo- and biocatalytic approaches. The application we target is Nuclear Magnetic Resonance study of large proteins. NMR is widely used in studying the structure and dynamics of proteins in solution. However, it is inherently limited by the upper size limit, where fast relaxation contributes to complex and uninterpretable spectra. Selective isotopic labelling (2H, 15N, 13C) of proteins using designer amino acid and sugars (and their precursors thereof) can improve this limit, and also enables use of specialised relaxation-optimised NMR techniques such as methyl-TROSY [1]. Particularly, biocatalysis can offer benefits such as mild reaction conditions, high inherent selectivities, and biodegradable catalysts, while chemo-catalysts can be exploited at selective steps for their high productivities and where biocatalytic scope remains limited. Previous work in the Vincent group has established NAD+ cofactor deuteration using [NiFe] hydrogenase enzymes, which can be further applied in redox cascades (fig. 1) for the synthesis of various isotopically labelled chemicals [2], [3]. Further research in this area would enable more practical, routine use of NMR-labelled designer proteins, and contribute towards areas encompassing sustainable manufacturing, biochemical discoveries, and development of novel therapeutics and pharmaceuticals. |
| References: | [1] Ollerenshaw, J. E., Tugarinov, V. & Kay, L. E. Magn. Reson. Chem., 2003, 41, 843-852. [2] Rowbotham, J. S., Ramirez, M. A., Lenz, O., Reeve, H. A. & Vincent, K. A. Nat. Comms., 2020, 11, 1454. [3] Rowbotham, J. S., Nicholson, J., Ramirez, M., Urata, K., Todd, P., Karunanithy, G., Lauterbach, L., Reeve, H., Vincent, K. ChemRxiv., Cambridge: Cambridge Open Engage., 2022. 10.26434/chemrxiv-2022-82tz0 |
| Figures: | 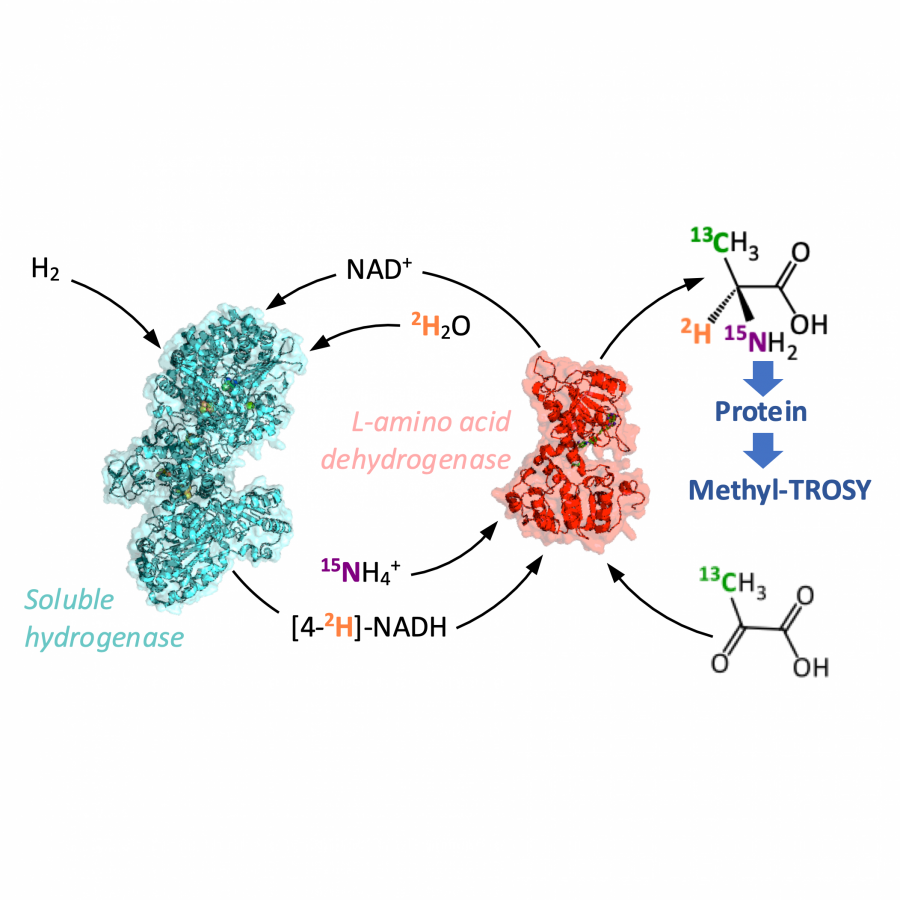 Figure 1 Reaction scheme for the synthesis of isotopically labelled L-alanine through use of deuterated NAD2H cofactor, generated in situ |
#836 | A BIOCATALYTIC PROCESS FOR MILDER BLUE DENIM DYEING |
|
| Presenting author: | Gonzalo BIDART of DTU |
| Other authors: | Natalia PUTKARADZE of DTU David TEZE of DTU Leila LO LEGGIO of KU Sumesh SUKUMARA of DTU Ólafur ÖGMUNDARSON of UNIVERSITY OF ICELAND Anna-Mamusu SESAY of DESIGNSKOLEN KOLDING Charlotte JANSEN of DTU Katrine QVORTRUP of DTU Folmer FREDSLUND of DTU Ditte WELNER of DTU |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Techno-Economic assessment / Life-cycle assessment / Rational engineering |
| Purpose: | Indigo is the most used dye for blue denim worldwide(1). Its synthesis and the dyeing process require chemical steps that are environmentally damaging, including the use of reducing agents and alkali for indigo solubilization. Different approaches aim at replacing the harmful processes with ecologically attractive alternatives, but the economic and social aspects of sustainability are often overlooked, resulting in poor implementation(2,3,4). The glycosyltransferase PtUGT1 adds a glucose moiety to the reactive indigo precursor indoxyl to form indican, preventing spontaneous oxidation and keeping the dye-precursor soluble(5). Forming indigo directly in the yarn through indican dyeing is a promising route that uses mild conditions(5). Indican eliminates the requirement for reducing agents while still ending as indigo, the only known molecule yielding the unique hue of blue denim. However, currently there isn’t a source of bulk indican. To efficiently leverage indican in denim dyeing, two processes must be developed that have environmental benefits over the conventional method while still being affordable: a) a production process & b) a dyeing process (Fig. 1A). Here, we address these two aspects, considering the three dimensions of sustainability, i.e., 1) economic viability, 2) environmental performance, and 3) social impacts. To make biocatalytic indican synthesis economically feasible, we performed a Techno-Economic Assessment (TEA), which pinpointed that the main parameters to decrease reaction costs would be to implement a UDP-glucose recycling system, replace the buffer for water, and to increase substrate concentration above 80 mM. Unfortunately, PtUGT1 is inactive at this substrate concentration (Fig. 1B). Leveraging the structural information of PtUGT1 obtained by X-ray crystallography (PDB ID: 5nlm)(2), we therefore rationally designed 108 mutants to increase enzyme stability to tolerate the required high substrate concentration. As a result, we have developed several active PtUGT1 variants with up to 15°C increase in melting temperature (TmB) (Fig. 1C), which correlated with increased chemo-stability and a strong reduction in substrate deactivation at the required concentrations. This allowed the biocatalytic synthesis of indican from up to 100 mM indoxyl with a 65% yield (Fig. 1B). Further, TEA, social sustainability, and comparative Life Cycle Assessment (LCA) of indican dyeing processes showed that, either enzymatic dyeing or a newly discovered and developed photolytic method, would outperform the current indigo dyeing process in 15 or 14 of 18 impact categories, including global warming, terrestrial ecotoxicity, marine ecotoxicity, and ozone depletion, among others (Fig. 2). Overall, our results emphasize the power of integrating technical and sustainability aspects from early stages of biocatalytic process development. |
| References: | [1] Balfour-Paul, J. Indigo (Firefly Books, 2011). [2] Bozic, M.; Kokol, V. Dye. Pigment. 2008, 76 (2), 299-309. [3] Berry, A et. al. J. Ind. Microbiol. Biotechnol. 2002, 28 (3), 127-133. [4] Rai, S.; Saremi, R.; Sharma, S.; Minko, S. Green Chem. 2021, 23 (20), 7937-7944. [5] Hsu TM et. al. Nat Chem Biol. 2018 Mar;14(3):256-261. |
| Figures: | 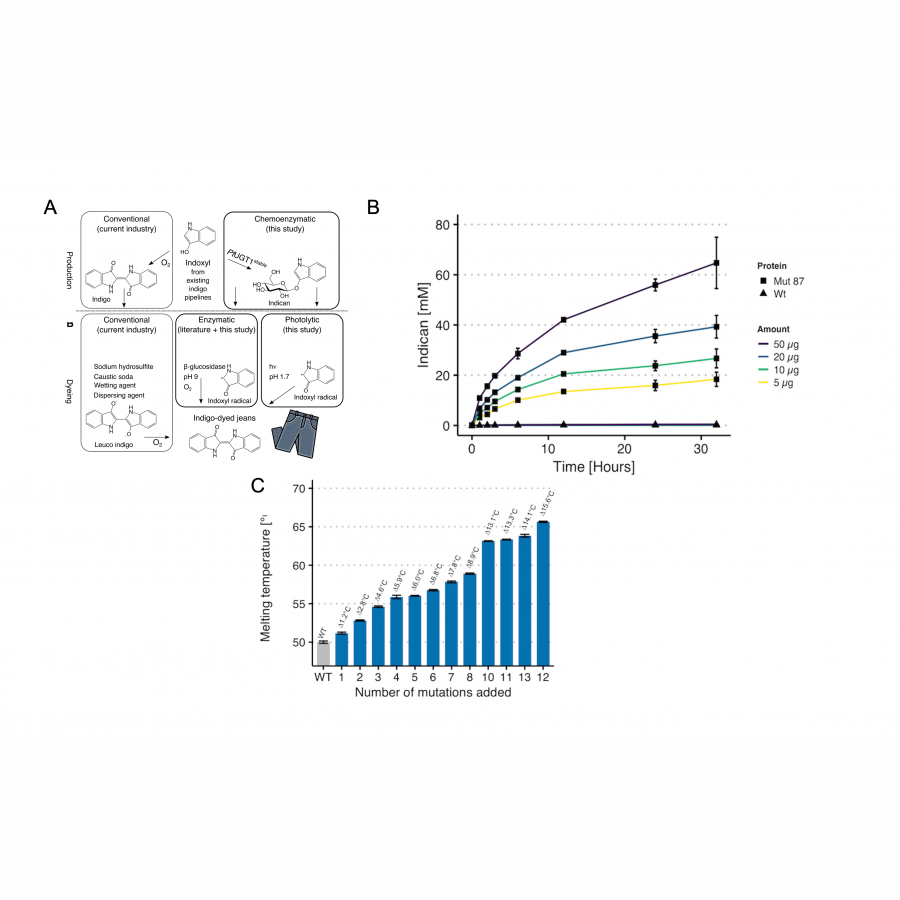 Figure 1 A) Overview of indigo and indican production and dyeing processes. B) Kinetics of indican synthesis using 100 mM indoxyl-acetate as substrate. C) DSF comparing TmB of PtUGT1 WT and designed mutants. 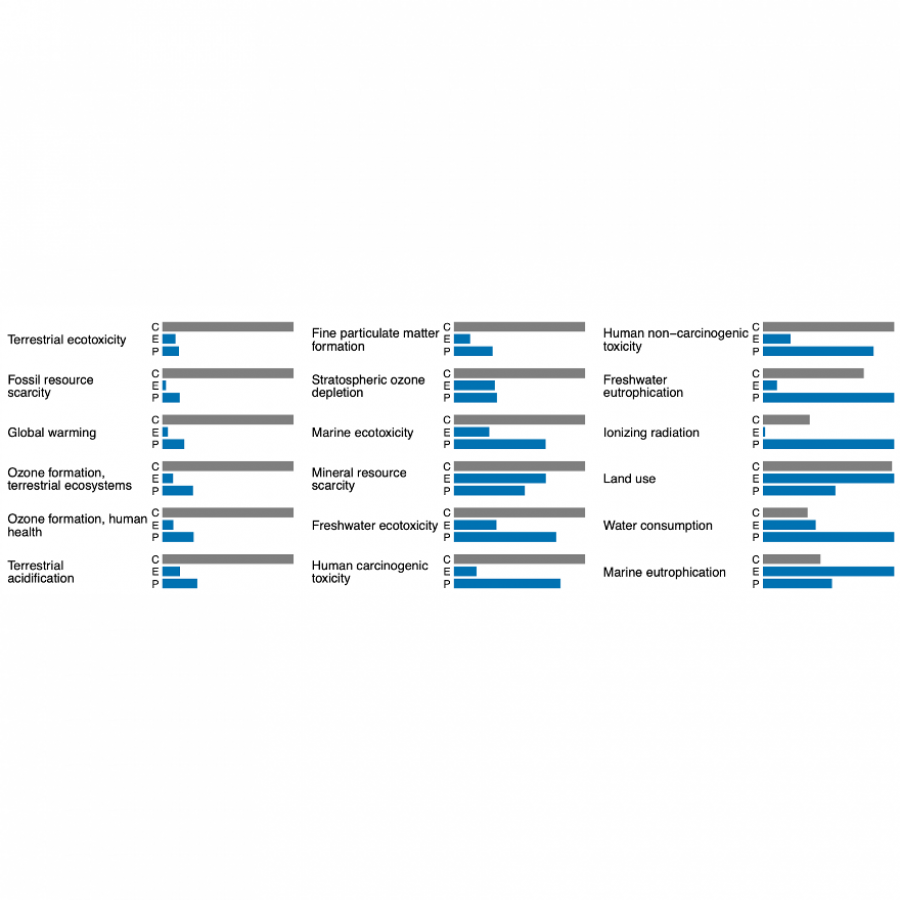 Figure 2 LCA midpoints (C = conventional indigo dyeing, E = enzymatic indican dyeing, P = photolytic indican dyeing) |
#839 | Novel triple mutant of an extremophilic glycosyl hydrolase enables the rapid synthesis of thioglycosides |
|
| Presenting author: | Lauriane PILLET of UNIVERSITY OF BERN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymatic thioglycosides synthesis / Enzyme engineering / Extremophilic enzyme / Sustainable biocatalytic processes |
| Purpose: | Due to their their low susceptibility to hydrolysis, thioglycosides are highly interesting molecules that have been used as enzyme inhibitors and in carbohydrate-based therapeutics. A plethora of chemical synthesis to access thioglycosides have been reported, however, these procedures are complex, offer limited stereochemical control and often require non-sustainable reagents [1]. Selective and sustainable biosynthetic methods relying on mutant forms of glycosidases which have had their catalytic residues replaced by neutral ones have been reported. While thiooligosaccharides have been synthetized via these strategies [2], enzymatic syntheses of thioglycoconjuguates are scarce. Moreover, attempts to translate these elegant proof-of-concepts to industrial processes are often plagued by the poor resistance of enzymes to the organic solvents that are often required to dissolve thiols. In order to expand the range of reaction conditions suitable for biocatalytic preparation of thioglycosides, we selected a β-glycosyl hydrolase (GH1; EC 3.2.1.21) from Halothermothrix orenii (HorGH1), an extremophilic enzyme with a high tolerance to extreme temperatures, pHs and organic solvents. We previously engineered this enzyme towards thioglycosidase activity and highlighted the key role of an arginine residue (M299R mutation) in the recognition of thioglycosides [3]. Herein, we present the first example of an extremophilic glycosyl hydrolase engineered towards thioglycosynthase activity with a novel combination of mutations [4]. Among the 8 HorGH1 variants resulting from a combination of the E166A (acid/base residue), the E354G (nucleophilic residue) and the M299R mutation, the triple mutant HorGH1 M299R/E166A/E354G gave access to a range of high-value thioglycosides with exquisite stereoselectivity and good to excellent conversions (61-93%). Aside from being easy to handle and cofactor independent, this robust catalyst remained active for 48 hours despite the presence of 30 % DMSO. Overall, this works expands the repertoire of mutant glycosidases available for thioglycoside synthesis and provides an innovative, safe, green and profitable synthetic route for the construction of S-glycosidic linkages. |
| References: | [1] K. Pachamuthu, R. R. Schmidt. Synthetic routes to thiooligosaccharides and glycopeptides. Chem Rev, 2006, 106, 160-187. [2] M. Jahn, H. Chen, J. Müllegger, J. Marles, R. A. J. Warren, S. G. Withers. Thioglycosynthases: double mutant glycosidases that serve as scaffolds for thioglycoside synthesis. Chem. Commun., 2004, 4, 274-275. [3] N. Almulhim, N. R. Moody, F. Paradisi. Engineering novel S-glycosidase activity into extremo-adapted beta-glucosidase by rational design. Appl. Microbio.l Biotechnol., 2020, 104, 4407-4415. [4] L. Pillet, D. Lim, N. Almulhim, A. I. Benítez-Mateos, F. Paradisi. Novel triple mutant of an extremophilic glycosyl hydrolase enables the rapid synthesis of thioglycosides. Chem. Commun., 2022, 58, 12118-12121. |
| Figures: | 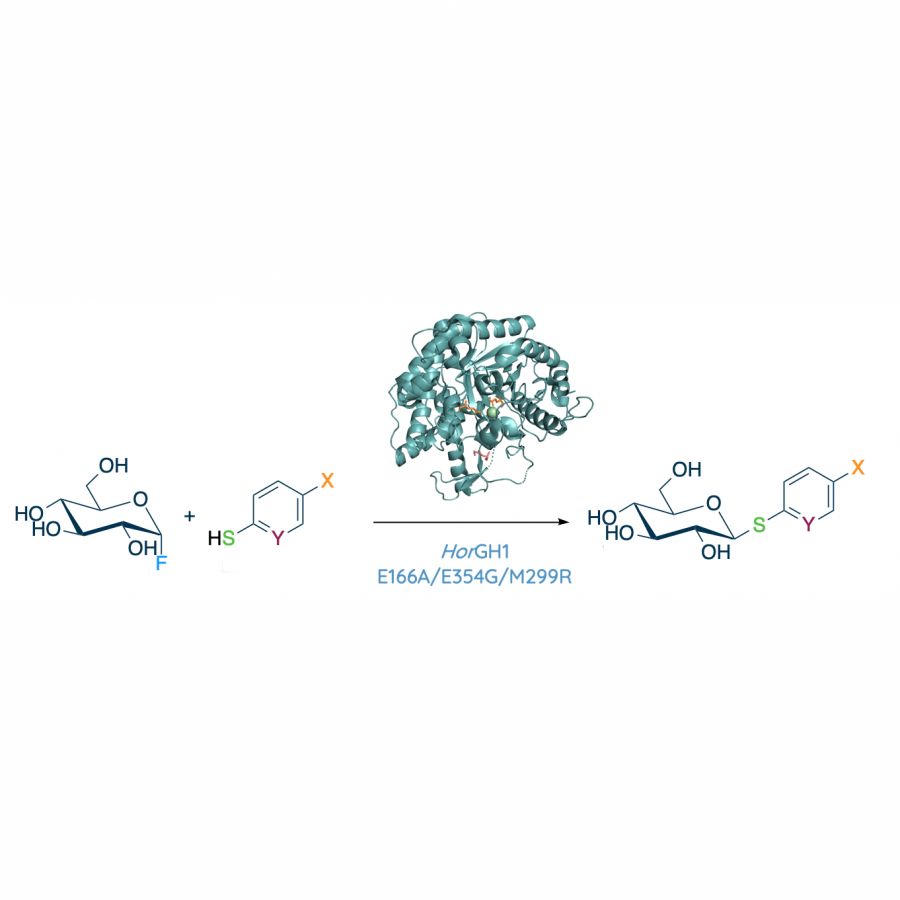 Thioglycosides synthesis with the triple mutant HorGH1 M299R/E166A/E354G A novel triple mutant of an extremophilic glycosyl hydrolase allowed the stereoselective, efficient, and sustainable synthesis of still elusive thioglycosides. |
#840 | Biocatalysis in drug design: Engineered RedAm to access chiral building block with multiple stereocenters |
|
| Presenting author: | Arnau RUE of UNIVERSITY OF MANCHESTER |
| Other authors: | Martin HAYES of ASTRAZENECA Nicholas TURNER of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / RedAm / Semi-rational mutagenesis / Drug design |
| Purpose: | The pharmaceutical industry is constantly looking for innovative ways to access bioactive compounds with new structure-activity relationships (SAR), this has increased the demand for novel building blocks in drug design. A wide variety of these compounds allows to cover a big spectrum of different drug properties such as increased permeability, potency enhancement or different selectivity. Complex molecules are key to expand compound libraries, however chiral compounds with multiple stereocenters can be particularly difficult to access by traditional organic synthesis. Biocatalysis can be a powerful tool to cover this space since it can access new synthetic methodologies and it allows to control the stereoselectivity of the reaction. Moreover, its inherent ability to produce natural products derivates, which tend to naturally interact with protein drug targets, is highly desirable. Herein, we report a biocatalytic process to access the desired enantiomer of a building for drug discovery, this molecule has multiple chiral centers and proved to be hardly accessible by organic synthesis. The first step aimed to identify active RedAms, however none of identified hits produced the desired enantiomer. Therefore, IR-09 was engineered following a semi-rational mutagenesis approach, which was designed around the principle that diastereoselectivity and enantioselectivity of the enzyme are based on the orientation of the ligand in the active site. A crystal structure of IR-09 was obtained and target residues for site directed mutagenesis were identified. Finally, variants W204A and W204S were able to access the desired enantiomer (S,S,S). These results suggested that residue W2404 was key to switch selectivity, hence site saturation mutagenesis was performed. New variants which could also produce the (S,S,S)-enantiomer were identified. For example, W204R exhibited the highest selectivity yielding 45% e.r.. which is very close to the theoretical maximum e.r. (50%), since the starting material is a racemic mixture. Evolution of this enzyme not only allowed to switch the stereoselectivity but also to incorporate stereospecificity for the substrate. This is the case of variant W204G, which has preference for the (S,S) starting material, thus performing selective reductive amination and concomitant kinetic resolution of the substrate. Furthermore, this biocatalytic step can be incorporated into a chemoenzymatic cascade to enable the production of the primary amine. Finally, the evolved RedAm proved to retain high levels of conversion (>90%) on preparative scale, therefore being an interesting backbone for further evolution in case the drug progresses to process development. |
| References: | Devine, P. N.; Howard, R. M.; Kumar, R.; Thompson, M. P.; Truppo, M. D.; Turner, N. J. Extending the Application of Biocatalysis to Meet the Challenges of Drug Development. Nat. Rev. Chem. 2018, 2 (12), 409-421. https://biotrans2023.livescience.io/# Fryszkowska, A.; Devine, P. N. Biocatalysis in Drug Discovery and Development. Curr. Opin. Chem. Biol. 2020, 55, 151-160. Aleku, G. A.; France, S. P.; Man, H.; Mangas-Sanchez, J.; Montgomery, S. L.; Sharma, M.; Leipold, F.; Hussain, S.; Grogan, G.; Turner, N. J. A Reductive Aminase from Aspergillus Oryzae. Nat. Chem. 2017, 9 (10), 961-969. |
| Figures: | 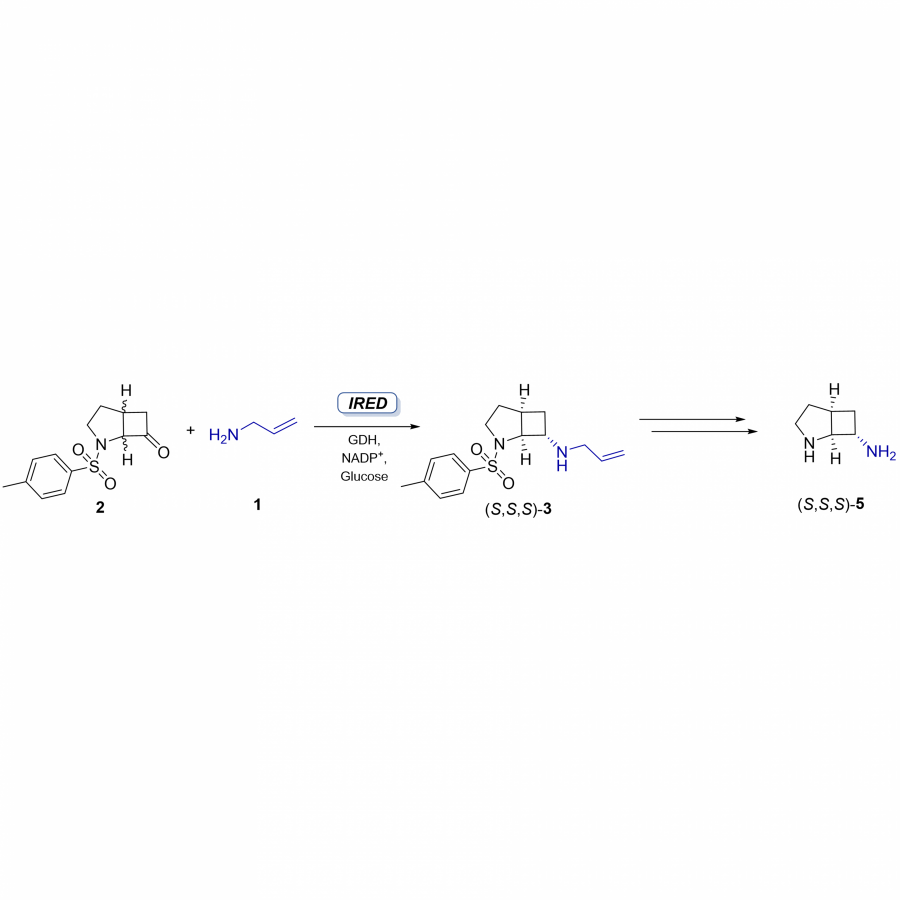 Synthetic route to key building block Enzymatic reductive amination of ketone 2 with allylamine (1) to provide (S,S,S)-3, which can be easily converted to key component (S,S,S)-5. 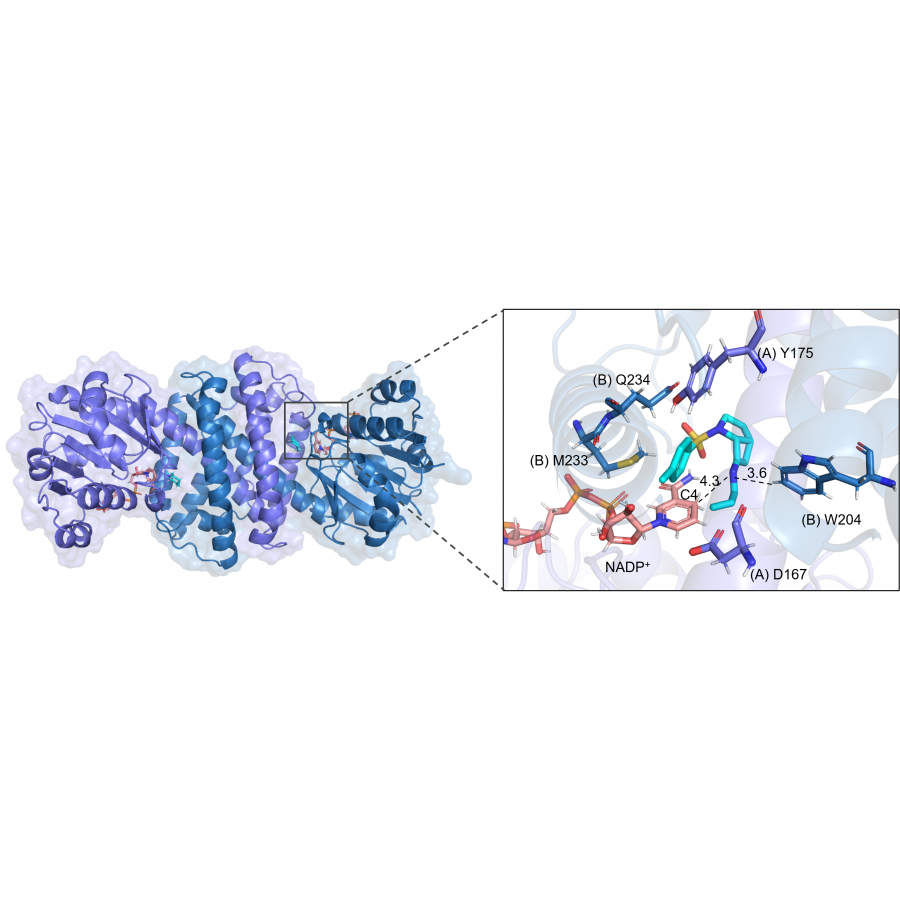 IR-09 crystal structure and active site IR-09 crystal structure and closer look to the active site with ligand and targeted residues for mutagenesis. |
#847 | Designing in vitro enzymatic cascades for the synthesis of Amaryllidaceae alkaloids |
|
| Presenting author: | Daniel CASTELLANO GARRIDO of UNIVERSITY COLLEGE LONDON |
| Other authors: | Helen HAILES of UNIVERSITY COLLEGE LONDON Jack JEFFRIES of UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biosynthetic Pathway / Amaryllidaceae Alkaloid / Enzyme engineering / In-silico modelling |
| Purpose: | Designing in vitro enzymatic cascades for the synthesis of Amaryllidaceae alkaloids Daniel Castellano Garrido, Prof John Ward, Dr Rebecca Roddan, Dr Yu Wang Dr Maria Bawn, Dr Jack Jeffries and Prof Helen Hailes Department of Chemistry and Department of Biochemical Engineering, University College London "Amaryllidaceae alkaloids are plant medicinal compounds that have been used as natural remedies for centuries in various cultures. The structural complexity of these compounds makes them challenging to produce through chemical synthesis at scale. Therefore, new more environmentally friendly alternatives are being explored to produce these alkaloids. Some examples include the metabolic engineering of plants or in vivo heterologous expression of the biosynthetic pathway in model organisms like yeast [1], while others focus on in vitro biosynthetic pathway assembly by designing modular enzymatic cascades that lead to rapid and selective product formation [2]. This research focuses on the later approach since it allows better control of the individual components of the reaction and provides flexibility with regards to the product analogues that can be chosen as targets, rather than being limited to the natural product. This approach has been particularly successful with the benzylisoquinoline alkaloids (BIAs) family [3], however, the Amaryllidaceae alkaloids remain relatively unexplored. In this work, the first-committed enzyme of the Amaryllidaceae biosynthetic pathway, norbelladine synthase [4], has been coupled to other enzymes in a cascade with the aim to generate an in vitro platform for the production of novel alkaloids. Furthermore, in-silico analysis of the central enzyme from the BIAs pathway, norcoclaurine synthase revealed significant sequence homology with norbelladine synthase and rational mutagenesis has been performed to explore their specificities." Bibliography 1. Rahmat E, Kang Y. Yeast metabolic engineering for the production of pharmaceutically important secondary metabolites. Appl Microbiol Biotechnol [Internet]. 2020 Jun 1 [cited 2023 Mar 15];104(11):4659–74. 2. Subrizi F, Wang Y, Thair B, Méndez-Sánchez D, Roddan R, Cárdenas-Fernández M, et al. Multienzyme One-Pot Cascades Incorporating Methyltransferases for the Strategic Diversification of Tetrahydroisoquinoline Alkaloids. Angew Chemie Int Ed [Internet]. 2021 Aug 16 [cited 2022 Jun 7];60(34):18673–9. 3. Roddan R, Sula A, Méndez-Sánchez D, Subrizi F, Lichman BR, Broomfield J, et al. Single step syntheses of (1S)-aryl-tetrahydroisoquinolines by norcoclaurine synthases. Commun Chem 2020 31 [Internet]. 2020 Nov 13 [cited 2021 Aug 14];3(1):1–10. 4. Singh A, Massicotte MA, Garand A, Tousignant L, Ouellette V, Bérubé G, et al. Cloning and characterization of norbelladine synthase catalyzing the first committed reaction in Amaryllidaceae alkaloid biosynthesis. BMC Plant Biol [Internet]. 2018 Dec 7 [cited 2021 Jan 4];18(1):338. |
| References: | 1. Rahmat E, Kang Y. Yeast metabolic engineering for the production of pharmaceutically important secondary metabolites. Appl Microbiol Biotechnol [Internet]. 2020 Jun 1 [cited 2023 Mar 15];104(11):4659-74. 2. Subrizi F, Wang Y, Thair B, Méndez-Sánchez D, Roddan R, Cárdenas-Fernández M, et al. Multienzyme One-Pot Cascades Incorporating Methyltransferases for the Strategic Diversification of Tetrahydroisoquinoline Alkaloids. Angew Chemie Int Ed [Internet]. 2021 Aug 16 [cited 2022 Jun 7];60(34):18673-9. 3. Roddan R, Sula A, Méndez-Sánchez D, Subrizi F, Lichman BR, Broomfield J, et al. Single step syntheses of (1S)-aryl-tetrahydroisoquinolines by norcoclaurine synthases. Commun Chem 2020 31 [Internet]. 2020 Nov 13 [cited 2021 Aug 14];3(1):1-10. 4. Singh A, Massicotte MA, Garand A, Tousignant L, Ouellette V, Bérubé G, et al. Cloning and characterization of norbelladine synthase catalyzing the first committed reaction in Amaryllidaceae alkaloid biosynthesis. BMC Plant Biol [Internet]. 2018 Dec 7 [cited 2021 Jan 4];18(1):338. |
#852 | Clean Enzymatic Production of Ursodeoxycholic Acid Enabled by a Newly Identified NADH-dependent 7β-Hydroxysteroid Dehydrogenase |
|
| Presenting author: | Chunxiu LI of EAST CHINA UNIVERSITY OF SCIENCE AND TECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ursodeoxycholic acid / 7β-Hydroxysteroid dehydrogenase / NADH dependency / Cofactor regeneration |
| Purpose: | 7β-Hydroxysteroid dehydrogenases (7β-HSDHs) play an important role in the enzymatic synthesis of ursodeoxycholic acid (UDCA), which is a value-added compound with a range of pharmacological activities. Since the cofactor NADH is much cheaper than NADPH, the production of UDCA using an NADH-dependent 7β-HSDH has better prospects than that using an NADPH-dependent 7β-HSDH. However, a major bottleneck in UDCA biosynthesis is the poor catalytic activity of the current NADH-dependent 7β-HSDHs. In this work, a new and natively NADH-dependent Rs7β-HSDH from Roseococcus sp. was identified, which showed a high specific activity of 63.3 U mg−1protein toward 7-oxo-lithocholic acid, with a catalytic efficiency (kcat/KM) of 515 mM–1 s–1. In a preparative biotransformation (100-mL scale) using Rs7β-HSDH and an NAD+-dependent 7α-HSDH, with O2/NOX and HCOO−/FDH systems for the regeneration of cofactors (NAD+ and NADH), 25 mM chenodeoxycholic acid was completely converted to UDCA in one-pot two-step cascade, with an 80% isolated yield and a total turnover number (TTN) of 6586 for Rs7β-HSDH. The environmental factor (E-factor) of this process was 8.37 when water was excluded, much lower than those obtained from other processes reported so far, indicating a great potential of this Rs7β-HSDH for more sustainable and cleaner enzymatic production of UDCA. |
| References: | [1] A. Stiehl, Ann. Med. 1994, 26, 345-349. [2] M. Sackmann, J. Pauletzki, U. Aydemir, J. Holl, T. Sauerbruch, J. Hasford, G. Paumgartner, Hepatology. 1991, 14, 1136-41. [3] X. Zhang, D. D. Fan, X F. Hua, T. Zhang, Bioprocess. Biosyst. Eng. 2019, 42, 1537-1545. [4] T. Kanazawa, A. Shimazaki, T. Sato, T. Hoshino, Proc. Jpn. Acad. 1954, 30, 391-392. [5] H. Hofmann, Acta Chem. Scand. 1963, 17, 173-186. [6] P. S. Dangate, C. L. Salunke, K. G. Akamanchi, Steroids. 2011, 76, 1397-1399. [7] W. Chen, D. H. Hu, Z. L. Feng, Z. P. Liu, Steroids. 2021, 172, 108870. [8] X. L. He, L. T. Wang, X. Z. Gu, J. X. Xiao, W. W. Qiu, Steroids.2019, 140,173-178. [9] J. X. Shen, D. D. Dong, Z. F. Wang, J. F. Wan, X. J. Cao, Sci Rep. 2021, 11, 16273. [10] H. P. Li, Z. N. You, Y. Y. Liu, G. W. Zheng, H. Gong, Y. M. Mo, N. Zhu, Y. P. Bai, J. H. Xu, ACS Sustain. Chem. Eng. 2021, 10, 456-463. [11] T. Eggert, D. Bakonyi, W. Hummel, J. Biotechnol. 2014, 191, 11-21. |
| Figures: | 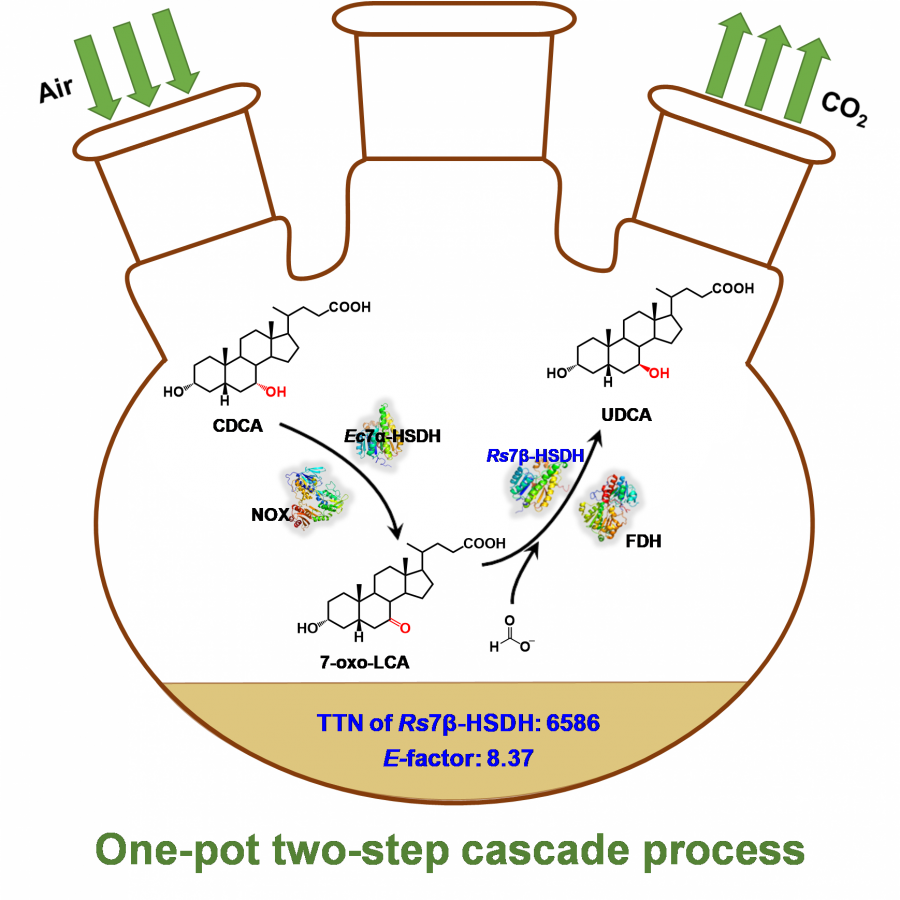 Abstract graphic One-pot synthesis of ursodeoxycholic acid using dual NAD-dependent HSDHs, NOX and FDH for cofactor regeneration makes this cascade process green and clean. |
#863 | Optimized synthesis of plant lignans via chromosomal integration of a multi-enzyme cascade in E. coli |
|
| Presenting author: | Ronja KNÖFEL of HEINRICH-HEINE-UNIVERSITÄT |
| Other authors: | U. Joost LÜLF of HEINRICH-HEINE-UNIVERSITÄT Philipp A. BECHTOLD of HEINRICH-HEINE-UNIVERSITÄT Vlada B. URLACHER of HEINRICH-HEINE-UNIVERSITÄT |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Plant lignans / E. coli / Multi-enzyme cascades / Chromosomal integration |
| Purpose: | The lignans (-)-deoxypodophyllotoxin and (-)-podophyllotoxin are precursors in the synthesis of the chemotherapeutic etoposide [1]. To overcome their limited availability in the natural source - the endangered plant Sinopodophyllum hexandrum – alternative routes have been suggested, including chemo-enzymatic synthesis [2,3] and the heterologous production in tobacco leaves [4]. The genes coding for the enzymes involved in the biosynthetic pathway from (+)-pinoresinol to (-)-deoxypodophyllotoxin in S. hexandrum has been deciphered [5]. In our previous studies, we introduced the heterologous biosynthesis of lignans in Escherichia coli [1, 6]. The cascade included eight genes from four plants which were cloned in several plasmids. To ensure the redox cofactor availability, the cascade was divided between two E. coli cells [1]. Generally, the use of multiple plasmids suffers from plasmid-instability, reduced cell growth, higher costs due to the need for e.g. antibiotics and high cell-to-cell variability. In this study, we integrated the genes encoding the four-step cascade from (+)-pinoresinol to (-)-pluviatolide, an important cross-road compound of lignans and precursor of (-)-deoxypodophyllotoxin synthesis, into the E. coli chromosome. Pluviatolide synthesis in resting and growing recombinant cells was analyzed for both episomal and chromosomal expression systems. Energy and carbon sources were compared and the effect of cell permeabilization on product titers investigated. Product analysis was done by LC/MS. Our results revealed that the biotransformation with resting cells yielded similar quantities of the product (-)-pluviatolide, independently on the type of gene expression. In contrast, growing cells with chromosomally integrated foreign genes performed better which led to doubled pluviatolide titers. Generally, the addition of glucose or glycerol had a positive impact on biotransformation. While glycerol improved the productivity of non-permeabilized resting cells, permeabilized cells performed best after glucose supplementation. In both cases up to 99% (+)-pinoresinol were converted in (-)-pluviatolide. In growing cells, the biotransformation was limited by cell death after 24 hours, but the addition of glucose as carbon and energy source led to further cell growth and yielded 99% (-)-pluviatolide. |
| References: | [1] Decembrino, D., et al., Microb. Cell Fact 2021, 20, 183 [2] Li, J., et al., Angew Chem Int Ed Engl 2019, 58, 11657-11660 [3] Lazzarotto, M., et al., Angew Chem Int Ed Engl 2019, 58, 8226-8230 [4] Schultz, B. J., et al., J. Am. Chem. Soc. 2019, 141, 19231-19235 [5] Lau, W.; Sattely, E. S., Science 2015, 349 , 1224-1228 [6] Decembrino, D. et al. ACS Synth Biol 2020, 9 , 3091-3103 |
#865 | Repertoire of thermostable chimeras engineered by SCHEMA-RASPP for the degradation of bio-based plastics. |
|
| Presenting author: | DIANELIS TOLEDO MONTERREY of ICP-CSIC |
| Corresponding author: | MIGUEL ALCALDE of ICP-CSIC |
| Other authors: | IVAN MATELJAK of EVOENZYME MIKEL DOLZ of CSIC JAVIER VIÑA-GONZÁLEZ of EVOENZYME |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Chimeragenesis / SHEMA-RASPP / Plastic degradation / Thermostability |
| Purpose: | Cutinases (EC 3.1.1.74) are serine esterase enzymes belonging to the superfamily of α/β hydrolases that catalyze the hydrolysis of the ester bond. These enzymes are very attractive for their implementation in industrial processes such as cotton scouring and plastic degradation [1,2]. The increasing interest in cutinases has dramatically enhanced the portfolio of available enzymes (from bacterial and fungus sources) with a variety of features ranging from high thermostability to the ability to hydrolyze different plastic derivatives [3]. From this perspective, it would be worthwhile to combine the properties of the individual enzymes to perform a more efficient degradation of plastics. Enzyme chimeragenesis is a powerful engineering tool aimed at obtaining robust enzymes with enhanced thermostability and broader substrate scope [4]. Among the most successful strategies to generate chimeric proteins, the SCHEMA-RASPP algorithm, developed by the Frances Arnold group [5], has been applied to many enzymes systems, including P450s, β-lactamases, cellulases, arginases, channel rhodopsins and laccases. SCHEMA yields chimeric proteins by homologous recombination of different parental types and complemented by RASPP generates libraries by minimizing 3D disruption of the enzyme for a range of mutations [6]. Here, we present a repertoire of chimeras employing as parental types three cutinases orthologs with 55% sequence identity and diverse biochemical features. Our SCHEMA-RASPP library comprised 7 SCHEMA blocks and 2187 possible combinations. From the family of functional chimeras we preliminary identified 14 designs with striking improvements in thermostability and substrate promiscuity, opening up a venue for future protein engineering enterprises. |
| References: | [1] gururaj, p., khushbu, s., monisha, b., selvakumar, n., chakravarthy, m., gautam, p., & d. nandhini, g. prep. biochem. biotechnol. 2020, 51(6), 550-561. [2] c. knott, b., erickson, e., d. allen, m., e. gado, j., graham, r., l. kearns, f., pardo, i., topuzlu, e., j. anderson, j., p. austin, h., dominick, g., w. johnson, c., a. rorrer, n., j. szostkiewicz, c., copié, v., m. payne, c., l. woodcock, h., s. donohoe, b., t. beckham, g., & e. mcgeehan, j. proc. natl. acad. sci. u. s. a. 2020, 117(41), 25476-25485. [3] martinez, a., & maicas, s. 2021. catalysts, 11(10). [4] heinzelman, p., d. snow, c., wu, i., nguyen, c., villalobos, a., govindarajan, s., minshull, j., & h. arnold, f. proc. natl. acad. sci. u. s. a. 2009, 106(14), 5610-5615. [5] a. smith, m., & h. arnold, f. methods mol. biol. 2014, 1179, 335-343. [6] mateljak, i., rice, a., yang, k., tron, t., & alcalde, m. acs synth. biol. 2019, 8(4), 833-843. |
| Figures: | 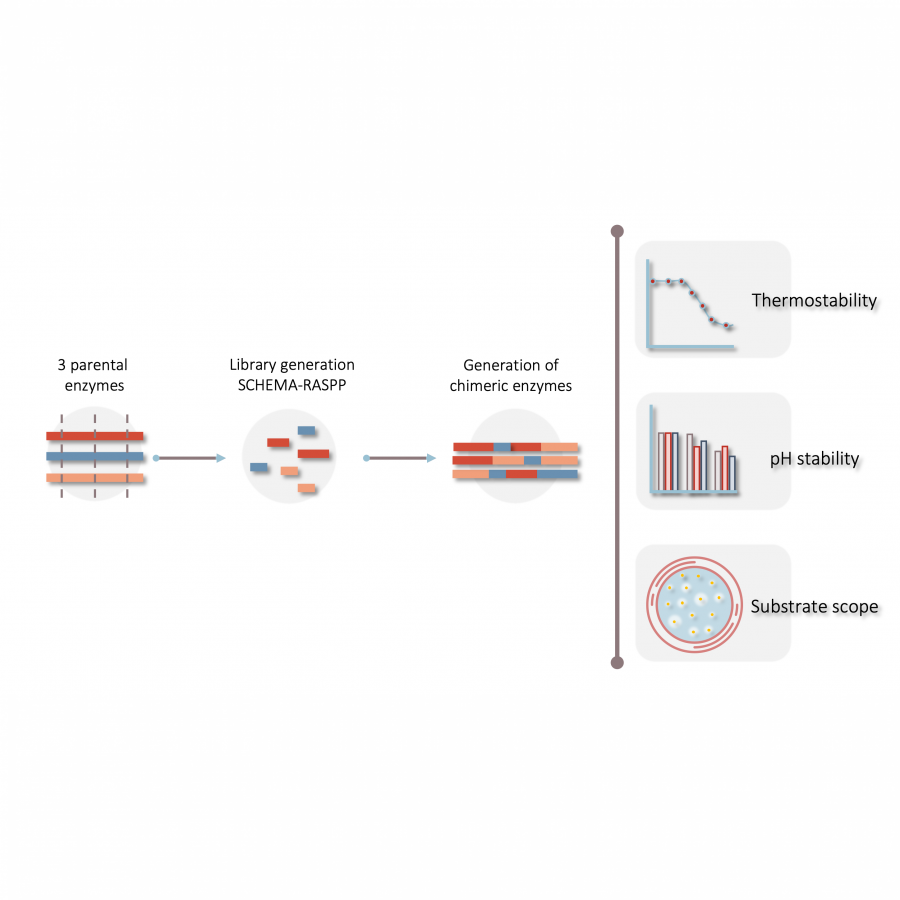 Graphical abstract Graphical abstract of chimeric enzymes generation and their further characterization. |
#866 | Enzymatic synthesis of sugar-based surfactants: for which properties? |
|
| Presenting author: | David GUIEYSSE of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Other authors: | Etienne SEVERAC of TOULOUSE BIOTECHNOLOGY INSTITUTE Chrystel FAURE of LABORATOIRE DE CHIMIE ET BIOLOGIE DES MEMBRANES ET NANO-OBJETS Fernando LEAL CALDERON of LABORATOIRE DE CHIMIE ET BIOLOGIE DES MEMBRANES ET NANO-OBJETS Magali REMAUD-SIMEON of TOULOUSE BIOTECHNOLOGY INSTITUTE Claire MOULIS of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | GH70 alpha-transglucosylases / glucosurfactant / glucodiversification / glucoconjugates |
| Purpose: | The global market of surfactants is one of the largest commodity markets in the world accounting for more $40 billion in 2020 and is projected to reach $52 billion in 2025[1]. Most of the surfactants are petro-sourced and synthesized by chemical transformation. The surfactant industry is increasingly interested in developing more environmentally friendly products, with market share increasing every year, especially with the growth of the biotech industries. This demand for biosurfactants is driving the industry to move away from synthetic surfactants to more sustainable alternatives. One way to achieve this is to develop new bio-based surfactants known for their biodegradability and low toxicity. Surfactants obtained from agroressources using enzymes are indeed increasingly in demand. In this context, enzymatic transglucosylation allowing the transfer of glycosidic motifs onto natural lipids can open access to a great diversity of products with a controlled hydrophilic-lipophilic balance [2]. GH70 alpha-transglucosylases that naturally catalyse the formation of a wide variety of glucose polymer from sucrose, a cheap and abundant glycosyl donor, are very good enzyme candidates to perform such reactions. Indeed, they are known to be quite efficient in transferring glucosyl moieties from sucrose onto non-natural acceptors, including different types of oligosaccharides, polyol, or polyphenol for example [3]. Our presentation will focus on the potential of these enzymes for the production of novel glucosylated lipid derivatives synthesized from sucrose and alkyl-glucosides or hydroxylated fatty acids of different sizes. We will show how our collection of GH70 transglucosylases comprising polymerases with different linkage specificities (alpha-1,2; alpha-1,3; alpha-1,4 and alpha-1,6) can be used to produce a very diverse library of new glucosurfactants. We will discuss their emulsifying, antimicrobial and stimuli-responsive properties. |
| References: | [1] C. Farias, F. Almeida, I. Silva, Thais Souza et al. Production of green surfactants: Market prospects. Electron. J. Biotechnol., 2021, 51, 28. [2] C. Moulis, D. Guieysse, S. Morel, E. Séverac, M. Remaud-Siméon. Natural and engineered transglycosylases: Green tools for the enzyme-based synthesis of glycoproducts. Current Opinion in Chemical Biology, 2021, 61, 96. [3] M. Molina, G. Cioci, C. Moulis, E. Séverac, M. Remaud-Siméon. Bacterial α-Glucan and Branching Sucrases from GH70 Family: Discovery, Structure-Function Relationship Studies and Engineering. Microorganisms, 2021 9 (8), 1607. |
#868 | DFF: a bio-based cross-linker for the immobilization of enzymes on both conventional and renewable carriers |
|
| Presenting author: | Chiara DANIELLI of UNIVERSITY OF TRIESTE |
| Other authors: | Luuk VAN LANGEN of VIAZYM B.V. Lucia GARDOSSI of UNIVERSITY OF TRIESTE |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | immobilization / crosslinker / bio-based / |
| Purpose: | Glutaraldehyde, a dialdehyde widely used as cross-linker for enzyme immobilization, possesses a widely documented toxicity.[1] Moreover, it has a very complex behavior in water solution: thirteen different forms have been identified, and it is still unclear which is responsible for enzyme immobilization.[2] Finally, it is industrially synthesized starting from fossil sources.[3] The objective of the present study is to find a renewable and potentially safer substitute for glutaraldehyde for enzyme immobilization. A bio-based dialdehyde has been systematically compared with glutaraldehyde, to determine whether their performances as enzyme cross-linkers and the stability of the final enzymatic preparations are comparable. The two dialdehydes were compared by immobilizing glucoamylase, a starch-hydrolyzing enzyme, on an amino-functionalized methacrylic carrier. The resulting enzyme preparations were analyzed in activity assays, as well as by a continuous flow experiment, to determine the recovered activity, the stability of the preparation under industrially relevant conditions and thus compare the performance of the two dialdehydes as cross-linkers. The novel cross-linker and glutaraldehyde had very similar performances in terms of recovered activity and stability of the immobilized enzyme preparations, evidencing the applicability of the new, bio-based molecule for enzyme immobilization in replacement of glutaraldehyde. The applicability of DFF was verified also studying the immobilization of different hydrolases on a lignocellulosic material. The resulting formulations, besides being fully bio-based, benefit from the lower toxicity of DFF which is less volatile than GA, easy to handle and has the additional advantage of reacting according to clear and simple reaction mechanisms. The latter feature enables its easier dosage as crosslinking agent while minimizing the chemical routes that might cause toxic effects. |
| References: | [1] https://www.atsdr.cdc.gov/sites/peer_review/tox_profile_glutaraldehyde.html [2] I. Migneault et al., BioTechniques, 2004, 37, 790-802 [3] C. Kohlpaintner et al., in Ullman's Encyclopedia of Industrial Chemistry, Wiley, 2013 |
#872 | Engineering of a bacterial cytochrome P450 monooxygenase for the synthesis of (-)-podophyllotoxin |
|
| Presenting author: | Alessa LAPPE of HEINRICH-HEINE UNIVERSITÄT |
| Other authors: | Mirco KEILHAMMER of PHILIPPS UNIVERSITÄT Joost LUELF of HEINRICH-HEINE UNIVERSITÄT Vlada B. URLACHER of HEINRICH-HEINE UNIVERSITÄT |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | P450 / protein engineering / (-)-podophyllotoxin / |
| Purpose: | The lignan (-)-podophyllotoxin is a natural plant precursor for the synthesis of the chemotherapeutics teniposide and etoposide, but its limited supply is dependent on the endangered plant Podophyllum hexandrum [1]. Therefore, the biosynthetic pathway towards (-)- podophyllotoxin was reconstituted in Escherichia coli [1]. An exception still is the unidentified enzyme catalyzing the last step of this cascade, the hydroxylation of ( ) deoxypodophyllotoxin to (-)-podophyllotoxin [1]. To overcome this boundary a cytochrome P450 monooxygenase (P450) was identified to catalyze the hydroxylation of (-)-deoxypodophyllotoxin to (-)-podophyllotoxin [2]. Still, the chemoselectivity is quite low and the reaction yields several products besides (-)-podophyllotoxin [2]. P450s are well described for their ability to introduce oxygen into non-activated C-H bonds of a vast range of chemically diverse compounds in the presence of molecular oxygen [3]. Their ability to catalyze regio- and stereoselective oxidations of complex molecules under mild conditions outcompetes many chemical catalysts [4] and makes them the right candidate to selectively hydroxylate complex compounds like (-)-deoxypodophyllotoxin. To achieve this reaction the two required electrons for the activation of molecular oxygen are delivered by NAD(P)H via redox partner proteins [4]. To extend activity or chemoselectivity of P450s, many enzymes were characterized over the last decades and optimized for specific applications using rational design and directed evolution methods [4]. In this work, we aimed to improve activity and regio- and stereoselectivity of a bacterial P450 towards the production of (-)-podophyllotoxin by means of rational protein design and saturation mutagenesis. Therefore, 12 residues in the active site identified from literature and docking studies were chosen for site-directed mutagenesis. The resulting mutants where analyzed in an implemented medium-throughput screening based on the whole cell conversion in E. coli in 96 deepwell plates and a LC/MS high-throughput screening. An identified mutant may allow to extend the enzyme cascade for the selective production of (-)-podophyllotoxin in E. coli. |
| References: | [1] D. Decembrino et al. Microb. Cell Fact. 2021, 20, 183 [2] D. Decembrino, 2021, Dissertation, Institut für Biochemie, Heinrich-Heine Universität Düsseldorf [3] V. B. Urlacher, M. Girhard, Trends Biotechnol. 2019, 37, 887-892 [4] Z. Li et al. J. Biol. Chem., 2020, 295, 833-849 |
#873 | Switchable UPO biocatalysis by genetically encoded photosensitizers |
|
| Presenting author: | Dominik HOMANN of MLU HALLE, INSTITUT FÜR CHEMIE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / unspecific peroxygenases / / |
| Purpose: | Unspecific peroxygenases (UPOs) can oxyfunctionalise a broad set of substrates only requiring hydrogen peroxide as a co-substrate. High turnover numbers, stabilities and excellent selectivities render UPOs exciting enzymes for C–H activations. Major challenges of UPOs – hetereologous expression, regioselective transformations and haem-bleaching – were intensive subject to different research projects.[1] Herein we report on a new approach to avoid haem-bleaching and thus inactivation of the enzyme. A fusion construct – photosensitizer, linker, UPO – was successfully designed an expressed in Pichia Pastoris. Photochemical as well as chemical optimisation followed and a broad substrate scope was screened. The PhotUPOs showed TONs in the range of a few thousand and ee-values above 99 %. This system adds an easy and switchable biophotocatalytic access to oxyfunctionalised C-H bonds. |
| Figures: | 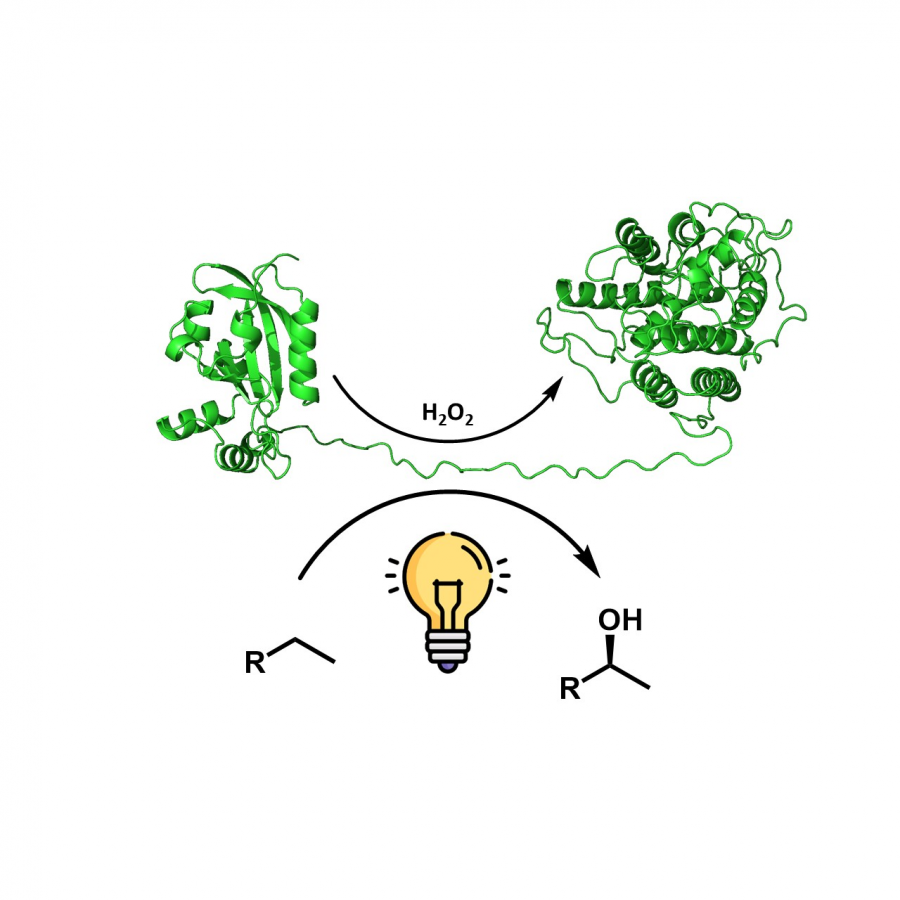 Figure 1: Principle of PhotUPO, utilizing a genetically encoded photosensitizer to fuel the UPO. . |
#875 | Biocatalysis for the valorisation of food-processing waste: the case of soapstock from seed oil refining |
|
| Presenting author: | Davide TESSARO of POLITECNICO DI MILANO |
| Other authors: | Elisabetta BRENNA of POLITECNICO DI MILANO Fabio PARMEGGIANI of POLITECNICO DI MILANO Valeria DE FABRITIIS of POLITECNICO DI MILANO Francesca TENTORI of POLITECNICO DI MILANO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Circular economy / Lipases / Epoxidation / Amidation |
| Purpose: | According to the United Nations' Food and Agricultural Organization (FAO), global food production will rise by more than 70% by 2050 to feed 9.1 billion people. Since significant amounts of waste and byproducts are inevitably produced throughout the agri-food chain during the processing and transformation of the products, their disposal is expected to have an increasingly negative impact on the environment and the economy. Therefore, the recovery and valorization of food waste are becoming key objectives in the framework of minimizing the environmental impact and improving the sustainability of the whole sector. We believe that, in this context, biocatalysis, relying on proteins purposely evolved to work with high efficiency on natural compounds, may play a major role in the transformation and valorization of many byproducts coming from the food industry. In particular, in the latest years, we devoted our attention to the seed oil refining process where we were able to apply biocatalytic tools to different byproducts (gums, soapstock, spent bleaching earth) employed as raw materials for the synthesis of valuable molecules. In this presentation, we will focus on the chemo-enzymatic treatment of soapstock. Soapstock is one of the most abundant by-products of vegetable oil refinement and consists of a sticky alkaline emulsion containing approximately 50% water, 10% sodium salts of fatty acids, 10% triglycerides and small percentages of partially hydrolyzed lipids. Herein we provide two examples where the relatively abundant oleic acid coming from HOSO (High Oleic sunflower Oil) soapstock was employed as the starting compound for the synthesis of valuable compounds. In both cases, the preliminary step was a lipase-mediated splitting of the soapstock affording a mixture of fatty acids (80-87% oleic acid). Then, this intermediate material was employed in two different syntheses. 1) In the first process, the self-epoxidation of oleic acid upon lipase-mediated perhydrolysis in the presence of hydrogen peroxide afforded the corresponding epoxide, which was subsequently hydrolyzed to the diol derivative and, finally, oxidized to commercially valuable azelaic and pelargonic acids (Figure 1). Epoxidation and glycol cleavage were optimized through a statistical approach and implemented under continuous-flow conditions to increase yields and productivity. [1,2] 2) In the second application, a lipase promoted the condensation of oleic acid with ethanolamine to provide the corresponding amide in good yield and excellent chemical purity (Figure 2). The use of a packed-bed reactor in continuous flow mode permitted to improve the spacetime yield of the reaction and the catalyst productivity. [3] |
| References: | [1] Brenna, E., Colombo, D., Di Lecce, G., Gatti, F. G., Ghezzi, M. C., Tentori, F., Tessaro D., Viola, M., 2020, Molecules, 25(8), 1882-1892. [2] Casali, B., Brenna, E., Parmeggiani, F., Tentori, F., Tessaro, D., 2022, Green Chemistry, 24(5), 2082-2093. [3] Brenna, E., De Fabritiis, V., Parmeggiani, F., Tentori, F., Tessaro, D., 2023, ACS Sustainable Chemistry & Engineering, 2764-2772. |
| Figures: | 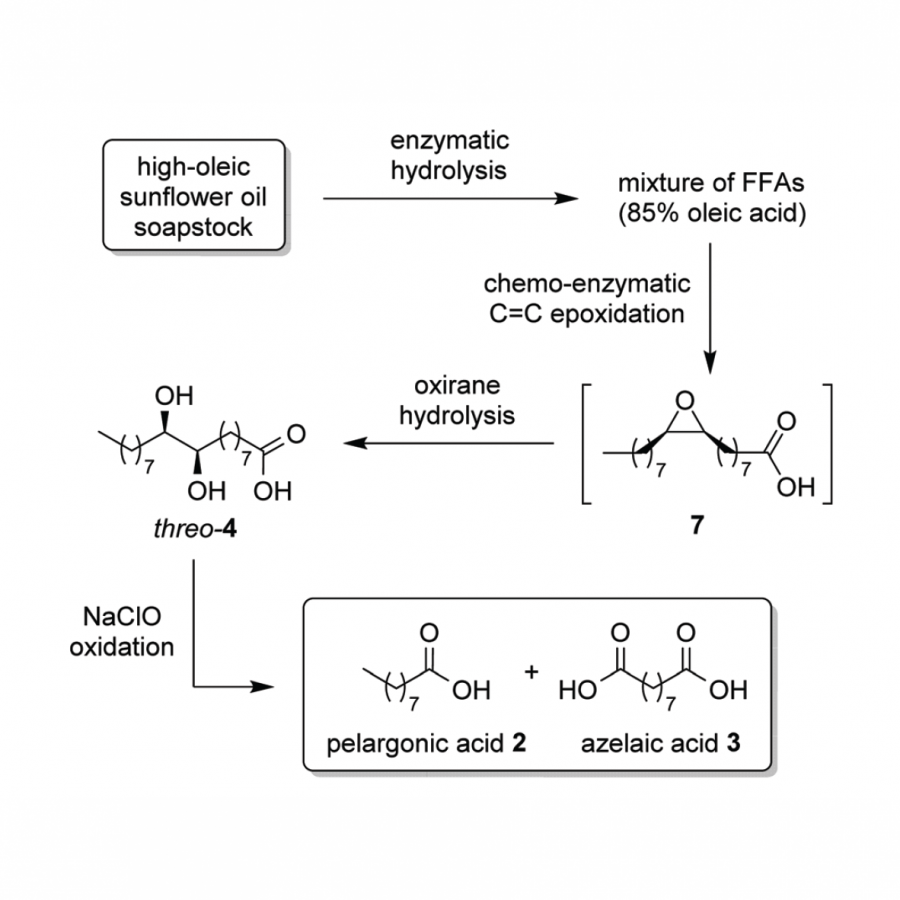 The first process Chemo-enzymatic synthesis of azelaic and pelargonic acids from soapstock  A second process From soapstock to oleyl ethanolamide |
#877 | Discovery enzyme-substrate interactions via numerical representations strategies and machine learning algorithms |
|
| Presenting author: | David MEDINA of UNIVERSIDAD DE MAGALLANES |
| Other authors: | Roberto URIBE-PAREDES of UNIVERSIDAD DE MAGALLANES Alvaro OLIVERA-NAPPA of UNIVERSIDAD DE CHILE Marcelo NAVARRETE of UNIVERSIDAD DE MAGALLANES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Machine learning algorithms / Numerical representations / enzyme-substrate affinity prediction / Protein languange models |
| Purpose: | Several computational models have been proposed to predict the affinity between an enzyme and its substrate. In most cases, structural information of both the enzyme and the substrate, together with molecular dynamics, are used as input for constructing predictive systems based on complex deep learning architectures. This strategy hampers its widespread application due to the high computational cost involved. In addition, in many cases, obtaining the secondary structure of the enzymes requires the use of computational systems for its prediction, adding up to the need of computation. Methods based on linear representation can address this issue. Representation via amino acid sequence for enzymes and SMILE for substrates have provided promising results, that nevertheless lack generalization capability and only allow classification recognition systems, limiting the application for the estimation of affinity between enzyme and substrate. This work explores alternatives to solve the efficient affinity prediction between an enzyme and a substrate. To this aim, we collected all the reactions reported in the KEGG database. Next, we build a data set of the affinities between all the enzymes and the substrates through virtual screening. This step enabled the calculation of a numerical value representing affinity. Lastly, we explored computational strategies for the numerical representation of the enzymes, substrates, and the interaction complex. Since our overall strategy aims to save computational power we included linear information, i.e., amino acid sequences and SMILES. Different amino acid codings were explored, including methods based on physicochemical properties, applications of transformations through convolutions, like the Fast Fourier Transform, and strategies based on pre-trained models via natural language processing methods. In the case of SMILES, methods based on learning numerical representations such as Mol2Vec, graph junction tree, or similar were explored. The interaction complexes were approached using concatenation methods, non-linear convolution, or combination strategies. First, the concatenation strategies were explored to build the dataset by combining the different methods explored for enzymes and substrate and building the predictive models using classic machine learning algorithms and deep learning architectures, like convolutional neural networks (CNN) and long short-term memory (LSTM). In this case, the best performance was 0.35 for the combination of secondary structure-property to represent the enzymes and Mol2Vec for representing the smiles with a random forest algorithm. Then, the convolutional or combination methods were explored, employing PCA strategies and Kernel-PCA techniques for all combinations of representation between enzyme and substrate representation. In this case, the best performance was achieved by the application of PCA techniques to the dataset built by the esm1b pre-trained model (in the case of enzymes) and Mol2Vec (in the case of SMILES) in the predictive model trained using LSTM architectures, achieving a performance of 0.56 Pearson coefficient. The model developed with natural language strategies, LSTM, and linear combinations strategies achieved better results than the concatenation methods, demonstrating synergy between the NLP and LSTM architectures. The generated model has been developed for usability purposes to correlate mutational changes in enzymes with the affinity between substrates. In addition, this model allows simulating interactions, which facilitates the discovery of new reactions between enzymes and substrates, becoming a relevant and advantageous strategy for landscape navigation and the discovery of new interest molecules. In future work, the model will be the improvement of the architecture and compare the results with methods available in the literature. |
#879 | Turning a hyperthermostable lactonase into a proficient phosphotriesterase for the decontamination of organophosphorus chemicals |
|
| Presenting author: | Pauline JACQUET of GENE&GREENTK |
| Corresponding author: | David DAUDÉ of GENE&GREENTK |
| Other authors: | Mikael ELIAS of UNIVERSITY OF MINNESOTA Eric CHABRIÈRE of AIX MARSEILLE UNIV / IHU-MÉDITERRANÉE INFECTION David DAUDÉ of GENE&GREENTK |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Phosphotriesterase / Hyperthermostable / Organophosphorus chemicals / Detoxification |
| Purpose: | Organophosphorus (OP) chemicals are neurotoxic compounds developed as pesticides (such as chlorpyrifos or malathion) and chemical warfare nerve agents (CWNA, such as sarin or VX). These compounds are responsible of worldwide pollution and represent a serious terrorist threat. Developing safe and efficient decontamination strategies against OP poisoning is of outmost interest and OP-degrading enzymes have gained special attention [1]. In that context, we focused on the enzyme SsoPox, a hyperthermostable phosphotriesterase-like lactonase, isolated from the extremophilic archaeon Saccharolobus solfataricus. This enzyme is highly robust and constitutes a promising candidate for engineering strategies. Considering its structural similarities with bacterial phosphotriesterases, SsoPox was engineered for increasing its capacity to degrade OP chemicals. Combinatorial approaches were used to increase catalytic performances and various improved variants were obtained. These variants carried from two to seven mutations and up to 2210-fold improvement were achieved [2,3]. Among these, two variants with tremendous degradation capacity towards OP were further considered for detoxification purposes. Two animal models were implemented to evaluate the protective effect of SsoPox variants against OP poisoning. Variant SsoPox-αsD6, improved against numerous pesticides (including chlorpyrifos, malathion or diazinon), was able to decrease toxicity and enhance survival in planarian flatworms exposed to acute poisoning and to protect them from long term effects upon chronic exposure [4,5]. This enzyme was further immobilized in a filtration device that may find interest for decontaminating toxic effluents [6]. Another variant, SsoPox-IIIC1, enhanced on ethyl-paraoxon and CWNA analogues, was encapsulated in nanoreactors made of polyethylene glycol−polypropylene sulfide membrane. These nanoreactors containing SsoPox were used to detoxify ethyl-paraoxon in mice in both prophylaxis and post-exposure treatments [7]. |
| References: | [1] Jacquet, P. et al. Current and emerging strategies for organophosphate decontamination: special focus on hyperstable enzymes. Environmental Science and Pollution Research 23, 8200-18 (2016). [2] Jacquet, P. et al. Rational engineering of a native hyperthermostable lactonase into a broad spectrum phosphotriesterase. Scientific Reports 7, (2017). [3] Jacquet, P et al. Changes in Active Site Loops Conformation Relates to a Transition From Lactonase to Phosphotriesterase. in preparation. [4] Poirier, L. et al. Enzymatic degradation of organophosphorus insecticides decreases toxicity in planarians and enhances survival. Scientific Reports 7, (2017). [5] Poirier, L., Plener, L., Daude, D. & Chabriere, E. Enzymatic decontamination of paraoxon-ethyl limits long-term effects in planarians. Sci Rep 10, 3843 (2020). [6] Poirier, L. et al. Evaluation of a robust engineered enzyme towards organophosphorus insecticide bioremediation using planarians as biosensors. Chemico-Biological Interactions 306, 96-103 (2019). [7] Pashirova, T. et al. Enzyme Nanoreactor for In Vivo Detoxification of Organophosphates. ACS Appl. Mater. Interfaces 14, 19241-19252 (2022). |
#880 | Polyphosphate kinases: the perfect enzymes for phosphorylation? |
|
| Presenting author: | Jennifer ANDEXER of UNIVERSITY OF FREIBURG, INSTITUTE OF PHARMACEUTICAL SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Polyphosphate kinases of the family 2 (PPK2) have become popular biocatalysts for the regeneration of adenosine 5´-triphosphate (ATP), as well as the biocatalytic production of nucleotides. Based on phylogenetic analyses PPK2s have been assigned to three different classes I, II, and III that catalyse the phosphorylation of ADP, AMP, and both nucleotides, respectively. We used combinations of these enzymes successfully for the phosphorylation of nucleotides, e.g. in biomimetic systems for the regeneration of cofactors such as S adenosylmethionine and coenzyme A; as well as for the production of nucleotide triphosphates (NTPs) from non-physiological nucleosides. By analysing three-dimensional structures from all classes of PPKs and a range of biochemical assays using physiological and non-physiological substrates, we are working on the elucidation of the PPK2s’ molecular mechanism and substrate discrimination. This includes the analysis of polyP molecules with varying chain lengths and nucleoside derivatives such as adenosine 5´-tetra- und pentaphosphate. Inorganic polyphosphate (polyP) is an abundant, inexpensive and stable phosphate donor, and PPK2s can be used for the phosphorylation of AMP as well as ADP. Nevertheless, during our efforts to optimise the PPK2-catalysed reactions for the biocatalytic production of NTPs, we could never reach full conversion to the corresponding NTP. An investigation of the thermodynamic equilibrium of these enzymes in comparison with family-1 PPKs and literature data from other kinases shows that the thermodynamic equilibrium of the PPK reaction is located less far on the NTP side than it is the case for other kinases; this can be traced back to the phosphate transfer potential of the corresponding phosphate donors. The results raise the question how suitable PPKs are for biocatalytic applications, including syntheses of NTPs and NTP regeneration systems. |
| References: | K. Motomura, R. Hirota, M. Okada, T. Ikeda, T. Ishida, A. Kuroda, Appl. Environ. Microbiol. 2014, 80, 2602-2608. J. N. Andexer, M. Richter, ChemBioChem 2015, 16, 380-386. M. Tavanti, J. Hosford, R. C. Lloyd, M. J. B. Brown, ChemCatChem 2021, 13, 3565-3580. L. Gericke, D. Mhaindarkar, L. Karst, S. Jahn, M. Kuge, M. K. F. Mohr, J. Gagsteiger, N. V. Cornelissen, X. Wen, S. Mordhorst, H. J. Jessen, A. Rentmeister, F. P. Seebeck, G. Layer, C. Loenarz, J. N. Andexer, 2022, biorxiv 2022.09.26.509380. S. Mordhorst, A. Maurer, D. Popadic, J. Brech, J. N. Andexer, ChemCatChem. 2017, 9, 4164-4168. S. Mordhorst, J. Siegrist, M. Müller, M. Richter, J. N. Andexer, Angew. Chem. Int. Ed. 2017, 56, 4037-4041. S. Mordhorst, J. Singh, M. Mohr, R. Hinkelmann, M. Keppler, H. J. Jessen, J. Andexer, ChemBioChem 2019, 20, 1019-1022. D. Popadić, D. Mhaindarkar, M. H. N. D. Thai, H. C. Hailes, S. Mordhorst, J. N. Andexer, RSC Chem. Biol. 2021, 2, 883-891. M. Keppler, S. Moser, H. J. Jessen, C. Held, J. N. Andexer, Beilstein J. Org. Chem. 2022, 18, 1278-1288. |
| Figures: | 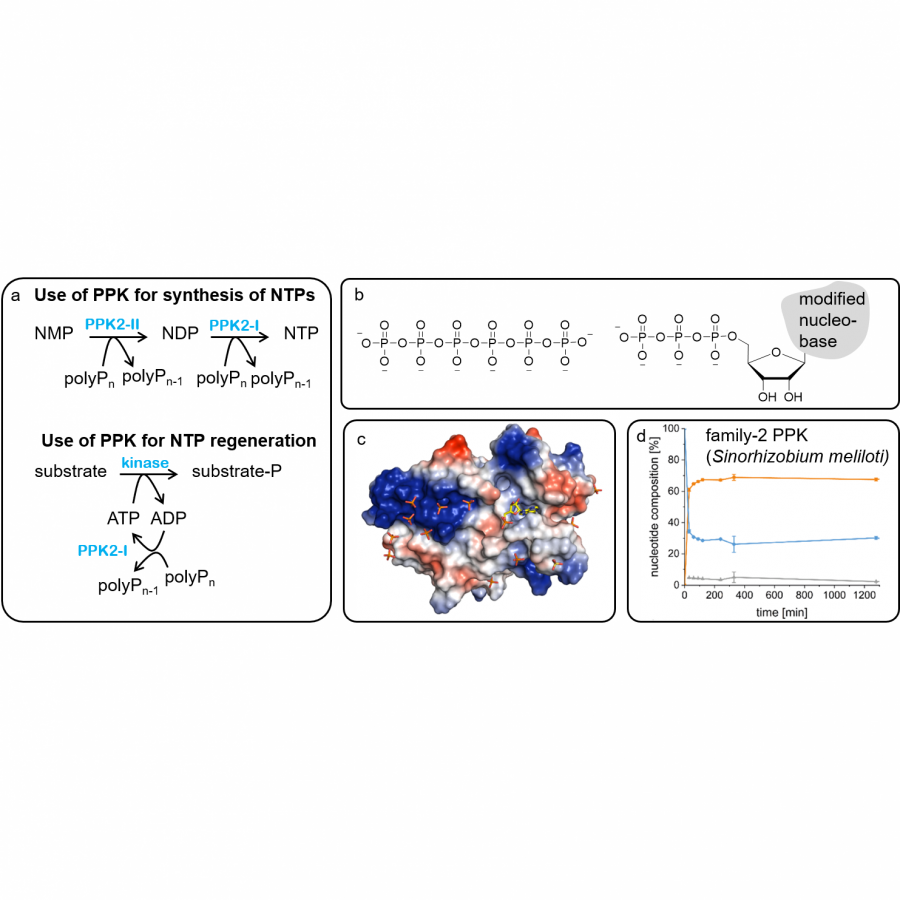 Family-2 polyphosphate kinases. a: Use of PPK2s for eiter biocatalytic synthesis of NTPs or ATP regeneration coupled to another kinase reaction; b: chemical structures of polyP and an NTP.; c: three-dimensional structure of a class II PPK2 with co-crystallised substrate; d: thermodynami |
#883 | Immobilisation of oleate hydratase on solid supports |
|
| Presenting author: | Keiko OIKE of DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY |
| Corresponding author: | Peter-Leon HAGEDOORN of DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Rob SCHOEVAART of CHIRALVISION B. V. Frank HOLLMANN of DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY Ulf HANEFELD of DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Immobilisation / Water addition / Fatty acid hydratase / Biorenewable |
| Purpose: | Oleate hydratase (Ohy) is an FAD dependent enzyme that catalyzes the hydration of unsaturated fatty acids forming hydroxy-fatty acids.[1] This catalytic activity makes this enzyme a potential catalyst for valorisation of biorenewable fatty acids into products for the chemical and polymer industry. However, the solubility of fatty acids in aqueous media is strongly limited and high amounts of co-solvents are necessary for solubilisation, which are reported to deactivate the enzyme.[2] In this regard, we studied the immobilisation of the Ohy from Rhodococcus erythropolis on commercially available solid supports. Immobilisation is a powerful technique to improve the performance of enzymes in biotechnological processes. The carrier binding separates the enzyme from the reaction medium and prevents protein aggregation, thus, often leading to higher stability.[3] However, there is only one report of a successful covalent immobilisation of Ohy on chitosan.[4] We used a screening kit[5] to test carriers (spherical polymeric beads) with different interaction types: a) covalent, b) adsorption, c) cationic, d) anionic and e) His Ni2+. The immobilisation process was evaluated by determination of the protein concentration in the supernatant before and after immobilisation as well as with activity measurements. We screened 18 different carriers from ChiralVision[5] and achieved a maximum activity recovery of approximately 30 % compared to the free enzyme, which is a good value taking into account that enzymes are often (partly) deactivated during the immobilisation process. Subsequently, we characterised the immobilised Ohy and studied its stability against different co-solvents and higher temperature. Leaching of the immobilised enzyme into the supernatant and storage stability were investigated as well. Lastly, we conducted recycling experiments with the immobilised enzyme. In conclusion, we report the first successful immobilisation of Ohy on solid carriers by ionic interaction. |
| References: | [1] P.-L. Hagedoorn, F. Hollmann, U. Hanefeld, Appl. Microbiol. Biotechnol. 2021, 105, 6159-6172. [2] J. Löwe, H. Gröger, Catalysts 2020, 10, 287. [3] R. A. Sheldon, A. Basso, D. Brady, Chem. Soc. Rev. 2021, 50, 5850-5862. [4] A. Todea, A. Hiseni, L. G. Otten, I. W. C. E. Arends, F. Peter, C. G. Boeriu, J. Mol. Catal. B Enzym. 2015, 119, 40-47. [5] ChiralVision, Product list 2022, https://chiralvision.com/products/immobeads, accessed on 15th March 2023. |
| Figures: | 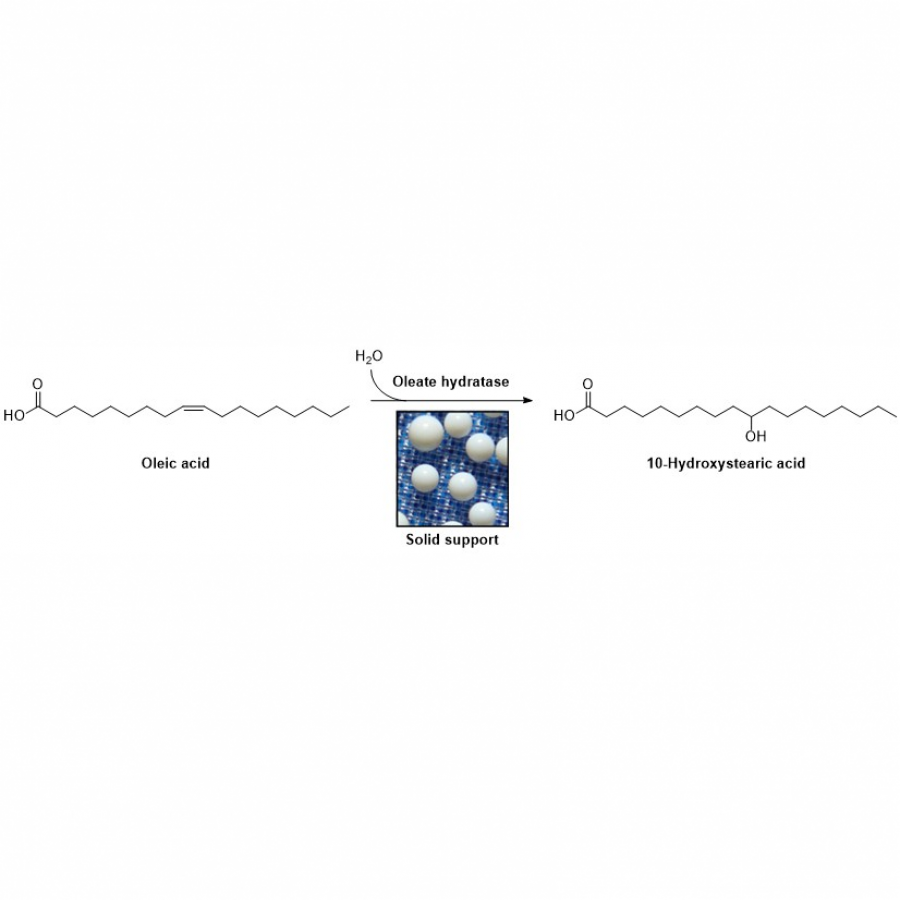 Figure 1 Hydration of oleic acid catalysed by immobilised oleate hydratase. |
#885 | Self-Sufficient Heterogeneous Biocatalysts in continuous flow asymmetric synthesis of β-Hydroxy esters |
|
| Presenting author: | Daniel ANDRÉS-SANZ of CENTRO DE INVESTIGACIÓN COOPERATIVA EN BIOMATERIALES CIC BIOMAGUNE |
| Other authors: | Fernando LÓPEZ-GALLEGO of CENTRO DE INVESTIGACIÓN COOPERATIVA EN BIOMATERIALES CIC BIOMAGUNE Ainho MAIZ of CENTRO DE INVESTIGACIÓN COOPERATIVA EN BIOMATERIALES CIC BIOMAGUNE |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cofactor Recycling / Industrial Biocatalysis / Beta-Hydroxy esters / Flow biocatalysis |
| Purpose: | β-Hydroxy esters find applications in the pharmaceutical, food and polymer industries for the synthesis of functional products. While conventional synthetic methods rely on chemical catalysis or whole-cell biosynthesis, the asymmetric reduction of β-ketoesters using cell-free enzymes presents a viable approach for the production of enantiomerically pure β-hydroxy esters. [1] However, the unbearable economic costs associated with enzymes and cofactors calls for the development of approaches to maximize their utilization. Herein, we report the development of two Self-Sufficient Heterogeneous Biocatalyst (SS-HBs) for the enantiodivergent and cost-effective synthesis of β-hydroxy esters from β-keto esters . Firstly, we carried out an analysis of the reductive activity of two enzymes: a thermophilic (S)-3-hydroxybutyryl-CoA dehydrogenase from Thermus thermophilus HB27 (TtHBDH) [1] and an (R)-Ketoreductase from Lactobacillus kefir (LkKRED) [2] against various β-keto esters. Subsequently, we immobilized them onto macroporous agarose beads and coated them with different cationic polymers to stabilize the quaternary structure of the enzymes and to co-immobilize the required cofactors. As a result, we managed to obtain two SS-HBs that did not require any exogenous supply of NAD(P)H, as both biocatalysts were able to catalyze their own cofactor recycling through 2-propanol oxidation. We constructed two packed bed reactors and determined the optimal operating conditions by computational simulations and evaluating its productivity at different substrate concentrations (50 - 1000 mM) and residence times. The behavior of the operational stability of the SS-HBs was tested at the optimal conditions, resulting in a Space-time yield (STY) of 55 g L-1 h-1 in the synthesis of enantiopure ethyl 3-(R)-hydroxybutyrate for 6 days with enzyme and NADPH TTN of 350000 and 10300 respectively. In the continuous synthesis of ethyl 3-(S)-hydroxybutyrate we obtained a STY between 49-27 g L-1 h-1 in 2 days with enzyme and cofactor TTN of 146000 and 2980 respectively. A deeper analysis revealed that the loss of activity was caused by thermal decomposition of NADH under operational conditions which was resolved by removing and replacing the cofactor, thus restoring maximum productivity. In essence, the development of robust and self-sustaining biocatalysts allows for reducing the NAD(P)H costs for the production of β-Hydroxy esters. In this case, from 200 € g-1 to less than 0.07€ g-1 for of ethyl 3-(S)-hydroxybutyrate cell-free production. Consequently, this technology enables the use of these biocatalysts in industrial settings, achieving the necessary cost-effectiveness benchmarks for efficient processes. |
| References: | [1] Orrego, A. H.; Andrés-Sanz, D.; Velasco-Lozano, S.; Sanchez-Costa, M.; Berenguer, J.; Guisan, J. M.; Rocha-Martin, J.; López-Gallego, F. Catal. Sci. Technol. 2021, 11, 3217- 3230, [2] A. Weckbecker, W. Hummel, Biocatal. Biotransform. 2006, 24, 380-389. [3] Bowie, James U. et al.Trends in Biotechnology, Volume 38, Issue 7, 766 - 778 |
| Figures: | 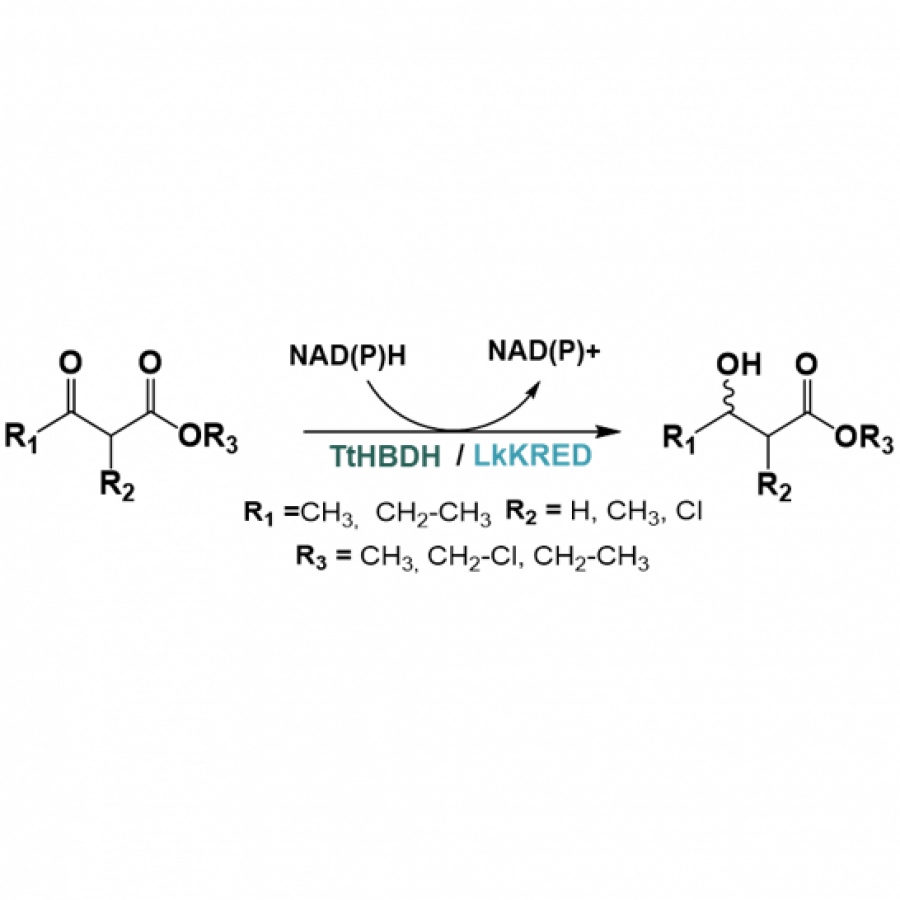 Figure 1 Reaction scheme of the asymmetric reduction of different beta-ketoesters by the S-selective NADH-dependent TtHBDH or R-selective NADPH dependent LkKRED 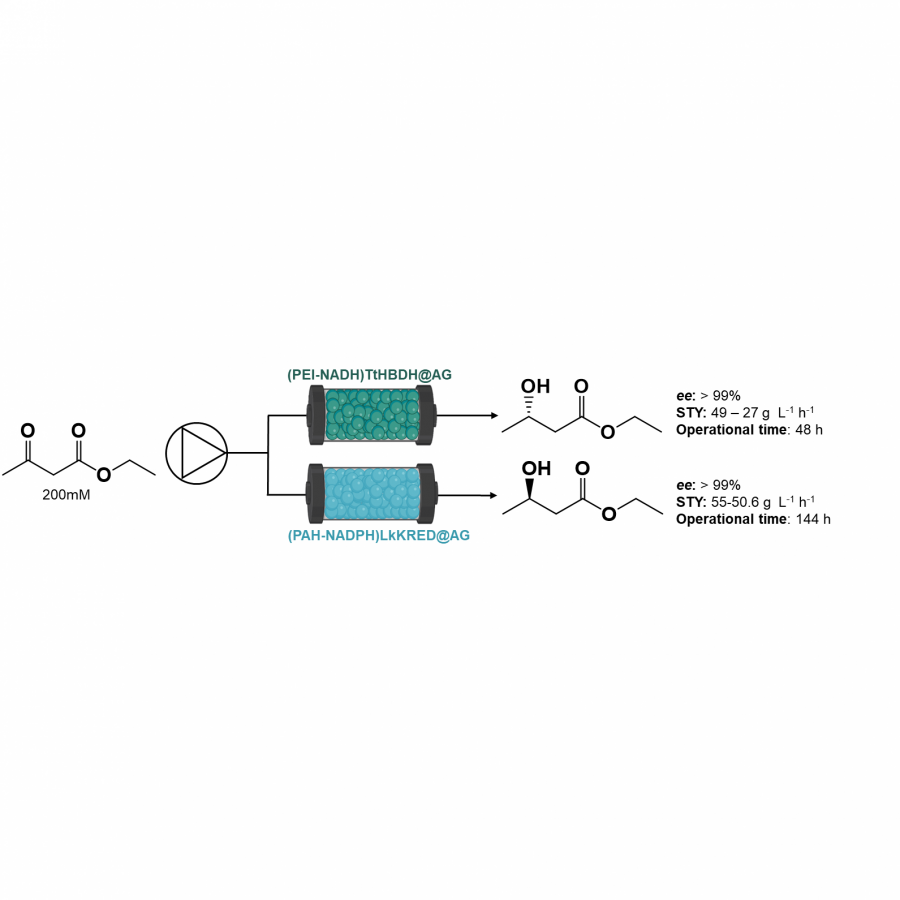 Figure 2 Reaction set-up for Self-Sufficient Heterogeneous Biocatalyst packed in Packed Bed Reactors |
#898 | Application of the C3-Methyltransferase StspM1 as Catalyst for the Formation of the Pyrroloindole motive in Natural Product Synthesis |
|
| Presenting author: | Mona HAASE of HHU DÜSSELDORF; INSTITUTE OF BIOORGANIC CHEMISTRY |
| Other authors: | Benoit DAVID of INSTITUTE OF BIO- AND GEOSCIENCES, IBG-4: BIOINFORMATICS; FORSCHUNGSZENTRUM JÜLICH Holger GOHLKE of INSTITUTE OF BIO- AND GEOSCIENCES, IBG-4: BIOINFORMATICS; FORSCHUNGSZENTRUM JÜLICH; HHU DÜSSELDORF; INSTITUTE FOR PHARMACEUTICAL & MEDICINAL CHEMISTRY Diana AMARIEI of HHU DÜSSELDORF; INSTITUTE OF BIOORGANIC CHEMISTRY Marcel SCHATTON of HHU DÜSSELDORF; INSTITUTE OF BIOORGANIC CHEMISTRY Jörg PIETRUSZKA of HHU DÜSSELDORF; INSTITUTE OF BIOORGANIC CHEMISTRY; FORSCHUNGSZENTRUM JÜLICH; INSTITUTE OF BIO- AND GEOSCIENCES IBG-1: BIOTECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Methylation / Indole / Preparative biocatalytic reaction / |
| Purpose: | The "Magic Methyl Effect" is a well-known phenomenon in medicinal chemistry, which describes that selective methylation of potential active drugs can boost their biological activity [1]. Using conventional chemical methods, methylation is still challenging regarding regio- and stereoselectivity [2]. In this context, the development of a suitable methylation method is an important task to produce active substances selectively and effectively. Having a look into nature, methyltransferases are used for methylation and to create interesting frameworks in a stereoselective manner [3]. In this work, the methyltransferase StspM1 is used for the diastereoselective methylation of diketopiperazines to form the pyrroloindole structural motive, which is widely present in different natural products like physostigmine, nocardioazine B and lansai B [4,5,6]. Besides the experimentally analyses of the substrate acceptance, the protein 3D structure was modelled and the binding mode of different DKP substrates and methylated products was predicted leading to interesting insides of the substrate binding and conversion within the binding pocket. For the synthetic utility of the methyltransferase, a cofactor recycling of SAM [7] was successfully implemented and the reaction conditions were optimized via a design of experiment approach. With this new established protocol, enzymatic methylation is feasible in a preparative scale with very good yields. |
| References: | [1] C.S. Leung et al., J. Med. Chem. 2012, 55, 4489-4500. [2] G.-J. Mei et al., Chem. Soc. Rev. 2021, 50, 5985-6012. [3] A. W. Struck et al., ChemBioChem 2012, 13, 2642-2655. [4] A. D. Borthwick, Chem. Rev. 2012, 112, 3641-3716. [5] H. Li et al., Chem. Commun. 2019, 55, 8390-8393. [6] a) Schneider et al., Angew. Chem. Int. Ed. 2021, 60, 23412-23418; b) D. A. Amariei et al., ACS Catal. 2022, 12, 14130-14139. [7] Liao, C. et al, Nat. Catal, 2019, 2(8), 696-701. |
| Figures: | 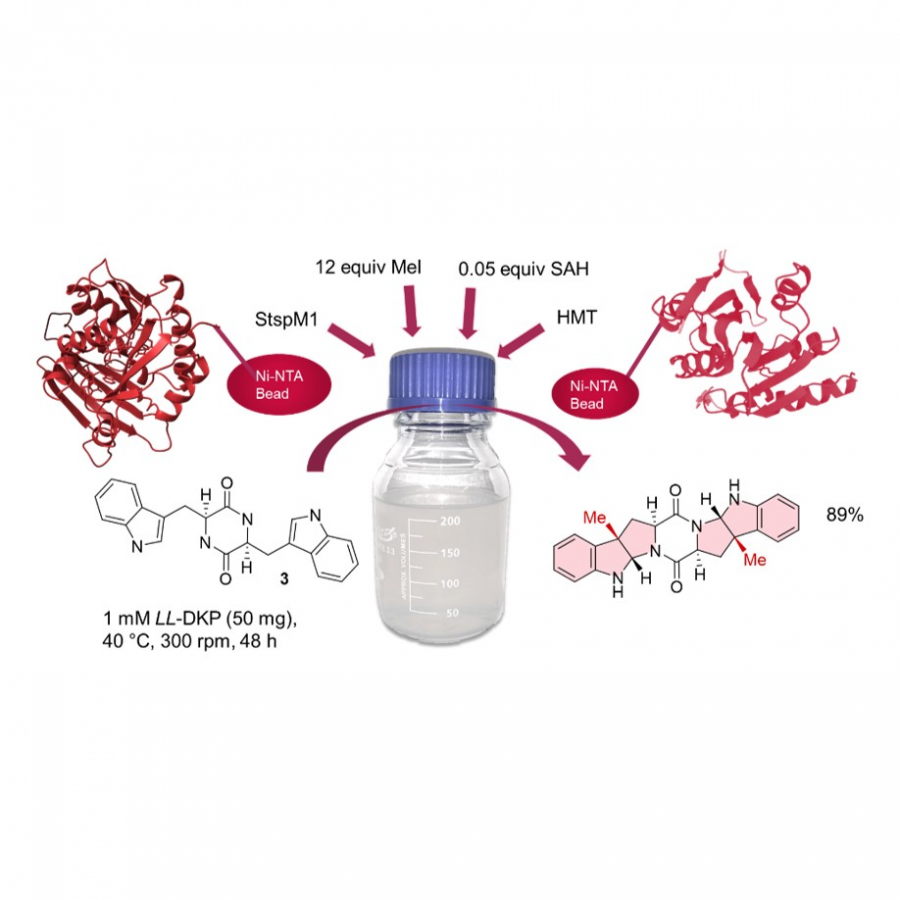 Biocatalytic methylation of LL-DKP Application of StspM1 (immobilized) in preparative scale using a recycling system with the halide methyltransferase (HMT) and S-adenosyl homocysteine (SAH) to methylate the substrate diketopiperazine (LL-DKP). |
#901 | Production of indigo in the cyanobacterium Synechocystis sp. 6803 by expression of a flavin-containing monooxygenase |
|
| Presenting author: | GIOVANNI LOPRETE of UNIVERSITY OF PADOVA |
| Corresponding author: | Elisabetta BERGANTINO of UNIVERSITY OF PADOVA |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Whole-cell biocatalysis / Indigo dye / Synthetic biology / Synechocystis |
| Purpose: | Production of indigo in the cyanobacterium Synechocystis sp. 6803 by expression of a flavin-containing monooxygenase Giovanni Loprete1, Caterina Martin2, Marco W. Fraaije3, Elisabetta Bergantino1 1 Department of Biology, University of Padova, Italy. 2 GECCO Biotech, Groningen, The Netherlands. 3 Molecular Enzymology, University of Groningen, Groningen, The Netherlands. Indigo is one of the most produced dyes in the world; except for being famous for the colouring of denim, it has a wide variety of other uses. It is currently produced by chemical processes putting a heavy burden on the environment [1]. To answer the demand for clean synthesis routes, biotechnological processes are being developed based on the employment of enzymes. Flavin-dependent monooxygenases (FMOs), naphthalene dioxygenases or cytochrome P450 are explored as biocatalysts in different configurations, i.e. as isolated (engineered) recombinant enzymes or in whole-cell biotransformations using recombinant expression in E. coli [2]. These oxidative enzymes catalyse the oxidation of indole to indoxyl, resulting in indigo formation. In turn, the substrate indole can be produced, together with pyruvate and ammonia, from L-tryptophan by employing tryptophanase (TRP). To our knowledge, the highest yield reported in the literature for such two-step system is 1,7 g of indigo per liter of culture grown for 3,5 days in a medium containing 2 g/L of l-tryptophan [2]. The use of isolated enzymes to drive redox reactions is hampered by their need of highly expensive redox cofactors such as NADPH. Cofactor regeneration is one of the main bottlenecks for the application of such biocatalysts at an industrial scale [3]. Recently, the possibility to use the photosynthetic apparatus of cyanobacteria for NADPH (re)generation has emerged. Among all cyanobacteria, Synechocystis sp. PCC6803 is progressively attracting interest as cell factory for the production of commodity chemicals, fuels and value-added products by essentially consuming carbon dioxide and water using light as energy source. Synechocystis strains expressing different enzymes have already been successfully tested in biotransformations in whole-cell configurations [4]. We have produced a Synechocystis strain constitutively expressing the FMO from Methylophaga aminisulfidi¬vorans (mFMO). Grown in batch and fed with 0.12 g/L indole as substrate, this strain produced 0.10 g/L of indigo. Noteworthy, indigo was found to be present in the supernatant in the form of small, dark blue agglomerates attached to the flasks. As stated above, our interest is to carry out the two-step cascade starting from L-tryptophan and to enhance the yield of indigo. We have therefore produced a second Synechocystis strain expressing both enzymes, TRP and mFMO, either encoded on an operon or expressed as a bifunctional fusion enzyme [2]. We will report on the results and the progress of this work. |
| References: | [1] Mocquard, le Lamer, Fabre, Mathieu, Chastrette, Vitrai and Vandenbossche (2022). Dyes and Pigments. 207, 110675. [2] Fabara and Fraaije (2020). Applied Microbiology and Biotechnology. 104, 925. [3] Böhmer, Köninger, Gómez-Baraibar, Bojarra, Mügge, Schmidt, Nowaczyk and Kourist (2017). Catalysts. 7, 240. [4] Malihan-Yap, Grimm and Kourist (2022). Chemie Ingenieur Technik. 11, 1628-1644. |
#902 | Unveiling the hidden side of brown-rot wood decay in anoxia |
|
| Presenting author: | Robert ROELLIG of BBF (INREA) MARSEILLE |
| Other authors: | Jean-Guy BERRIN of BBF (INREA) MARSEILLE |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | brown-rot wood decay / Fenton chemistry / anoxic fermentation / carbohydrate-active enzymes |
| Purpose: | Global forests are a large and persistent carbon sink [1] with fungal brown-rot wood decay playing a key role in the carbon cycle by participating in organic matter turnover [2]. For decades, this destructive decaying process was claimed to mainly rely on Fenton chemistry initiating the efficient degradation of plant cell wall polysaccharides [3]. However, the importance of O2 in this microbial degradation process has been overlooked. In this study, we designed fungal fermentation to study the effect and influence of O2 on this wood-decaying process. We developed a methodological set-up that enabled us to follow fungal growth, enzyme secretion, and biomass degradation using the brown rot fungi model organisms. Using O2 gradients and defined O2 concentrations (0 – 20.9%), we uncovered an anoxic lifestyle of Fomitopsis pinicola, which was able to grow on softwood in the absence of O2. Applying complementary biochemical techniques and deep proteomic analysis, we confirmed the presence of Fenton markers in the O2 growth condition and revealed its mechanism of wood degradation in the absence of O2. Strikingly, anoxic conditions induced the secretion of a wide range of cellulases and hemicellulases targeting both glucuronoxylan and galactomannan. In conclusion, we unveiled fungal wood decay under anoxic conditions highlighting, in a brown-rot fungus, the importance of carbohydrate-active enzymes for polysaccharide degradation. These results challenge the established dogma of Fenton chemistry-dependent brown-rot decay. We are convinced that this work will open new opportunities for industrial biocatalysis and anaerobic biotransformation applications. |
| References: | [1] Pugh et al.: Role of Forest Regrowth in Global Carbon Sink Dynamics. Proc. Natl. Acad. Sci. 2019, 116 (10), 4382-4387. [2] Floudas et al.: The Paleozoic Origin of Enzymatic Lignin Decomposition Reconstructed from 31 Fungal Genomes. Science 2012, 336 (6089), 1715-1719. [3] Martinez et al.: Genome, Transcriptome, and Secretome Analysis of Wood Decay Fungus Postia Placenta Supports Unique Mechanisms of Lignocellulose Conversion. Proc. Natl. Acad. Sci. 2009, 106 (6), 1954-1959. |
| Figures: |  The impact of O2 levels on the degradation of pine by brown-rot decayers The OxyMiST project is funded by a six-year grant from the Novo Nordisk Foundation, Grant number NNF20OC0059697. |
#906 | The problem of non-uniqueness in determining the parameters of enzyme kinetics for biocatalytic reactions |
|
| Presenting author: | Igor PLAZL of UNIVERSITY OF LJUBLJANA |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme kinetic parameters / biocatalytic reaction / / |
| Purpose: | The problem of non-uniqueness in determining the parameters of enzyme kinetics for biocatalytic reactions Mitja Lakner1 and Igor Plazl2,3* 1"Independent Scholar", Ljubljana, Slovenia 2Faculty of Chemistry and Chemical Technology, University of Ljubljana, Ljubljana, Slovenia Process intensification of the development of the biocatalytic process in the microreactor system requires the accurate determination of the parameters of the kinetic model of enzyme-catalyzed biotransformation. In general, the biocatalytic reaction is assumed to obey Michaelis and Menten kinetics. To determine the kinetic parameters, experimental measurements are usually performed in a batch system under conditions where the overall reaction rate is not affected by transport phenomena or biocatalyst concentration. As the complexity of the system and the nature of the biotransformations, e.g., reversible reaction, single or multi substrate reactions, competitive or noncompetitive inhibition, increased, Michaelis and Menten kinetics were further extended to describe multi substrate reactions with complex reaction behavior. And following the kinetic model, e.g., a ping-pong bi-bi mechanism, results in the number of parameters that need to be precisely determined for a given bioprocess. in this paper we deal with the problem of determining the kinetic parameters of enzyme kinetics with a larger number of parameters. A mathematical proof of the problem of non-uniqueness and, consequently, of the expected problems in determining the unique values of the parameters for the selected enzyme-catalysed reverse reaction of biotransformation is given. |
#907 | Enzymatic recovery of terephthalic acid from PET-PE multilayer materials using crude glycosylated PETase |
|
| Presenting author: | Daan M. VAN VLIET of WAGENINGEN FOOD & BIOBASED RESEARCH |
| Other authors: | Rick H.A.M. VAN DE VONDERVOORT of WAGENINGEN FOOD & BIOBASED RESEARCH Antoine P.H.A. MOERS of WAGENINGEN FOOD & BIOBASED RESEARCH Marc W.T. WERTEN of WAGENINGEN FOOD & BIOBASED RESEARCH Wouter TEUNISSEN of WAGENINGEN FOOD & BIOBASED RESEARCH Ulphard THODEN VAN VELZEN of WAGENINGEN FOOD & BIOBASED RESEARCH Mattijs K. JULSING of WAGENINGEN FOOD & BIOBASED RESEARCH Tom A. EWING of WAGENINGEN FOOD & BIOBASED RESEARCH |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | PETase / hydrolysis / recycling / plastics |
| Purpose: | Over the last 70 years, plastics have become a ubiquitous, indispensable part of society, but also a large waste stream. While about one-third of plastic waste is recycled, the remaining two-thirds is landfilled or incinerated, leading to unwanted accumulation in nature or CO2 emissions. One of the main types of plastic is polyethylene terephthalate (PET), which is a polyester consisting of ester-linked terephthalic acid and ethylene glycol. Although re-use or mechanical recycling is possible for products such as PET bottles, these solutions are not feasible for many other types of PET waste, such as common multilayer packaging materials. Recycling of PET within multilayer waste could be achieved by enzymatic hydrolysis and recovery of the monomers, making use of the intrinsic selectivity of enzymes. Within the EU-funded Horizon 2020 research project “ENZYCLE”, we aim to develop an enzymatic PET recycling process and to demonstrate the industrial feasibility at pilot scale. We selected two enzymes with high reported PETase activity: leaf-compost cutinase (LCC) and a recently described polyester hydrolase (PHL7). His6-tagged enzymes were heterologously expressed in the yeast Pichia pastoris, increasing their thermostability through glycosylation, and purified through affinity chromatography. At 70°C and pH 8 maintained by sodium phosphate buffer, the purified PETases efficiently hydrolysed amorphous PET film (40-50 mg) at a rate comparable to that of non-glycosylated LCC produced in Escherichia coli (1.2-1.4 mg PET µg-1 PETase d-1). We then explored the use of crude PETase in the form of P. pastoris culture filtrate for techno-economical purposes. The PETase activity of crude PHL7 was unaffected, whereas crude LCC showed partial PETase inhibition by an unknown component. Next, amorphous PET film hydrolysis was tested at 10 g scale (10 g/L) in pH-controlled bioreactors using crude PETase with and without sodium phosphate buffer, and monitored by HPLC analysis of monomer concentrations and by weight loss measurement of residual PET. Performance of crude LCC was unaffected by sodium phosphate concentration, whereas crude PHL7 required a high concentration (1 M) of sodium phosphate for thermostability at >60°C. Using crude LCC at 70°C, 98% of the PET was hydrolyzed in 43 hours at a constant rate (5.6 g L-1 d-1). We next tested two types of PET-polyethylene multilayer packaging materials (>96% w/w PET), which could both also be hydrolyzed almost completely (>96%) at 65°C in 92 hours, without any pretreatment required. Incubation at 70°C caused the PET in these materials to crystallize, lowering the final extent of hydrolysis. The substrate loading was raised to 200 g/L without loss of performance. Produced terephthalic acid was recovered by precipitation through addition of H2SO4 and filtration with yields of >77%, with the only contaminants being traces of mono(2-hydroxyethyl)terephthalic acid and 2 mol% isophthalic acid, which is commonly present in PET as co-monomer. Residual PET-polyethylene material still contained a significant fraction of PET (>35% w/w), which may be removable through hydrolysis with strong acid or base, enabling mechanical recycling of the polyethylene fraction. In conclusion, we designed a promising enzymatic process for back-to-monomer recycling of PET from multilayer packaging material using crude LCC produced in P. pastoris as biocatalyst. Future efforts within the project will include further testing of the robustness, a techno-economic analysis, and upscaling to pilot scale. |
#908 | Improvement of amino ester hydrolase (AEH) via protein engineering, machine learning, and process engineering |
|
| Presenting author: | Andreas BOMMARIUS of GEORGIA INSTITUTE OF TECHNOLOGY |
| Other authors: | Colton LAGERMAN of GEORGIA INSTITUTE OF TECHNOLOGY Emily JOE of GEORGIA INSTITUTE OF TECHNOLOGY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | amino ester hydrolase / stabilization / machine learning / protein engineering |
| Purpose: | Amino ester hydrolase (AEH) catalyzes the synthesis of semisynthetic beta-lactam antibiotics and is a potential alternative to Pen G acylase (PGA). While AEH is more active and selective than PGA towards the synthesis of targets with (R)-phenylglycyl side chains than PGA, its substrate specificity is limited, its biophysical behavior is complex, and it is unstable at temperatures > 25oC. We set out to improve the utility of AEH by improving its overall stability. While we had succeeded in stabilizing wild-type AEH from Xanthomonas campestris via site-directed mutagenesis [1], we now employ machine-learning based techniques, such as FireProt [2] and PROSS [3], to improve its thermal stability. In addition, we employed differential scanning fluorimetry (DSF) for enzyme unfolding, back reflection to study AEH aggregation, and analytical ultracentrifugation (AUC) to study AEH’s oligomericity behavior [4] and improve its deactivation kinetics [5]. We found that mutating increasingly larger numbers of residues, up to 30% of the residues, led to dramatic stabilization, as measured by DSF via the melting temperature Tm (Figure 1). However, most variants were found to be inactive. Only the (re) discovery of a Ca2+-binding site in X. campestris AEH reliably recovered activity, ranging from small to sizable [6]. The development of AEH towards a biocatalyst useful in large-scale synthesis serves as an example of the need to explore various techniques and pathways and not just to rely on a single development path, whether via machine learning or experimental protein engineering. |
| References: | [1] JK Blum, WD Ricketts, AS Bommarius, J. Biotechnol. 2012, 160, 214-221 [2] D Bednar, K Beerens, E Sebestova, J Bendl, S Khare, R Chaloupkova, Z Prokop, J Brezovsky, D Baker, J Damborsky, PLOS Comput. Biol. 2015, 11(11), e1004556 [3] A Goldenzweig, M Goldsmith, SE Hill, O Gertman, P Laurino, Y Ashani, O Dym, T Unger, S Albeck, J Prilusky, RL Lieberman, A Aharoni, I Silman, JL Sussman, DS Tawfik, SJ Fleishman, Mol. Cell. 2016, 63(2), 337-346 [4] AA Caparco‡, BR Bommarius‡, L Ducrot, JA Champion, C Vergne-Vaxelaire, AS Bommarius, submitted [5] CE Lagerman‡, JK Blum‡, TA Rogers‡, MA Grover, RW Rousseau, AS Bommarius, submitted [6] CE Lagerman, EA Joe, AS Bommarius, to be submitted |
| Figures: | 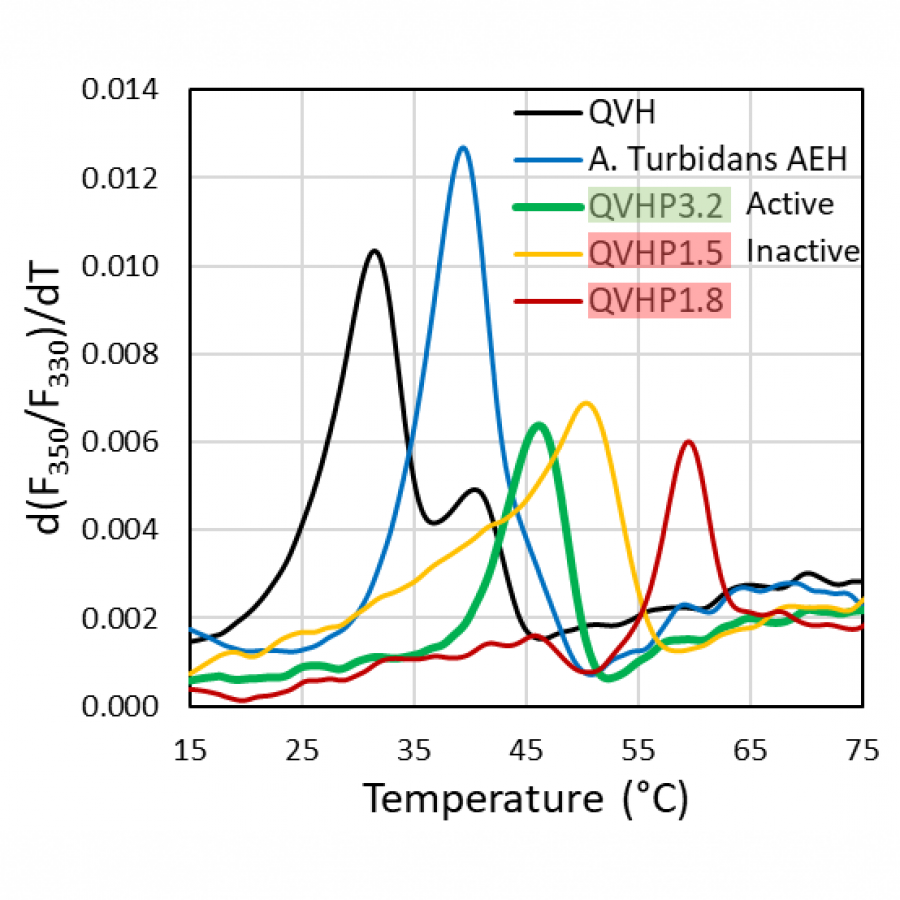 Melting temperature of AEH variants measured by Differential Scanning Fluorimetry (DSF.) Melting temperatures indicated by peaks |
#909 | Engineering of a thermostable tranketolase for larger substrate scope: focus on benzaldehyde derivatives |
|
| Presenting author: | Jules PERRARD of TU DARMSTADT |
| Other authors: | Mireia SALVADO PAU of BASF SE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Transketolase / Aromatics / Carboligation / Screening |
| Purpose: | Carbon-Carbon bond formation is one of the most attractive topics in organic synthesis. To overcome its limitations, the synthesis of C-C bonds using enzyme catalysis is a promising alternative, which can be achieved using a wide range of carboligases. For this purpose, we have further developed the thermostable transketolase from Geobacillus stearothermophilus (TKgst) as catalyst. The wild-type TKgst has proven to be useful in the irreversible addition of a two-carbon moiety to an alpha-hydroxyaldehyde when hydroxypyruvate (HPA) is used as a donor substrate [1]. However, the range of acceptors that can be used is the bottleneck of such a reaction. We focused our efforts to enhance the ability of TKgst to accept aromatic substrates such as benzaldehyde and its derivatives (figure 1). Within this scope, two libraries of a single amino acid mutation located in the active site, F435 and L191, have been created and screened for activity. This work would allow us to investigate the impact of these two residues on the catalytic activity of the enzyme. An in-depth activity and selectivity of the best mutants from these libraries is currently being further investigated. |
| References: | [1] Abdoul-Zabar, J.; Sorel, I.; Hélaine, V.; Charmantray, F.; Devamani, T.; Yi, D.; de Berardinis, V.; Louis, D.; Marlière, P.; Fessner, W.-D. and Hecquet, L., Adv. Synth. Catal., 2013, 355, 116-128. https://doi.org/10.1002/adsc.201200590 |
| Figures: | 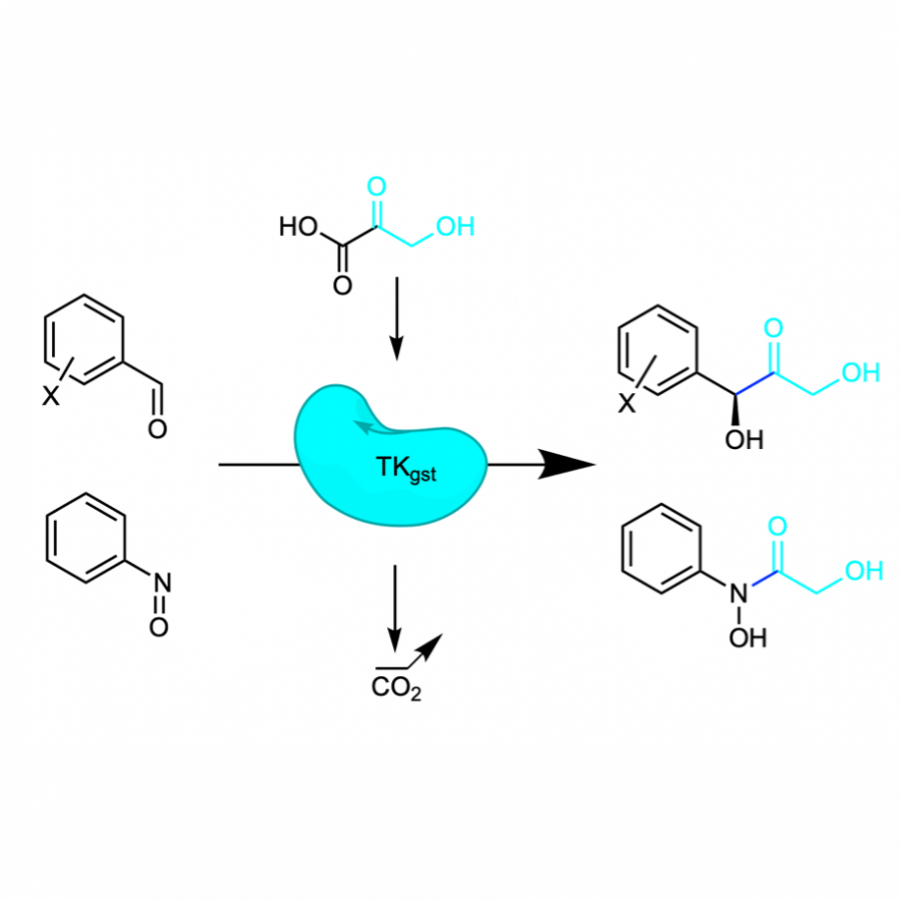 figure 1. Irreversible reaction of TK towards benzaldehyde and analogs. X=NO2, Br, F, CO2H |
#911 | NucLib: A chemical library of non-canonical (deoxy-) ribonucleotides produced by a versatile enzyme cascade. |
|
| Presenting author: | Christoph LODERER of TU DRESDEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ribonucleotide reductase / Non-canonical nucleotides / Nucleotide modfying enzymes / Enzyme cascades |
| Purpose: | Ribonucleotides and their corresponding deoxyribonucleotides are central to a myriad of biological processes such as storage and expression of genetic information, energy metabolism and signal transduction. Due to the biological omnipresence of this class of compounds, analogues of the canonical nucleotides are widely used to manipulate biological systems, leading to application in fields ranging from medicine to synthetic biology. The actual mode of action relies highly on the form of the respective nucleotide in terms of 3’-oxygenation and phosphorylation level. In the project NucLib, we aim for the establishment of a versatile and flexible enzymatic cascade for the biosynthesis all relevant forms of non-canonical nucleotides. This includes the ribose and deoxyribose forms as nucleoside mono-, do-, and triphosphates. In its core, the cascade consist of the enzymes phosphoribosyl transferase(1), polyphosphate kinase(2, 3) and ribonucleotide reductase(4, 5). We investigate the substrate specificities of the enzymes for the conversion of non-canonical substrates and improve the promiscuity by enzyme engineering. We also investigate the possibilities to produce each relevant compound either separately or in one complete mixture for each non-canonical nucleotide. The cascade was already successfully applied for the biosynthesis of 2Cl-adenosine triphosphate, the active metabolite of the anti-cancer drug Mavenclad®(6). Part of the cascade was used to synthesize the active metabolites of the drug 5-Aminoimidazole-4-carboxamide ribonucleotide (AICAR)(7). Further extension of the substrate scope will enable the creation of a chemical library of non-canonical (deoxy-) ribonucleotides to evaluate and facilitate their biotechnological application. |
| References: | (1) Esipov, R. S., Abramchik, Y. A., Fateev, I. V., Konstantinova, I. D., Kostromina, M. A., Muravyova, T. I., Artemova, K. G., and Miroshnikov, A. I. 2016, Acta Naturae. 8, 82-90. (2) Mordhorst, S., Singh, J., Mohr, M. K. F., Hinkelmann, R., Keppler, M., Jessen, H. J., and Andexer, J. N. 2019, ChemBioChem. 20, 1019-1022. (3) Motomura, K., Hirota, R., Okada, M., Ikeda, T., Ishida, T., and Kuroda, A. 2014, Appl. Environ. Microbiol. 80, 2602-2608. (4) Larsson, K. M., Jordan, A., Eliasson, R., Reichard, P., Logan, D. T., and Nordlund, P. 2004, Nat. Struct. Mol. Biol. 11, 1142-1149. (5) Loderer, C., Holmfeldt, K., and Lundin, D. 2019, PeerJ. 7, e6700. (6) Frisch, J., Marsic, T., and Loderer, C. 2021, Biomolecules. 11, 3, 346. (7) Eltoukhy, L., and Loderer, C. 2022, ChemBioChem, 23, e202100596. |
| Figures: | 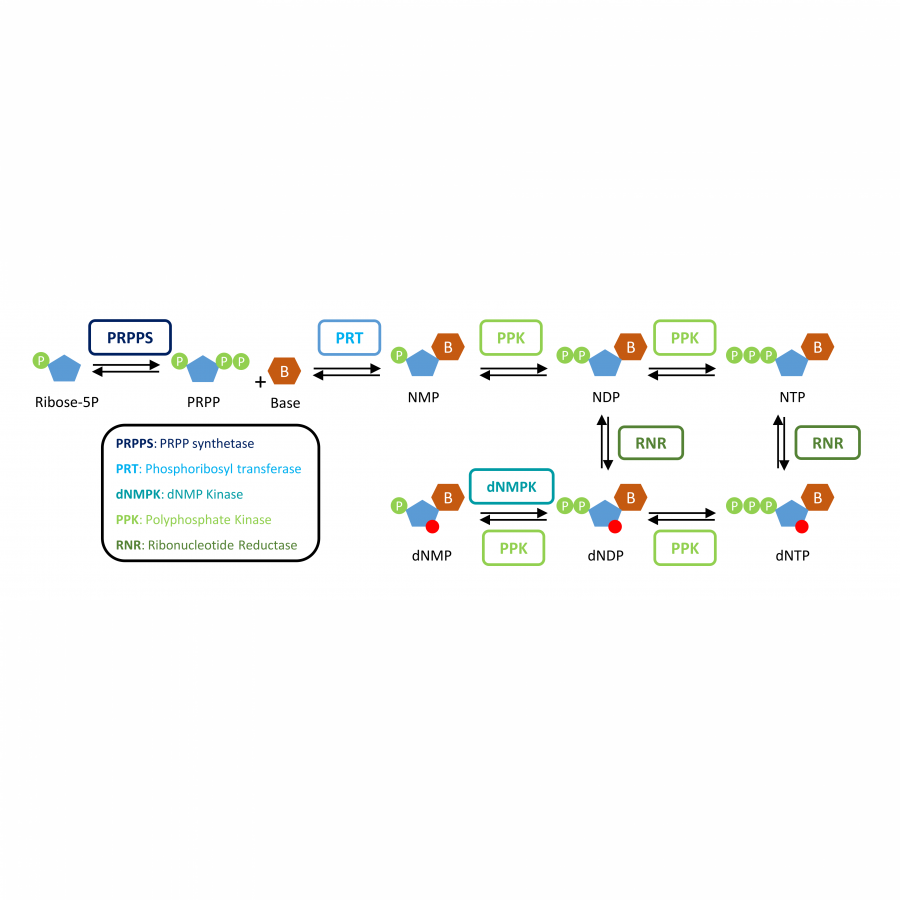 Figure 1: NucLib Cascade: Concept of the enzymatic cascade for biosynthesis of non-canonical (deoxy-) ribonucleotides for the creation of the NucLib chemical library. |
#912 | Microfluidics-based preparation of nano-sized cross-linked enzyme aggregates |
|
| Presenting author: | Tadej MENEGATTI of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY |
| Other authors: | Polona ŽNIDARŠIČ PLAZL of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cross-linked enzyme aggregate / enzyme immobilization / flow biocatalysis / membrane microreactor |
| Purpose: | Enzyme immobilization is a promising strategy for improving enzyme stability, activity, and reusability1. Cross-linked enzyme aggregates (CLEAs) are a popular immobilization approach that offers several advantages, including simple preparation, cost-effectiveness, and high enzyme loading 2. However, the quality and stability of CLEAs can be limited by the batch mode of preparation, resulting in non-uniform and unstable aggregates with low retained activity. In this study, we present a novel system for preparing uniform and stable CLEAs of nanosize using omega transaminase (ω-TA) as a model enzyme. Our system involves a continuous microflow tubing reactor with controlled flow rates of precipitant and crosslinker that allows for the formation of highly uniform and stable CLEAs. The resulting nanosize CLEAs exhibit enhanced stability compared to conventional CLEAs prepared in batch mode. The physicochemical properties of the nano-size CLEAs, including size distribution, morphology, and stability, were characterized. The nanosize CLEAs showed a narrow size distribution with an average hydrodynamic radius of about 80 nm and a high uniformity in terms of 0,17 polydispersity index Furthermore, the nanosize CLEAs demonstrated excellent stability under various conditions, including pH, temperature, and storage. Specifically, the nanosize CLEAs retained 87% activity compared to the free enzyme and showed improved stability at higher temperatures. To further demonstrate the utility of our nano-size CLEAs, we developed a membrane microreactor composed of the CLEAs immobilized onto porous membrane support. Microreactors offer many advantages over traditional batch reactors, such as increased mass and heat transfer rates, improved control over reaction conditions, and decreased enzyme deactivation. The membrane microreactor with immobilized nanoCLEA showed stable and continuous enzymatic activity over a prolonged period of operation, with a significant increase in operational stability compared to the immobilized free enzyme. Our results demonstrate the potential of our novel system for preparing uniform and stable nano-size CLEAs and subsequent use of these CLEAs in membrane microreactors for improved operational stability. This system can be applied to other enzymes and scaled up for industrial biocatalysis, offering a promising approach for improving the efficiency and sustainability of various biocatalytic processes. |
| References: | 1. Žnidaršič-Plazl, P. Biocatalytic process intensification via efficient biocatalyst immobilization, miniaturization, and process integration. Curr. Opin. Green Sustain. Chem. 32, 100546 (2021). 2. Sheldon, R. CLEAs, combi-CLEAs and ‘smart’ magnetic CLEAs: Biocatalysis in a bio-based economy. Catalysts 9, 261 (2019). |
| Figures: | 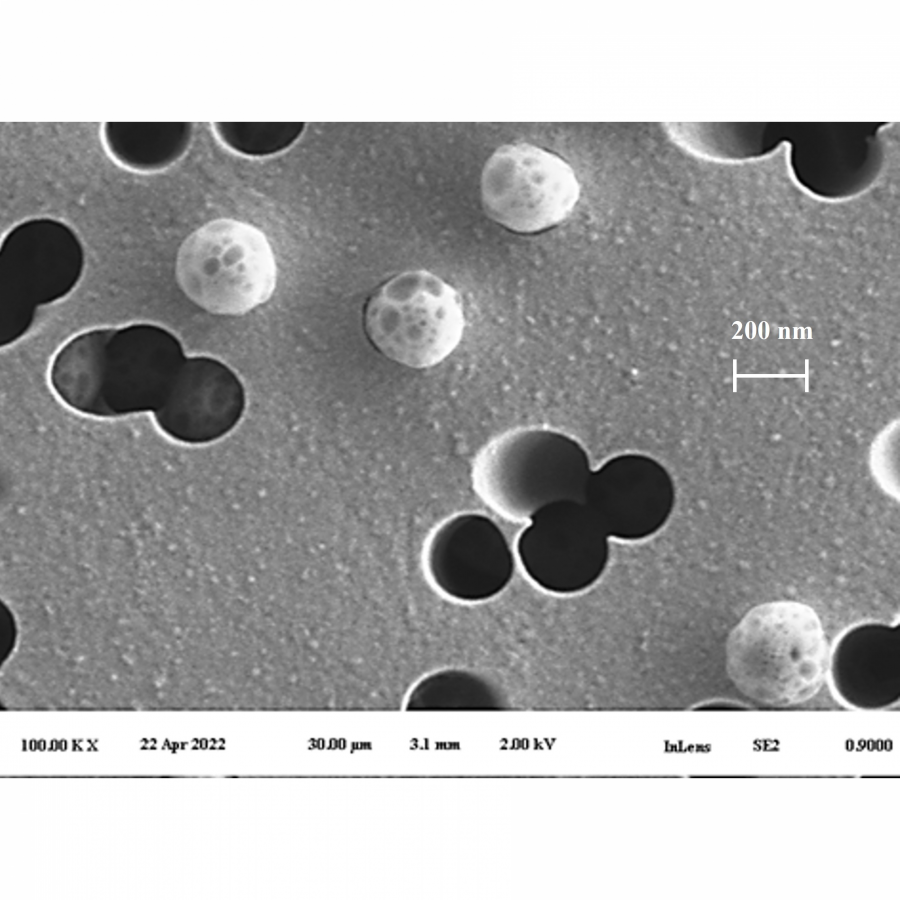 SEM image SEM image of nano-CLEA particles 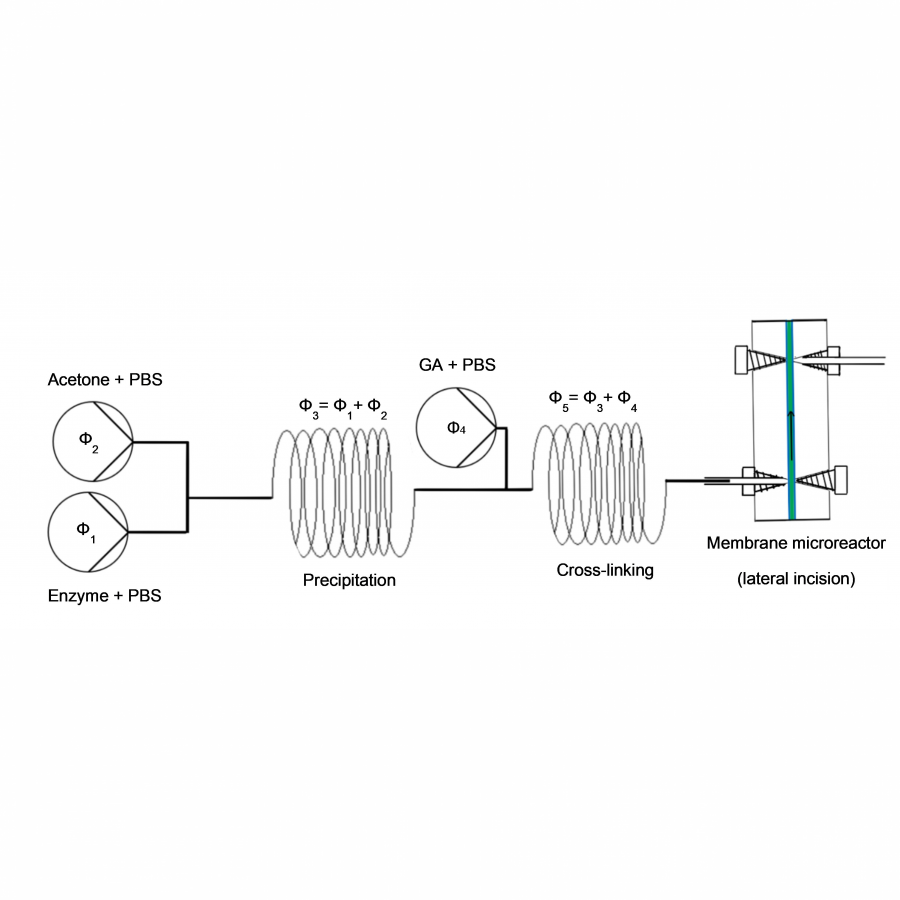 Experimental set-up The microfluidic system with 3 inlets for ATA-CLEAs generation and their in situ immobilization in a membrane microreactor. |
#914 | A high-throughput characterization strategy of a genetic toolbox for the improvement of cyanobacteria as hosts for whole-cell biocatalysis |
|
| Presenting author: | Julia JODLBAUER of TU VIENNA |
| Other authors: | Christian WALTL of TU WIEN Matthias SCHMAL of TU WIEN Florian RUDROFF of TU WIEN |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cyanobacteria / genetic toolbox / high-throughput / whole-cell biocatalysis |
| Purpose: | Cyanobacteria are promising and, quite literally, green candidates for microbial production platforms. Their photoautotrophic metabolism enables a sustainable production system by depending solely on water, carbon dioxide, and sunlight. They have also been shown to be valuable in whole-cell biocatalysis, producing large amounts of the energy-rich cofactor NADPH and oxygen in situ, which is of interest for the functionality of numerous industrially applied enzymes [1]. Despite these significant advantages, applicability was limited so far by remaining challenges. One of the biggest challenges is still to obtain sufficient expression levels of recombinantly expressed proteins within the cyanobacterial host. Furthermore, their photoautotrophic cultivation limits high-throughput screening possibilities, which also slows down progress within the field. To push cyanobacteria further to its limits, it is, therefore, necessary to improve the capacities and understanding of the host system. Within this work, we aim to tackle these challenges and improve cyanobacteria as hosts for biocatalysis. (i) We built a genetic toolbox to improve enzyme yields within the host. (ii) We developed a characterization strategy to analyze the genetic regulatory elements in a one-pot approach. To exemplify the strategy, we applied three enzymes to improve their performance in cyanobacteria. As enzymes, we investigated a ketoreductase (LfSDR1M50) [2], an enoate reductase (YqjM) [3], and a Baeyer-Villiger monooxygenase (CHMOAcineto) [4]. The genetic toolbox is built up of 12 different promoters and 21 ribosome binding sites, which were designed to be an extension of the cyanobacterial MoClo Kit, CyanoGate [5]. To screen and characterize all expression constructs in a one-pot manner, we fused a GFP tag C-terminally to the investigated enzymes. Like this, the GOI-specific expression libraries could be analyzed for their expression levels according to their fluorescence and, by flow cytometry, sorted from low-expression strains to high-expression strains. The genetic context of the sorted cells was then analyzed by deep sequencing. As a result, we got an in-depth characterization of the individual regulatory elements under different genetic contexts (LfSDR1M50-GFP, YqjM-GFP and CHMOAcineto-GFP). A broad range of different expression levels could be achieved. Here, the promoter, as well as the RBS, have a significant impact. Interestingly, by only varying the RBS, we reach a similar scope of various expression levels compared to solely varying the promoter. With this study, we therefore emphasize that RBSs are often underrated when designing genetic constructs despite this significant impact. However, as demonstrated previously [6], we see that the efficiency of an RBS varies strongly among different genetic contexts (GOIs), making it more challenging to predict a priori. Therefore, this study aims to bring further insights into RBS performance and underline the current shortage of understanding. To summarize, these results underline the untapped potential and the importance of further improving expression systems in cyanobacteria. Here, with our study, we present a promising approach to exploit further potential to find improved expression strains and whole-cell biocatalysts. |
| References: | [1] j. jodlbauer et al., trends biotechnol.,2021, 39(9), 875-889. [2] j. guo et al., ChemCatChem, 2018, 10(23), 5496-5504. [3] t. b. fitzpatrick et al., J. Biol. Chem., 2003, 278(22), 19891-19897. [4] h. r. mansouri et al., 2022, 12, 11761-11766. [5] r. vasudevan et al., 2019, Plant Physiol., 180(1), 39-55. [6] k. thiel et al., Microb. Cell Fact., 2018, 17(1), 1-12. |
| Figures: | 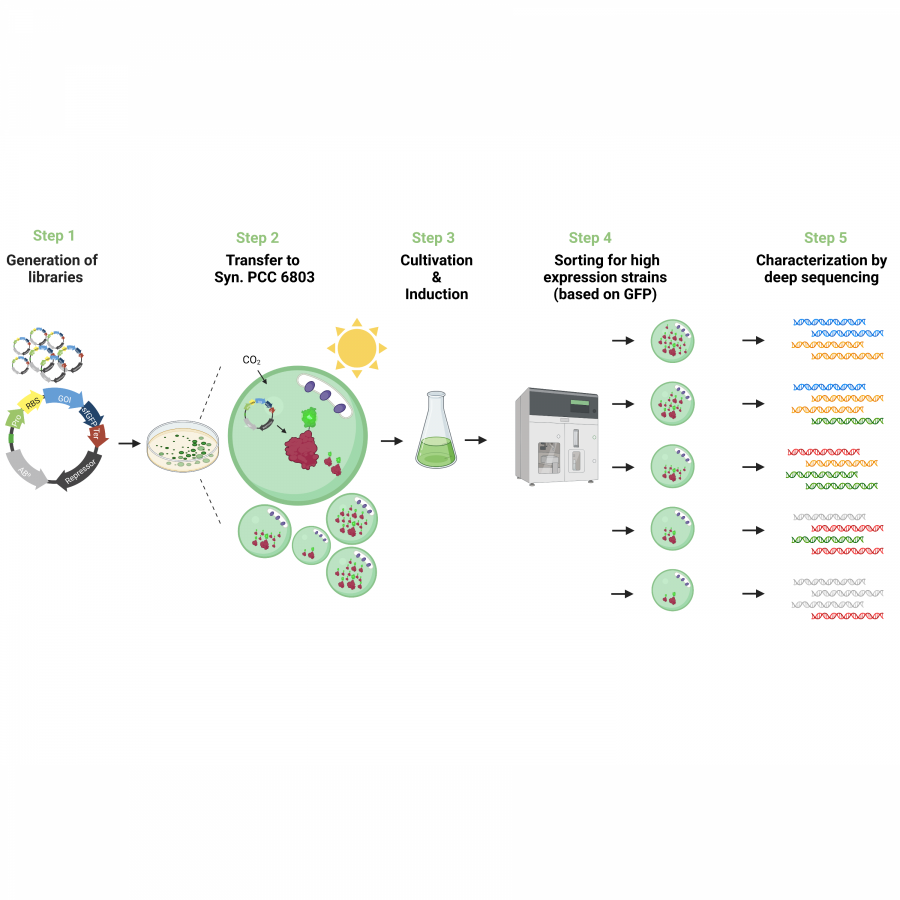 Characterization strategy of a genetic toolbox to improve gene expression in cyanobacteria An expression library with different combinations of RBSs and promoters is generated. A C-terminal GFP fusion tag allows the screening and analysis of the expression of a gene of interest (GOI) by FACS and subsequent deep sequencing. |
#917 | From smelly sulphur substrates to fragrant flavours – enzymatic synthesis of L methionine analogues and their usage for alkylation |
|
| Presenting author: | Michael MOHR of ALBERT-LUDWIGS-UNIVERSITÄT, FREIBURG |
| Corresponding author: | Jennifer N. ANDEXER of ALBERT-LUDWIGS-UNIVERSITÄT, FREIBURG |
| Other authors: | Raspudin SALEEM-BATCHA of ALBERT-LUDWIGS-UNIVERSITÄT, FREIBURG |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | unnatural amino acids / SAM analogues / enzymatic alkylation / enzyme cascade |
| Purpose: | Regio- and stereoselective alkylation of small molecules can impact their physicochemical and pharmaceutical properties.[1,2] Highly selective methyltransferases (MTs) are promising tools for the transfer of alkyl chains onto various substrates.[1,3,4] S-adenosyl-L-methionine (SAM) analogues are utilised for the alkylation and can either be generated from L-methionine analogues and ATP by an L methionine adenosyltransferase (MAT) or by using the analogues in combination with 5´-chloro adenosine and the SAM chlorinase SalL.[5,6] So far most of the commercially unavailable L-methionine analogues have to be chemically synthesised.[7] We explored an alternative approach, following the enzymatic synthesis of L-ethionine. The simplest modified L-methionine analogue can be synthesised from L-homocysteine and ethanthiol using the O-acetyl-L-homoserine sulfhydrolase from S. cerevisiae (ScOAHS). Investigation on the substrate scope of ScOAHS revealed a promiscuous enzyme. Starting from L homocysteine and organic thiols, different L methionine analogues with newly introduced functionalities at the transferable thioether residue could be synthesised. Based on these results we expanded our selective alkylation cascade,[3] using the O MT from Rattus norvegicus with the substrate protocatechuic aldehyde. We successfully transferred alkyl chains onto the substrate and thereby synthesised new derivatives of the flavour vanillin (s. Figure 1).[8] |
| References: | [1] A.-W. Struck, M. L. Thompson, L. S. Wong, J. Micklefield, ChemBioChem 2012, 13, 2642-2655. [2] H. Schönherr, T. Cernak, Angewandte Chemie International Edition 2013, 52, 12256-12267. [3] J. Siegrist, S. Aschwanden, S. Mordhorst, L. Thöny‐Meyer, M. Richter, J. N. Andexer, ChemBioChem 2015, 16, 2576-2579. [4] S. Mordhorst, J. Siegrist, M. Müller, M. Richter, J. N. Andexer, Angewandte Chemie International Edition 2017, 56, 4037-4041. [5] F. Michailidou, N. Klöcker, N. V. Cornelissen, R. K. Singh, A. Peters, A. Ovcharenko, D. Kümmel, A. Rentmeister, Angewandte Chemie International Edition 2021, 60, 480-485. [6] T. D. Davis, S. Kunakom, M. D. Burkart, A. S. Eustaquio, Methods Enzymol 2018, 604, 367-388. [7] S. Singh, J. Zhang, T. D. Huber, M. Sunkara, K. Hurley, R. D. Goff, G. Wang, W. Zhang, C. Liu, J. Rohr, S. G. Van Lanen, A. J. Morris, J. S. Thorson, Angew. Chem. 2014, 126, 4046-4050. [8] M. K. F. Mohr, R. Saleem-Batcha, J. N. Andexer, manuscript in preparation |
| Figures: | 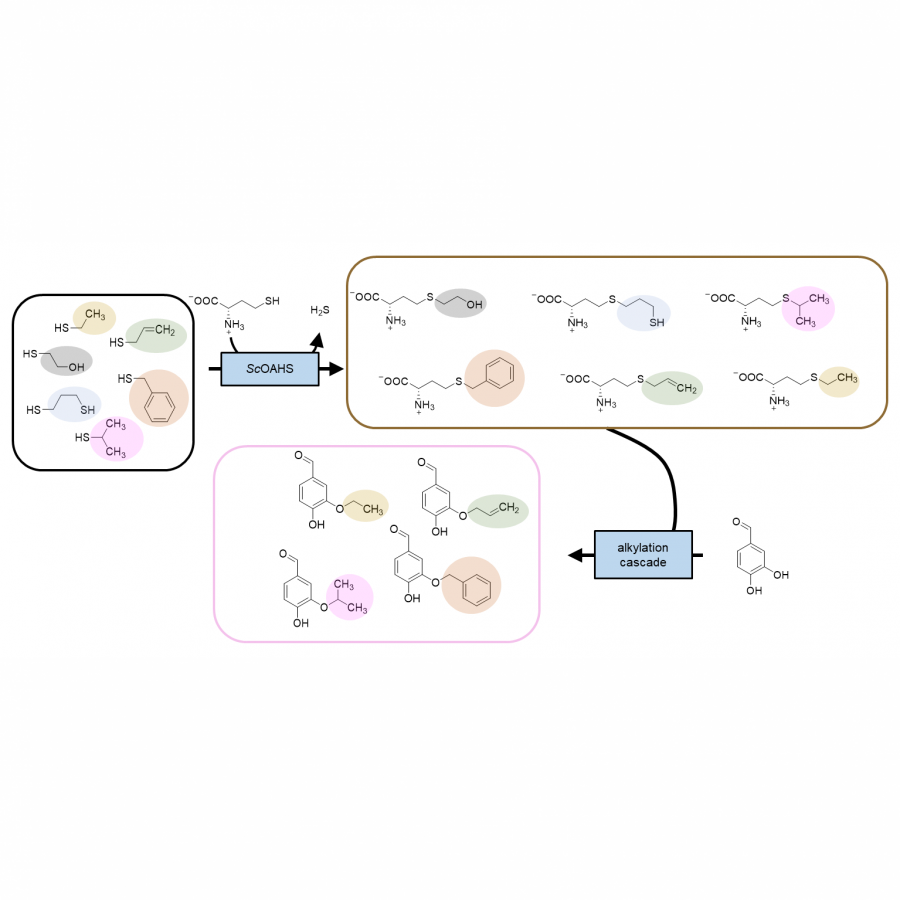 Figure 1 Utilisation of sulphur substrates for alkylation. Organic thiols (black box) are used by ScOAHS with L-homocysteine to synthesise the L-methionine analogues (blue box). When this set up is combined with the alkylation cascade enzymes the utilised RnCOMT t |
#918 | Biocatalytic hydronitration of α,β-unsaturated carboxylic acids |
|
| Presenting author: | Matteo ALEOTTI of UNIVERSITY OF GRAZ |
| Other authors: | Hannah DREISBACH of UNIVERSITY OF GRAZ Rémi CORLAY of UNIVERSITY OF GRAZ Mélanie HALL of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymatic hydronitration / α,β-unsaturated carboxylic acids / Protein engineering / Nitrosuccinate lyase |
| Purpose: | The nitro functionality (-NO2) is one of the most versatile groups in chemistry. Indeed, due to its strong electron-withdrawing properties, it confers exceptional reactivity in a myriad of synthetic reactions. Moreover, nitro compounds are notorious precursors to amines, and they are widely exploited as chemotherapeutic agents, prodrugs, explosives and as building blocks for dyes and polymers.[1] Despite the synthetic difficulties related to the introduction of the nitro group, such as the requirement for high temperatures and mixtures of strong acids, and the poor selectivity of the synthesis, this reaction is still exploited on industrial scale due to a lack of milder approaches. A few enzymes however can handle the nitro moiety in the context of natural metabolic pathways under mild conditions.[2] Thus, biocatalysis may provide a suitable alternative to perform selective nitration reactions under environmentally friendly conditions. In this work, the enzymatic nitration of α,β-unsaturated carboxylic acids by the nitrosuccinate lyase (CreD)[3] was investigated. We could demonstrate that the enzyme can catalyze the hydronitration of fumaric acid on 50 mM substrate loading with high catalytic efficiency (TTN up to 20,000). In order to expand the substrate scope to a variety of conjugated carboxylic acids, and due to the substrate selectivity of the enzyme for fumaric acid, the re-design of the active site is currently being explored and will be discussed, along with recent findings on the promiscuity within this enzyme family. |
| References: | [1] N. Ono, The Nitro Group in Organic Synthesis, Wiley VCH, New York, 2001. [2] A. J. Waldman, T. L. Ng, P. Wang, E. P. Balskus, Chem Rev 2017, 117, 5784-5863. [3] Y. Katsuyama, Y. Sato, Y. Sugai, Y. Higashiyama, M. Senda, T. Senda, Y. Ohnishi, FEBS J 2018, 285, 1540-1555. |
| Figures: | 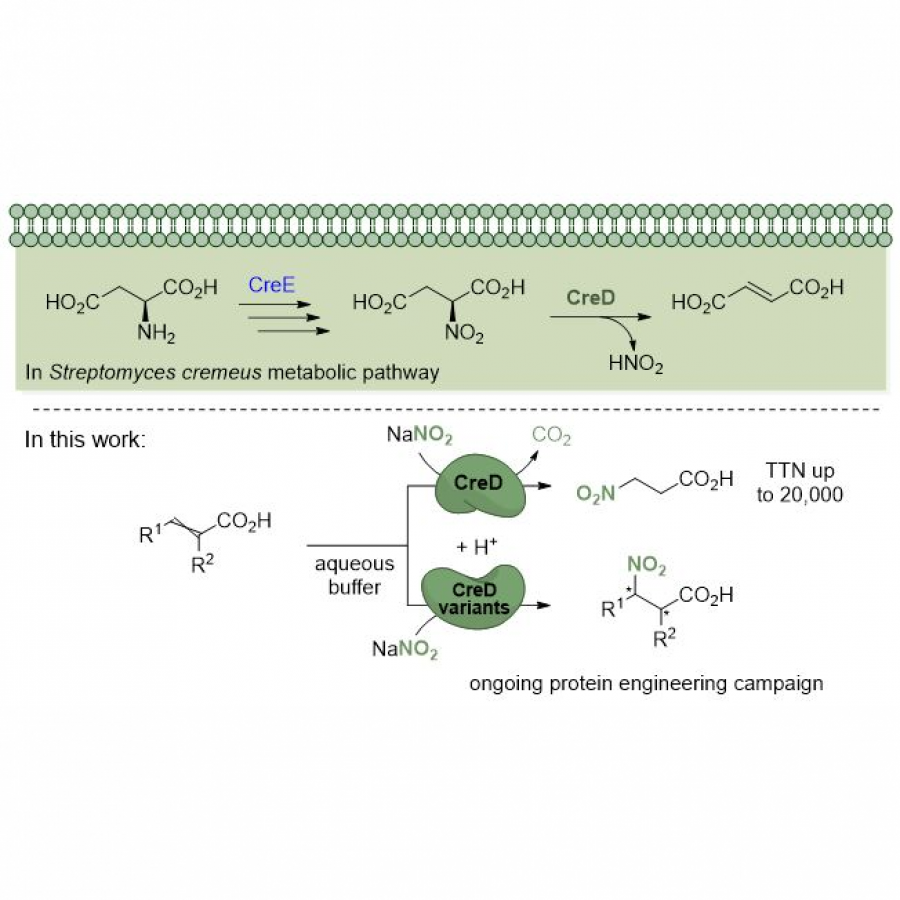 Figure 1: Occurrence of the nitrosuccinate lyase activity of CreD in Streptomyces cremeus (above) and enzymatic strategies for the hydronitration of unsaturated carboxylic acid using CreD (below). |
#922 | Exploring the co-immobilization of multi-enzyme system to scale-up step-wise biotransformations |
|
| Presenting author: | Francisco Javier ARCOS of CIC BIOMAGUNE |
| Corresponding author: | Fernando LÓPEZ-GALLEGO of CIC BIOMAGUNE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flow biocatalyst / Multi-enzyme systems / smart co-substrates / |
| Purpose: | The co-immobilization of multi-enzyme systems offers the opportunity to design smart biocatalysts to carry out stepwise biotransformations. However, some aspects related to sequential enzymatic reactions such as the kinetic parameters of the system, the spatial distribution and the orientation of the enzymes on the support surface, must be optimized to maximize the efficiency of the process. In this work, we set out to develop a biocatalyst capable of transforming aliphatic diols into ω-hydroxy acids in the same reactor through a 5-enzyme system. For this purpose, we evaluated the effect of spatial distribution by designing several combinations, testing them in batch, reaching in the first one up to 65% conversion of the final product (1). To immobilize that system, we have previously designed a tri-funtional carrier to assamble a biocatalyst thorugh different chemistries. Two spatial distributions were tested, resulting the one biocatalyst where the 5 enzymes were co-immobilized the most efficient. Despite promising results, no longer than 50% was achieved. We explored the change of localization of ine of the enzyme of the system, resulting an effective increase in the biocatalyst productivity. In addition, we increased the operational stability of the system by a post-immobilization step in which the immobilized enzymes were cross-linked with a polycationic polymer. This more robust biocatalyst was implemented in batch, reaching a conversion up to 80% of the final product. When biocatalyst was applied in flow-through process, presented a espected limitation since regeneration system is a oxygen-dependent reaction. To overcome the oxygen limiting factor, we employed a smart co-substrate for in situ oxygen generation for a successful flow reaction. In a flow rate of 10 μl/min and a 20 mM concentration of main substrate (1,5-Pentanediol), a conversion up to 75% of the final product (5-hydroxypentanoic acid) was achieved, whereas no product was detected by no smart co-substrate application. Overall, this work has allowed us to improve our knowledge in co-immobilized enzyme systems to scale up multi-enzyme transformations. We observed a positive effect by having all enzymes located in the same vessel compared to those separated under flow reaction conditions. Furthermore, we solved an intrinsic problem of employment of oxygen-depedent enzymes in flow. Overall, we demostrated that complex multi-enzymatic reactions can be a reality in the application in flow system through a rational design. |
| References: | [1] Velasco‐Lozano, S., Santiago‐Arcos, J., Grazia Rubanu, M., & López‐Gallego, F. (2022). Cell‐Free Biosynthesis of ω‐Hydroxy Acids Boosted by a Synergistic Combination of Alcohol Dehydrogenases. ChemSusChem, 15(9). https://doi.org/10.1002/cssc.202200397 [2] Santiago-Arcos, J., Velasco-Lozano, S., & López Gallego, F. (2022). Multi-enzyme co-immobilization on tri-heterofunctional supports. Biomacromolecules. https://doi.org/10.26434/chemrxiv-2022-0d7h2 |
| Figures: | 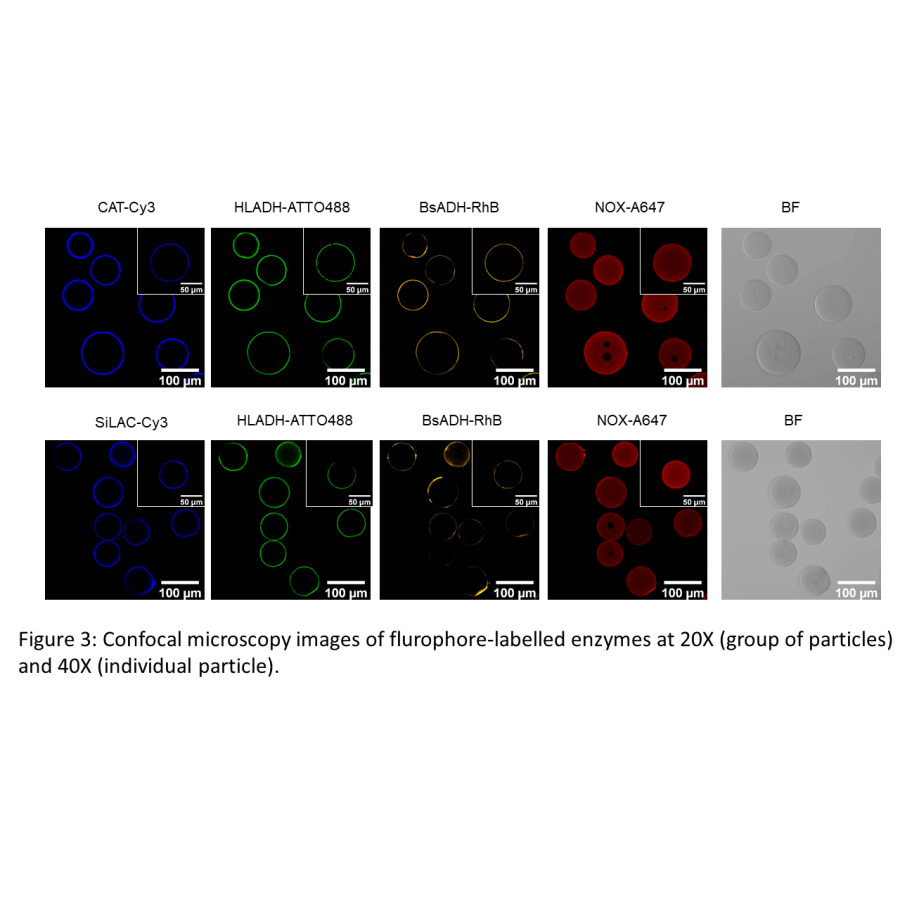 Spatial localization of each enzyme involve in the cascade Confocal microscopy images of flurophore-labelled enzymes at 20X (group of particles) and 40X (individual particle). 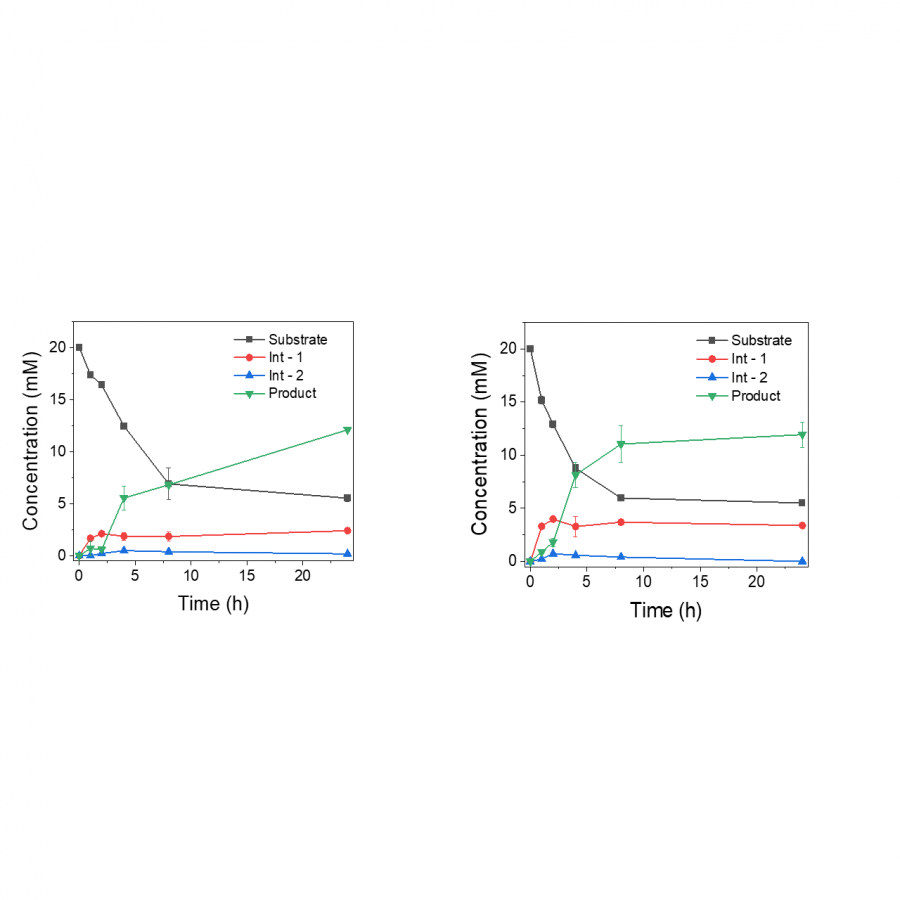 Study of both distributions under batch conditions Kinetic at 24 hours of each spatial distribution systems: Distribution 1 (on the left) and Distribution 2 (on the right). Rxn mix: 10 mM 1,5-Pentanediol; 1 mM NAD+, 0.15 mM FAD; T(P) 100 mM pH 8
|
#923 | Aromatic hydroxylation of Trans-cinnamic acid performed by the P450 Cinnamate-4-hydroxylase: A QMMM study |
|
| Presenting author: | Sónia SANTOS of NORTHUMBRIA UNIVERSITY |
| Corresponding author: | Warispreet SINGH of NORTHUMBRIA UNIVERSITY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysts / Hydroxylation / Aromatic / Lignin |
| Purpose: | Being part of the plant metabolic system, the Cinnamate 4-hydroxylase (C4H) enzyme belongs to the CYP73A family of the cytochrome P450 monooxygenase known for being a crucial biocatalyst. C4H is responsible for the hydroxylation of trans-cinnamic acid into p-coumaric acid (Scheme 1). This enzyme can have an impact on structure, development, and defense, which makes in-depth knowledge of the mechanism and interaction with the substrate during the catalytic reaction important.1 Playing different and impactful roles in plant’s life, this enzyme must be properly studied in complex with their natural substrates. Trans-cinnamic acid was docked, into the active center of the C4H enzyme followed by comprehensive MD simulations and QM/MM calculations. However, unlike the hydroxylation of alkyl substrates in which the mechanism is initiated by the abstraction of the allylic hydrogen by the Iron-oxo complex, the case of aromatics is not so straightforward. Based on our QM/MM calculations, we propose to different conformations are achieved during the transition state of the aromatic hydroxylation. After a tetrahedral sigma complex formation2, the reaction proceeds through a protonated porphyrin intermediate or a direct rearrangement in comparison to the alternative epoxide and ketone pathways for aromatic hydroxylation. |
| References: | [1] B. Zhang, K. M. Lewis, A. Abril, D. R. Davydov, W. Vermerris, S. E. Sattler and C. Kang, Plant Physiol., 2020, 183, 957-973. [2] C. M. Bathelt, A. J. Mulholland and J. N. Harvey, J. Phys. Chem. A, 2008, 112, 13149-13156. |
| Figures: | 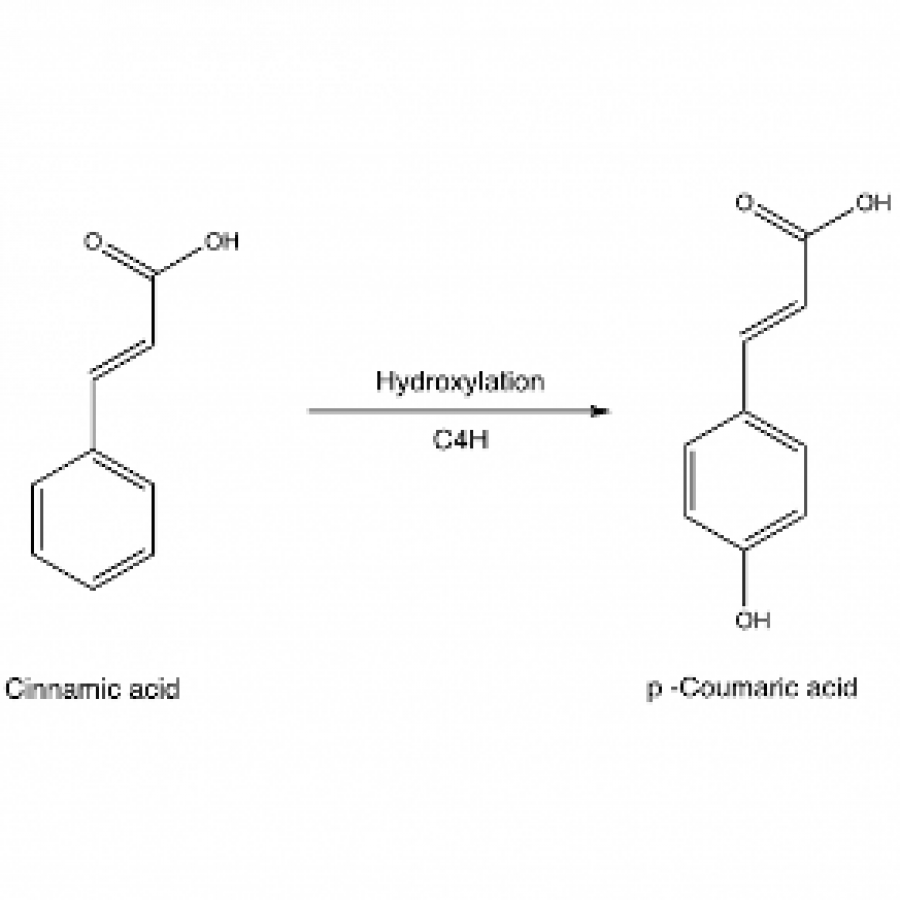 Scheme 1 Hydroxylation of Trans-cinnamic acid into р-coumaric acid by the P450 enzyme C4H. |
#926 | Establishing an in vivo cascade in cyanobacteria for light-driven redox biocatalysis on gram scale |
|
| Presenting author: | Adrian TÜLLINGHOFF of HELMHOLTZ-CENTRE FOR ENVIRONMENTAL RESEARCH (UFZ) |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photo-biotechnology / Biocatalytic cascade / Redox biocatalysis / Light-driven biocatalysis |
| Purpose: | Establishing an in vivo cascade in cyanobacteria for light-driven redox biocatalysis on gram scale Cyanobacteria are ideal host organisms for truly sustainable whole-cell biocatalysis, as both reduction equivalents and O2 are provided in vivo via water oxidation with light as sole energy source. 1 This attracted the coupling of redox reactions, especially oxygenases, to the photosynthetic light reaction via NADPH and O2 in recombinant cyanobacteria. Introducing heterologous reactions into microbial hosts often suffers from reactant toxicity. Based on a recombinant Synechocystis sp. PCC 6803 strain harboring a Baeyer-Villiger monooxygenase (BVMO)2, we could implement the first artificial light-driven redox cascade for the conversion of cyclohexanone to the polymer building block 6-hydroxyhexanoic acid. BVMO and lactonase co-expression, both from Acidovorax sp. CHX100, enabled this two-step conversion with an activity of up to 63.1 ±1.0 U gCDW-1 without accumulating inhibitory ε-caprolactone. Thereby, one of the key limitations of biocatalytic reactions, i.e., reactant inhibition or toxicity, was overcome. In 2 L stirred-tank-photobioreactors, the process could be stabilized for 48 h, forming 23.50 ± 0.84 mM (3.11 ± 0.12 g L-1) 6-HA. The high specificity enabling a product yield (YP/S) of 0.96 ± 0.01 mol mol-1 and the remarkable biocatalyst-related yield of 3.71 ± 0.21 g6-HA gCDW-1 illustrate the potential of producing this non-toxic product in a synthetic cascade. The fine-tuning of the energy burden on the catalyst was found to be crucial, which indicates a limitation by the metabolic capacity of the cells possibly being compromised by biocatalysis-related reductant withdrawal. Product balancing revealed that up to 16% of intracellular reductant can sustainably be branched off without inflicting biocatalyst growth. This study shows the feasibility of light-driven biocatalytic cascade operation in cyanobacteria and highlights respective metabolic limitations and engineering targets. |
| References: | (1) Toepel et al., 2023, Curr. Opin. Biotechnol. 80, 102892. (2) Tüllinghoff et al., 2022, Front. Catal. 1, 780474. |
| Figures: | 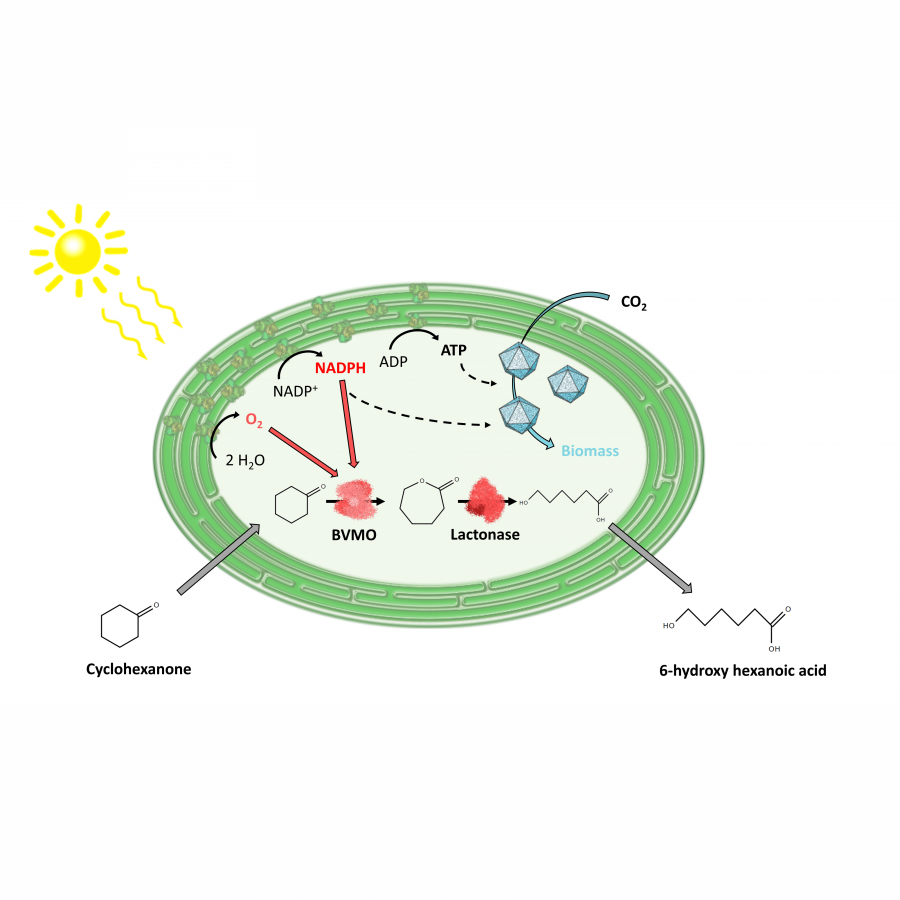 Light-driven redox biocatalysis in cyanobacterial whole-cells via an in vivo cascade With light as only enery source, reduction equivalents and O2 derived from water splitting are utilized by heterologously expressed Bayer-Villiger-moonooxygenase and lactonase to convert cyclohexanone via caprolacton to 6-hydroxyhexanoic acid. |
#932 | Photobiocatalytic decarboxylation of fatty acids in flow – optimization and application in a two-step reaction sequence |
|
| Presenting author: | Stefan SIMIC of UNIVERISTY OF GRAZ |
| Other authors: | Isabella KROSCHEL of UNIVERISTY OF GRAZ Christoph WINKLER of UNIVERISTY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photobiocatalysis / Flow / Reaction sequence / Immobilization |
| Purpose: | The combination of photo- and biocatalysis has provided unique solutions for biocatalysis by both direct and indirect supply of photoinduced electrons to redox enzymes.[1-5] Fatty-acid photodecarboxylase from Chlorella variabilis (CvFAP), an FAD-dependent photoenzyme, was first described in 2017 and has since shown promising results for the production of biofuels.[6-11] Currently, the most common form of biofuel is biodiesel, however, multiple properties of biodiesel limit its practical applicability.[12] As a viable alternative to biodiesel, drop-in biofuels consist of aliphatic compounds, thereby having higher caloric value and similar properties to those of fossil fuels. Despite this, the chemocatalytic production of drop-in biofuels demands harsh and energy-intensive conditions.[13] In this regard, biocatalytic methodology employing CvFAP for biofuel production would enable comparatively mild reaction conditions and circumvent side-product formation, although the photostability of CvFAP remains a key limiting factor for its future applications.[14] Despite the benefits of performing light-dependent reactions in continuous flow,[15] utilization of CvFAP in flow remained unprecedented until recently.[16-17] Herein the CvFAP-catalyzed photodecarboxylation of palmitic acid in continuous flow under homogeneous and heterogeneous catalysis conditions is described. A custom-made flow setup employing fluorinated ethylene propylene tubes was used. Under homogeneous conditions up to 65% yield was achieved after 10 min residence time, amounting to a space-time yield of 5.7 g·L-1·h-1, representing the highest pentadecane productivity of any CvFAP-catalyzed process to date. Additionally, a two-step reaction sequence involving lipase-catalyzed hydrolysis of triolein and subsequent CvFAP-catalyzed decarboxylation of the resulting oleic acid, was performed in flow. This represents the first instance of a two-reaction sequence involving CvFAP in continuous flow. |
| References: | [1] W. Harrison, X. Huang, H. Zhao, Acc. Chem. Res. 2022, 355-359. [2] S. H. Lee, D. S. Choi, S. K. Kuk, C. B. Park, Angew. Chem., Int. Ed. 2018, 57, 7958-7985. [3] J. A. Maciá Agulló, A. Corma Canós, H. García Gómez, Chem. Eur. J. 2015, 21, 10940-10959. [4] Y. Peng, Z. Chen, J. Xu, Q. Wu, Org. Process Res. Dev. 2022. [5] L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist, W. Kroutil, ACS Catal. 2019, 9, 4115-4144. [6] M. Amer, E. Z. Wojcik, C. Sun, R. Hoeven, J. M. Hughes, M. Faulkner, I. S. Yunus, S. Tait, L. O. Johannissen, S. J. Hardman, Energy Environ. Sci. 2020, 13, 1818-1831. [7] S. Bruder, E. J. Moldenhauer, R. D. Lemke, R. Ledesma-Amaro, J. Kabisch, Biotechnol. Biofuels 2019, 12, 1-13. [8] X. Guo, A. Xia, F. Li, Y. Huang, X. Zhu, W. Zhang, X. Zhu, Q. Liao, Energy Convers. Manage. 2022, 255, 115311. [9] Y. Ma, X. Zhang, W. Zhang, P. Li, Y. Li, F. Hollmann, Y. Wang, ChemPhotoChem 2020, 4, 39-44. [10] B.-S. Chen, Y.-Y. Zeng, L. Liu, L. Chen, P. Duan, R. Luque, R. Ge, W. Zhang, Renewable Sustainable Energy Rev. 2022, 158, 112178. [11] W. Xu, Y. Chen, D. Li, Z. Wang, J. Xu, Q. Wu, Molecular Catalysis 2022, 524, 112261. [12] R. R. Monteiro, S. S. da Silva, C. L. Cavalcante Jr, F. M. T. de Luna, J. M. Bolivar, R. S. Vieira, R. Fernandez-Lafuente, Biotechnol. Adv. 2022, 108045. [13] S. Karatzos, J. S. van Dyk, J. D. McMillan, J. Saddler, Biofuels, Bioproducts and Biorefining 2017, 11, 344-362. [14] B. Lakavath, T. M. Hedison, D. J. Heyes, M. Shanmugam, M. Sakuma, R. Hoeven, V. Tilakaratna, N. S. Scrutton, Anal. Biochem. 2020, 600, 113749. [15] C. Sambiagio, T. Noël, Trends Chem. 2020, 2, 92-106. [16] L. A. Benincá, A. S. França, G. C. Brêda, R. A. Leão, R. V. Almeida, F. Hollmann, R. O. de Souza, Molecular Catalysis 2022, 528, 112469. [17] S. Simić, M. Jakstaite, W. T. Huck, C. K. Winkler, W. Kroutil, ACS Catal. 2022, 12, 14040-14049. |
| Figures: | 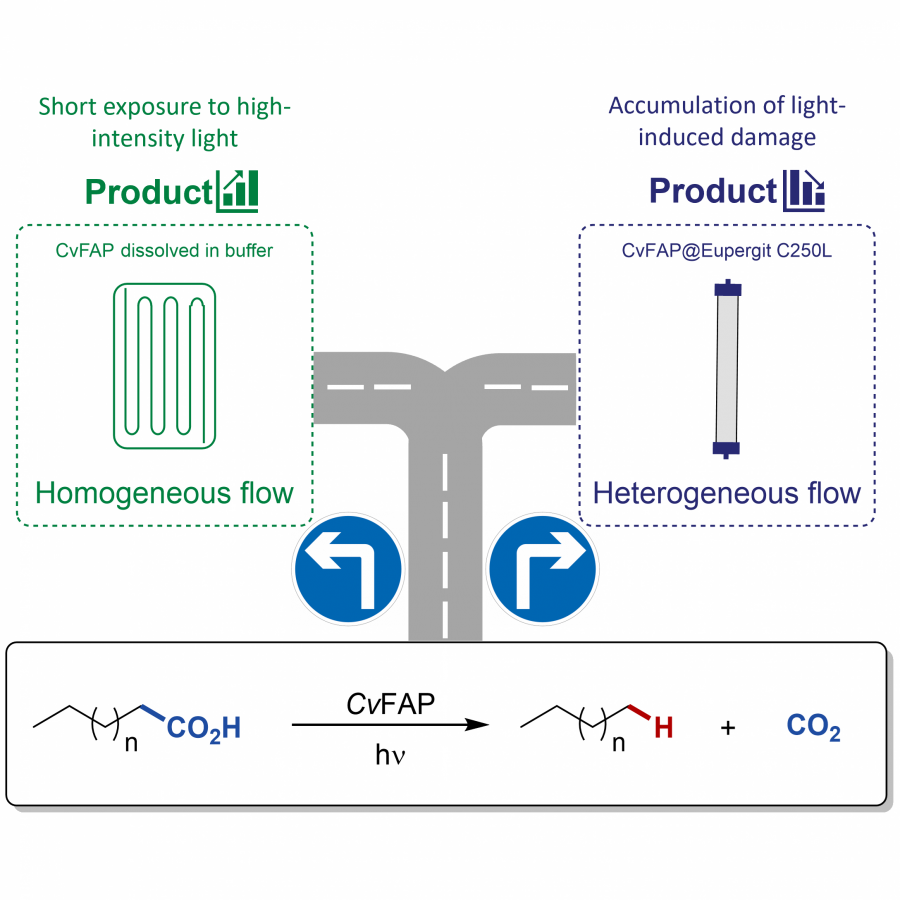 CvFAP-catalyzed photodecarboxylation of fatty acids in homogeneous and heterogeneous flow |
#933 | Application of bacterial thermostable carbonic anhydrases in amine scrubbing for highly efficient CO2 capture from biogenic emissions |
|
| Presenting author: | IO ANTONOPOULOU of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Other authors: | ULRIKA ROVA of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY PAUL CHRISTAKOPOULOS of BIOCHEMICAL PROCESS ENGINEERING, DIVISION OF CHEMICAL ENGINEERING, LULEÅ UNIVERSITY OF TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | carbon capture / amine scrubbing / carbonic anhydrase / industrial emissions |
| Purpose: | The recently released report by the Intergovernmental Panel on Climate Change (IPCC) leaves no doubt about the urgency to dramatically cut greenhouse gas emissions. In order to stay on track to limit earth’s warming to 1.5°C, emissions need to be cut by 45% by 2030, compared to 2019 levels. To reach these goals, Bioenergy with Carbon Capture and Utilization and Storage (BECCUS) is now openly discussed as a solution to decrease net CO2 emissions, as well as a potential technology to move away from fossil carbon by using CO2 as an alternative feedstock for renewable chemicals and fuels. A promising and novel alternative to the current state of the art for post-combustion CO2 capture is the use of the enzyme carbonic anhydrase (CA) (1). CA is one of the fastest enzymes in nature, catalyzing the hydration of CO2 to bicarbonate, which is the limiting step of CO2 absorption. CA is a biocatalyst that can be employed alone in aqueous media to capture CO2, or it can be used as additive in traditional chemical absorption processes, such scrubbing with amine or potassium carbonate solutions, or even the accelerated weathering of alkaline materials that can act as co-sequestrating agents. Aim of this work is to evaluate the potential of novel carbonic anhydrases, such as an evolved CA from Desulfovibrio vulgaris (2,3), to act for improving the rate, efficiency and, in cases, the energy use in carbon capture. We screened five bacterial carbonic anhydrases for their performance as additive in amine-based CO2 capture using methyldiethanolamine (MDEA). The reaction conditions, such as the CO2 absorption temperature, MDEA concentration, enzyme load, effect of CO2 concentration in the gas and effect of inhibitors (NOx, SOx) concentration in the gas, were optimized, targeting to highest productivity and most economic process. The enzymes were also subjected to 10 consecutive absorption-desorption cycles to identify their capability of reuse in terms of stability towards desorption temperatures (80oC) and the highly alkaline amine environment. By developing competitive routes for sequestrating CO2, a realistic pathway opens towards its storage or use as a renewable feedstock. Use includes the production of building block chemicals, such as organic acids via microbial processes, or use of sequestrated carbonates as a liming agent in agriculture. Circular use of materials and in particular CO2 that is of the main gases responsible for global warming, will aid towards a circular bioeconomy but also tackle the challenge of achieving a climate neutral society, aligning with other emerging solutions such as electrification and improving energy efficiency in industrial processes. This work is supported by the H2020 funded project VIVALDI (Grant agreement ID: 101000441). |
| References: | 1. De Oliveira Maciel, A., Rova, U., Christakopoulos, P., Antonopoulou I. (2022). Carbonic anhydrase to boost CO2 sequestration: Improving carbon capture utilization and storage (CCUS). Chemosphere, 299, 134419. 2. Alvizo, O., Nguyen, L.J., Savile, C.K. et al. (2014). Directed evolution of an ultrastable carbonic anhydrase for highly efficient carbon capture from flue gas. PNAS, 111(46), 16436-16441. 3. Sjöblom, M., Antonopoulou, I., Giménez, I.G., de Oliveira Maciel, A., Khokarale, S.G., Mikkola, J.-P., Rova, U., Christakopoulos, P. (2020). Enzyme-assisted CO2 absorption in aqueous amino acid ionic liquid amine blends. ACS Sustainable Chemistry & Engineering, 36(8), 13672-13682. |
| Figures: | 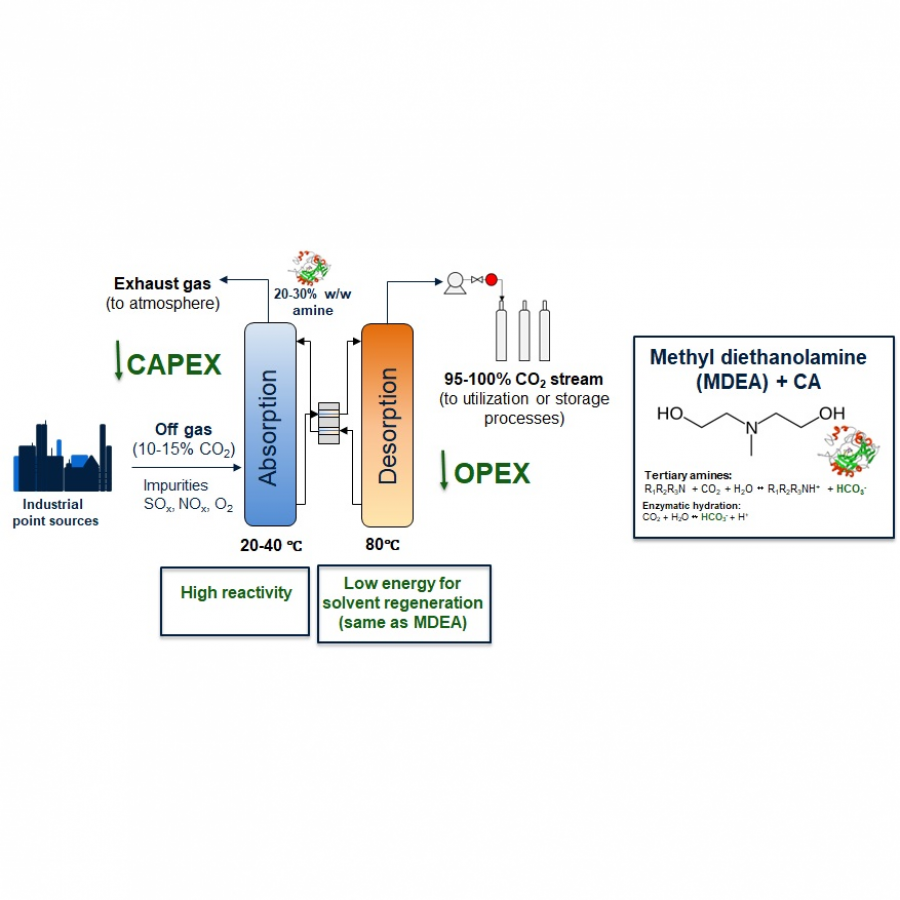 Enzyme-assisted CO2 capture by amine scrubbing Use of CA enhances the CO2 absorption step while maintaining the good desorption properties of MDEA |
#936 | NEW REACTIVITIES FOR TRANSALDOLASE FROM THERMOPLASMA ACIDOPHILUM |
|
| Presenting author: | Massimo DI MASCIO of UNIVERSITÄT GÖTTINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cascade reaction / phosphosugars / / |
| Purpose: | The enzyme transaldolase from Thermoplasma acidophilum (TacTAL) is a thermostable protein that physiologically converts D-fructose 6-phosphate (F6P, the donor) and D-erythrose 4-phosphate (E4P, the acceptor) into D-sedoheptulose 7-phosphate (S7P) and D-glyceraldehyde 3-phosphate (G3P) [1]. It is possible to convert the enzyme from a transaldolase to an aldolase by mutating two amino acids; in fact, while the wild-type TacTAL cannot split the F6P in G3P and dihydroxyacetone, the double mutant E60Q/F132Y can do so [2]. In this study, the affinity of TacTAL (wild-type and “aldolase” variant) for different 5-carbon sugars as substrates was studied using different techniques such as steady-state kinetics, HPLC, mass spectrometry and circular dichroism. Using protein crystallography, the way the enzyme binds these compounds was studied, and the interactions between the substrates and some important residues were identified. The enzymatic production of various 8- and 7-carbon-atom sugars from F6P and/or 5-carbon-atom sugars was demonstrated by GC-MS. Finally, an enzymatic cascade process was developed to maximise the yield of the transaldolase reactions, in which F6P is used as a donor substrate. In this process, glyceraldehyde-3-phosphate (generated when F6P is cleaved) is converted to glycerol-3-phosphate using NADH and the two enzymes: triose-phosphate isomerase (TIM) and sn-glycerol-3-phosphate:NAD+ 2-oxidoreductase (G3PDH). This reaction converts NADH into NAD+. Finally, NAD+ is regenerated using aldehyde dehydrogenase (ADH) from Saccharomyces cerevisiae and isopropanol as substrate. In this last step, ADH converts isopropanol to acetone and NAD+ to NADH by regenerating the cofactor. The results of this study provide a better understanding of how TacTAL works. In the future, it will be possible to improve the enzyme's reactivity towards the compounds tested in this research using rational design. Finally, by optimising the cascade reaction for the production of sugars with 8- and 7-carbon atoms, it might be possible to scale up the reaction increasing the final quantity of the desired product. |
| References: | [1] A. Lehwess-Litzmann, P. Neumann, C. Parthier, S. Lüdtke, R. Golbik, R. Ficner, K. Tittmann, N. Chem. Bio, 2011, 7, 678-684. [2] V. Sautner, M. Friedrich, A. Lehwess-Litzmann, K. Tittmann, Biochemistry, 2015, 54, 4475-4486. |
| Figures: | 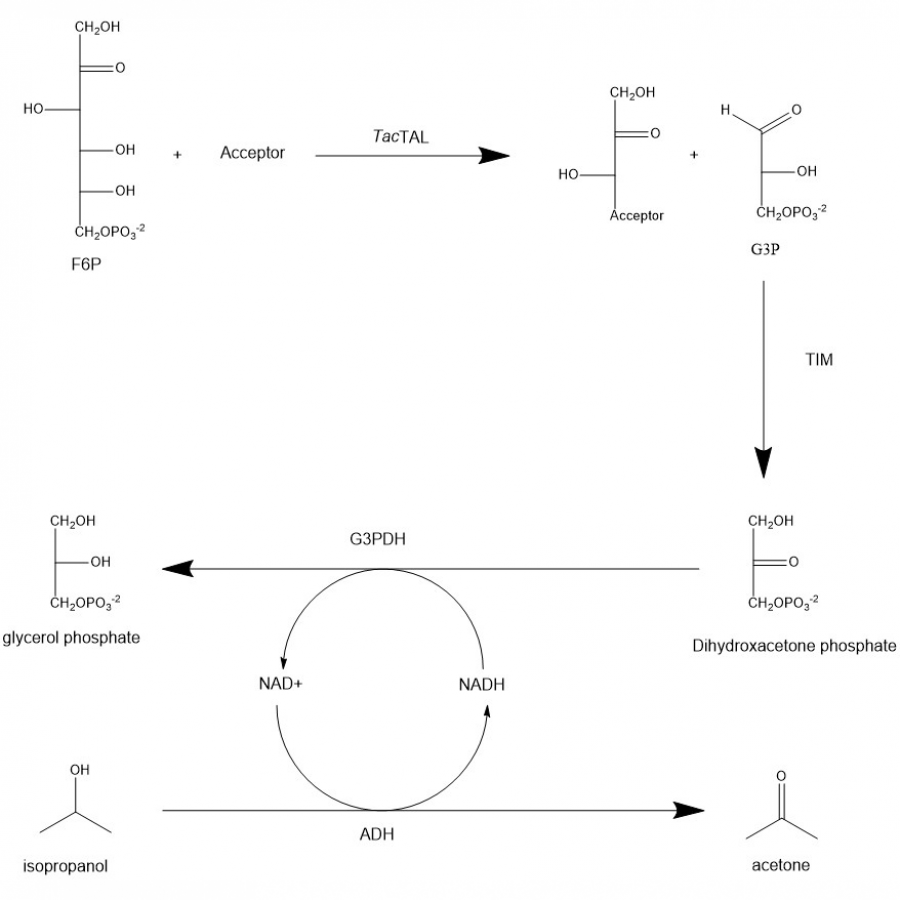 Cascade reaction Cascade reaction, the cofactor NADH needed to eliminate Dihydroxyacetone phosphate is regenerated by the ADH reaction using isopropanol. |
#940 | Enhancement of subunit contacts in human dipeptidyl-peptidase I |
|
| Presenting author: | Liza ULČAKAR of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA |
| Other authors: | Marko NOVINEC of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA Milena STOJKOVSKA DOCEVSKA of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA Nina VARDA of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | dipeptidyl-peptidase I / oligomerization / cooperativity / protein engineering |
| Purpose: | The association of enzymes to form oligomeric structures is widespread in nature. The formation of homooligomers is a structural and functional advantage over the monomeric form, as it usually results in increased stability and can often lead to more complex regulation of activity [1]. A common functional property of many oligomeric proteins is cooperative substrate binding, which results from communication between substrate binding sites on different subunits. This allosteric information is transmitted through the complex in the form of conformational changes of different proportions [2]. Despite its benefits, not all homooligomeric enzymes exhibit cooperative behavior. For example, mammalian dipeptidyl-peptidase I (DPPI) is a homotetramer that belongs to a family of mostly monomeric enzymes, namely the papain-like cysteine proteases (PLPs). In humans, its most evident physiological function, in addition to nonspecific protein turnover, is the activation of granule-associated serine proteases in immune cells [3]. DPPI is synthesized as a preproenzyme that associates to form dimers. During processing into the mature, tetrameric enzyme, the internal propeptide is excised and each monomer is cleaved into three non-covalently bound polypeptide chains, i.e., the exclusion domain and the heavy and the light chains [4]. The resulting tetrameric complex is a dimer of dimers with two sets of isologous subunit interactions, namely, the head-to-tail and lateral interactions [5]. Despite initial findings of cooperativity in this enzyme, these results could not be reproduced by us or by other research groups [6–8]. Furthermore, we performed a computational analysis of the communication pathways between active sites on different subunits of DPPI and concluded that there are no cooperative communication pathways in the protein. In DPPI, all subunits, as well as domains within each subunit, are linked by non-covalent interactions. Therefore, their association is reversible, and they will inevitably dissociate when sufficiently diluted. Thus, over a certain concentration range, an equilibrium exists between different oligomeric forms. The goal of our research group is to develop variants of DPPI with increased affinity between subunits by strengthening the interactions at subunit interfaces, thereby minimizing the probability of subunit dissociation. Moreover, we believe that enhanced interaction between specific amino acid residues could lead to the emergence of a communication pathway and thus cooperativity between subunits. In addition to developing improved enzymes, our research will advance the state-of-the art on the evolution of cooperativity in oligomeric proteins. This knowledge can then be applied to the design of oligomeric and cooperative enzymes from monomers. In order to engineer a cooperative DPPI, we have been using rational and semirational protein engineering approaches. By calculating the correlation energies and the distances between interacting amino acid residues on the contact surfaces, we determined that the lateral interactions are weaker than the head-to-tail interactions [9]. We then aimed to enhance the lateral interactions by introducing disulfide bonds, hydrophobic interactions and salt bridges into the contact surface. However, these mutant variants remain to be characterized in the future [9]. Unfortunately, rationally introduced mutations have an unpredictable effect on the folding ability of DPPI. Therefore, we opted for a semirational approach by randomly mutating predefined interaction hotspots on the contact surfaces. The resulting library is then screened using the commercially available bacterial two-hybrid system BACTH [10]. Experiments are underway to identify DPPI variants with increased affinity for association. |
| References: | [1] d. s. goodsell, a. j. olson. annu. rev. biophys. biomol. struct. 2000, 29, 105-153. [2] g. g. hammes, s. j. benkovic, s. hammes-schiffer. biochemistry. 2011, 50, 48, 10422-10430 [3] c. t. n. pham, t. j. ley. immunology. 1999, 96, 8627-8632. [4] a. s. lamort, y. hamon, c. czaplewski, et al. int. j. mol. sci. 2019, 20(19), 4747. [5] d. turk, v. janjic, i. stern, m. podobnik, et al. embo j. 2001, 20(23), 6570-6582. [6] m. rebernik, b. lenarcic, m. novinec. protein expr. purif. 2019, 157, 21-27. [7] m. rebernik, t. snoj, m. klemencic, m. novinec. arch. biochem. biophys. 2019, 675, 108121. [8] v. santilman, m. jadot, f. mainferme. eur. j. cell biol. 2002, 81(12), 654-663. [9] b. cigic, r. h. pain. eur. j. biochem. 1999, 264(3), 944-951. [10] n. varda. dipl. delo. 2021. [11] a. battesti, e. bouveret. methods. 2012, 58(4), 325-334. |
#941 | Designed out-of-active-site mutations in human oxidosqualene cyclase modulate the activation entropy and enthalpy of the cyclization reaction |
|
| Presenting author: | David HUETING of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Per-Olof SYRÉN of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Abstract Cholesterol and steroids in general, are essential in all complex life forms. Cholesterol precursors are synthesized from (S)-2,3-oxidosqualene via lanosterol with the aid of the catalyst human oxidosqualene cyclase (hOSC). Due to the essential role of cholesterol in health and disease and large implication an imbalance can have, the enzymes involved in the synthesis of cholesterol have gained increased attention. To understand the mechanism behind the catalysis is essential to understanding the processes and help in understanding the ins and outs of imbalanced cholesterol. The enzyme involved in synthesizing lanosterol from oxidosqualene, has been well studied and the reaction mechanism has been resolved via QM/MM. However, in these types of enzymes the catalysis and mechanism is depending on two major energetic driving forces, entropy and enthalpy. In this enzyme the reaction is mainly driven by entropy. We hypothesized that the entropy of the reaction is provided by the water molecules present in the enzyme and that the nature of the reaction can be changed. We intended to change the driving force of the reaction to enthalpy via water tunnel analysis, MD simulations and site directed mutagenesis. We aimed to block of tunnels found within the protein with bulky substrates to alter the accessibility of water in the active site. MD simulations uncovered that in some variants the tunnels were indeed closed and a reduced number of water molecules was around the active site. The reaction kinetics were tested using Eyring transition state theory. With this technique, we can extract the entropic and enthalpic contribution in the catalysis of the enzyme. Indeed what we see is that the changed water contribution in the active site and via these tunnels have an immense impact on the entropy and enthalpy of the reaction. We observed a complete change of entropy of as much as 49 kcal/mol at 300K. The enthalpy changed with similarly high values of 37.5 kcal/mol. Additionally we tested computational methods to predict variants based on sequence and on provided tunnel blocking mutations. Some of the variants designed had an increased activity of 5-6x. A different subset of variants had an altered temperature dependance, where catalysis within the enzyme went faster at lower temperatures. We were able to engineer enzymes to have a completely different energetic profile for catalysis, increasing our understanding of these types of enzymes and enabling a novel way to engineer enzymes for increased activity at high and low temperatures. |
#944 | Co-engineering of hemicellulases from a xylan PUL for improved deconstruction of plant biomass |
|
| Presenting author: | Ahmed KHAMASSI of TOULOUSE BIOTECHNOLOGY INSTITUTE / INSTITUT DE PHARMACOLOGIE ET DE BIOLOGIE STRUCTURALE |
| Other authors: | Thomas ENJALBERT of INSTITUT DE PHARMACOLOGIE ET DE BIOLOGIE STRUCTURALE Sandra PIZZUT of TOULOUSE BIOTECHNOLOGY INSTITUTE Sophie BOZONNET of TOULOUSE BIOTECHNOLOGY INSTITUTE Estelle BONNIN of UR 1268 BIA, BIOPOLYMERS INTERACTIONS ASSEMBLIES Cédric MONTANIER of TOULOUSE BIOTECHNOLOGY INSTITUTE Samuel TRANIER of INSTITUT DE PHARMACOLOGIE ET DE BIOLOGIE STRUCTURALE Claire DUMON of TOULOUSE BIOTECHNOLOGY INSTITUTE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Xylanase / Xylosidase / Enzyme engineering / Structure-Function Characterization |
| Purpose: | Valorization of Lignocellulosic (LC) biomass is a major economic and environmental challenge. Indeed, LC biomass is a renewable and inexhaustible carbon source on Earth, considered as a key raw material to drive a sustainable economy and reduce our carbon footprint. This biomass is mainly composed of a complex matrix of cellulose and hemicellulose polysaccharides surrounded by lignin, an aromatic polymer [1]. Xylan is the main hemicellulosic polysaccharide in cereals and plays a crucial role in plat cell wall integrity [2]. This polysaccharide is composed of a linear chain of D-xylopyranoside units with β-1,4 linkages that can be substituted by L-arabinose and D-glucuronic acid. Microorganisms have evolved to deconstruct LC and have developed a large set of glycoside hydrolases (GHs), acts in combination with carbohydrate esterases (CE) and lytic polysaccharide monooxygenases (LPMO), all classified in the CAZy database, in order to entangle this complex network and metabolize this carbon source (fig.1) [3]. For the deconstruction of xylan, several main chain depolymerizing GHs, endo-1,4-β-D-xylanases and β-D-xylosidases, are required in combination with debranching enzymes, such as α-L-arabinofuranosidases and/or carbohydrate esterases. GHs have been shown to act in synergy to deconstruct LC and increase the yield of saccharification [4]. However, the molecular determinants that influence enzyme synergy remain unclear [3,4]. In bacteroidetes, these enzymes are often encoded by fine-tuned gene clusters co-regulated for detection, transport and hydrolysis of a dedicated polysaccharide, and called Polysaccharide Utilization Locus (PUL) [5]. Moreover, PUL-encoded GHs were shown to work in synergy and optimize the deconstruction of polysaccharides. In order to investigate the molecular mechanisms involved in enzyme synergy, we “co-engineered” a xylanase and a xylosidase belonging to a xylanolytic PUL [6], meaning we submitted them to a simultaneous cycle of random mutagenesis and screened for pairs exhibiting better arabinoxylan degradation (fig.2). Here, we will present this strategy that allowed the screening of 24 000 mutants and identification of six hits harboring improved activities toward soluble xylan and wheat bran. In order to explain the differences observed in activity and synergy, we will also present the biochemical and structural characterization of the most interesting mutants. This work aims to provide new insights to a better understand enzyme synergy while selecting improved mutants to better valorize lignocellulosic biomass. |
| References: | [1] Li, T. & Takkellapati, S. The current and emerging sources of technical lignins and their applications. Biofuels, Bioproducts and Biorefining 12, 756-787 (2018). [2] Heinonen, E., Henriksson, G., Lindström, M. E., Vilaplana, F. & Wohlert, J. Xylan adsorption on cellulose: Preferred alignment and local surface immobilizing effect. Carbohydrate Polymers 285, 119221 (2022). [3] Khamassi A., Dumon C. Enzyme synergy for plant cell-wall polysaccharide degradation. Essays in Biochemistry (2023) In press. [4] Van D, J. S. & Pletschke, B. I. A review of lignocellulose bioconversion using enzymatic hydrolysis and synergistic cooperation between enzymes--factors affecting enzymes, conversion and synergy. Biotechnol Adv 30, 1458-1480 (2012). [5] Grondin, J. M., Tamura, K., Déjean, G., Abbott, D. W. & Brumer, H. Polysaccharide Utilization Loci: Fueling Microbial Communities. J. Bacteriol. 199, e00860-16, /jb/199/15/e00860-16.atom (2017). [6] Bastien, G. et al. Mining for hemicellulases in the fungus-growing termite Pseudacanthotermes militaris using functional metagenomics. Biotechnol Biofuels 6, 78 (2013). |
| Figures: | 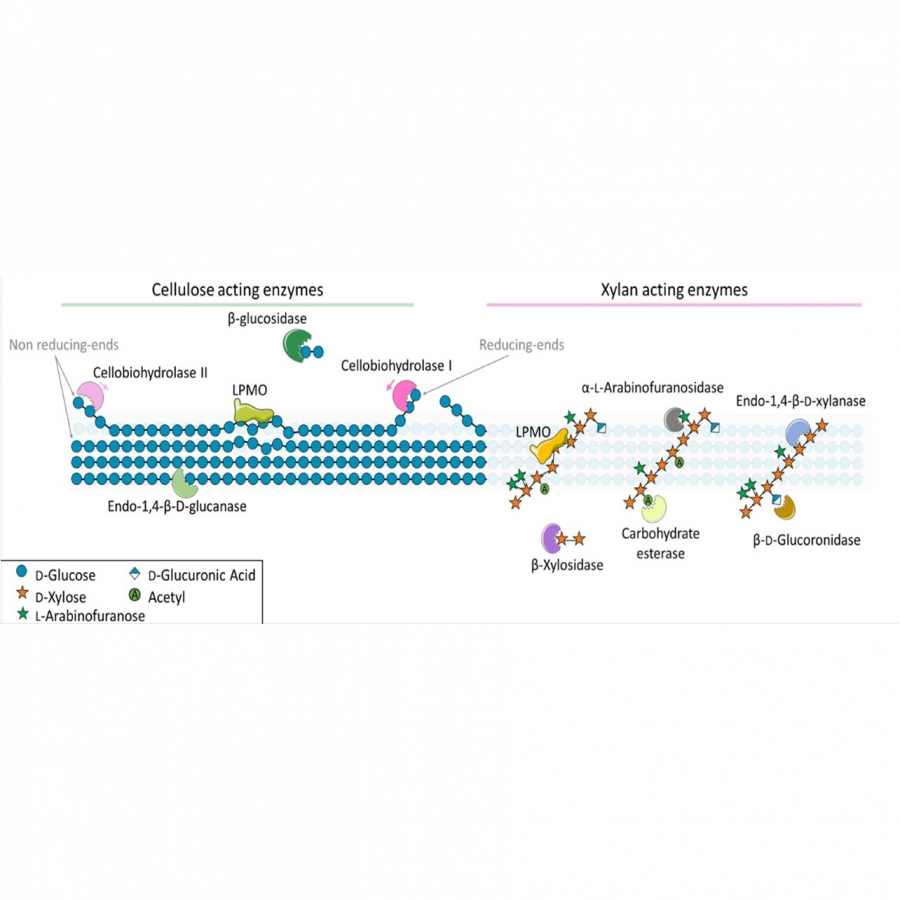 Fig. 1 : Schematic representation of enzyme activities degrading cellulose and xylan On the left, enzymes acting on cellulose polymers and β-glucosidase acting on cellobiose. On the right, schematic representation of xylan polymers with several ramifications, enzymes acting on xylan and β-xylosidase hydrolyzing xylobiose [3]. 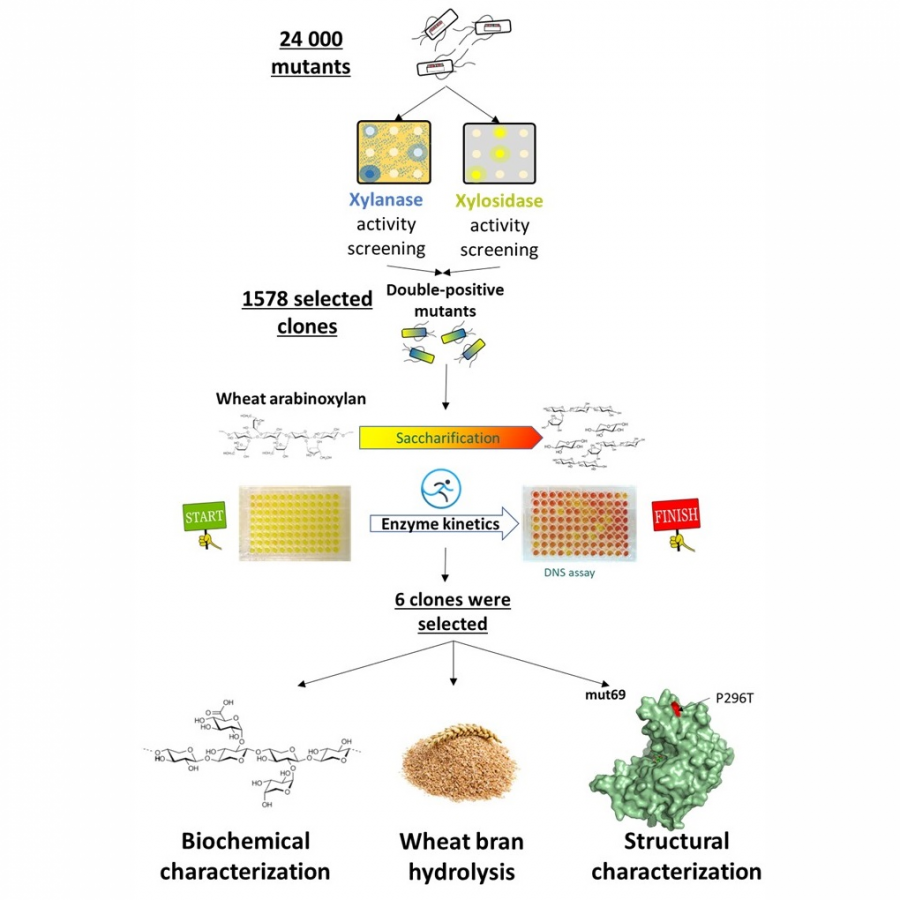 Schematic representation of the co-engineering strategy, performed to screen xylanase-xylosidase mutant library Main steps from the creation of the mutant library to the structure-function characterization of the xylanase-xylosidase positive clones were shown. |
#950 | Halohydrin dehalogenase-catalysed biotransformation in micellar media |
|
| Presenting author: | Petra ŠVACO of RUĐER BOŠKOVIĆ INSTITUTE |
| Other authors: | Maja MAJERIĆ ELENKOV of RUĐER BOŠKOVIĆ INSTITUTE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | halohydrin dehalogenase / micellar media / substrate inhibition / enzyme stability |
| Purpose: | Halohydrin dehalogenases (HHDHs) are enzymes that catalyse the formation and conversion of epoxides [1]. They have been widely used to transform epoxide into an β-substituted alcohol, often resulting in optically pure products [2]. However, the application of HHDHs in aqueous media, in which reactions are usually performed, is limited by the low solubility of hydrophobic substrates, their hydrolytic instability and the presence of numerous inhibitions [3]. Engineering the reaction medium can lead to significant improvements in terms of process efficiency, ultimately affording higher product yields. In this work, micellar media were introduced as an alternative for enantioselective biosynthesis employing HHDHs. Three different non-ionic surfactants (TPGS-750-M, Brij® C10 and TWEEN®) were evaluated for the effect on the activity and stability of HHDH from Agrobacterium radiobacter AD1 (HheC) in form of cell-free extract and whole cells. Whole cells showed greater stability in 2% TPGS-750-M compared to the cell-free extract under the same conditions. This non-ionic surfactant with hydrophobic core forms micelles in water which serve as a medium for biocatalysis, but also as a reservoir for substrates and products [4]. To test if this feature may provide decreased substrate inhibition of HheC. The influence of different epoxide concentrations was compared for the azidolysis of 2-benzyl-2-methyloxirane in buffer and 2% TPGS-750-M. A considerable shift in maximum of enzyme activity towards higher substrate concentrations (from 10 to 50 mM) is observed in 2% TPGS-750-M, compared to the one achieved in a buffer. We also found that HheC in micellar media containing TPGS-750-M in a higher concentration (4 and 6%), has a better capacity to tolerate higher concentration of tested epoxide. These results suggest a promising effect of micellar media on biocatalytic properties of HheC. |
| References: | [1] Hasnaoui-Dijoux, G., Majerić Elenkov, M., Lutje Spelberg, J.H., Hauer, B., Janssen, D.B., ChemBioChem. 2008, 9, 1048-1051. [2] Findrik Blažević, Z., Milčić, N., Sudar, M., Majerić Elenkov, M., Adv Synth Catal. 2021, 363, 388-410. [3] Milčić, N., Sudar, M., Dokli, I., Majerić Elenkov, M.M., Findrik Blažević, Z., React Chem Eng. 2023, 8, 673-686. [4] Cortes-Clerget, M., Akporji, N., Zhou, J., Gao, F., Guo, P., Parmentier, M., Gallou, F., Berthon, J.-Y., Lipshutz, B.H., Nat Commun. 2019, 10, 2169. |
| Figures: | 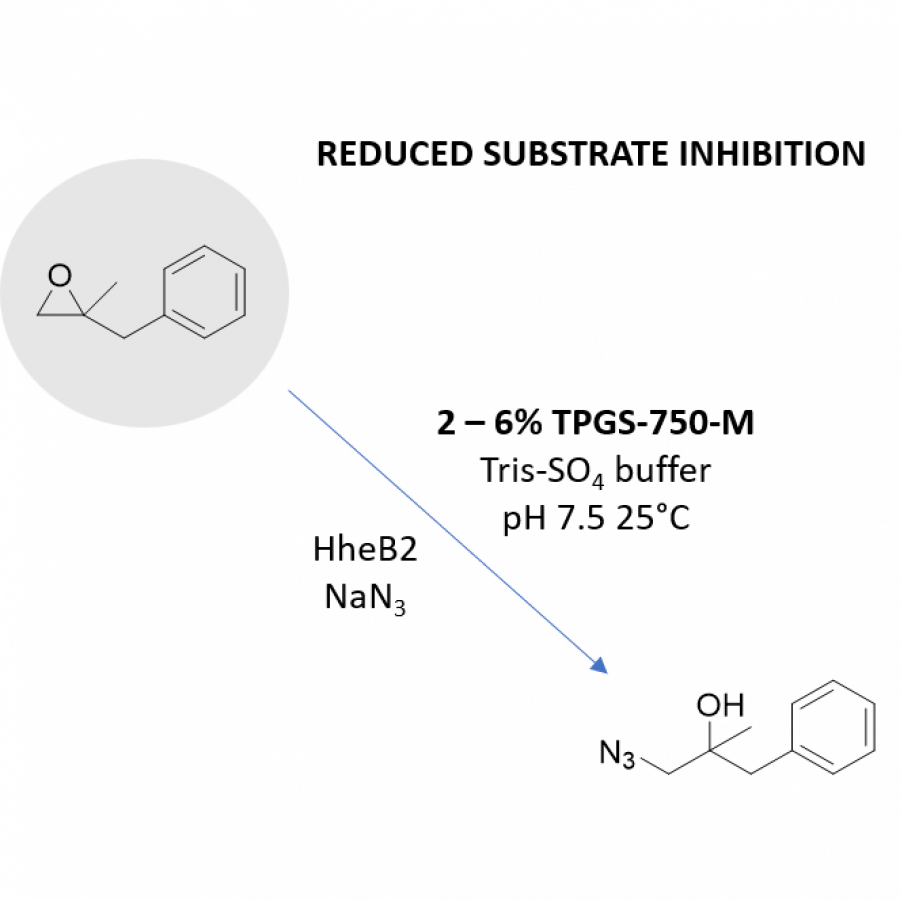 Halohydrin dehalogenase-catalysed biotransformation in micellar media Selected reaction for evaluation of minimizing substrate inhibition of HheB2 in micellar media. |
#952 | A Unique Cyclic Diphosphoglycerate Synthase enzyme for Extremolyte Production in Thermus thermophilus |
|
| Presenting author: | Jennifer LITTLECHILD of UNIVERSITY OF EXETER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Extremolyte / Cyclic diphosphoglycerate synthase / Synthetic Pathway / Thermophilic host |
| Purpose: | The primative microorganism Methanothermus. fervidus was first isolated in 1981 by Setter and has a genome coding for only 1311 proteins and 50 RNA genes. It is smallest sequenced genome observed for a free-living organism. This anaerobic hyperthermophilic archaeon produces energy through the reduction of carbon dioxide with hydrogen to produce methane. It accumulates an unusual cyclic form of 2,3 phosphoglycerate for protein stabilisation at temperatures up to 97 degrees C. The thermophilic bacteria Thermus thermophilus has been used as a host for expression for the two enzyme cascade for synthesis of the so called small molecule extremolyte cDPG which has healthcare and cosmetic applications [1]. The enzymes used in this synthetic pathway, 2-phosphoglycerate kinase (2PGK) and cyclic diphosphoglycerate synthetase (cDPGS) have been over-expressed in Escherchia coli to study them both biochemically and structurally. Both enzymes have low sequence identity to other known enzymes available in the Protein Data Base. The cDPGS structure has been solved to high resolution using seleno-methionine due to no molecular replacement models being available. It has also been solved in complex with 2,3-diphosphoglycerate and ADP Mg2+ showing the conformational changes that occur on substrate binding [2]. The structure appears unique with only one domain showing homology to other known proteins that have unrelated function. The structure of the other enzyme, 2PGK is currently being solved and is remote regarding homologues currently known. This leads the way for other thermophilic proteins to be used for whole cell synthesis of related small molecules with industrial applications. |
| References: | [1] De Rose, S. A., Finnigan, W., Harmer, N. J., Littlechild, J. A. (2021). Production of the Extremolyte Cyclic 2,3-Diphosphoglycerate Using Thermus thermophilus as a Whole-Cell Factory . Front. Catal. 1:80341 [2] De Rose, S. A., Isupov, M., Worthy, H., Harmer, N. J., Littlechild, J. A. Structural characterisation of cyclic 2,3-diphosphoglycerate synthetase from Methanothermus fervidus.in preparation |
| Figures: |  Figure 1 Structural superimposition of the cDPGS monomer with its three closest structural homologues as reported by the DALI server. From left to right: Aquifex aeolicus tetraacyldisaccharide 4′-kinase (red ribbon), Klebsiella pneumoniae urease accessory protein  Figure 2 Cartoon illustration of the ADP and 2,3 DPG bound to cDPGS. The two monomeric subunits are shown in cyan and green. ADP and 2,3 DPG are shown in ball-and-stick representation (carbon, green; oxygen, red; nitrogen, blue; phosphorus, pink). The Mg2+ atom i |
#953 | Biocatalytic cascades for the synthesis of amines with two stereogenic centres |
|
| Presenting author: | Francesco MUTTI of UNIVERSITY OF AMSTERDAM |
| Other authors: | Tanja KNAUS of UNIVERSITY OF AMSTERDAM Maria CORRADO of UNIVERSITY OF AMSTERDAM |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalytic cascades / chiral amines / oxidoreductases / transferases |
| Purpose: | Chiral amines are obtained through different enzymatic and chemo-enzymatic methods comprising kinetic resolution, dynamic kinetic resolution and deracemisation of racemic mixture, and asymmetric synthesis from prochiral substrates. These processes are performed using a variety of enzymes, namely hydrolases, transaminases, ammonia lyases, amine oxidases, imine reductases/reductive aminases, amine dehydrogenases, engineered cytochromes P450, etc. Therefore, structurally diverse chiral amines are accessible nowadays using biocatalysis; however, in most cases, only amines possessing one stereogenic centre are generated. Chiral amines with more than one stereogenic centre are found in many biologically active compounds but are more challenging to synthesise; these amines could be obtained by developing biocatalytic cascades. Early examples are the biocatalytic synthesis of enantioenriched 1,2 amino-alcohols having two stereogenic centers by starting from an aldehyde and pyruvate and combining a carboligase with a transaminase,[1] or by starting from a diketone and combining an alcohol dehydrogenase with a transaminase.[2] Another option is the asymmetric transamination of an α-hydroxy ketone.[3] In this context, we have recently reported a sequential multi-enzymatic route for the formal chemo-, regio- and stereoselective aminohydroxylation of β-methylstyrene that consumes only dioxygen, ammonia and formate, and generates carbonate as the only by-product.[4] This cascade comprised enantioselective epoxidation and hydrolysis followed by a hydride-borrowing alcohol amination (Fig. 1, A and B1). This was also the first study in which the regioselectivity of the dual-enzyme hydride-borrowing alcohol amination was investigated and exploited in preparative scale. In this way, β-methylstyrene was converted into (1R,2R) and (1S,2R)-phenylpropanolamine in 59–63% isolated yields, and up to >99.5 : <0.5 d.r. and e.r. In another study, we developed a variation of the above-mentioned cascade by combining an alcohol dehydrogenase, a transaminase and an alanine dehydrogenase for the second stereo- and regio-selective alcohol amination step (Fig. 1, A and B2).[5] Thus, again, 1,2-amino alcohols with two stereogenic centres were obtained with e.r. and d.r. up to >99.5 : <0.5 and analytical yields up to 95%. Notably, this was the first report on an enzymatic method that enabled to obtain all the four possible phenylpropanolamine stereoisomers in excellent enantio- and diastereo-selectivity. With the aim of expanding the accessible chemical space for chiral amines by using biocatalysis, we investigated a cascade that combined ene-reductases with imine reductases/reductive aminases to enable the conversion of α,β-unsaturated ketones into primary, secondary, and tertiary amines containing two stereogenic centres and in very high chemical purity (up to >99%), diastereomeric ratio, and enantiomeric ratio (up to >99.8 : <0.2), (Fig. 2).[6] Compared with previously reported strategies,[7] our strategy could synthesise two, three, or even all four of the possible stereoisomers of the amine products while precluding the formation of side-products. Furthermore, ammonium or alkylammonium formate buffer was used as the only additional reagent since it acted both as an amine donor and as a source of reducing equivalents. |
| References: | [1] T. Sehl et al. Angew. Chem. Int. Ed. 2013, 52, 6772-6775. [2] T. Sehl et al. Green Chem. 2014, 16, 3341-3348. [3] J. D. Zhang et al. J. Biotechnol. 2019, 290, 24-32. [4] M. L. Corrado et al., Green Chem. 2019, 21, 6246-6251. [5] M. L. Corrado et al. ChemBioChem 2021, 22, 2345-2350. [6] T. Knaus et al. ACS Catal. 2022, 12, 14459-14475. [7] a) D. Monti et al. ChemCatChem 2015, 7, 3106-3109; b) L. Skalden et al. Tetrahedron 2016, 72, 7207-7211; c) E. P. J. Jongkind et al. ChemCatChem 2021, 14, e2021015; d) T. W. Thorpe et al. Nature 2022, 604, 86-91. |
| Figures: |  Multi-enzymatic synthesis of 1,2 amino alcohols with two stereogenic centres. Conversion of styrenes into 1,2-amino alcohols possessing two stereogenic centres via biocatalytic cascades comprising monooxygenases, epoxy hydrolases, alcohol dehydrogenases, transaminases, amine dehydrogenases.  Multi-enzymatic synthesis of amines with two stereogenic centres. Conversion of alpha,beta-unsaturated ketones into amines possessing two stereogenic centres via biocatalytic cascades comprising ene reductases, amine dehydrogenases, imine reductases. |
#958 | A Novel ‘Split-gene’ transketolase from the hyper-thermophilic bacterium Carboxydothermus hydrogenoformans: structure and biochemical characterisation |
|
| Presenting author: | Paul JAMES of NORTHUMBRIA UNIVERSITY |
| Other authors: | Jennifer LITTLECHILD of UNIVERSITY OF EXETER Misha ISUPOV of UNIVERSITY OF EXETER Simone antonio DE ROSE of UNIVERSITY OF EXETER |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ‘split-gene’ transketolase / thermal stability / Industrial applications / hyperthermophilic |
| Purpose: | The genes encoding the two parts of a novel split transketolase were identified from the genome of the hyperthermophilic Carboxydothermus hydrogenoformans. These have been cloned and over-expressed in Escherichia coli before being reconstituted and purified by size exclusion chromatography. This novel enzyme is the first TK to be reconstituted and characterised both biochemically and structurally with the enzyme active using hydroxypyruvate as the ketol donor and different aldehyde acceptors. The reconstituted active alpha2beta2 tetrameric enzyme has been characterised biochemically and its structure determined to high resolution. The activity of this enzyme has been monitored using the non-natural commercially interesting reaction using hydroxypyruvate as the ketol donor and a range of aldehydes including glycolaldehyde, butylaldehyde and cyclohexane aldehyde as the acceptor substrate. This reaction is used for the synthesis of a range of unusual sugars of interest for the pharmaceutical industries and proceeds to 100% completion due to the release of the product carbon dioxide This novel reconstituted transketolase is thermally stable with no loss of activity after incubation for 1 hour at 70 ºC and is stable after 1-hour incubation with 50 % of the organic solvents methanol, ethanol, isopropanol, DMSO, acetonitrile and acetone. In the presence of the cofactor thiamine pyrophosphate and calcium, the crystals of the reconstituted alpha2beta2 tetrameric transketolase allowed the structure to be determined to 1.4 Å resolution. The ability of thiamine pyrophosphate (TPP) enzymes such as transketolase to form carbon-carbon bonds is a valuable feature used in the synthesis of high value chiral compounds and there is an interest to extend the range of substrates and products of these reactions. 1-Deoxy-D-xylulose 5-phosphate synthase (DXP synthase) is another TPP-dependent catalyst which transfers a two-carbon unit from pyruvate onto specific aldose D-glyceraldehyde 3-phosphate but is able to use the cheaper and more stable pyruvate as the ketol donor. A structural comparison of the split Carboxythermus transketolase, the Escherichia coli full length transketolase and the DXP synthase has been carried out in an attempt to rationalise the substrate specificity differences between the three enzymes. This reconstituted ‘split-gene’ enzyme is the first example of a split transketolase to be fully characterised allowing its potential for industrial biocatalysis to be evaluated. |
| References: | James, P., Isupov, M.N., De Rose, S.A., Sayer, C., Cole, I.S., and Littlechild, J.A. (2020). A 'Split-Gene' Transketolase From the Hyper-Thermophilic Bacterium Carboxydothermus hydrogenoformans: Structure and Biochemical Characterization. Front Microbiol 11, 592353. doi: 10.3389/fmicb.2020.592353. Littlechild, J., Turner, N., Hobbs, G., Lilly, M., Rawas, A., and Watson, H. (1995). Crystallization and preliminary X-ray crystallographic data with Escherichia coli transketolase. Acta Crystallogr D Biol Crystallogr 51, 1074-1076. doi:10.1107/S0907444995005415. |
| Figures: | 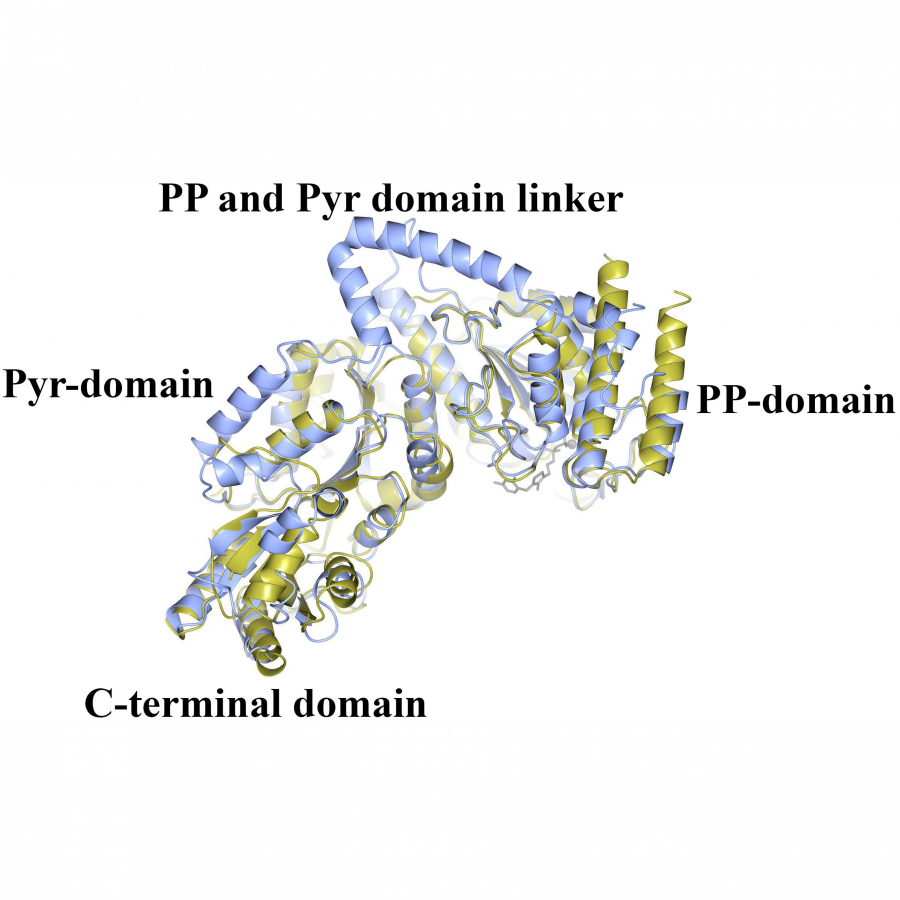 A comparison of the structure of ChTK αβ heterodimer A comparison of the structure of ChTK αβ heterodimer (gold) superimposed onto the monomer of EcTK (ice blue) showing the equivalent positions of the PP-domain, the Pyr-domain and the C-terminal domain. The linker between the PP and Pyr domains is present 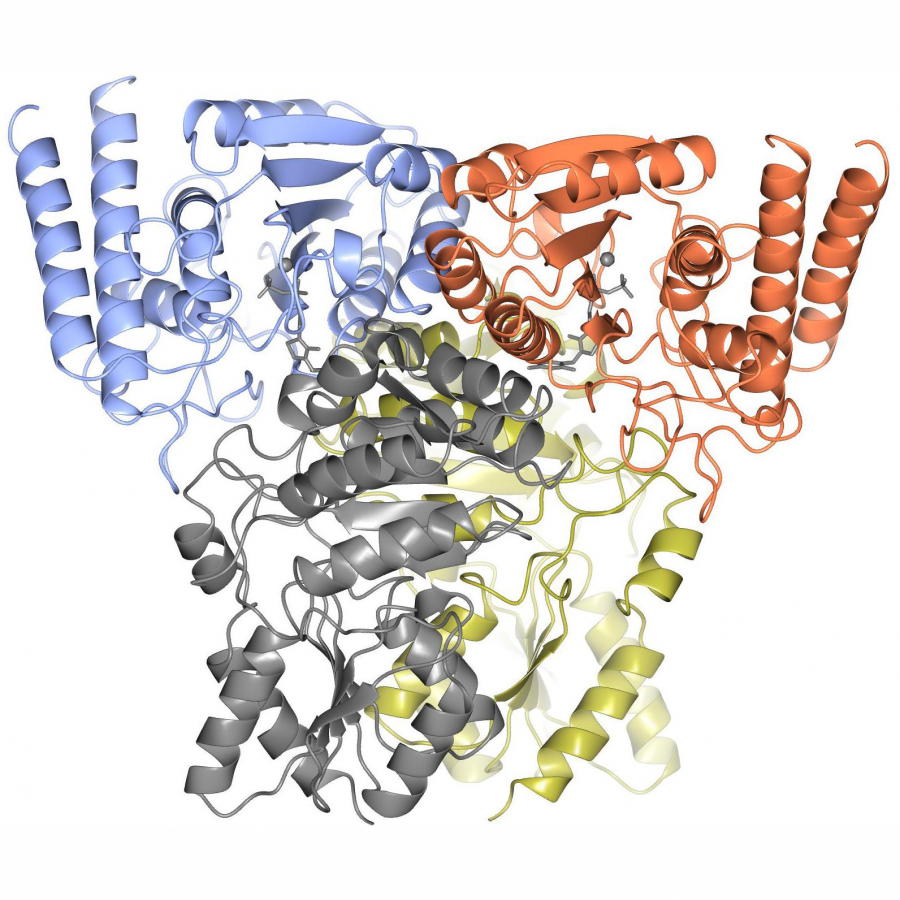 The overall structure of ChTK-F heterotetramer The overall structure of ChTK-F heterotetramer is shown as a cartoon model with the ChTK-N monomers shown in blue and red and the ChTK-C monomers shown in gray and gold with the Ca2+ and TPP cofactor shown as stick models in gray. |
#959 | Expanding the enzyme universe: Incorporation of functional secondary amines through stop codon suppression |
|
| Presenting author: | Ivana DRIENOVSKA of VRIJE UNIVERSITEIT AMSTERDAM |
| Other authors: | Alejandro GRAN SCHEUCH of VU AMSTERDAM Elisa BONANDI of VU AMSTERDAM |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | non-canonical amino acids / artificial enzymes / biocatalysis / new-to-nature reactivities |
| Purpose: | Enzymes are immensely powerful catalysts. Their unmatched rate accelerations and impressive selectivities have inspired chemists to unlock their catalytic potential in industrial applications. This industrial biotechnology revolution is of great importance since enzymes can represent an environmentally friendly, “green’’ alternative to multi-step chemical syntheses, which often require harsh conditions, utilize toxic reagents and solvents, and show poor atom economy [1]. Notably though, natural enzymes are only able to catalyze a fraction of the reactions routinely employed by synthetic chemists. As a result, creating designer biocatalysts with the ability to efficiently catalyze transformations that cannot be found in nature’s repertoire remains a long-standing goal in chemical biology [2]. Efforts toward this goal typically consider only canonical amino acids in the initial design process. However, natural amino acids contain limited functional groups covering relatively limited chemistry and as a result, posttranslational modifications and cofactors are often required for proteins to gain their full functionality. In this study, a panel of ncAAs with functional secondary amines was designed, synthesized, and characterized, and a library of designer enzymes was prepared by incorporating these newly prepared ncAAs into an evolvable protein scaffold. (Figure 1) The catalytic properties of the resulting artificial enzymes were evaluated using the Michael addition of nitromethane to cinnamaldehyde. Variants displayed increased conversion and were able to access (S)-product with modest enantioselectivity. These findings highlight the potential of ncAAs in expanding the reaction scope of designer enzymes and provide a promising avenue for the development of more efficient and versatile biocatalysts. |
| References: | [1] R. Wohlgemuth, Cur. Opin. Biotechnol., 2010, 21, 713-724. [2] D. Hilvert, Annu. Rev. Biochem. 2013, 82, 447-470. [3] I. Drienovská, G. Roelfes, Nat. Catal., 2020, 3, 193-202. |
| Figures: | 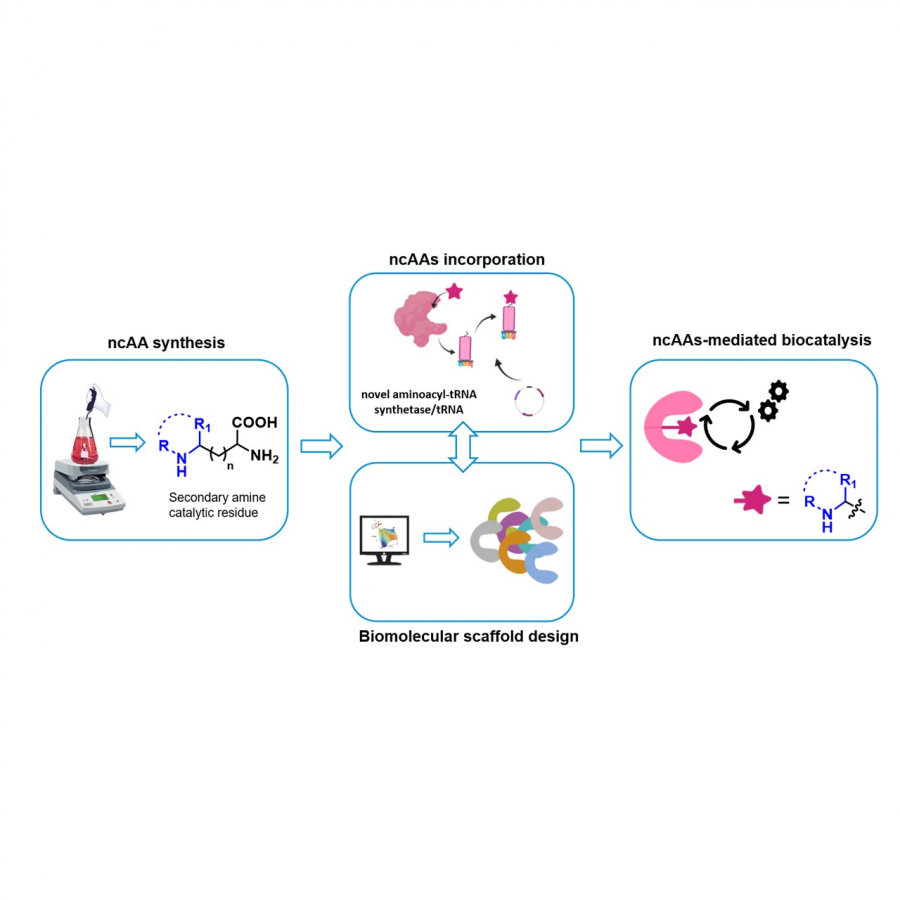 Figure 1. Schematic representation of the presented project focused on the investigation of various secondary amine-based non-canonical amino acids as catalytic residues. |
#964 | First Aid for Medicinal Chemists: Bioprospecting Cytochrome P450 Enzymes to Diversify Natural Product |
|
| Presenting author: | Nico FESSNER of UNIVERSITY OF FREIBURG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cytochrome P450 Enzymes / Natural product diversification / White-rot fungi / Enzyme discovery |
| Purpose: | Cytochrome P450 enzymes (P450s) can selectively oxidise highly functionalised molecules in a synthetic late-stage fashion via the activation of inert C-H bonds, making them ideal for medicinal chemists to diversify natural products in drug discovery processes (Figure 1) [1]. White-rot fungi are an attractive source for bioprospecting novel P450s because their extraordinary lifestyle of metabolising lignin and detoxifying xenobiotics is enabled by a diverse enzymatic portolio [2]. We noticed that the P450ome of the white-rot fungus Polyporus arcularius revealed an unusual enrichment of the sparingly characterised family CYP5035. The efficient heterologous expression of a small CYP5035 library in Pichia pastoris allowed identifying multifunctional P450s with activities towards several different natural product classes [3] and up-scaling of the whole-cell biotransformations to analyse their catalytic potential in several follow-up studies [4-6]. Recently, we also showed that human P450 enzyme 3A4, a key player in any drug discovery process, is highly efficient at diversifying a wide range of different natural product classes to a similar extent as its model substrate testosterone [7]. Expressed in P. pastoris, P450 3A4 diversified six steroids, alkaloids and terpenes to an unprecedented degree at a preparative 100-mg scale, allowing the identification of a total of 31 authentic human metabolites, many for the first time [8]. Therefore, eukaryotic P450s can act as an efficient first aid kit for medicinal chemists to diversify natural products for drug discovery purposes [1,9]. |
| References: | [1] Fessner, N.D., ChemCatChem 2019, 11, 2226-2242. [2] Syed, K.; Shale, K.; Pagadala, N.S.; Tuszynski, J., PLoS One 2014, 9, e86683. [3] Fessner, N.D.; Nelson, D.R.; Glieder, A., Appl. Microbiol. Biotechnol. 2021, 105, 6779-6792. [4] Fessner, N.D.; Grimm, C.; Kroutil, W.; Glieder, A., Biomolecules 2021, 11, 1708. [5] Fessner, N.D.; Weber, H.; Glieder, A., Biochem. Biophys. Res. Commun. 2022, 595, 35-40. [6] Fessner, N.D.; Weber, H.; Glieder, A., European J. Org. Chem. 2022, 2022, e202101436. [7] Fessner, N.D.; Srdič, M.; Weber, H.; Schmid, C.; Schönauer, D.; Schwaneberg, U.; Glieder, A.. Adv. Synth. Catal. 2020, 362, 2725-2738. [8] Fessner, N.D.; Grimm, C.; Srdič, M.; Weber, H.; Kroutil, W.; Schwaneberg, U.; Glieder, A., ChemCatChem 2022, 14, e202101564. [9] Fessner, N.D.; Badenhorst, C.P.S.; Bornscheuer, U.T., ChemCatChem 2022, 14, e202200156. |
| Figures: | 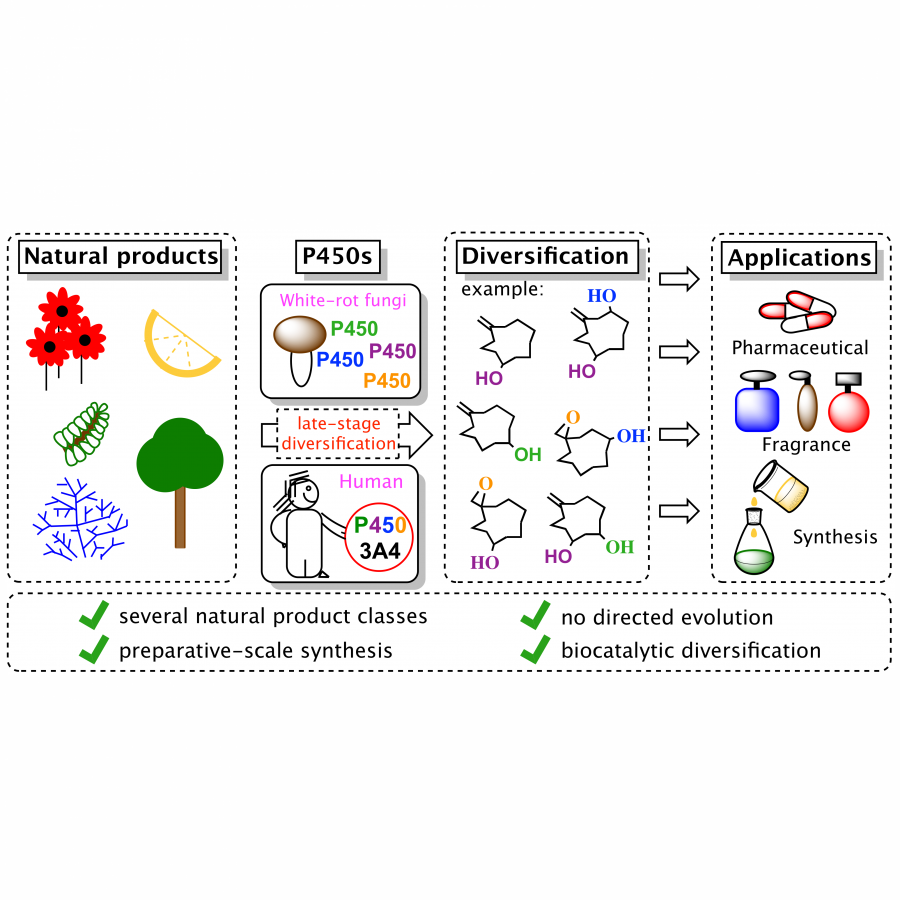 P450s - Synthetic First Aid for Chemists P450s can serve as First Aid for Chemists for the synthesis and diversification of natural products in drug discovery. We used white-rot fungal and human P450s to demonstrate their efficiency for this task. |
#967 | Screen-printed electrodes for the characterization of lignocellulose degrading enzymes |
|
| Presenting author: | Marjorie OCHS of UNIVERSITÉ CLAUDE BERNARD LYON 1 |
| Other authors: | Issa FALL of UNIVERSITÉ DE REIMS CHAMPAGNE ARDENNE, INRAE, FARE, UMR A 614, CHAIRE AFERE Bastien DOUMÈCHE of UCBL Sofiene ABDELLAOUI of UNIVERSITÉ DE REIMS CHAMPAGNE ARDENNE, INRAE, FARE, UMR A 614, CHAIRE AFERE Caroline RÉMOND of UNIVERSITÉ DE REIMS CHAMPAGNE ARDENNE, INRAE, FARE, UMR A 614, CHAIRE AFERE Harivony RAKOTOARIVONINA of UNIVERSITÉ DE REIMS CHAMPAGNE ARDENNE, INRAE, FARE, UMR A 614, CHAIRE AFERE |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | electrochemistry / lignin / oxidases / lignocellulosic biomass |
| Purpose: | Oxidative enzymes are a class of biocatalysts that catalyzes the oxidation of their substrate by electron transfers. Some of them are active on lignocellulosic biomass and can be used in biorefineries. For example, lytic polysaccharide monooxygenases (LPMO) can depolymerize crystalline cellulose and participate to reduce the cell wall recalcitrance while lignolytic enzymes can convert lignin into bio-based phenolic compounds[1-2]. Despite they offer the advantages to be bio-based, specific and active in mild conditions, enzymes need controlled pH and temperature to be efficient, that may be incompatible with industrial processes. One solution is to modify and/or discover new biocatalysts together with the development of measurement methods of the enzyme activity within industrial conditions. Classical methods for the characterization of enzymatic activities usually require soluble artificial substrates bearing chromophores for spectrophotometric measurements. However, when polymers are used as substrate, these methods are not relevant (soluble and small artificial substrate vs natural substrate in heterogeneous media) and other methods such as liquid chromatography are preferred. Unfortunately, there are time and money consuming. This technological lock hinders the knowledge and consequently the development of biocatalysts efficient on solid substrate. For these reasons, an efficient method for a high throughput measurement of enzymatic activities is of interest. Electrochemistry represents an alternative method suitable for the measurement of electron transfers that occur in enzymatic reactions. Since several years, we are developing 96-well electrochemical methods dedicated to high-throughput screening[3-6]. In this work we describe the use of screen-printed electrodes for the characterization of oxidases involved in the degradation of lignocellulosic biomass. |
| References: | [1] W. T. Beeson,V. Vu, E. A. Span, C. M. Phillips, M. A. Marletta; Annu. Rev. Biochem. 2015, 84:923-46. [2] JC. Sigoillot, JG. Berrin, M. Bey, L. Lesage-Meessen, A. Levasseur, A. Lomascolo, E. Record, E. Uzan-Boukhris, Advances in Botanical Research, 2018, 61, 263-308. [3] S. Abdellaoui, M. Bekhouche, A. Noiriel, R. Henkens, C. Bonaventura, L.J. Blum, B. Doumèche, Chem. Commun. 2013,49, 5781-5783. [4] S. Abdellaoui, A. Noiriel, R. Henkens, C. Bonaventura, L.J. Blum, B. Doumèche, Anal. Chem. 2013, 85, 3690-3697. [5] C. Aymard, C. Bonaventura, R. Henkens, C. Mousty, L. Hecquet, F. Charmantray, L.J. Blum, B. Doumèche, ChemElectroChem 2017, 4, 957-966. [6] C.M.G. Aymard, M. Halma, A. Comte, C. Mousty, V. Prévot, L. Hecquet, F. Charmantray, L.J. Blum, B. Doumèche, Anal. Chem. 2018, 90, 9241-9248. |
#969 | Size reduction of Sesquiterpene cyclase BcBOT2 via site directed mutagenesis and further protein optimizations for future NMR studies |
|
| Presenting author: | Trang NGUYEN of LEIBNIZ UNIVERSITY HANOVER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | BcBOT2 is a sesquiterpene cyclase from the fungus Botrytis cinerea and has the potential to convert farnesyl diphosphate and its derivatives into a structurally more complex sesquiterpene such as presilphiperfolan-8ß-ol. Since only homology models are available so far, the aim is therefore to achieve a structural elucidation of this protein in solute form with the help of NMR spectroscopic methods. Due to its initial size of 47 kDa, the enzyme had to be reduced in size by removing parts of the N- and C-terminus via site-directed mutagenesis. For this purpose, a mutant with an unchanged enzymatic activity could be generated, which would come to a size of 37.6 kDa by the additional removal of the 6x-His tag. Since triple labelling (15N, 13C, 2H-labelling) is required in this case, optimization experiments regarding protein expression, solubility and stability were carried out before the actual enrichment. |
#973 | Designing an enzyme for sustainable propylene production |
|
| Presenting author: | Pamela TORRES-SALAS of REPSOL S.A |
| Other authors: | MARC GARCIA-BORRAS of UNIVERSIDAD DE GIRONA MIGUEL ALCALDE of INSTITUTO DE CATÁLISIS ENRIQUE ESPI of REPSOL PATRICIA GOMEZ of EVOENZYME |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sustainable polymers / propylene / enzyme directed evolution / volatile fatty acids |
| Purpose: | Repsol is a global multi-energy provider. We strive to drive the evolution towards a decarbonization energy model, and with this ambition in mind we have set ourselves the goal of reaching a net zero emissions company by 2050. Accordingly, Repsol have initiated a transformation process that will enable us to turn our businesses into multi-energy hubs, which in the short term will be able to process alternative feedstocks to generate fuels and materials with a low carbon footprint. Thinking about this, we have explored new renewable raw materials that allow us to obtain more sustainable polymers. Hereby, an alternative route for the synthesis of propylene has been designed. Propylene would be polymerized to polypropylene. This polymer belongs to the group of polyolefins and is used in a vast variety of applications including food packaging, textiles, laboratory equipment, automotive components and transparent films. Volatile fatty acids (VFAs) from controlled anaerobic digestion were identified as a potential renewable raw material. From these VFAs, a potential enzymatic route that allows the decarboxylation of butyric acid to obtain propylene has been identified. After performing a screening of several enzymes, we have chosen one of them, which showed very low enzyme activity (1%). Thus, a directed evolution pathway was designed to improve its activity. Firstly, a computational study was accomplished to identify promising positions into the sequence. Afterwards, an experimental study was carried out to implement them in the laboratory, leading to improvements in the enzyme activity, although we are still far away from an industrial application. Nevertheless, we will show other Repsol’s circular economy initiatives nearly industrial. |
#980 | Biosynthesis of valuable hydroxyl compounds via C=O/C-H asymmetric oxidoreductive reactions: Engineering of enzymatic selectivity |
|
| Presenting author: | Yao NIE of JIANGNAN UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Hydroxyl compounds / Asymmetric biosynthesis / Ketoreductases / Hydroxylases |
| Purpose: | Selective synthesis of valuable hydroxyl compounds has become as an attractive field due to the increasing demands of pharmaceuticals and biologically active natural products. In the pharmaceutical industry, hydroxyl compounds including chiral cyclic and heterocyclic alcohols and hydroxyl derivatives can serve as intermediates of many chiral drugs such as flossonol, (−)- prosopinine, (R)-ladostigil, sertraline, fosamprenavir, sulopenem, ezetimibe, aprepitant, and ibrutinib, etc. To date, the main strategies to selectively produce hydroxyl compounds are chemical and enzymatic methods. Most chemical methods apply complex chiral catalysts for asymmetric redox, addition, and ring opening reactions under harsh conditions. In turn, most enzymatic methods use alcohol dehydrogenase/reductases to catalyze C=O bond asymmetric reduction, P450 enzymes or dioxygenases for C−H bond asymmetric hydroxylation, and lipases to catalyze C−O bond asymmetric hydrolysis [1,2]. In this work, we highlight the development of the strategies for enzymes catalyzing selective synthesis of various valuable hydroxyl compounds, with focus on the asymmetric reduction of C=O bonds by ketoreductases [3,4] and the asymmetric hydroxylation of C−H bonds by hydroxylases [5], involving identification and engineering of the functional enzymes and construction of the enzymatic systems. These works present green and powerful strategies for the selective synthesis of valuable hydroxyl compounds. |
| References: | [1] Wu, L., Qin, L., Nie, Y., Xu, Y., Zhao, Y.L.,Biotechnol. Adv. 2022, 54, 107793. [2] Qin, L., Wu, L., Nie, Y., Xu, Y., Catal. Sci. Technol. 2021, 11(8), 2637-2651. [3] An, J., Nie, Y., Xu, Y., Crit. Rev. Biotechnol. 2019, 39(3), 366-379. [4] Nie, Y., Wang, S.S., Xu, Y., Luo, S.G., Zhao, Y.L., Xiao, R., Montelione, G.T., Hunt, J.F., Szyperski, T., ACS Catal. 2018, 8(6), 5145-5152. [5] Wu, L., An, J., Jing, X., Chen, C.-C., Dai, L., Xu, Y., Liu, W., Guo, R.-T., Nie, Y., ACS Catal. 2022, 12(19), 11586-11596 |
#983 | ENZYME-PROMOTED PROMISCUOUS TRANSFORMATIONS OF IMINES AND THEIR ANALOGS: NEW RESULTS |
|
| Presenting author: | Piotr KIEŁBASIŃSKI of CENTRUM BADAŃ MOLEKULARNYCH I MAKROMOLEKULARNYCH POLSKIEJ AKADEMII NAUK |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme promiscuity / nitrones / iminium salts / addition of nitromethane to the C=N bond |
| Purpose: | Presentation of new achievements in the application of enzyme catalytic promiscuity ENZYME-PROMOTED PROMISCUOUS TRANSFORMATIONS OF IMINES AND THEIR ANALOGS: NEW RESULTS Piotr Kiełbasiński, Małgorzata Kwiatkowska, Ignacy Janicki Centrum Badań Molekularnych i Makromolekularnych PAN, Sienkiewicza 112, 90-363 Łódź E-mail: piotr.kielbasinski@cbmm.lodz.pl Enzyme catalytic promiscuity is the ability of a single enzyme active site to catalyze several chemical transformations, among them those which are different from natural.[1,2] We recently reported on the enzyme–catalyzed nucleophilic addition of nitromethane to aldimines (the aza-Henry reaction)moted addition of nitromethane to nitrones Then we turned to iminium sa which led to synthetically useful β-nitroamines. However, to our great disappointment, this reaction proved to be non-stereoselective.[3] Looking for other solutions we resorted to the use of various imine analogues. Thus, we first investigated addition of nitromethane to a number of nitrones. Only one of them, shown in Scheme 1, proved reactive; However, both the yields and enantiomeric excess were moderate. Scheme 1. Enzyme prolts. Their reaction with nitromethane led, besides the desired adduct, to various unexpected by-products, among them 2-nitroethenylarenes resulting from the retro-Michael elimination of the ammonium salt (Scheme 2). Scheme 2. Enzyme promoted addition of nitromethane to iminium salts Futher investigations aimed at the suppression of the consecutive reaction are in progres. The results will be discussed. The investigations were financed by the Polish National Research Centre (NCN), grant OPUS 2016/23/B/ST5/02443 for P.K. Bibliography [1] Busto, E.; Gotor-Fernandez, V.; Gotor, V. Hydrolases: catalytically promiscuous enzymes for conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, 39, 4504-4523. [2] Madalińska, L.; Kwiatkowska, M.; Cierpiał, T.; Kiełbasiński, P. Investigations on enzyme catalytic promiscuity: the first attempts at a hydrolytic enzyme-promoted addition of nucleophiles to α,β-unsaturated sulfinyl acceptors. J. Mol. Catal. B: Enzym. 2012., 81, 25-30. [3] Janicki, I.; Łyżwa, P.; Kiełbasiński, P. The first enzyme-promoted addition of nitromethane to imines (aza-Henry reaction). Bioorg. Chem. 2020, 94, Article No. 103 377 |
| References: | [1] Busto, E.; Gotor-Fernandez, V.; Gotor, V. Hydrolases: catalytically promiscuous enzymes for conventional reactions in organic synthesis. Chem. Soc. Rev. 2010, 39, 4504-4523. [2] Madalińska, L.; Kwiatkowska, M.; Cierpiał, T.; Kiełbasiński, P. Investigations on enzyme catalytic promiscuity: the first attempts at a hydrolytic enzyme-promoted addition of nucleophiles to α,β-unsaturated sulfinyl acceptors. J. Mol. Catal. B: Enzym. 2012., 81, 25-30. [3] Janicki, I.; Łyżwa, P.; Kiełbasiński, P. The first enzyme-promoted addition of nitromethane to imines (aza-Henry reaction). Bioorg. Chem. 2020, 94, Article No. 103 377 |
| Figures: | 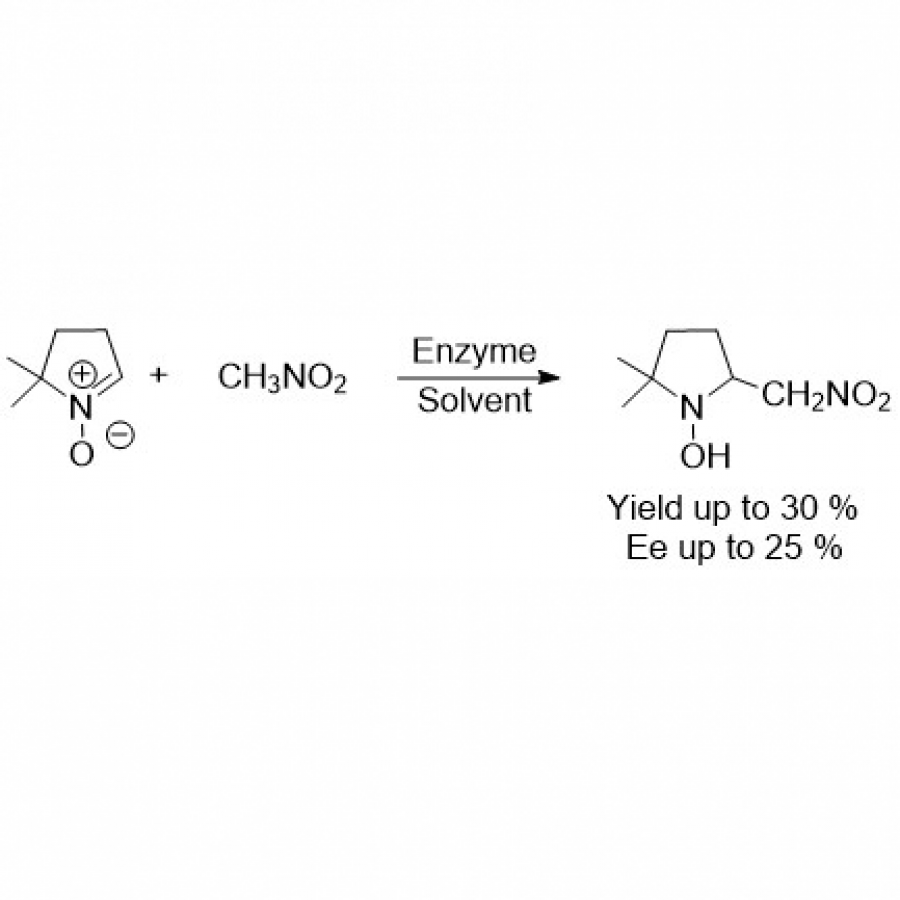 Scheme1 Scheme 1. Enzyme promoted addition of nitromethane to nitrones 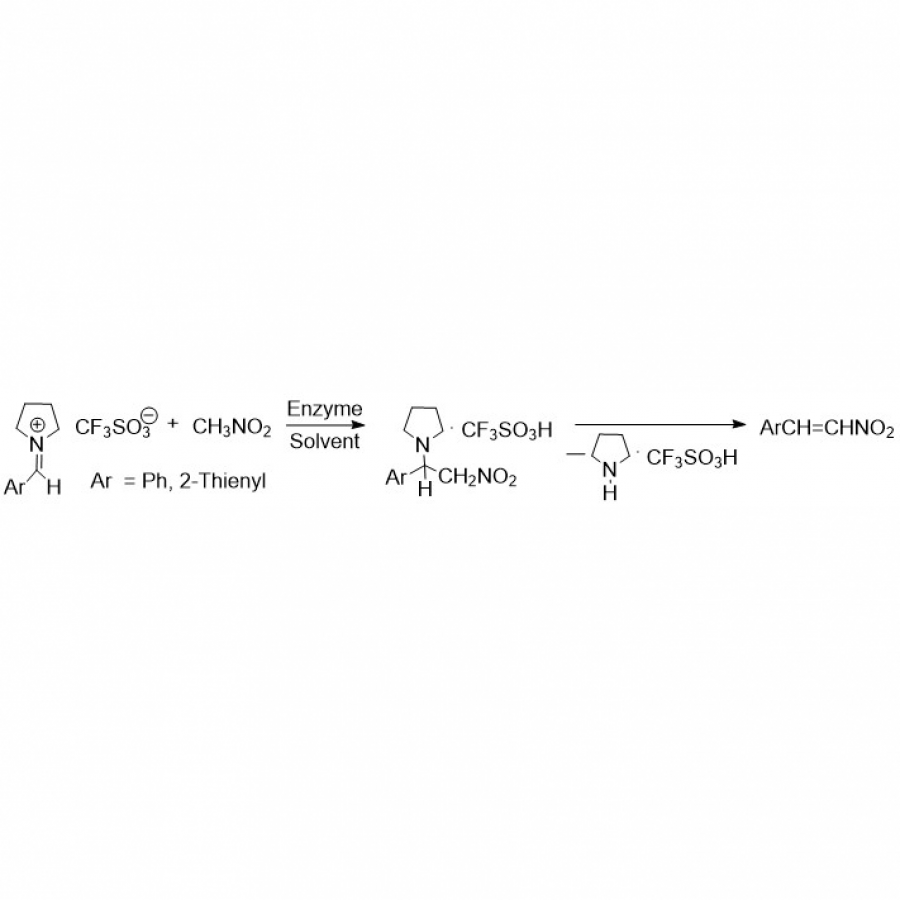 Scheme 2 Scheme 2. Enzyme promoted addition of nitromethane to iminium salts |
#994 | Engineering Glucose-Sensitive Galactose Oxidase for Stability at High Temperatures: A Fusion of Directed Evolution and Computational Methods |
|
| Presenting author: | Merve KESER of ICP-CSIC |
| Corresponding author: | Miguel ALCALDE of ICP-CSIC |
| Other authors: | David GONZALEZ PEREZ of ICP-CSIC Eva GARCIZ RUIZ of ICP-CSIC Valeria RISSO of UNIVERSITY OF GRANADA Juan BAUTISTA CRESPO of EYOWN TECHNOLOGIES S.L. Sarel FLEISHMAN of WEIZMANN INSTITUE Roland LUDWIG of BOKU VIENNA Roman KITTL of DIRECTSENS GMBH Ivan MATELJAK of EVOENZYME |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | galactose oxidase / Ancestral sequence reconstruction / thermostability / engineering |
| Purpose: | Frances Arnold group's pioneering research led to a significant breakthrough in the conversion of galactose oxidase into glucose-6-oxidase (Glu-6-Ox), demonstrating regioselectivity not found in nature[1]. Since then, researchers have delved into the substrate promiscuity of Glu-6-Ox, which spans oligo- and polysaccharides to primary and secondary alcohols. This promiscuity has potential applications in biocatalysis and organic synthesis. However, due to the harsh conditions of industrial applications, thermostable enzymes are highly sought after. Machine learning and artificial intelligence have made a breakthrough in protein engineering, and with algorithms that are ever more precise in predicting enzyme properties, combining wet lab-directed evolution and these computational tools allows for the streamlining of enzyme engineering. In this study, we showcase the synergistic effects of computational algorithms, such as ancestral sequence reconstruction and atomistic and phylogenetic design calculations, and classical directed evolution to develop a functional thermostable Glu-6-Ox suitable for organic synthesis in industrial applications. |
| References: | [1] L. Sun, T. Bulter, M. Alcalde, I. P. Petrounia, and F. H. Arnold, “Modification of galactose oxidase to introduce glucose 6-oxidase activity,” ChemBioChem, vol. 3, no. 8, pp. 781-783, 2002. |
| Figures: | 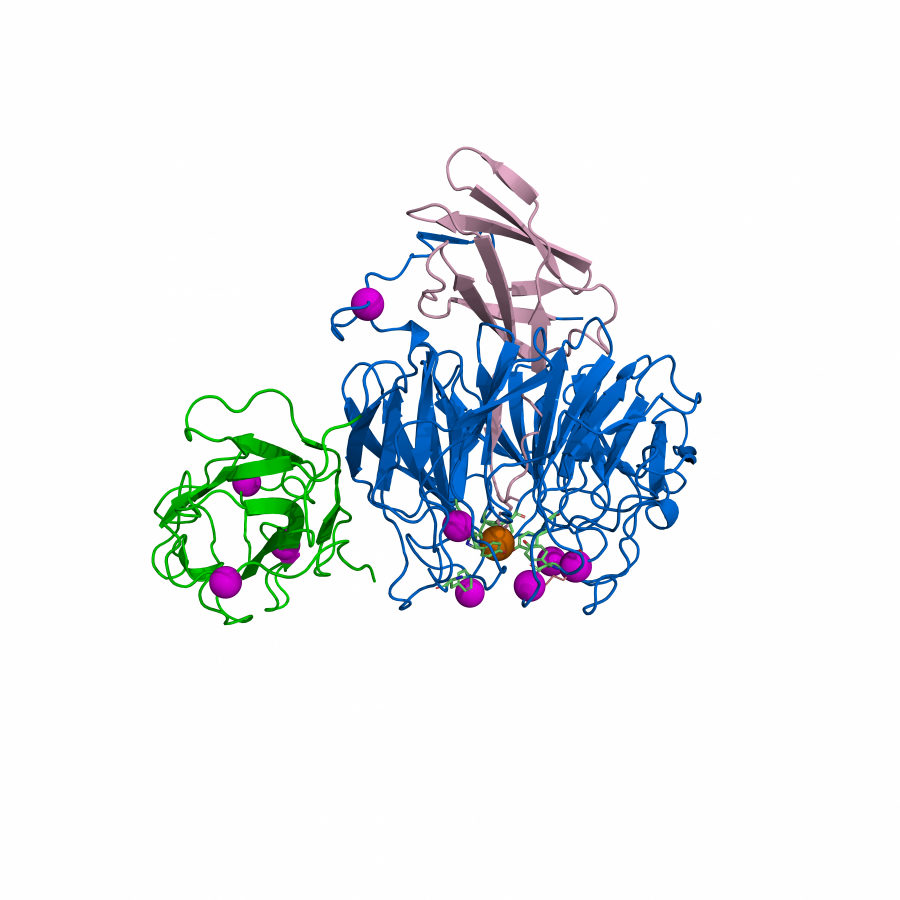 RQW Variant The RQW variant engineered by the Francis Arnold group has 9 mutations that facilitate expression in E.coli and sensitivity to glucose as a substrate. 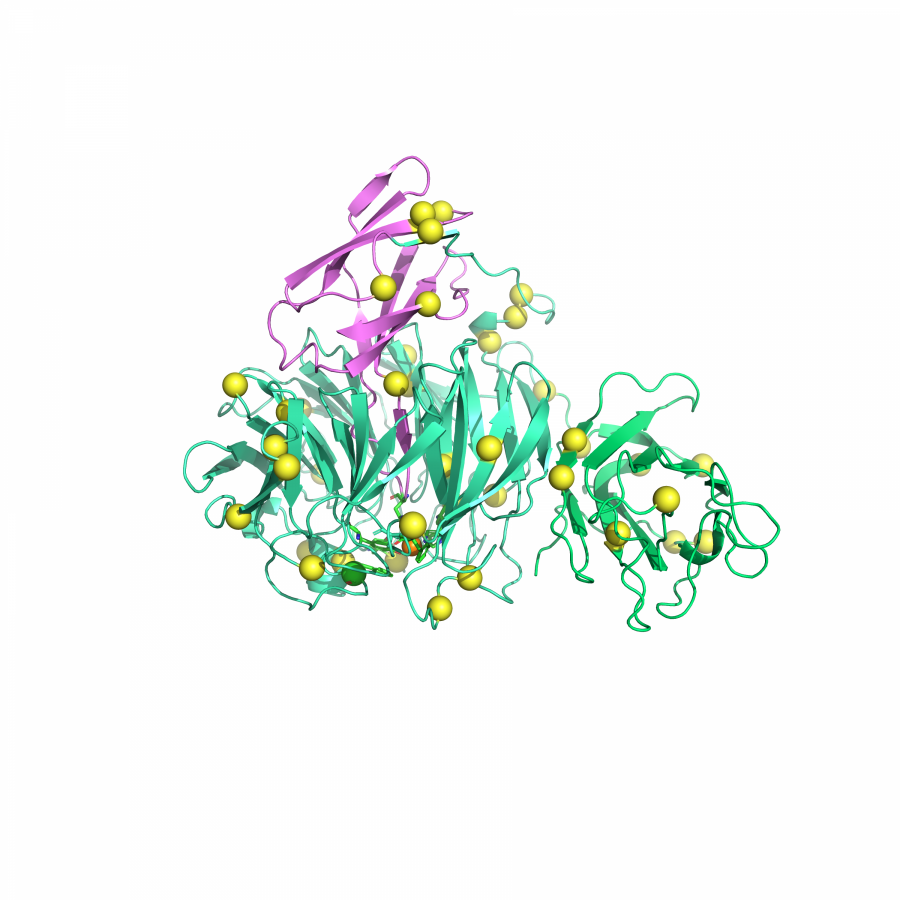 Final variant Sarlac yellow spheres: Final mutations acquired through directed evolution and computational campaigns for increased thermostability |
#997 | Comparison of the biocatalytic performances of immobilized nitrilase in milli-scale reactors for continuous production of nicotinic acid |
|
| Presenting author: | Maïté MICHAUD of UNIV. GRENOBLE ALPES, CEA, LITEN, DTCH |
| Other authors: | Carine VERGNE-VAXELAIRE of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV. EVRY, UNIV. PARIS-SACLAY Véronique DE BERARDINIS of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV. EVRY, UNIV. PARIS-SACLAY Anne ZAPARUCHA of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV. EVRY, UNIV. PARIS-SACLAY Guillaume NONGLATON of UNIV. GRENOBLE ALPES, CEA, LETI, DTBS Zoé ANXIONNAZ-MINVIELLE of UNIV. GRENOBLE ALPES, CEA, LITEN, DTCH |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flow processes / Wall enzyme immobilization / Process intensification / Bioreactor |
| Purpose: | Continuous bioreactor context Enzyme immobilization strategies are being actively investigated by the scientific community. In addition to the obvious interest of re-use of the biocatalyst, it is also essential for process intensification with integration of continuous operations [1]. Flow catalysis is indeed highly coveted by industrials to maximize the productivity but represent a major challenge. In this regard, the pharmaceutical industry has expressed its willingness to move from their traditional batch production to a continuous operating mode [2]. Promising technologies with high scaling potential due to their maturity are packed bed reactors (PBR) and wall immobilized enzyme reactors. Miniaturization at microscale for better mixing conditions have been the privileged option for wall immobilized enzyme reactors [3]. However, sizing up and numbering up should be combined to mitigated the complexity and cost of the heating and pumping apparatus at industrial scale [4]. With the reactor size increase, the geometry need to be optimized to maintain the mixing efficiency. Experimental results In this context, we immobilized a commercial nitrilase (specific activity measured at 1.1 U/mg) in a milli-scale reactor (i.e. characteristic dimension of 1mm) shaped with a pillar geometry. The reactor surface is coated with a thin layer of silica microbeads (i.e. total volume loss less than 10%) for higher loading capacity. The biocatalytic performances are compared to those obtained with a milli-size PBR [5]. For fair comparison between the two technical solutions, the biocatalytic performances are assessed once the outlet conversion is around 50%. In the milli-pillar reactor, the specific productivity is higher than in the PBR (Table 1) meaning that the ratio of useful immobilized enzymes is higher. However, the total turnover is not as high as the one obtained with the PBR because of sharper decrease of the outlet conversion with time (Figure 1). This difference in the immobilized enzyme stability may be explained by the change in the immobilization strategy (i.e. protocol, bead size and bead chemical composition). The kinetic impact of these choices will be investigated for cleared understanding of the different behaviours obtained with each technology. It has to be noted that using a milli-structured reactor allows to reduce the pumping power (i.e theoritical presure drop at least 50% higher for PBR than those measured with milli-structured reactor). This operating characteristic can be one of the fundamental criteria for choosing a technology, especially when process scaling-up is considered. Geometry optimization with scale-up perspectives Change in reactor structure will be experimentally studied by applying similar immobilization protocol and operating conditions to a zigzag milli-reactor (fluid volume of 0.51 mL). Computational fluid dynamics (CFD) using Comsol software are in progress and aim at combining mass transfer, laminar flow and Michaelis-Menten surface reaction equations. The objective is to understand the influence of the reactor characteristic dimensions as well as of the internal structure modifications on the outlet conversion. Optimum geometry choices based on modelling results would allow testing an experimental multi-stage up-scaling. The final goal of the project is to treat a pilot-scale throughput of 1 L/h. |
| References: | [1] J.M. Bolivar et al., Chem. Soc. Rev., 2022, 51, 6251-6290. [2] P. Zhang et al., Chem. Eur. J., 2018, 24 (11), 2776-2784. [3] D. Valikhani et al., ChemCatChem, 2017, 9 (1), 161-166. [4] S.G. Newman et al., Green Chem., 2013, 15 (6), 1456-1472. [5] C. Teepakorn et al., Biotechnol.J., 2021, 16, 2100010. |
| Figures: | 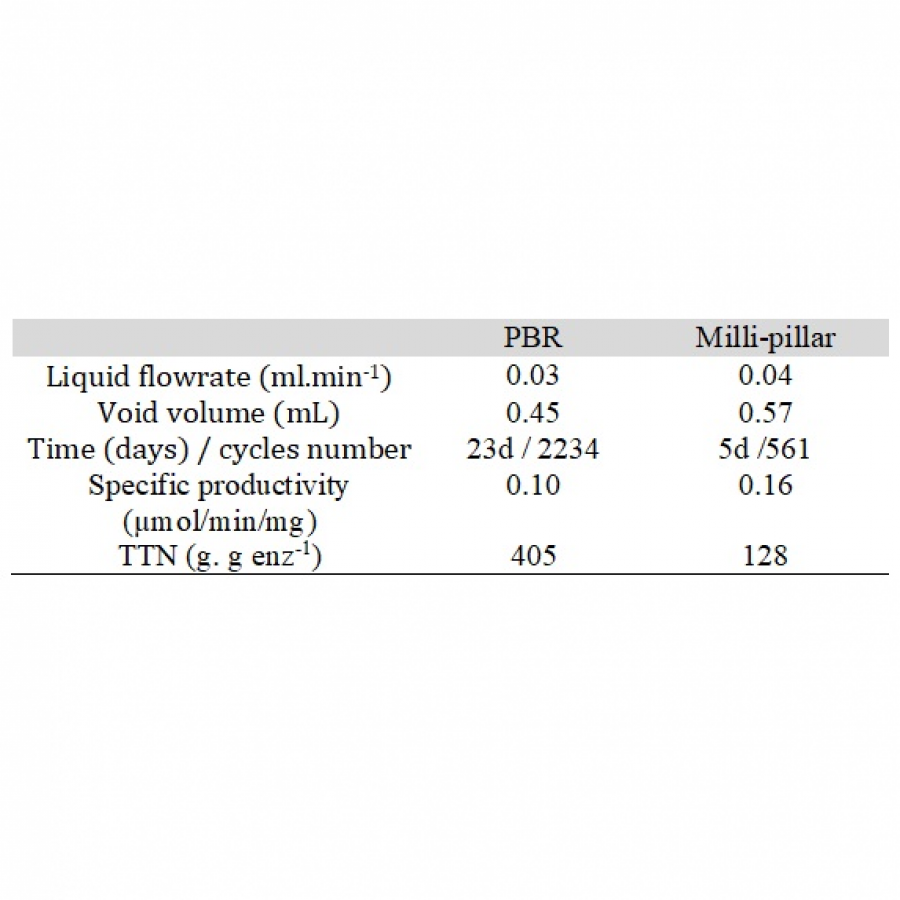 Table 1 Biocatalytic performances of the PBR and surface coated milli-pillar reactor - Calculated metrics once the outlet conversion is at 50% 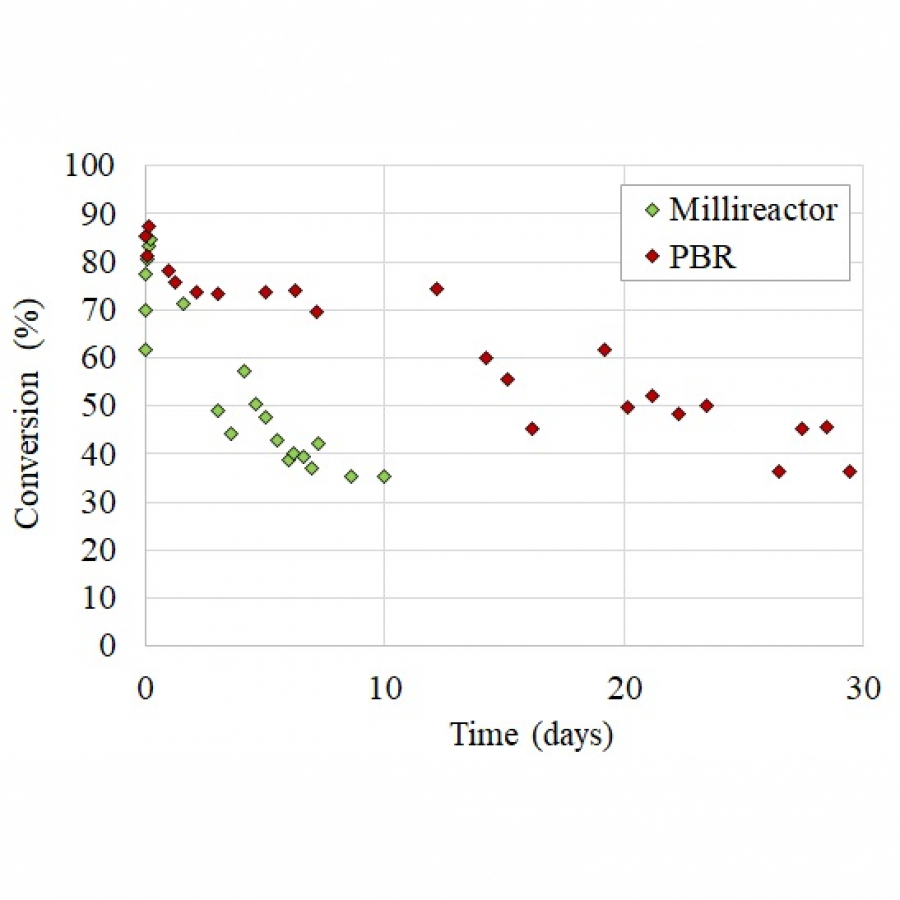 Figure 2 Biocatalytic conversion of the PBR and milli-pillar shaped reactor over time |
#999 | Vanillyl Alcohol Oxidase of Diplodia corticola – Insights into an Ether-cleaving Oxidase |
|
| Presenting author: | Nils WEINDORF of RHUR-UNIVERSITY BOCHUM |
| Other authors: | Daniel EGGERICHS of RUHR-UNIVERSITY BOCHUM Natalie WELZEL of RUHR-UNIVERSITY BOCHUM Dirk TISCHLER of RUHR-UNIVERSITY BOCHUM |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | vanillyl alcohol oxidase / ether cleavage / flavoproteins / |
| Purpose: | Vanillyl alcohol oxidases (VAOs) are members of the VAO/PCMH-flavoprotein superfamily, or more precisely, the subgroup of 4-phenol oxidases, whose members can be hypothesized to be related to the degradation of naturally occurring aromatic compounds.[1] Within this subgroup, VAOs represent oxidases that are found as a fungal counterpart to the bacterial eugenol oxidases (EUGOs) and 4-ethyl phenol oxidases (4EPOs). To date, three members of this subgroup have been characterized, the VAO of Penicillium simplicissimum, the EUGO of Rhodococcus jostii RHA1, and the 4EPO of Gulosibacter chungangensis. [2]–[4] It has been shown that all three of these enzymes harbor great promiscuity with regard to their substrate scope and reactivity, however, differences can be found, which may be related to structural differences. Among the oxidation of benzylic alcohols, γ-hydroxylations, deaminations, dehydrogenations and hydroxylations are performed by these enzymes. PsVAO has also been described to catalyze the cleavage of benzyl ethers, while this has not been done for RjEUGO and Gc4EPO. For PsVAO, benzyl ethers have also been proposed to be the natural substrates, as the expression of the vao gene in P. simplicissimum is described to be induced by 4-hydroxy benzyl methyl ether.[2] Structural data of the known enzymes and studies to elucidate the active site of these enzymes, showed key residues in the active site that are responsible for observed activities, and also for engineering of PsVAO to change its chemoselectivity in hydroxylation reactions.[4]–[9] Although PsVAO showed high promiscuity and greater activities compared to RjEUGO and Gc4EPO, no other VAOs have yet been characterized due to difficulties in their heterologous production and purification, yielding dysfunctional enzymes, with their covalently bound FAD-cofactor being trapped in an intermediate oxidation state. However, due to the diversity of the natural habitats of different VAO hosts, there is valid reason to expect the discovery of VAOs that differ from PsVAO, further broadening the promiscuity of this enzyme family. Based on VAOs active sites, and with the incentive to find a VAO, showing improved capabilities for cleaving ethers, as they are the most abundant bond in lignin,[10] and are also challenging for chemical breakdown, requiring harsh conditions, a phylogenetic comparison of putative vao genes was performed. This revealed one promising residue, E466 in the VAO of Diplodia corticola. Other VAOs feature a cysteine at that respective position. The homology model and autodocking hinted at E466 being positioned close to the ether-oxygen of putative substrates (see Figure 1). Heterologous production (9.1 mg/l broth) of DcVAO, using a pET expression system, was possible in E. coli NiCo 21 (DE3) with the help of the pKJE7 chaperone, promoting correct folding.[11] We investigated the substrate scopes of PsVAO and DcVAO, using an end-point kinetic determining the produced hydrogen peroxide. DcVAO catalyzed the same reactions that have been described for PsVAO, and showed e.g., the same preference for hydroxylation reaction, compared to dehydrogenation reactions. A notable difference between PsVAO and DcVAO could be found for performing the ether cleavage of vanillyl ether derivates, where for DcVAO activities of up to 4.0 ± 0.1 U/mg could be observed, while PsVAO only showed activities of up to 1.8 ± 0.1 U/mg. As we suspected E466 in DcVAO to be responsible for increased activities, the point mutants E466C, containing the cysteine found in other VAOs and E466L, containing the leucine found in EUGO were created. DcVAO E466L showed a reduced activity on the respective ether to 0.8 ± 0.1 U/mg. The E466C variant showed an activity of 2.5 ± 0.1 U/mg. These findings hint at E466 of DcVAO being responsible for the improved ability to perform ether cleavages. |
| References: | [1] T. A. Ewing, M. W. Fraaije, A. Mattevi, and W. J. H. van Berkel, “The VAO/PCMH flavoprotein family,” Arch Biochem Biophys, vol. 632, pp. 104-117, 2017, doi: 10.1016/j.abb.2017.06.022. [2] E. de Jong, W. J. H. van B. Berkel, R. P. van der Z. Zwan, and J. A. M. de B. Bont, “Purification and characterization of vanillyl-alcohol oxidase from Penicillium simplicissimum,” Eur J Biochem, vol. 657, pp. 651-657, 1992. [3] J. Jin, H. Mazon, R. H. H. Van Den Heuvel, D. B. Janssen, and M. W. Fraaije, “Discovery of a eugenol oxidase from Rhodococcus sp. strain RHA1,” FEBS Journal, vol. 274, no. 9, pp. 2311-2321, 2007, doi: 10.1111/j.1742-4658.2007.05767.x. [4] L. Alvigini et al., “Discovery, biocatalytic exploration and structural analysis of a 4-ethylphenol oxidase from Gulosibacter chungangensis,” ChemBioChem, pp. 1-10, 2021, doi: 10.1002/cbic.202100457. [5] R. H. H. Van Den Heuvel, M. W. Fraaije, and W. J. H. Van Berkel, “Direction of the reactivity of vanillyl-alcohol oxidase with 4-alkylphenols,” 2000. doi: 10.1016/S0014-5793(00)01992-X. [6] T. A. Ewing et al., “Two tyrosine residues, Tyr-108 and Tyr-503, are responsible for the deprotonation of phenolic substrates in vanillyl-alcohol oxidase,” Journal of Biological Chemistry, vol. 292, no. 35, pp. 14668-14679, Sep. 2017, doi: 10.1074/jbc.M117.778449. [7] A. Mattevi, M. W. Fraaije, A. Mozzarelli, L. Olivi, A. Coda, and W. J. H. Van Berkel, “Crystal structures and inhibitor binding in the octameric flavoenzyme vanillyl-alcohol oxidase: The shape of the active-site cavity controls substrate specificity,” Structure, vol. 5, no. 7, pp. 907-920, 1997, doi: 10.1016/S0969-2126(97)00245-1. [8] R. H. H. Van Den Heuvel, M. W. Fraaije, M. Ferrer, A. Mattevi, and W. J. H. Van Berkel, “Inversion of stereospecificity of vanillyl-alcohol oxidase,” 2000. doi: 10.1073/pnas.160175897 [9] Q. T. Nguyen, G. de Gonzalo, C. Binda, A. Rioz-Martínez, A. Mattevi, and M. W. Fraaije, “Biocatalytic properties and structural analysis of eugenol oxidase from Rhodococcus jostii RHA1: A versatile oxidative biocatalyst,” ChemBioChem, pp. 1359-1366, 2016, doi: 10.1002/cbic.201600148. [10] J. Ralph, C. Lapierre, and W. Boerjan, “Lignin structure and its engineering,” Current Opinion in Biotechnology, vol. 56. Elsevier Ltd, pp. 240-249, Apr. 01, 2019. doi: 10.1016/j.copbio.2019.02.019. [11] K. Nishihara, M. Kanemori, M. Kitagawa, H. Yanagi, and T. Yura, “Chaperone coexpression plasmids: Differential and synergistic roles of DnaK-DnaJ-GrpE and GroEL-GroES in assisting folding of an allergen of japanese cedar pollen, Cryj2, in Escherichia coli,” 1998. doi: 10.1128/aem.64.5.1694-1699.1998 |
| Figures: | 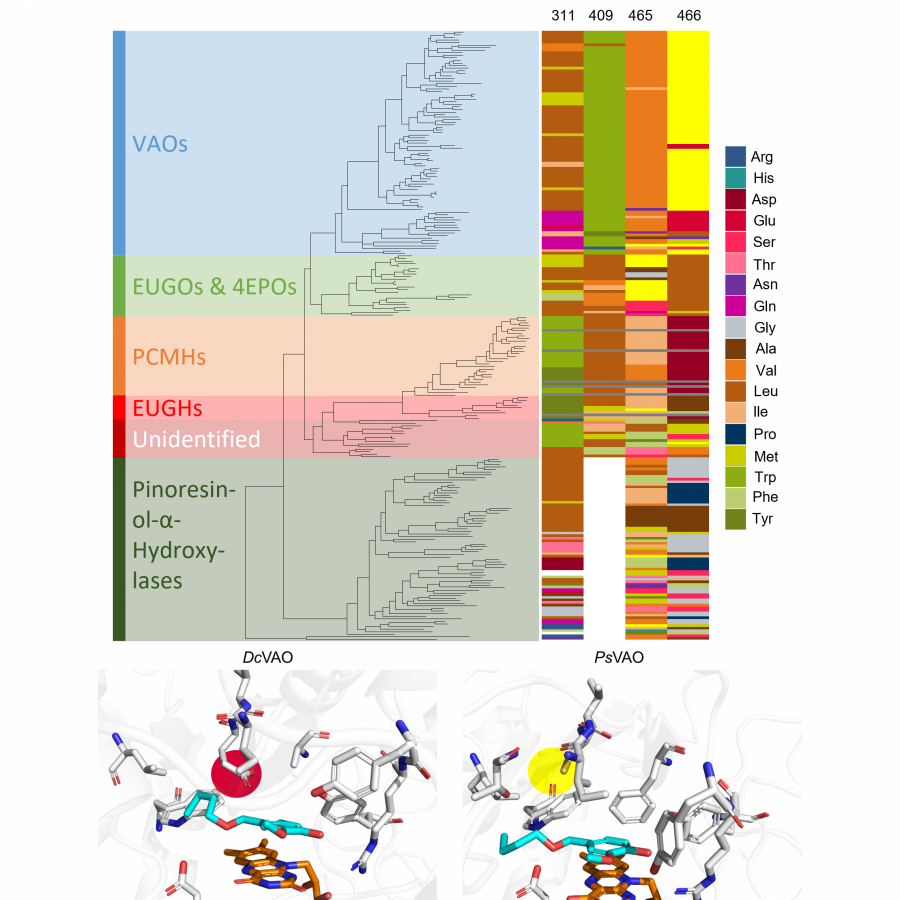 Phylogenetic comparison of the 4-phenol oxidizing enzymes of the VAO/PCMH-flavoprotein superfamily. Highlighted are four positions (DcVAO numbering) that interact with the p-subsituent of phenolic substrates. Within the VAOs, position 466 stands out, as DcVAO features a glutamate instead of a cysteine, which may affect ether cleavages. 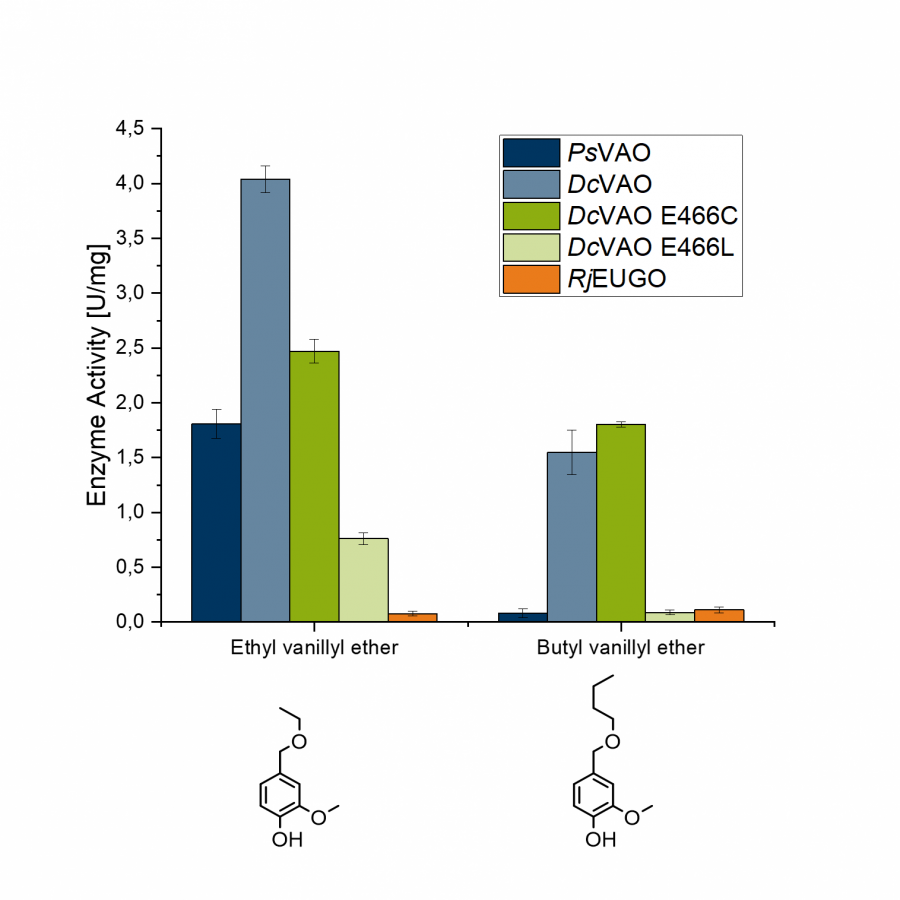 Comparison of enzyme activities on ether substrates. Activities for vanillyl alcohol oxidases (VAOs) of Penicillim simplicissimum and Diplodia corticola, two mutants of DcVAO at position 466, and eugenol oxidase (EUGO) of Rhodococcus jostii RHA1, determined using a xylenol-orange-based assay. |
#1005 | Enzymatic Decarboxylation Facilitated by Local-Oriented Electric Field |
|
| Presenting author: | Chenghua ZHANG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY CHINESE ACADEMY OF SCIENCES |
| Corresponding author: | Xiang SHENG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY CHINESE ACADEMY OF SCIENCE |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | decarboxylase / reaction mechanism / local electric field / density functional theory |
| Purpose: | Decarboxylation of the aromatic carboxylic acids is an important but challenging reaction in the biocatalytic synthesis of many valuable compounds and has thus attracted great attentions. Enzymatic decarboxylation reaction usually requires cofactor and metal ion for the activity. For example, the decarboxylation of 3,4-dihydroxybenzoic acid by the decarboxylase AroY requires the prenylated flavin mononucleotide (prFMN) as the cofactor[1] and ortho-benzoic acids decarboxylases from the amidohydrolase superfamily needs Mn2+/Mg2+ for the activities[2]. However, the recently characterized gallic acid decarboxylase (AGDC1) is capable of catalyzing the cofactor- and metal-independent non-oxidative decarboxylation of 3,4,5-trihydroxybenzoic acid and 3,4-dihydroxybenzoic acid.[3] In the present study, by using quantum chemical cluster approach, the detailed reaction mechanism of AGDC1 is revealed. Interestingly, it is demonstrated that the build-in local electric field (LEF) in the enzyme is important for the reaction activity. Two lysine residues adjacent to the binding pocket are proven to be the key residues in inducing the LEF. The strengths of the LEFs in AGDC1 and AroY are calculated and compared. It is shown that the LEF in AGDC1 is much stronger than that in AroY. Our findings provide deep insights into the cofactor- and metal-independent enzymatic decarboxylation reaction, and the obtained information are helpful in the mechanism-guided protein engineering of the enzymes. |
| References: | [1] S.E. Payer, S.A. Marshall, N. Barland, X. Sheng, T. Reiter,et al. Angew. Chem. Int. Ed. 2017, 56, 13893-13897. [2] X. Sheng, F. Himo, Comput. Struct. Biotechnol. J. 2021, 19, 3176-3186. [3] M. Zeug, N. Markovic, C.V. Iancu, J. Tripp, M. Oreb, J-Y. Choe, Sci. Rep. 2021, 11, 3056. |
| Figures: | 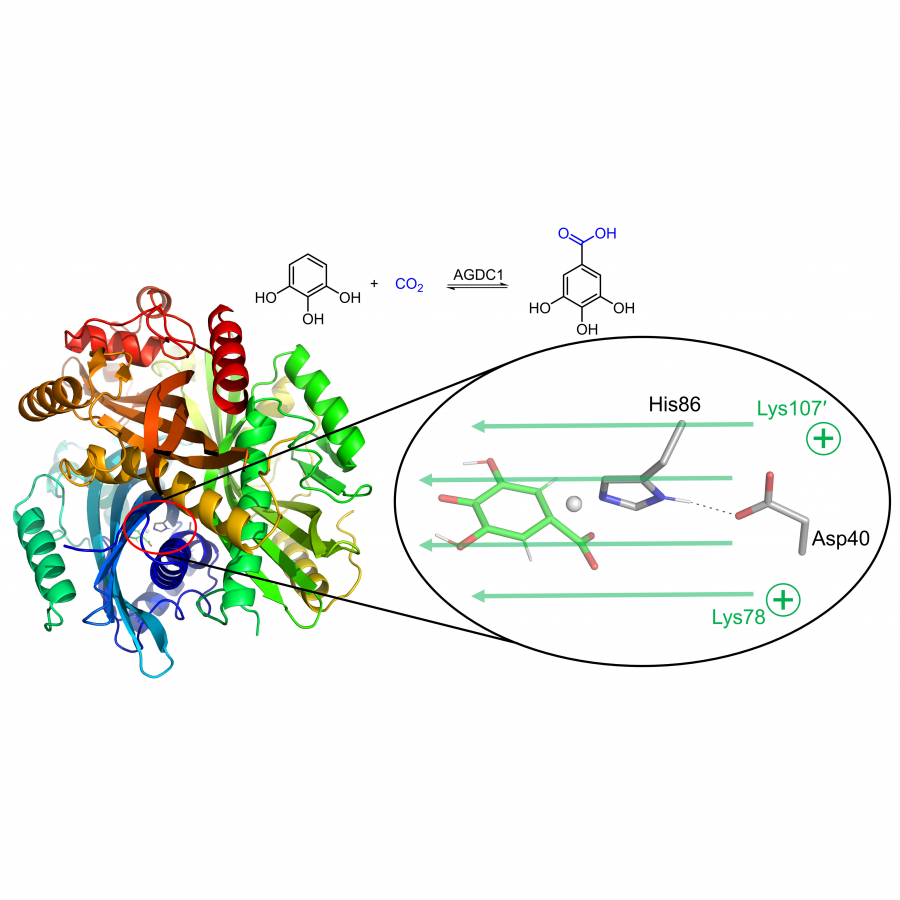 The cofactor- and metal-independent decarboxylation reaction of gallic acid decarboxylase facilitated by local-oriented electric field. Figure 1 |
#1006 | NOVEL ARYLSULFOTRANSFERASES FOR SULFATION OF (POLY)PHENOLS |
|
| Presenting author: | Kateřina VALENTOVÁ of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Other authors: | Katerina BRODSKY of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Barbora PETRÁNKOVÁ of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Lucie PETRÁSKOVÁ of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Pavla BOJAROVÁ of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | arylsulfotransferase / polyphenols / sulfation / enzyme catalysis |
| Purpose: | Sulfation is an important reaction in nature, and sulfated (poly)phenolic compounds are of interest as standards of mammalian phase II metabolites or pro-drugs. Due to their complex structure, a large number of structurally different metabolites, such as mono-, di- or trisulfates, can be formed. Their isolation from biological material is impractical; however, they can be synthesized in vitro. Nature has developed efficient sulfation tools using 3′-phosphoadenosine 5′-phosphosulfate (PAPS) as a sulfate donor for a variety sulfotransferases (SULTs, EC 2.8.2). In general, sulfotransferases catalyze the transfer of a sulfuryl group from a donor to an acceptor substrate. Most sulfation reactions in vivo are catalyzed by various SULTs. However, these typically intracellular enzymes are inherently unstable under in vitro conditions. The major bottleneck in SULT-catalyzed reactions is the need to use PAPS, which is extremely expensive and unstable (1). A more promising group of enzymes for potential use in the laboratory are PAPS-independent arylsulfotransferases (ASTs, EC 2.8.2.1) that use simple aromatic sulfate donors such as p-nitrophenyl sulfate (p-NPS). These enzymes of bacterial origin appear as an elegant synthetic tool for the "green" and sustainable sulfation of polyphenols. Moreover, they are easily expressed heterologously in E. coli. There are only a few arylsulfotrasferases that have been characterized and used for the sulfation of (poly)phenols. The best studied is the recombinant AST from Desulfitobacterium hafniense (2-8). In the present study, five potential recombinant arylsulfotrasferases were produced in E. coli, purified to homogeneity, and characterized. These enzymes were then tested in sulfation reactions with p-NPS as the donor and various phenolic compounds as acceptors: Flavonoids, flavones, phenolic acids, and even phenolic glycosides. The reactions were performed both on an analytical and preparative scale. The tested enzymes showed different substrate preferences, which helped us to find the optimal conditions for the preparation of sulfate derivatives of phenolic compounds. The products were detected by HPLC and some of them were isolated for NMR analysis. Obtaining sufficient amounts of sulfated compounds should allow us to characterize the human metabolites in terms of their stereochemistry and sulfation sites. In conclusion, enzymatic sulfation of polyphenols with bacterial ASTs is an effective method for producing metabolites identical to metabolic intermediates found in the human body. Supported by the Czech Health Research Council, grant number NU21-02-00135. 1. Purchartová, K., ChemCatChem 2015, 7, 3152-3162. 2. Valentová, K., Int. J. Mol. Sci. 2017, 18, 2231. 3. Valentová, K., Int. J. Mol. Sci. 2018, 19, 2349. 4. Begines, P., J. Agric. Food Chem. 2019, 67, 7281-7288. 5. Káňová, K., J. Agric. Food Chem. 2020, 68, 11197-11206. 6. Brodsky K., ChemSusChem 2022, 15, e202201253. 7. Kolaříková V., Int. J. Mol. Sci. 2022, 23, 15171. 8. Petrásková L., Int. J. Mol. Sci. 2022, 23: 5743. |
| References: | 1. Purchartová, K., ChemCatChem 2015, 7, 3152-3162. 2. Valentová, K., Int. J. Mol. Sci. 2017, 18, 2231. 3. Valentová, K., Int. J. Mol. Sci. 2018, 19, 2349. 4. Begines, P., J. Agric. Food Chem. 2019, 67, 7281-7288. 5. Káňová, K., J. Agric. Food Chem. 2020, 68, 11197-11206. 6. Brodsky K., ChemSusChem 2022, 15, e202201253. 7. Kolaříková V., Int. J. Mol. Sci. 2022, 23, 15171. 8. Petrásková L., Int. J. Mol. Sci. 2022, 23: 5743. |
| Figures: | 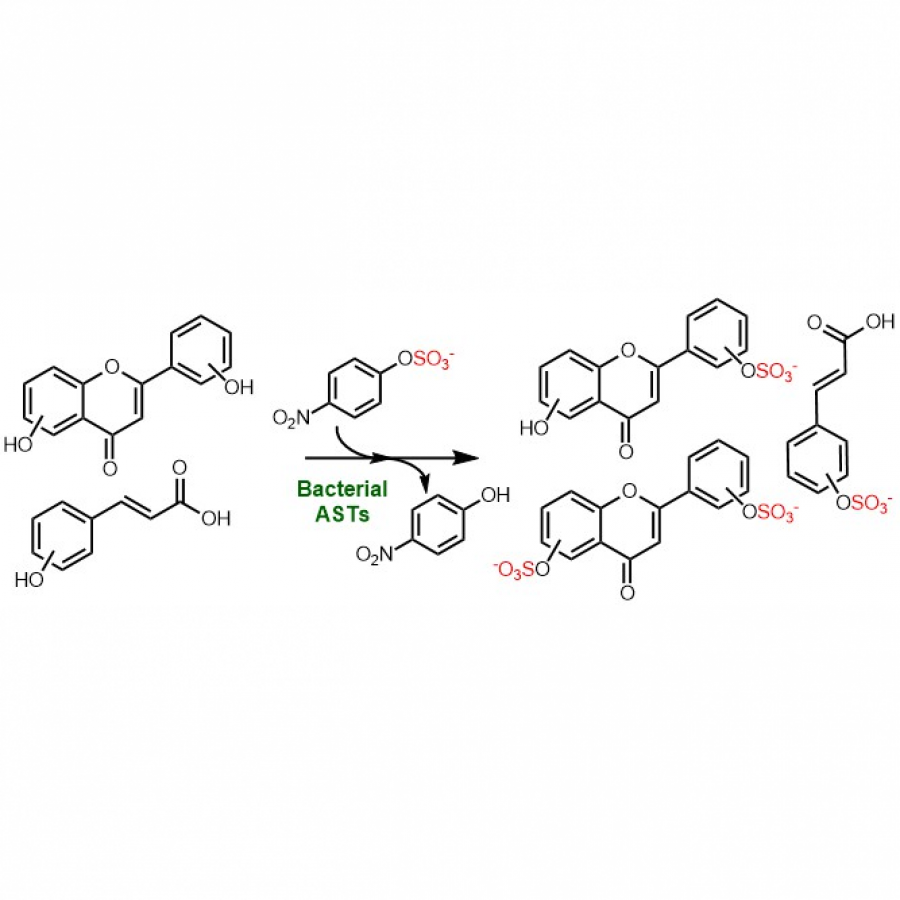 Sulfation of phenols by bacterial arylsulfotransferases AST - arylsulfotransfererase |
#1007 | Sustainable valorization of agro-food industry by-products for production of vanillin and vanillic acid |
|
| Presenting author: | Ewa SZCZEPAŃSKA of WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Filip BORATYŃSKI of WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Katarzyna PATEJUK of WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Witold PIETRZAK of WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Teresa OLEJNICZAK of WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | vanillin / natural flavours / by-products valorization / Solid-State Fermentation |
| Purpose: | Vanillin is the compound of great interest to the industry. It is used to augment and enhance the aroma and taste of food preparations, perfumes, and detergents. Vanillic acid, which is a derivative of vanillin, has many biological activities, which have been mainly used in the pharmaceutical industry as a precursor for the production of drugs and an antioxidant. Currently, 85% of the world's supply is vanillin synthesized chemically from guaiacol, and the remaining 15% is produced from lignin, either by extraction from vanilla seeds and microbial biosynthesis (<1%). [1] The key concept of modern industrial biotechnology is green chemistry, in particular the application of the principles of sustainable development and the implementation of the circular economy in production processes. In this view, biocatalysis and utilizing organic wastes and by-products are among the most promising green research areas for sustainable manufacturing of food ingredients, pharmaceutical intermediates, and fine chemicals. The main goal of this research was to screen microorganisms capable of releasing ferulic acid from raw materials such as brewery spent grain (BSG) and linseed oilcake (LOC) and then transforming it into phenolic compounds attractive for the industry, such as vanillin and vanillic acid (Figure 1). Strains were purchased from commercially available collections and have also been isolated by us from different environments. The tests were performed in Submerged Fermentation system (containing a liquid medium where the carbon source was replaced with selected by-product) and Solid-State Fermentation system using moistened agro-food industry by-products. The progress of the process was controlled over time by the use of chromatographic techniques. As a result, biocatalysts were divided into three groups: (1) effectively releasing ferulic acid from by-products, (2) biosynthesizing vanillic acid, and (3) vanillin from ferulic acid released from by-products. |
| References: | [1] W. Huang, C.Y. Du, J.A. Jiang, Y.F. Ji, Res. Chem. Intermed. 2013, 39 (6). |
| Figures: | 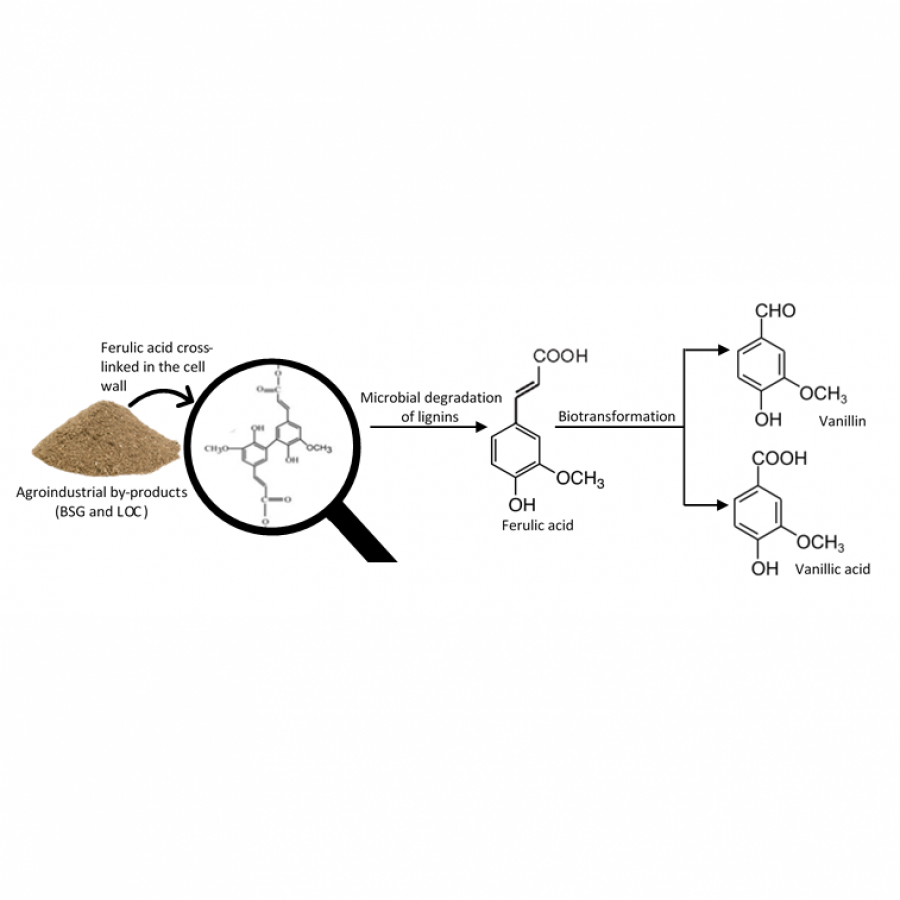 Figure 1. Production of phenolic compounds from agroindustrial by-products. |
#1011 | recycling cascades combining aldolases and transaminases for the highly selective synthesis of hydroxylated amino acids |
|
| Presenting author: | Thierry GEFFLAUT of ICCF-UNIVERSITÉ CLERMONT-AUVERGNE |
| Other authors: | Virgil HELAINE of ICCF-UNIVERSITÉ CLERMONT-AUVERGNE Lea GOURBEYRE of ICCF-UNIVERSITÉ CLERMONT-AUVERGNE Christine GUERARD HELAINE of ICCF-UNIVERSITÉ CLERMONT-AUVERGNE Marielle LEMAIRE of ICCF-UNIVERSITÉ CLERMONT-AUVERGNE Veronique DE BERARDINIS of GENOSCOPE-UNIVERSITE PARIS-SACLAY Jean-Louis PETIT of GENOSCOPE-UNIVERSITE PARIS-SACLAY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | aldolases / transaminases / recycling cascades / genome mining |
| Purpose: | The combination of aldolases (AL) and transaminases (TA) in bi-enzymatic cascades constitutes a straightforward approach to prepare 1,3-amino-alcohols with high stereoselectivity: a ketol, selectively obtained from achiral carbonyl compounds through AL-catalysis is further converted with high stereoselectivity into 1,3-amino alcohol by action of a TA in the presence of an amino donor substrate.[1,2] In the course of a project devoted to the discovery of new AL and TA from microbial diversity using a genome mining approach,[3,4] we have identified a set of AL and TA suitable for the synthesis of various valuable γ-amino alcohols. Moreover, we have developed a recycling cascade model, in which the nucleophilic substrate of AL is generated from the amino donor substrate of TA (see figure). This process thus brings the benefit of optimal atom economy. whereas the thermodynamically favoured aldolisation, affords an equilibrium shift for the transamination reaction thus solving the reversibility problem encountered with TA-catalysed reactions. Moreover We have also demonstrated that the dynamic nature of the recycling cascade affords a large increase in stereoselectivity through various ways that will be discussed. AL with complementary enantioselectivities were thus combined with L- or a D-α-TA to prepare in high yield a variety of 4-hydroxy-2-amino acids including all four stereomers of 4-hydroxy-glutamic acid, L-syn and D-syn-4-hydroxynorvaline and D-anti-4,5-dihydroxynorvaline. Furthermore, the stereocontrol afforded by the recycling cascade was extended to 3 chiral centers with the synthesis of the bioactive (2S,3R,4S)-4-hydroxy-isoleucine (Hil) as well as 3 new β-branched 4-hydroxyamino acids analogues of Hil. |
| References: | [1] C. Guérard-Hélaine, E. Heuson, M. Ndiaye,L. Gourbeyre, M. Lemaire, V. Hélaine, F. Charmantray, J.L. Petit, M. Salanoubat, V. De Berardinis, T. Gefflaut, Chem. Commun. 2017, 53 (39), 5465. [2] C. J. Moreno, K. Hernández, S.J. Charnok, S. Gittings, M. Bolte, J. Joglar, J. Bujons, T. Parella, P. Clapés, ACS Catal. 2021, 11 (8), 4660. [3] V. De Berardinis, C. Guérard-Hélaine, E. Darii, K. Bastard, V. Hélaine, A. Mariage, J.-L. Petit, N. Poupard, I. Sánchez-Moreno, M. Stam, T. Gefflaut, M. Salanoubat, M. Lemaire, Green Chem. 2017, 19 (2), 519. [4] E. Heuson, J.-L. Petit, A. Debard, A. Job, F. Charmantray, V. De Berardinis, T. Gefflaut, Appl. Microbiol. Biotechnol. 2016, 100, 397. |
| Figures: | 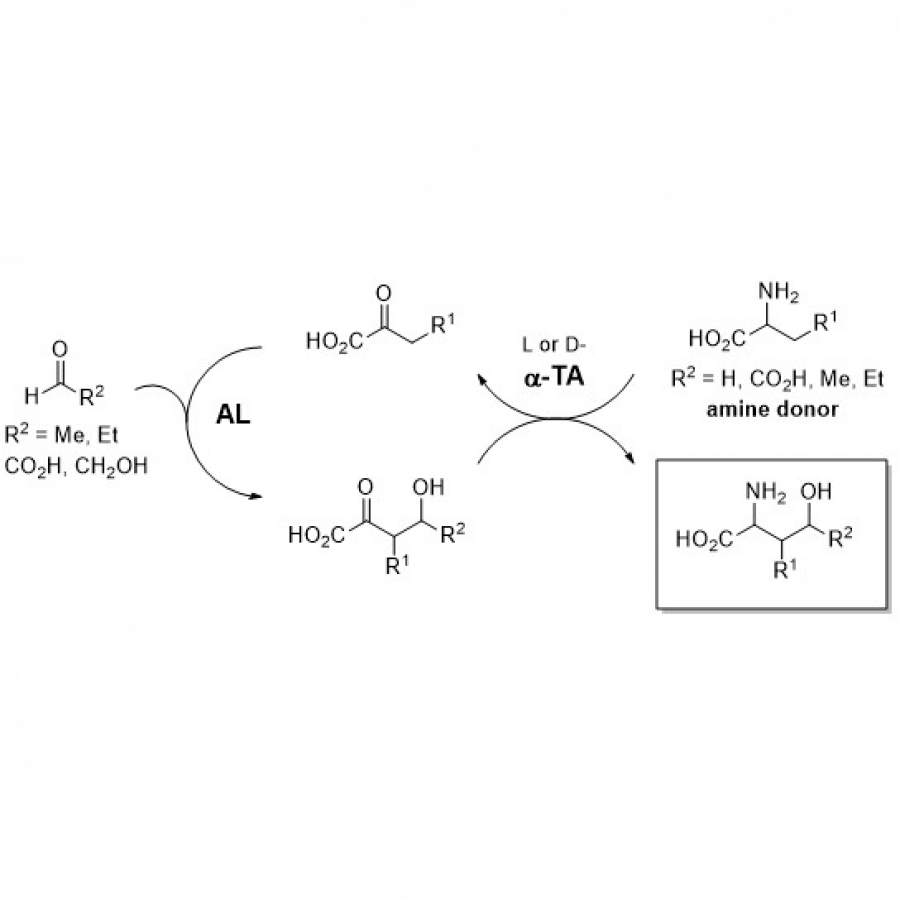 AL-TA recycling cascades A variety of 4-hydroxy-2-amino acids were prepared with high yield and selectivity. |
#1013 | Allostery in Purine Nucleoside Phosphorylases Revealed by a Combination of Protein Crystallography and Molecular Dynamics Simulations |
|
| Presenting author: | Aleksandra MARŠAVELSKI of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA |
| Other authors: | Boris GOMAZ of RUĐER BOŠKOVIĆ INSTITUTE ZORAN ŠTEFANIĆ of RUĐER BOŠKOVIĆ INSTITUTE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | allostery / Purine Nucleoside Phosphorylases / protein crystallography / molecular dynamics simulations |
| Purpose: | Enzymes purine nucleoside phosphorylases (PNPs) [1] have been chosen as a model system for investigating allostery due to several compelling reasons. First, PNPs play a critical role in the purine salvage pathway by catalyzing the synthesis of purine nucleotides. This pathway is essential for maintaining cellular homeostasis and is conserved across various organisms. Second, PNPs exist in different oligomeric forms, such as homohexamers in bacteria and homotrimers in higher organisms, providing an opportunity to study the influence of the oligomeric state on allosteric behavior. Third, the structural and functional diversity observed in PNPs, coupled with the presence of allosteric binding sites, suggests the involvement of intricate communication networks within the protein. Investigating the allosteric mechanisms in PNPs can provide valuable insights into the general principles of allostery and aid in the design of novel strategies for modulating enzyme activity. To fully comprehend the intricacies of allostery, which is fundamentally a dynamic phenomenon, it becomes imperative to move beyond the static structures obtained from X-ray crystallography and delve into the dynamic aspects using molecular dynamics simulations. Allosteric communication within proteins relies on non-covalent interactions between amino acids, and thus, unraveling the allosteric pathways necessitates an examination of the evolving interaction networks over time. Introducing time evolution through molecular simulations adds complexity that can only be managed by programmatic approaches capable of handling the vast quantities of data generated. In this context, machine learning methods emerged as invaluable tools, offering robust capabilities tailored specifically for processing and analyzing such voluminous datasets. By combining molecular dynamics simulations, programmatic data processing, and machine learning techniques, we started to uncover the intricate details of allosteric pathways in PNPs and gain deeper insights into the fundamental principles underlying protein function and regulation. This research is part of the ongoing project Allosteric communication pathways in oligomeric enzymes (ALOKOMP, https://alokomp.irb.hr/, financed by Croatian Science Foundation grant no. IP-2019-04-6764). |
| References: | [1] narczyk m., wojtyś m. i., leščić ašler i., žinić b., luić m., jagusztyn-krynicka e. k., štefanić z., bzowska a. J Enzyme Inhib Med Chem. 2022, 37, 1083-1097. |
| Figures: | 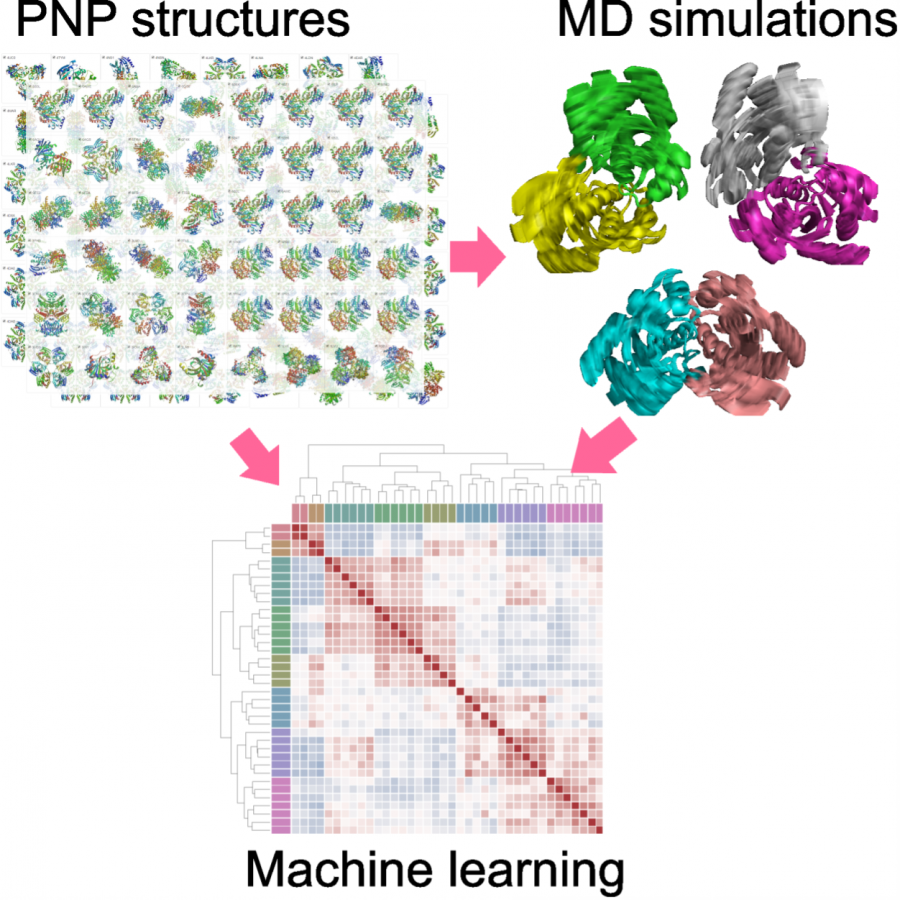 FIGURE 1 A scheme of the conducted research. |
#1014 | Photoenzymatic asymmetric [2+2] cycloaddition reaction |
|
| Presenting author: | Dongshan WU of PEKING UNIVERSITY |
| Corresponding author: | Xiaoguang LEI of PEKING UNIVRERSITY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photoenzymatic / Asymmetric [2 + 2] cycloaddition / Energy tansfer / Directed evolution |
| Purpose: | Cyclobutanes are important structural motifs in organic chemical synthesis, drug discovery and natural products. At present, the asymmetric [2+2] cycloaddition reaction is one of the most effective ways to obtain the chiral cyclobutane skeletons. Traditional methods mainly include the three strategies of polarized [2+2] cycloaddition, [2+2] cycloaddition catalyzed by transition metals and [2+2] cycloaddition catalyzed by photocatalysis with assist of chiral ligands. However, these reactions have some defects, such as limited substrate type, high temperature for thermodynamically driven reactions, expensive chiral catalyst or chiral ligand are needed for stereoselectivity control, and yield and stereoselectivity need to be further improved. On the other hand, enzymatic catalysis has been widely concerned because of its reaction efficiency and high selectivity, chemenzymatic synthesis has been widely concerned. More and more enzymatic reactions have been developed in industry and research. However, there are far fewer reaction types that can be realized by enzyme catalysis than traditional chemical reactions, and there are still many reaction types that cannot be realized by enzyme catalysis. Therefore, it is urgent to develop the hybrid catalytic function of enzymes to enable them to perform the non-natural catalytic function. In recent years, some unnatural photochemical reactions involving flavin-dependent enzymes have been reported. In this regard, we proposed the use of flavin-dependent enzymes combined with photochemistry to carry out intermolecular asymmetric [2+2] cycloaddition reactions. In order to realize the construction of multi-chiral substituted cyclobutanes with high stereoselectivity without expensive transition metals and chiral ligands under mild conditions. Further combine structural biology, photochemistry, and computational chemistry to study the reaction mechanism, with a view to discovering new reaction mechanisms and inspiring synthetic ideas. At present, through the screening of reaction conditions, with the help of a series of FAD dependent intermolecular Diels–Alderases and oxidases and FMN dependent ene reductases, we have achieved the photoenzymatic intermolecular/intramolecular [2+2] cycloaddition reaction . Through a series of spectral experiments and DFT calculations, we have basically confirmed that the mechanism of the reaction may be initiated by the triplet-triplet energy transfer between FAD/FMN and the substrate. This is the first case of natural photoenzymatic reaction based on energy transfer and the first case of photoenzymatic [2+2] cycloaddition |
| References: | 1. Lee-Ruff, E., Mladenova, G. Enantiomerically Pure Cyclobutane Derivatives and Their Use in Organic Synthesis. Chem. Rev. 103, 1449-1484 (2003). 2. Schuster, D. I., Lem, G., Kaprinidis, N. A. New Insights into an Old Mechanism: [2 + 2] Photocycloaddition of Enones to Alkenes. Chem. Rev. 93, 3-22 (1993). 3. Yoon, T. P. Photochemical Stereocontrol Using Tandem Photoredox-Chiral Lewis Acid Catalysis. Acc. Chem. Res. 49, 2307-2315 (2016). 4. Lia, J., Amatunia, A., Renata, H. Recent advances in the chemoenzymatic synthesis of bioactive natural products. Cur. Opin. Chem. Biol. 55, 111-118 (2020). 5. Hyster, T. K. Radical Biocatalysis: Using Non-Natural Single Electron Transfer Mechanisms to Access New Enzymatic Functions. Synlett 31, 248-254 (2020). 6. Sandoval, B. A., Hyster, T. K. Emerging strategies for expanding the toolbox of enzymes in biocatalysis. Cur. Opin. Chem. Biol. 55, 45-51 (2020). 7. Gao, L., Su, C., Du, X. X., Wang, R. S., Chen, S. M., Zhou, Y., Liu, C. W., Liu, X. J., Tian, R. Z., Zhang, L. Y., Xie, K. B., Chen, S., Guo, Q. Q., Guo, L. P., Hano,Y., Shimazaki, M., Minami, A., Oikawa, H., Huang, N., Houk, K. N., Huang, L. Q., Dai, J. Lei, X. G. FAD-dependent enzyme-catalysed intermolecular [4+2] cycloaddition in natural product biosynthesis. Nat. Chem. 12, 620-628 (2020). 8. Gao, L., Zou, Y., Liu, X. et al. Enzymatic control of endo- and exo-stereoselective Diels-Alder reactions with broad substrate scope. Nat Catal 4, 1059-1069 (2021). 9. Jiannan Zhao, J. J. L. Brosmer, J. L. J. Am. Chem. Soc. 139, 9807-9810 (2017) |
| Figures: | 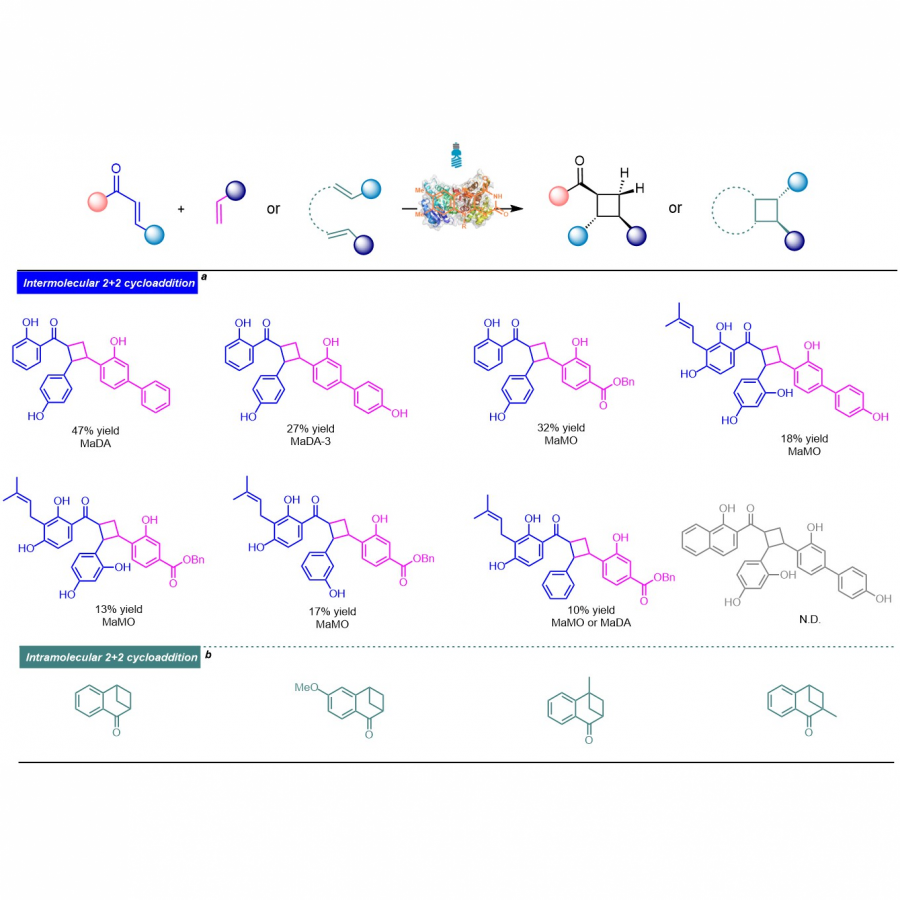 Photoenzymatic asymmetric [2+2] cycloaddition reaction a: The intermolecular 2+2 cycloaddition was catalyzed by Diels Alderases and oxidase reported previously
b: The intramolecular 2+2 cycloaddition was catalyzed by Ene reductase 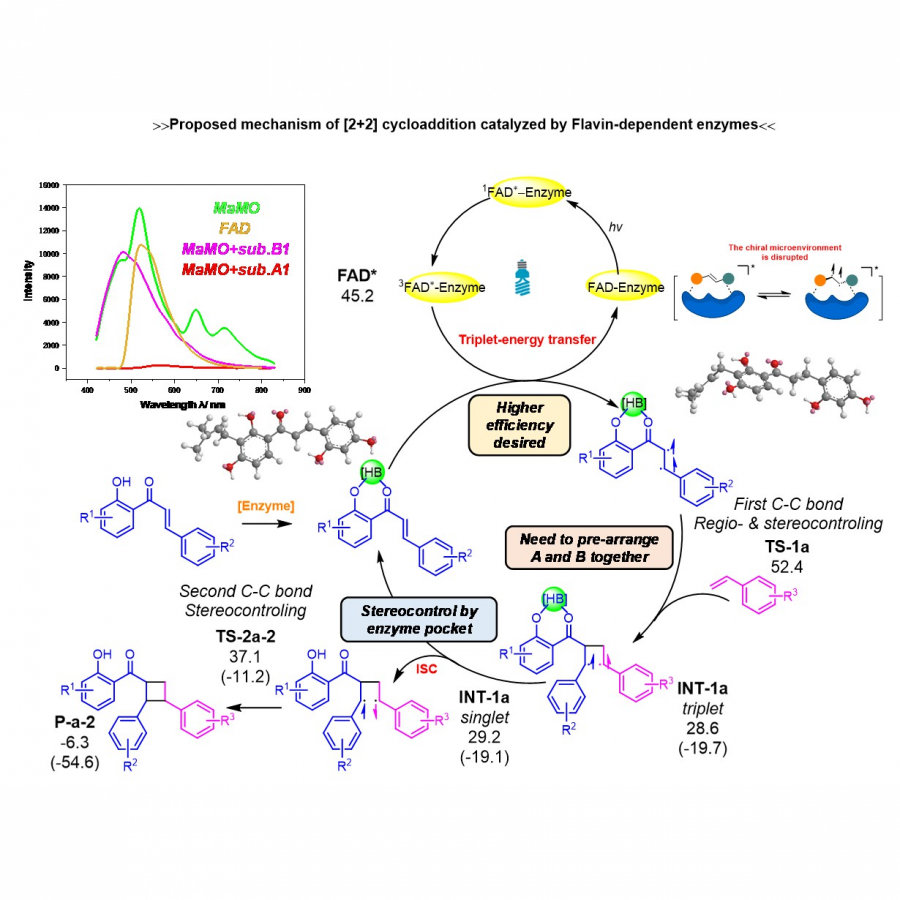 Proposed mechanism of [2+2] cycloaddition catalyzed by Flavin-dependent enzymes Mechanistically, our [2+2] cycloaddition proceeds via
energy transfer from the triplet flavins to the chalcone 1, which undergoes intermolecular cyclization to generate diradical intermediate 2. Then the cycloadduct 4 is constructed through ISC. |
#1020 | Biocatalytic Synthesis of Novel Fructooligosaccharides and Fructans by Levansucrase and Levanase from Discovery to Application |
|
| Presenting author: | Salwa KARBOUNE of MCGILL UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Levansucrase / Levanase / Carbohydrate / Glycosylation |
| Purpose: | The enormous potential of biocatalysis as a key technology for the sustainable synthesis of tailor-made oligo/polysaccharides and glycostructures is widely recognized. When compared to chemical routes, biocatalytic approaches offer excellent regio/stereoselectivity, mild operating conditions, higher product quality, and sustainability benefits. Carbohydrate-based biomolecules are essential compounds in innumerable biological functions. Their tailor-made synthesis, shaped on demand, is of high importance in various fields and industries, including food, pharmaceutical and chemical industries. Levansucrases (2,6-ß-fructan:6-ß-D-fructosyl transferase) have attracted much interest because of their ability to directly use the free energy of cleavage of sucrose to transfer the fructosyl group to an acceptor. We have pioneered the development of this glycosylation route based on levansucrase-catalyzed transfructosylation of sucrose for the synthesis of ß-(2-6)-oligolevans and levans. Furthermore, the synthesis of hetero- fructooligosaccharides by levansucrase was achieved in the presence of various acceptors. For efficient synthesis, we have successfully developed for the first time a novel bi-enzymatic system for the synthesis of ß-(2-6)-fructooligosaccharides of controlled size from sucrose, in which levansucrase mainly catalyzes the synthesis of levans, while levanase through its hydrolytic activity regulates the product molecular size and acceptor availability. As part of the proof of the applicability of the synthetic activity of levansucrase, we successfully used, for the first time, this enzymatic glycosylation to convert low-grade maple products into high value-added ones enriched with FOSs, oligolevans and levans and to produce functional sweetener, lactosucrose, from dairy by-products. The presentation will cover our progress from discovery of new levansucrase and levanase to developing strategies for improving their actions and to exploiting them in advanced applications. |
| References: | [1] Chen, L., Hill, A., Mariage, A., Petit, J.L., de Berardinis, V., Karboune, S. 2023. ACS Chem. Biol. 18, 3, 465-475 [2] Karboune, S., Seo, S., Li, M., Waglay, A., Lagacé, L. 2022. Food Chem, 382, 132355. [3] Bahlawan, R., Karboune, S. 2022. Process Biochem, 122, 248. [4] Hill, A., Chen, L., Mariage, A., Petit, J.L., de Berardinis, V., Karboune, S. 2019. Catal Sci Technol, 9, 2931. [5] Hill, A., Karboune, S., Mateo, C. 2017. Process Biochem, 61, 63. |
| Figures: | 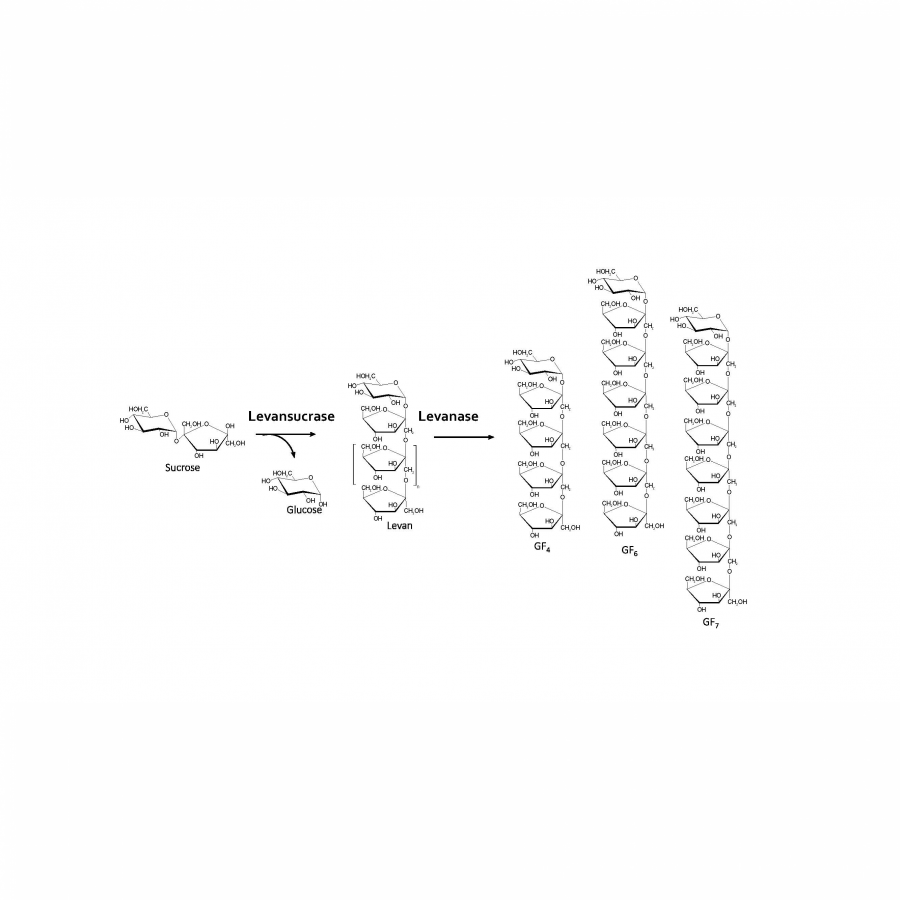 Combined use of Levansucrase and Levanase na |
#1021 | Steric hindrance modification-based substrate specificity evolution of amine dehydrogenases |
|
| Presenting author: | Xiaoqing MU of JIANGNAN UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Chiral amines / Amine dehydrogenases / Steric hindrance / Substrate specificity |
| Purpose: | Chiral amine compounds serve as valuable structural motifs in bioactive compounds, therapeutically useful molecules, and active pharmaceutical intermediates. Direct reductive amination of prochiral ketones is highly attractive in the synthesis of chiral amine, which was ranked second on the chemical reaction list of the most promising to challenge the pharmaceutical industry highlighted by the American Chemical Society. In recent decades, there has been increasing attention to the synthesis of chiral amines catalyzed by amine dehydrogenases (AmDHs) given that its sole expense is ammonia and that water is the only byproduct. However, the prochiral ketone acceptance of the existing natural and engineered AmDHs toolbox is limited, resulting in the synthetic application of AmDHs has remained challenging. Here, we highlight the development of strategies for the substrate specificity evolution of AmDHs based on the steric hindrance effect modification between the enzyme substrate-binding pocket and substrate molecular. These works provide a referable strategy for the divergent substrate acceptance evolution of AmDHs family members and other oxidoreductases with analogous substrate-binding pocket. |
| References: | [1] T. Wu, Y. Wang, N. Zhang, D. Yin, Y. Xu, Y. Nie, X. Mu, ACS Catalysis 2023, 13, 158-168. [2] X. Mu, T. Wu, Y. Mao, Y. Zhao, Y. Xu, Y. Nie, ChemCatChem 2021, 13, 5243-5253. [3] T. Wu, X. Mu, Y. Xue, Y. Xu, Y. Nie, Biotechnology for Biofuels 2021, 14, 207. [4] L. Yang, X. Mu, Y. Nie, Y. Xu, Metabolic Engineering 2021, 64, 122-133. [5] J. Li, X. Mu, T. Wu, Y. Xu, Bioresources and Bioprocessing 2022, 9, 33. |
#1024 | Discovery and rational mutagenesis of methionine sulfoxide reductase (MsrA) biocatalysts to expand the substrate scope of the kinetic resolution of chiral sulfoxides |
|
| Presenting author: | Silvia ANSELMI of UCL |
| Other authors: | Alexandra T. P. CARVALHO of ALMAC Angela SERRANO-SANCHEZ of UNIVERSITY OF KENT Jose L. ORTEGA-ROLDAN of UNIVERSITY OF KENT Jill CASWELL of ALMAC Iman OMAR of UCL Gustavo PEREZ-ORTIZ of KING’S COLLEGE LONDON Sarah M. BARRY of KING’S COLLEGE LONDON Thomas S. MOODY of ALMAC Daniele CASTAGNOLO of UCL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sulfoxides / methionine reductase / mutagenesis / mutant enzyme |
| Purpose: | Chiral sulfoxides are an important class of organic compounds that are often used in organic synthesis as chiral auxiliaries, synthons for C–C bond forming reactions, directing groups in C–H bond functionalisation and can partake in numerous other functionalisation reactions. Moreover, sulfoxides are widely found in pharmaceutically active ingredients such as the blockbuster drug esomeprazole. The most common biocatalytic approach to the synthesis of chiral sulfoxides rely on the use of oxidative enzymes such as monooxygenases or peroxygenases.[1] However, these can present some disadvantages when it comes to their application on large scale industrial production, such as low-yielding profiles, the use of expensive cofactors and the need of effective oxygenation throughout the reaction mixture.[1] A less common route to obtain asymmetric sulfoxides is the use of reductive enzymes, which, starting from a racemic mixture, can perform kinetic resolutions to afford single enantiomers. For example, Methionine sulfoxide reductases (Msrs) are enzymes known to reduce (S)-methionine sulfoxide to methionine in order to repair cellular damage by reactive oxygen species and have proved to be capable of reducing non-natural sulfoxides.[2,3] Therefore, following our interest in industrially applicable green methodologies for the synthesis of sulfur-containing compounds, herein we show how Methionine sulfoxide reductases A (MsrAs) were used for the development of a new biocatalytic protocol for the synthesis of (R)-sulfoxides at high substrate concentrations.[4] In this investigation, we identified the selective and robust scMsrA from Saccharomyces cerevisiae as hit enzyme from a panel of 15 MsrAs. A series of small alkyl aryl sulfoxides racemates were effectively reduced with excellent conversion and ee (>90%) using dithiothreitol (DTT) as the enzyme regenerating agent, a cheaper and low molecular weight dithiol substitute to thioredoxin. Moreover, with the aim to expand the substrate scope of MsrA biocatalysts, a library of mutant enzymes has been designed via rational mutagenesis utilising in silico docking, molecular dynamics, and structural NMR studies. The mutant enzyme MsrA33 was found able to catalyse the kinetic resolution of bulky sulfoxide substrates bearing non-methyl substituents on the sulfur atom with ees up to 99%, overcoming a significant limitation of the currently available MsrA biocatalysts. |
| References: | [1] J. Han, V. A. Soloshonok, K. D. Klika, J. Drabowicz and A. Wzorek, Chem. Soc. Rev., 2018, 47, 1307-1350. [2] M. A. Rahman, H. Nelson, H. Weissbach and N. Brot, J. Biol. Chem., 1992, 267, 15549-15551. [3] S. Anselmi, N. Aggarwal, T. S. Moody and D. Castagnolo, ChemBioChem, 2021, 22, 298-307. [4] S. Anselmi, A. T. P. Carvalho, A. Serrano-Sanchez, J. L. Ortega-Roldan, J. Caswell, I. Omar, G. Perez-Ortiz, S. M. Barry, T. S. Moody and D. Castagnolo, ACS Catal., 2023, Accepted, in press. |
| Figures: | 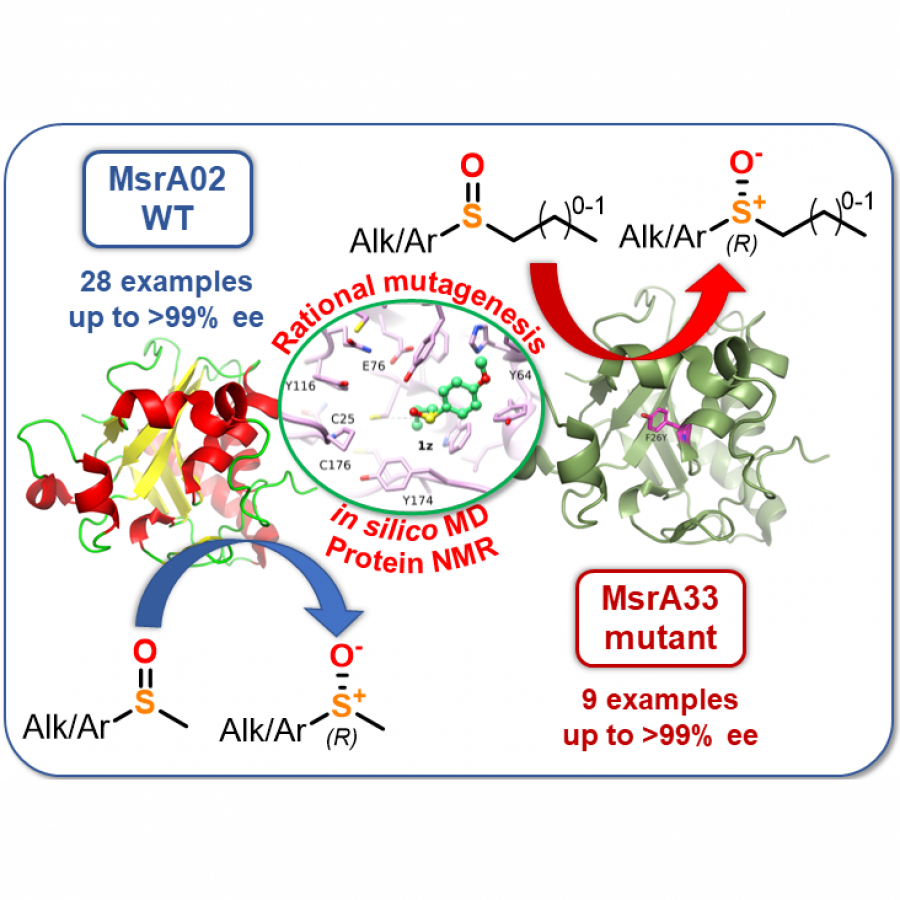 Graphical Abstract Discovery and Rational Mutagenesis of Methionine Sulfoxide Reductase Biocatalysts To Expand the Substrate Scope of the Kinetic Resolution of Chiral Sulfoxides |
#1026 | Novel thermostable aminotransferase from Streptomyces sp.: discovery and functional characterization |
|
| Presenting author: | Stefania PATTI of SCITEC-CNR |
| Other authors: | Michail ISUPOV of UNIVERSITY OF EXETER Simone Antonio DE ROSE of UNIVERSITY OF EXETER Marta VANONI of SCITEC-CNR Daniela UBIALI of UNIVERSITY OF PAVIA Daniela MONTI of SCITEC-CNR Jennifer LITTLECHILD of UNIVERSITY OF EXETER Erica Elisa FERRANDI of SCITEC-CNR |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transaminases / enzyme discovery / enzyme engineering / biocatalysis |
| Purpose: | Aminotransferases (ATAs, E.C. 2.6.1.x) are pyridoxal-5’-phosphate (PLP) dependent enzymes which catalyze the formation of primary chiral amines by transferring an amino group from an amine donor to a prochiral ketone (acceptor), thus generating a new stereogenic centre at the end of the reaction. ATAs are of great industrial interest for the production of enantiomerically pure chiral amines. In this study, a gene encoding for a putative transaminase was discovered in Streptomyces sp. BV333 employing a combined identification technique, which included functional screening and genome mining. The identified gene shared sequence similarities with thermostable ATA sequences, including those from Thermomicrobium roseii (43% identity) [1] and from hot spring metagenomes (B3-ATA, 41% identity) [2]. The corresponding enzyme (Sbv333-ATA) was successfully produced in Escherichia coli co-expressing GroES/GroEL chaperons. Interestingly, it was found that Sbv333-ATA was extremely thermostable, with a melting temperature (Tm) that was only marginally lower (85 °C) than those of the most thermostable transaminases previously reported (87–88 °C) [2]. Moreover, Sbv333-ATA displayed a broad substrate specificity, as far as the amino acceptor spectrum is concerned, and a remarkable activity in the transamination of β-ketoesters, which are rarely accepted by known ATAs [3]. Recently, new studies have been carried out to evaluate enzyme stability in the presence of organic (co)solvents. Sbv333-ATA proved to be stable in the presence of up to 20% (v/v) of the water-miscible-co-solvents methanol, ethanol, acetonitrile, dimethylsulfoxide, and in biphasic systems with petroleum ether, toluene and ethyl acetate as organic phase. Furthermore, new amine donors were evaluated as possible substrates for this ATA. This enzyme was also crystallized, and the high-resolution structures of both the native form and the complex with the inhibitor gabaculine were determined. Mutagenesis studies are being conducted to broaden the substrate scope of Sbv333-ATA. |
| References: | [1] S. Mathew, K. Deepankumarb, G. Shinc, E. Y. Hongd, B. Kimd, T. Chunge, H. Yun, RSC Adv. 2016, 6, 69257-69260 [2] E. E. Ferrandi, A. Previdi, I. Bassanini, S. Riva, X. Peng, D. Monti, Appl. Microbiol. Biotechnol. 2017, 101, 4963-4979 [3] E. E. Ferrandi, J. Spasic, L. Djokic, Y. Vainshtein, R. Senthamaraikannan et al., Catalysts 2021, 11, 919 |
#1027 | A mild and chemoselective CALB biocatalysed synthesis of sulfoxides exploiting the dual role of AcOEt as solvent and reagent |
|
| Presenting author: | Iman OMAR of UCL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sulfoxides / CALB / biocatalysis / |
| Purpose: | Sulfoxides are an important class of organic compounds that often are seen used in organic synthesis as chiral auxiliaries, synthons for C–C bond forming reactions, directing groups in C–H bond functionalisation and can partake in numerous other functionalisation reactions. Moreover, sulfoxides are widely found in pharmaceutically active ingredients such as the blockbuster drug omeprazole. Sulfoxides are commonly obtained through the oxidation of the corresponding sulfide precursor.1 However, the current methodologies adopted for their synthesis rely on the use of harsh chemicals such as nitric acid, hypohalites, peroxides and oxone, all of which present limited industrial use as can be shock sensitive and explosive, hence unsuitable for large scale production.2 Following our interest in the development of new and industrially applicable green methodologies for the synthesis of sulfur-containing drugs and drug-like synthons, herein we report a facile, chemoselective and scalable biocatalytic protocol for the synthesis of sulfoxides using immobilised Candida antarctica lipase B (CALB), a very robust enzyme which retains its activity in both aqueous and organic solvents, and AcOEt with a dual role of more environmentally friendly reaction solvent and enzyme substrate.3 A series of 27 sulfides that included alkyl, aryl, carbonyl and alkene bearing compounds as well as omeprazole were successfully oxidised in high yields and with excellent E-factors to the corresponding sulfoxides with little to no overoxidation by-products. In addition, a large-scale experiment starting from 3 g of sulfide substrate afforded the corresponding sulfoxide in excellent yield. Finally, a series of enzyme recyclability experiments were carried out to further confirm the industrial potentiality of the methodology. This method proves to be cost effective, robust and selective with few side-reactions. Furthermore, we show that the use of AcOEt as solvent and CALB substrate improves the industrial sustainability of the method, providing an overall greener methodology. |
| References: | 1. S. Anselmi, N. Aggarwal, T. S. Moody and D. Castagnolo, ChemBioChem, 2020, 22, 298-307. 2. G. Hughes and J. C. Lewis, Chem. Rev., 2018, 118, 1-3. 3. S. Anselmi, S. Liu, S. H. Kim, S. M. Barry, T. S. Moody and D. Castagnolo, Org. Biomol. Chem., 2021, 19, 156-161. |
| Figures: | 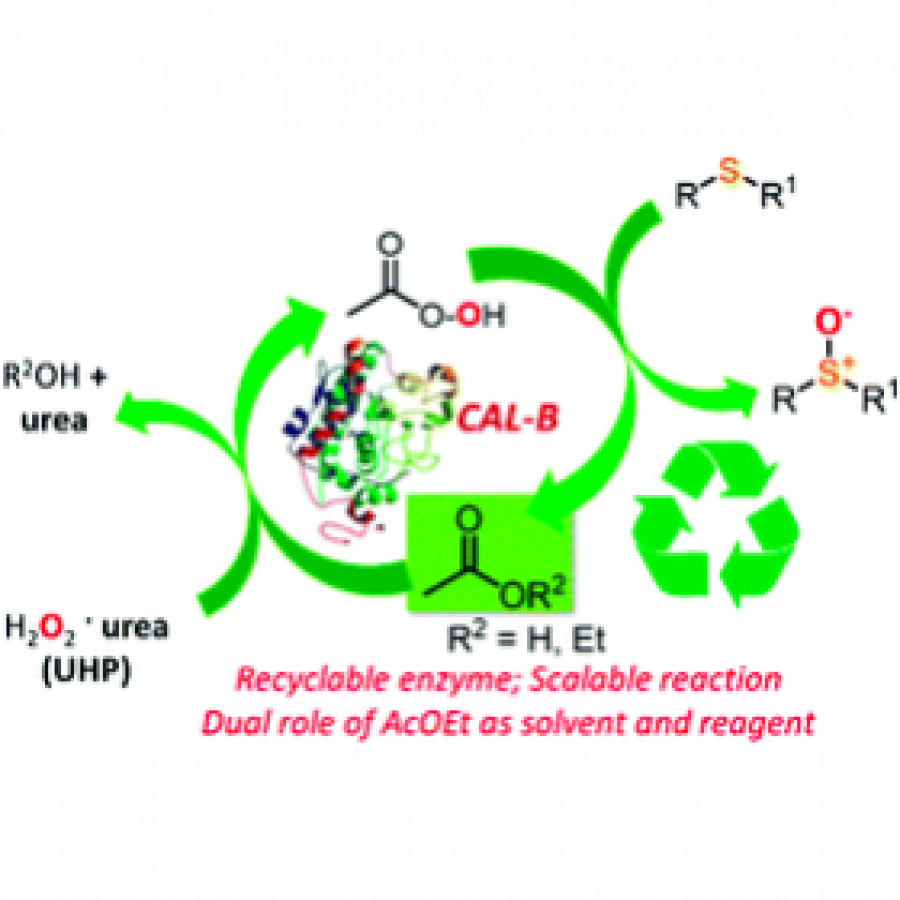 CALB for the biocatalytic synthesis of sulfoxides A mild, chemoselective and sustainable biocatalysed synthesis of sulfoxides has been developed exploiting CALB and using AcOEt with a dual role of more environmentally friendly reaction solvent and enzyme substrate. A series of sulfoxides, including the d |
#1030 | Self-assembly of His-tagged amine transaminase with functionalized nanoparticles |
|
| Presenting author: | Polona ZNIDARSIC-PLAZL of UNIVERSITY OF LJUBLJANA, FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY |
| Other authors: | Borut SKETA of UNIVERSITY OF LJUBLJANA, FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY Katja TRILER of UNIVERSITY OF LJUBLJANA, FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY Diana BALOGH-WEISER of BUDAPEST UNIVERSITY OF TECHNOLOGY AND ECONOMICS, DEPARTMENT OF ORGANIC CHEMISTRY AND TECHNOLOGY László POPPE of BUDAPEST UNIVERSITY OF TECHNOLOGY AND ECONOMICS, DEPARTMENT OF ORGANIC CHEMISTRY AND TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | amine transaminase / nanoparticles / immobilization / self-assembly |
| Purpose: | Nanomaterials have great potential as carriers for enzyme immobilization because they provide a very large specific surface area to which enzymes can be bound. Other advantages are good accessibility of the enzyme in the immobilized enzyme preparation and high mechanical strength [1]. The most commonly used nanomaterials for enzyme immobilization are carbon nanotubes, various magnetic nanoparticles, and silica nanoparticles [2]. In this work, the immobilization of His-tagged amine transaminase from the c-LEcta metagenomic library on functionalized silica nanoparticles was investigated. Nanoparticles with diameters of 250 and 500 nm with affinity linkers of different chain lengths containing amine groups were used [3]. Further complexation of cobalt, gadolinium, or iron ions on the surface of the nanoparticles allowed the immobilization of His-tagged enzyme via coordinative bonds. The immobilization yield and the obtained activity were compared, and the self-assembly of the particles and enzymes was inspected by scanning electron microscopy (SEM). The conversion of (S)-α-methylbenzylamine and sodium pyruvate to acetophenone and L-alanine was chosen as a model reaction for the evaluation of amine transaminase activity [4]. The highest observed immobilization yield of 77% and the highest immobilization efficiency of 44.8% were obtained with 250 nm particles with the longest of the linkers tested. Among the tested metal ions linked to the 250 nm nanoparticles, cobalt resulted in the highest immobilization yield and activity. The self-assembly was also confirmed by SEM. References: [1] M. Sharifi, M. J. Sohrabi, S. H. Hosseinali, A. Hasan, P. H. Kani, A. J. Talaei, A. Y. Karim, N. M. Qadir Nanakali, A. Salihi, F. M. Aziz, B. Yan, R. H. Khan, A. A. Saboury, M. Falahati, Int. J. Biol. Macromol., 2020, 143, 665–676. [2] M. Bajić, P. Žnidaršič-Plazl, M. Kingston, V. Hessel. General aspects of immobilized biocatalysts and their applications in flow. In: M. B. Nielsen (Ed.). Science of Synthesis: Knowledge Updates 2018/1. Stuttgart, New York: Georg Thieme. 2018, pp. 397-443 [3] G. Koplányi, E. Bell, Z. Molnár, G. Katona, P. L. Neumann, F. Ender, G. T. Balogh, P. Žnidaršič-Plazl, L. Poppe, D. Balogh-Weiser, ChemBioChem, 2023, e202200713. [4] T. Menegatti, P. Žnidaršič-Plazl, Front. Bioeng. Biotechnol., 2021, 9, 752064. |
| References: | [1] M. Sharifi, M. J. Sohrabi, S. H. Hosseinali, A. Hasan, P. H. Kani, A. J. Talaei, A. Y. Karim, N. M. Qadir Nanakali, A. Salihi, F. M. Aziz, B. Yan, R. H. Khan, A. A. Saboury, M. Falahati, Int. J. Biol. Macromol., 2020, 143, 665-676. [2] M. Bajić, P. Žnidaršič-Plazl, M. Kingston, V. Hessel. General aspects of immobilized biocatalysts and their applications in flow. In: M. B. Nielsen (Ed.). Science of Synthesis: Knowledge Updates 2018/1. Stuttgart, New York: Georg Thieme. 2018, pp. 397-443 [3] G. Koplányi, E. Bell, Z. Molnár, G. Katona, P. L. Neumann, F. Ender, G. T. Balogh, P. Žnidaršič-Plazl, L. Poppe, D. Balogh-Weiser, ChemBioChem, 2023, e202200713. [4] T. Menegatti, P. Žnidaršič-Plazl, Front. Bioeng. Biotechnol., 2021, 9, 752064 |
#1039 | One-step isolation and immobilization of His-tagged amine transaminase from cell lysate in a microreactor with functionalized nanofiber mat |
|
| Presenting author: | Borut ŠKETA of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA |
| Other authors: | Polona ŽNIDARŠIČ PLAZL of FACULTY OF CHEMISTRY AND CHEMICAL TECHNOLOGY, UNIVERSITY OF LJUBLJANA |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | amine transaminase / microreactor / immobilization / biotransformation |
| Purpose: | One-step isolation and immobilization of His-tagged amine transaminase from cell lysate in a microreactor with functionalized nanofiber mat Borut Šketa* and Polona Žnidaršič-Plazl Faculty of Chemistry and Chemical Technology, University of Ljubljana, Večna pot 113, SI-1000 Ljubljana, Slovenia *Email: borut.sketa@fkkt.uni-lj.si Microreactors are powerful tools for rapid development and intensification of biocatalytic processes. Their main advantages derive from their small dimensions, which allow very efficient mass and heat transport, controlled flow regimes and continuous process operation [1], To achieve long-term use of enzymes in the flow, biocatalysts are usually immobilized inside the microreactors, which also improves their operational stability [2,3]. Since enzyme purification significantly contributes to biocatalysts costs and process time, a one-step isolation and immobilization of the target proteins constructed with different types of affinity tags offers an excellent alternative [4]. In this work, His-tagged wild-type amine transaminase (ATA -wt) from the c-LEcta metagenome library was expressed as a soluble protein in Escherichia coli BL21(DE3). The cell lysate was centrifuged and used to immobilize the enzymes on functionalized nonwoven nanofiber membranes in a microreactor. The microreactor consisted of two poly(methyl methacrylate) plates and two polytetrafluoroethylene gaskets forming a hexagonal channel in which the nonwoven nanofiber mat was embedded between the two gaskets. The commercial polymeric nanofiber mat functionalized with Cu2+ ions (Tiss-IMAC-Cu from NanoMyP) was selected based on preliminary experiments with purified enzyme and offers very large surface for enzyme bonding. The retained enzyme activity, volumetric productivity (space-time yield, STY) of the microreactor, and stability were monitored in operando based on a model reaction using (S)-α-methylbenzylamine as amine donor, pyruvate as amine acceptor, and pyridoxal-5'-phosphate (PLP) as a cofactor that was present only in the immobilization step [5]. Immobilization of unpurified amine transaminase from the cell lysate in a microreactor yielded enzyme load of over 1000 U mL-1. By skipping the purification step, the amount of waste produced, and the time required for enzyme isolation, purification and immobilization were significantly reduced. A STY of over 20 U mL-1 was achieved at tested conditions. Over 90% of enzyme activity was retained after 4 days of continuous operation without exogenously added PLP. Keywords: amine transaminase, microreactor, immobilization, biotransformation References (1) Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K. V.; Woodley, J. M. Trends Biotechnol., 2015, 33, 302–314. (2) Sheldon, R. A.; Basso, A; Brady, D. Chem. Soc. Rev., 2021,50, 5850-5862. (3) Žnidaršič-Plazl, P. Curr. Opin. Green Sustain. Chem., 2021, 32, 100546. (4) Koplányi, G.; Bell, E.; Molnár, Z.; Katona, G.; Neumann, P.L.; Ender, F.; Balogh, G.T.; Žnidaršič-Plazl, P.; Poppe, L.; Balogh- Weiser, D. ChemBioChem 2023, e202200713 (5) Menegatti, T.; Žnidaršič-Plazl, P. Front. Bioeng. Biotechnol., 2021, 9, 752064. |
| References: | [1] Wohlgemuth, R.; Plazl, I.; Žnidaršič-Plazl, P.; Gernaey, K. V.; Woodley, J. M. Trends Biotechnol., 2015, 33, 302-314. [2] Sheldon, R. A.; Basso, A; Brady, D. Chem. Soc. Rev., 2021,50, 5850-5862. [3] Žnidaršič-Plazl, P. Curr. Opin. Green Sustain. Chem., 2021, 32, 100546. [4] Koplányi, G.; Bell, E.; Molnár, Z.; Katona, G.; Neumann, P.L.; Ender, F.; Balogh, G.T.; Žnidaršič-Plazl, P.; Poppe, L.; Balogh- Weiser, D. ChemBioChem 2023, e202200713 [5] Menegatti, T.; Žnidaršič-Plazl, P. Front. Bioeng. Biotechnol., 2021, 9, 752064. |
#1051 | Enhancing violacein production through adaptive laboratory evolution with the aid of biosensor |
|
| Presenting author: | Da-ae GWON of CHEMICAL ENGINEERING / POSTECH |
| Corresponding author: | Jeong Wook LEE of CHEMICAL ENGINEERING / POSTECH |
| Other authors: | Chang Ha WOO of SCHOOL OF INTERDISCIPLINARY BIOSCIENCE & BIOENGINEERING(I-BIO)/ POSTECH Donghyeon KIM of CHEMICAL ENGINEERING / POSTECH |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Violacein / Adaptive laboratory evolution / Metabolic engineering / Biosensor |
| Purpose: | Violacein has attractions due to its purple color, and it is highly antibacterial and antiviral. It is a naturally occurring secondary metabolite by microorganisms such as Chromobacterium violaceum and Janthinobacterium lividum. The development of natural violacein producers is limited due to their pathogenic nature and the insufficient genetic engineering tools available. As an alternative, researchers have used conventional model organisms such as Escherichia coli to construct heterologous violacein biosynthetic pathways. In addition, most studies so far have produced violacein using glucose or glycerol, and galactose has never been used as a carbon source. Galactose, which is commonly found in non-edible biomass such as macroalgae and dairy waste, is considered a promising carbon source for biochemical production. Therefore, we aim to engineer E. coli to produce violacein from galactose. In a rational approach, to improve target molecule production detailed understanding of pathways and regulatory systems was required. However, it is often difficult to engineer E. coli to overproduce the target product or intermediate in this way due to the complex biosynthetic pathways and regulatory mechanisms. In order to improve violacein production from galactose, we used the adaptive laboratory evolution (ALE) method, which is relatively less reliant on biological knowledge. We hypothesized that by increasing the production of the intermediate tryptophan, violacein would be produced more. We used the biosensor-assisted ALE to generate a population of cells capable of effective catabolism of galactose to tryptophan. While conducting the ALE, we employed a tryptophan-responsive biosensor to effectively enrich tryptophan-producing cells. By using tnaC, which is a leader peptide of tnaCAB operon, we created a tryptophan-responsive genetic device that induces tetA expression in response to tryptophan. With wild-type tnaC, the tryptophan-responsive sensor had a narrow operational range, so the wild-type sensor was engineered to extend it. The wild-type tnaC sensor was modified with D21T mutation to lower its affinity for tryptophan, resulting in a wider operational range and we used it during ALE. Then, we tried to screen cells overproducing tryptophan by gradually increasing selection pressure. After six passages, we transformed the violacein-producing plasmid and screened the darkest colony. Compared to the parent strain, the evolved strain showed a 2.7-fold higher concentration in flasks. Whole-genome sequencing revealed that the evolved strain accumulated several mutations that might be related to the production directly and indirectly. There was a notable missense mutation identified on the coding sequence of galR, which encodes a DNA-binding transcription factor that inhibits the transcription of operons involved in D-galactose transport and catabolism. It appears that the mutation occurs in the DNA binding domain of GalR, indicating that the altered amino acid may relieve the regulation of GalR-regulated genes. Due to the missense mutation in GalR, galactose catabolism may be affected and galactose utilization may be increased, resulting in increased violacein production. In summary, our study demonstrated that the use of biosensor-assisted ALE can enable the rapid and targeted evolution of strains to increase the production of violacein without the need for extensive genomic engineering. By avoiding the need to target specific genes, we were able to evolve the strain selectively and efficiently toward our desired outcome. Furthermore, the evolved strain also exhibited improved galactose utilization capabilities, which could potentially be leveraged for the production of other biochemicals such as tryptophan derivatives. Overall, these findings highlight the potential of ALE as a powerful tool for metabolic engineering and strain improvement. |
| References: | [1] Gwon, D et al. Int. J. Mol. Sci. 2021, 22, 6594. [2] Wang, T. et al. Nat Chem Biol. 2020, 16, 440 |
| Figures: | 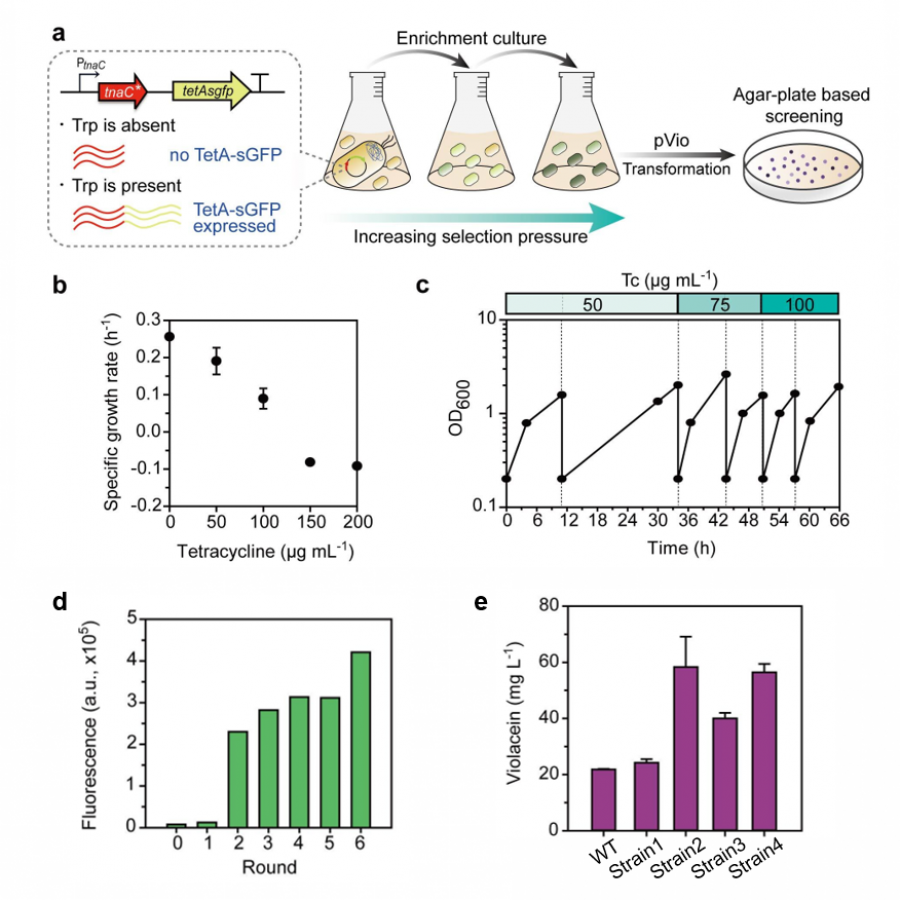 ALE with tryptophan-responsive biosensor (a) Schematic representation of ALE strategy for violacein production (b) Specific growth rate was measured to set the initial selection pressure. (c) Growth profile during ALE (d) Fluorescence of each round (e) Violacein production of several candidates 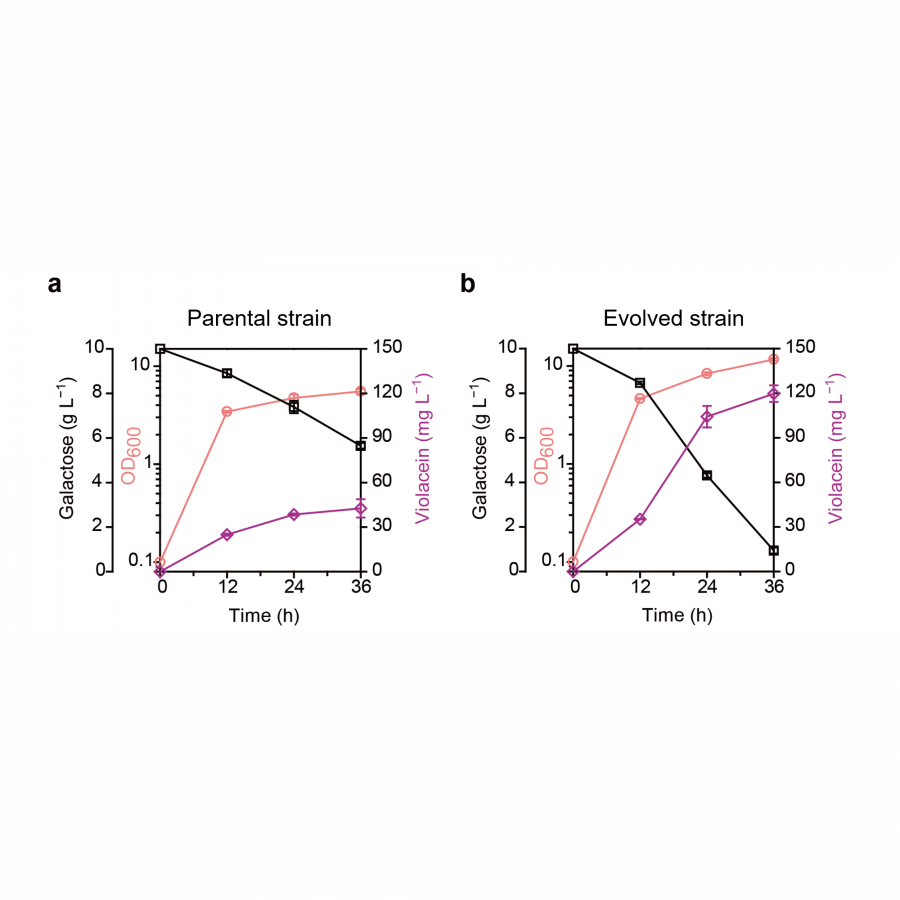 Fermentation profiles of parental and evolved strain Comparison between the (a) parental strain and (b) evolved strain in M9-YE with galactose during 36-h flask culture. Circle, OD600; rectangle, galactose; diamond, violacein. |
#1055 | From Farm to Table: New Strategies for Phosphate Recovery |
|
| Presenting author: | Johanna STOTZ of RWTH AACHEN UNIVERSITY, INSTITUTE OF BIOTECHNOLOGY |
| Corresponding author: | Ulrich SCHWANEBERG of RWTH AACHEN UNIVERSITY, INSTITUTE OF BIOTECHNOLOGY; DWI –LEIBNIZ INSTITUT FÜR INTERAKTIVE MATERIALIEN |
| Other authors: | Patrick LENZ of RWTH AACHEN UNIVERSITY, INSTITUTE OF BIOTECHNOLOGY Anna Joëlle RUFF of RWTH AACHEN UNIVERSITY, INSTITUTE OF BIOTECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | phytase / phosphate recovery / enzyme engineering / bioeconomy |
| Purpose: | Recently, we published a novel three-step production process to mobilize the phosphate from plant biomass waste streams and produce food-grade polyP [1]. In the first step of the process, the phosphate of plant biomass was mobilized using a phytase blend with a yield of 37.1 mg PO43-/g rye bran. The thereby produced soluble P-extract was then fed to starved Saccharomyces cerevisiae, which polymerize the phosphate to polyP. The polyP can then be extracted from the cells or applied directly in the form of polyP-rich yeast extract. Besides rye bran, further by-products of the food production process were successfully used for P-mobilization, such as deoiled seeds [2]. The obtained (bio-)phosphate has food-grade quality and thus may be incorporated into food preparations, for example as preservative, stabilizer, or texture improver (for instance for spreadability of cheese preparations or texture-improving property in sausage preparations). Such novel processes are needed for a sustainable resource management, since phosphorous is currently obtained exclusively through rock mining, even though it is a finite resource[3]. Within the P2Value project, the Institute of Biotechnology in cooperation with the Institute of Microbiology at RWTH Aachen University is focusing on phosphate mobilization from plant residues and by-products. Plants store phosphate in the form of phytic acid (inositol hexakisphosphate), which can hardly be metabolized by monogastric organisms [4]. Therefore, we aim to engineer phytases that can hydrolyze lower inositol-phosphates to further optimize the P-mobilization process [5, 6]. Furthermore, an enzymatic toolbox will be investigated for the valorization of novel biogenic waste streams. The enzyme production and P-mobilization will be further scaled up to take the first steps towards developing an industrial process. In this way, the (bio)phosphate extracted from food manufacturing side streams can be reintroduced into food preparations, contributing to sustainable phosphate management and a circular economy. With support from Federal Ministry of Food and Agriculture by decision of the German Bundestag, FKZ 2220NR170A |
| References: | [1] K.R. Herrmann, J. Fees, J.J. Christ, I. Hofmann, C. Block, D. Herzberg, S. Bröring, B. Reckels, C. Visscher, L.M. Blank, U. Schwaneberg, A.J. Ruff, Biotechnological production of food-grade polyphosphate from deoiled seeds and bran, EFB Bioeconomy Journal 3 (2023). [2] K.R. Herrmann, A.J. Ruff, U. Schwaneberg, Phytase-Based Phosphorus Recovery Process for 20 Distinct Press Cakes, ACS Sustainable Chemistry & Engineering 8(9) (2020) 3913-3921. [3] C. Alewell, B. Ringeval, C. Ballabio, D.A. Robinson, P. Panagos, P. Borrelli, Global phosphorus shortage will be aggravated by soil erosion, Nat Commun 11(1) (2020) 4546. [4] N. Widderich, N. Mayer, A.J. Ruff, B. Reckels, F. Lohkamp, C. Visscher, U. Schwaneberg, M. Kaltschmitt, A. Liese, P. Bubenheim, Conditioning of Feed Material Prior to Feeding: Approaches for a Sustainable Phosphorus Utilization, Sustainability 14(7) (2022). [5] B. Infanzon, K.R. Herrmann, I. Hofmann, S. Willbold, A.J. Ruff, U. Schwaneberg, Phytase blends for enhanced phosphorous mobilization of deoiled seeds, Enzyme Microb Technol 153 (2022) 109953. [6] K.R. Herrmann, I. Hofmann, D. Jungherz, M. Wittwer, B. Infanzon, S.N. Hamer, M.D. Davari, A.J. Ruff, U. Schwaneberg, Generation of phytase chimeras with low sequence identities and improved thermal stability, J Biotechnol 339 (2021) 14-21. |
#1057 | Enzyme catalysed enantiospecific hydrolysis of silyl ether bonds |
|
| Presenting author: | Lisa M. PICK of CHAIR OF MOLECULAR BIOTECHNOLOGY, TU DRESDEN |
| Other authors: | Marion B. ANSORGE-SCHUMACHER of CHAIR OF MOLECULAR BIOTECHNOLOGY, TU DRESDEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | silyl ether / protection group cleavage / enzyme discovery / enantiospecific catalysis |
| Purpose: | Reversible formation of silyl ethers is widely used in organic synthesis to protect hydroxyl functions from unwanted reactions.[1] Extension of the application of silyl ethers from mere protecting groups to the simultaneous kinetic resolution of racemic mixtures of alcohols has been an object of intensive research for many years.[2] Despite considerable advances with various chemo catalysts, however, stereospecificity is still not satisfactory. A solution might provide the use of enzyme catalysts, which due to their special active sites often enable high stereospecificity in addition of being a greener alternative to chemical catalysts. The ability of enzymes to mediate conversion of silyl ethers has been reported for quite some time[3], but mostly without (enantio-)specificity. We recently showed that literature-described lipase-mediated conversion of silyl ethers is unspecific and independent of its catalytic triad. Therefore, we were looking for enzymes specifically catalysing silyl ether conversion. Here we present two newly identified enzymes cleaving the silyl ether bond of TMS-protected 1-phenylethanol with opposite enantiospecificity. After purification of these proteins from their natural source, identifying them by in-gel digest and LC-MS/MS and heterologous expression in E.coli, we aimed to analyse their catalytic mechanism by introducing site-specific mutations. Thereby, we could identify a few amino acids leading to partly or complete loss of function. Comparison of both enzymes might further allow us to understand the origin of their enantiospecificity. Although their biological function remains unknown, homologous proteins seem to be widely distributed in nature, and by that give us a whole repertoire of possible enzymatic catalysts for silyl ether conversion. Moreover, they allow us to gain further understanding of the needs of enzymatic catalysts specifically acting on silyl ether bonds. |
| References: | [1] P. J. Kocienski, Protecting Groups, Georg Thieme Verlag, 2005. [2] J. Seliger, M. Oestreich, Chemistry - A European Journal 2019, 25, 9358-9365. [3] U. T. Bornscheuer, R. J. Kazlauskas, in Enzyme Catalysis in Organic Synthesis (Ed.: H. G. Karlheinz Drauz, Oliver May), Wiley-VCH Verlag GmbH & Co. KGaA, 2012, pp. 1693-1733. |
| Figures: | 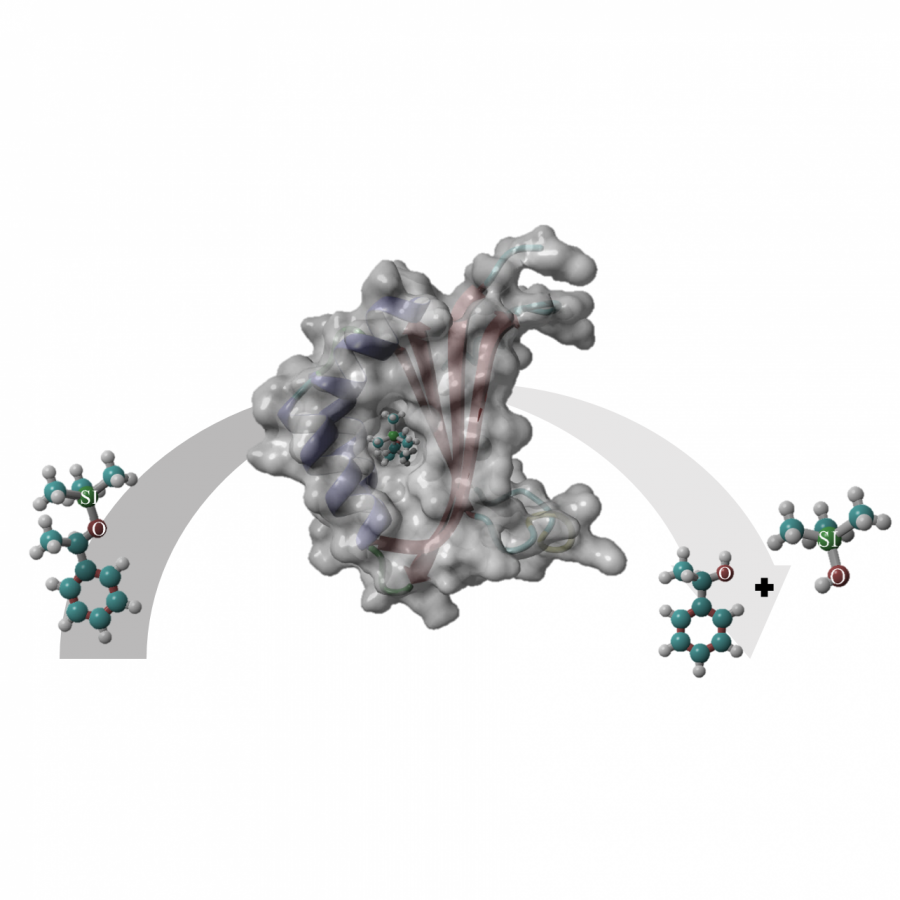 Enzymatic catalysed hydrolysis of trimethylsilyl-protected 1-phenylethanol. |
#1061 | Chemoselective Methyltransferases – Distinguishing between N- and O-Methylation |
|
| Presenting author: | Emely JOCKMANN of UNIVERSITY OF FREIBURG |
| Corresponding author: | Jennifer N. ANDEXER of UNIVERSITY OF FREIBURG |
| Other authors: | Fabiana SUBRIZI of UNIVERSITY COLLEGE LONDON Helen C. HAILES of UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chemoselective methyltransferases / S-adenosyl-l-methionine / enzyme cascade / |
| Purpose: | Methylation reactions are of great interest for pharmaceutical industry. The small carbon fragment can change the pharmacological functions of drugs in various ways. This phenomenon is described as the ‘magic methyl effect’. In industry, reagents such as highly toxic methyl iodide or dimethyl sulfate are used. As an alternative to traditional chemical methods, biocatalytic approaches present a sustainable route for the transfer of methyl groups.[1] Methyltransferases (MTs) catalyse methylations in different biosynthetic pathways. They transfer the methyl group from the cofactor S-adenosyl-L-methionine (SAM) onto different atoms chemo- and regioselectively. Regarding the (hetero) atom that receives the methyl group, MTs are divided into O-, N-, S- and C-MTs. The enzymatic reaction is based on an SN2 mechanism,[2] involving different enzyme residues to increase the nucleophilic character of the species attacking the electrophilic methyl group. There are several classification systems for MTs available. Joshi et al. introduced a system for O-MTs. One key difference between class I and II O-MTs is their dependance on metal ions. Class II enzymes do not require divalent cations and are often found in plant organisms catalysing reactions involved in secondary metabolism.[3] The classes are not strictly limited to O-MTs but also include N-MTs with related amino acid sequences. Hence, the anthranilic acid N-MT (ANMT) from Ruta graveolens (Rg) is considered as a class II member, just like its closest relatives caffeic acid O-MTs (CaOMTs).[4] Zubieta et al. identified a histidine in position 269 to be the catalytic base in CaOMT from Medicago sativa (Ms) deprotonating the hydroxyl group of the natural substrate.[5] RgANMT contains a histidine residue in the same position. Nevertheless, both enzymes are highly chemoselective and methylate different functional groups (Figure 1 b/c).[4] This indicates that the mechanisms of the enzymes or the involved residues must differ in some ways.[5] Here, we present an extended substrate scope for RgANMT and a CaOMT from Prunus persica (Pp). The substrates analysed contained both, an amino- and hydroxyl group and were methylated chemoselectively leading to a broad range of pure products. This offers a good starting point for upscaling biocatalytic reactions methylating more complex molecules with different nucleophiles.[6, 7] |
| References: | [1] H. Schoenherr, T. Cernak, Angew Chem Int Ed 2013, 52, 12256-12267. [2] R. W. Woodard, M. D. Tsai, H. G. Floss, P. A. Crooks, J. K. Coward, J. Biol. Chem. 1980, 255, 9124-9127. [3] C. P. Joshi, V. L. Chiang, Plant Mol Biol 1998, 37, 663-674. [4] B. Rohde, J. Hans, S. Martens, A. Baumert, P. Hunziker, U. Matern, Plant J 2008, 53, 541-553. [5] C. Zubieta, P. Kota, J.-L. Ferrer, R. A. Dixon, J. P. Noel, Plant Cell 2002, 14, 1265-1277. [6] H. Simon-Baram, S. Roth, C. Niedermayer, P. Huber, M. Speck, J. Diener, M. Richter, S. Bershtein, ChemBioChem 2022, 23, e202200162. [7] E. Jockmann, F. Subrizi, H. C. Hailes, J. N. Andexer, manuscript in preparation. |
| Figures: | 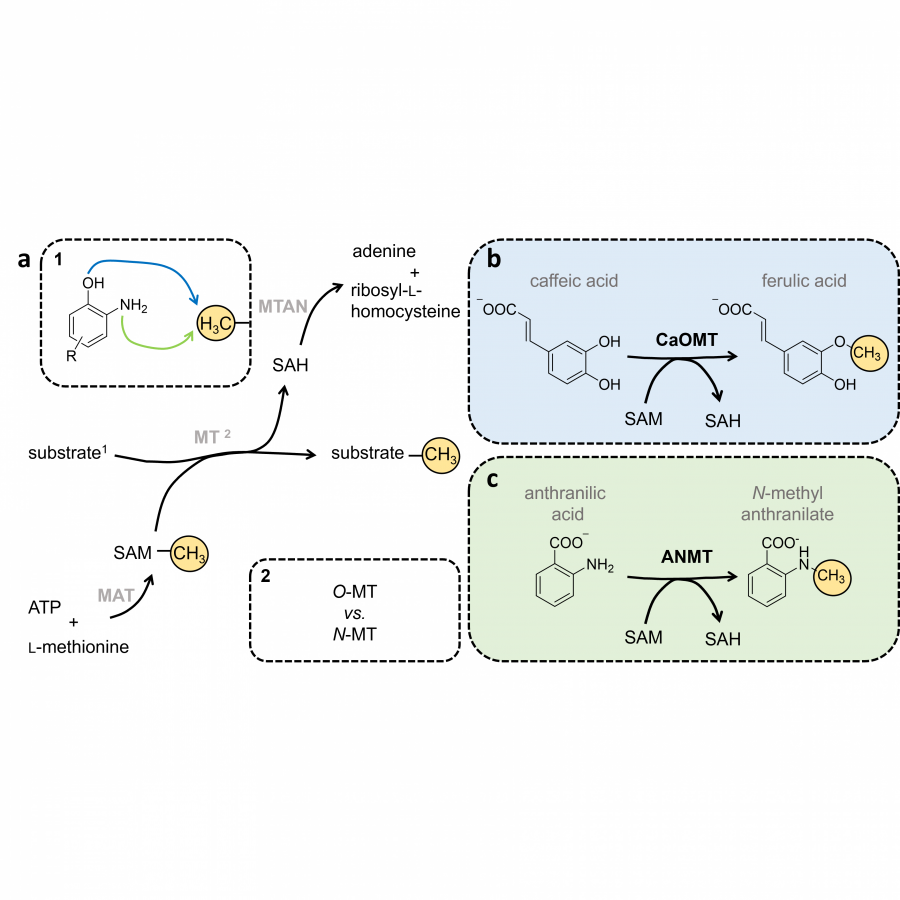 Three-enzyme cascade for methylation reactions a: Three-enzyme cascade used for methylation reaction. b: Natural methylation reaction of the caffeic acid O-MT (CaOMT) forming ferulic acid. c: Natural reaction of anthranilic acid N-MT (ANMT) methylating anthranilic acid in N-position. |
#1065 | P450 peroxygenases – regio- and stereoselective hydroxylation of fatty acids |
|
| Presenting author: | Klara BANGERT of UNIVERSITY GRAZ |
| Other authors: | Wolfgang KROUTIL of UNIVERSITY GRAZ Stefaan DE WILDEMAN of B4PLASTICS |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | P450 Peroxygenases / Fatty Acids / Regioselectivity / Stereoselectivity |
| Purpose: | P450 peroxygenases, a subgroup of the cytochrome P450s, allow a similar oxy-functionalization as P450 monooxygenases but are independent of reduced nicotinamide cofactors and rely on hydrogen peroxide only. [1] The mechanism relies on the hydrogen peroxide-shunt. [2] These peroxygenases can for instance be used for the biocatalytic α-functionalization of medium-chain fatty acids. Six potential enzyme candidates of the CYP152 family, P450Spα [3], P450CLA [4], P450Bsβ F79L/G290F [5], P450Exα [6], CYP152K6 [7] and P450Jα [8] were expressed, purified and the conversions of caproic acid (C6:0), caprylic acid (C8:0) and capric acid (C10:0) were investigated (Figure 1). Biotransformations with P450CLA, P450Spα, P450Exα, and CYP152K6 led to successful conversion giving the corresponding α-hydroxylated fatty acids. The most promising results were obtained with P450Spα whereby C8:0 and C10:0 were very well converted (>99% conv.) and a high percentage of α-hydroxylated products was achieved (for C8:0 and C10:0 89% and 85% α-hydroxylation, respectively). P450Exα, is an excellent candidate for the conversion of C6:0 (95% conv.) and showed high regioselectivity (70% α-hydroxylation). The main side-product was the β-hydroxylated product in the conversion of C8:0 and C10:0. All tested CYP152 are (S)-stereoselective. P450Spα, P450Exα, and CYP152K6 displayed high stereoselectivity for the conversion of C6:0, C8:0 and C10:0. In addition, P450Exα successfully converted the dicarboxylic acids nonane-1,9-dioic acid and decane-1,10-dioic acid exclusively to the corresponding monohydroxylated 2-hydroxynonane-1,9-dioic acid and 2-hydroxydecane-1,10-dioic acid, the latter with >99% conversion. This project has received funding from the European Union’s Horizon 2020 research and innovation program under Marie Skolodowska-Curie grant agreement ID: 956621. |
| References: | [1] Wang, Y.; Lan, D.; Durrani, R.; Hollmann, F., Curr. Opin. Chem. Biol. 2017, 37, 1-9. [2] Shoji, O.; Watanabe, Y., J. Biol. Inorg. Chem. 2014, 19, 529-539. [3] Matsunaga I., Kusunose E., Yano I., Ichihara K., Biochem., Biophys. Res. Commun. 1994, 201, 1554-1560. [4] Girhard M., Schuster S., Dietrich M., Duerre P., Urlacher V. B., Biochem. Biophys. Res. Commun. 2007, 362, 114-119. [5] Fujishiro T., Shoji O., Nagano H., Sugimoto H., Shiro Y., Watanabe Y., J. Biol. Chem. 2011, 286, 29941-9950. [6] Onoda H., Shoji O., Suzuk K., Sugimoto H., Shiro Y. and Watanabe Y., Catal. Sci. Technol. 2018, 8, 434-442. [7] Girvan H. M., Poddar H., McLean K. J., Nelson D. R., Hollywood K. A., Levy C. W., J. Inorg. Biochem. 2018, 188, 18-28. [8] Armbruster J., Steinmassl M., Mueller Bogota C. A., Berg G., Nidetzky B., Dennig A. Chem. - Eur. J. 2020, 26, 15910-15921. |
| Figures: | 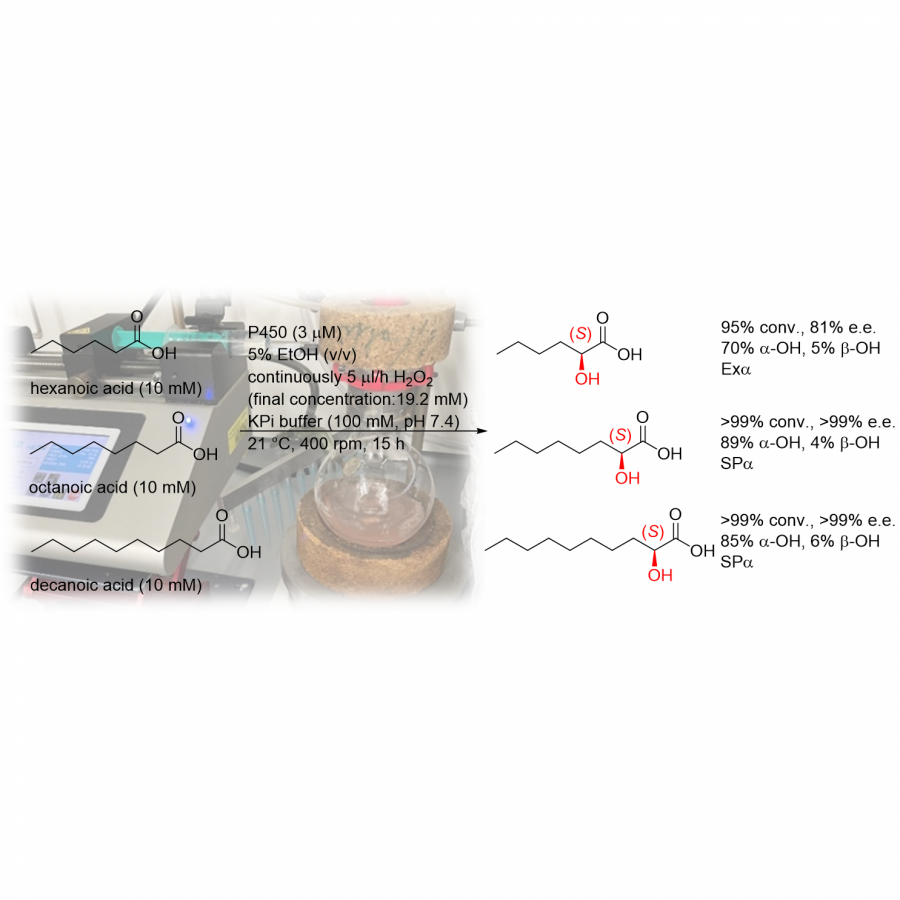 Figure 1. Conversion of C6:0, C8:0 and C10:0 with P450 peroxygenase to α-hydroxy acids Figure 1. Conversion of C6:0, C8:0 and C10:0 with P450 peroxygenase to α-hydroxy acids |
#1068 | The Mechanism of Action of Flavin-Dependent Halogenases |
|
| Presenting author: | Rhys BARKER of UNIVERSITY OF MANCHESTER |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | To rationally engineer the substrate scope and selectivity of flavin-dependent halogenases (FDHs), it is essential to first understand the reaction mechanism and substrate interactions in the active site. FDHs have long been known to achieve regioselectivity through an electrophilic aromatic substitution at C7 of the natural substrate Trp, but the precise role of a key active-site Lys residue remains ambiguous. Formation of hypochlorous acid (HOCl) at the co-factor-binding site is by direct reaction of molecular oxygen and a single chloride ion with reduced FAD and flavin hydroxide, respectively. HOCl is then guided 10 Å into the halogenation active-site. Lys79, located in this site, has been proposed to direct HOCl towards Trp C7 through hydrogen bonding or direct reaction with HOCl to form a -NH2Cl+ intermediate. Here, we present the most likely mechanism for halogenation based on MD simulations and active-site DFT ‘cluster’ models of FDH PrnA in complex with its native substrate L-tryptophan, hypochlorous acid and FAD co-factor. MD simulations with different protonation states for key active-site residues suggest that Lys79 directs HOCl through hydrogen bonding, which is confirmed by calculations of the reaction profiles for both proposed mechanisms. |
| References: | 1. Wolfenden, R.; Snider, M. J., The depth of chemical time and the power of enzymes as catalysts. Acc. Chem. Res. 2001, 34, 938-945. 2. Coin, I.; Beyermann, M.; Bienert, M., Solid-phase peptide synthesis: from standard procedures to the synthesis of difficult sequences. Nat. Protoc. 2007, 2, 3247-3256. 3. V, M., Roche's fuzeon challenge. Chem. Eng. News 2005, 83. 4. Dong, C. J.; Flecks, S.; Unversucht, S.; Haupt, C.; van Pee, K. H.; Naismith, J. H., Tryptophan 7-halogenase (PrnA) structure suggests a mechanism for regioselective chlorination. Science 2005, 309 (5744), 2216-2219. Poor, C. B.; Andorfer, M. C.; Lewis, J. C., Improving the stability and catalyst lifetime of the halogenase RebH by directed evolution. Chembiochem 2014, 15, 1286-1289. 5. Karabencheva-Christova, T. G.; Torras, J.; Mulholland, A. J.; Lodola, A.; Christov, C. Z., Mechanistic insights into the reaction of chlorination of tryptophan catalyzed by tryptophan 7-halogenase. Sci. Rep. 2017, 17395-17410 . 6. Ainsley, J.; Mulholland, A. J.; Black, G. W.; Sparagano, O.; Christov, C. Z.; Karabencheva-Christova, T. G., Structural insights from molecular dynamics simulations of tryptophan 7-halogenase and tryptophan 5-halogenase. ACS Omega 2018, 3, 4847-4859. 7. Yeh, E.; Cole, L. J.; Barr, E. W.; Bollinger, J. M.; Ballou, D. P.; Walsh, C. T., Flavin redox chemistry precedes substrate chlorination during the reaction of the flavin-dependent halogenase RebH. Biochemistry 2006, 45 , 7904-7912. Yeh, E.; Blasiak, L. C.; Koglin, A.; Drennan, C. L.; Walsh, C. T., Chlorination by a long-lived intermediate in the mechanism of flavin-dependent halogenases. Biochemistry 2007, 46, 1284-1292. 8. Barker, R. D.; Yu, Y.; De Maria, L.; Johannissen, L. O.; Scrutton, N. S. Mechanism of Action of Flavin-Dependent Halogenases. ACS Catalysis 2022, 12, 15352-15360. DOI: 10.1021/acscatal.2c05231. |
#1079 | Microorganisms as sources of bio-based functionalization of Polyethylene Glycol (PEG) |
|
| Presenting author: | Jan KHAN of TECHNISCHE UNIVERSITÄT DRESDEN |
| Other authors: | Thomas HEINE of TECHNISCHE UNIVERSITÄT DRESDEN Martin GEISLER of LEIBNIZ INSTITUTE OF POLYMER RESEARCH Anika KAUFMANN of LEIBNIZ INSTITUTE OF POLYMER RESEARCH Marion ANSORGE-SCHUMACHER of TECHNISCHE UNIVERSITÄT DRESDEN Julian THIELE of INSTITUTE OF CHEMISTRY, OTTO VON GUERICKE UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Synthetic polymers such as polyethylene glycol (PEG) are fundamental components of many functional materials. Their uses extend to drug delivery, pharmaceuticals, personal care, research, textiles and other fields. These polymers harbor potential for further improvement and novelty. Novel functionalities and applications of PEG can be achieved through the introduction of functional groups into its structural backbone. However, attempts at chemical synthesis strategies targeting internal functionalization of PEG have failed drastically. Microorganisms, such as bacteria and fungi, hold great potential for the biodegradability and modification of such polymers. Therefore, we propose a bio-based functionalization approach using enzymes from microbial sources as biocatalysts. This could lead the way towards internal functionalization of PEG. Additionally, the biodegradability and environmental sustainability of such polymer could be improved as a by-product of the project. |
#1080 | An enantiocomplementary photoenzyme for [2+2] cycloadditions |
|
| Presenting author: | Rebecca CRAWSHAW of UNIVERSITY OF MANCHESTER |
| Corresponding author: | Anthony GREEN of UNIVERSITY OF MANCHESTER |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photoenzyme / Genetic Code Expansion / Directed Evolution / Enzyme Engineering |
| Purpose: | The ability to program new modes of catalysis into proteins would allow the development of enzyme families with functions beyond those found in nature. To this end, genetic code expansion methodology holds particular promise, as it allows the site-selective introduction of new functional elements into proteins as noncanonical amino acid side chains. Here we exploit an expanded genetic code to develop a photoenzyme that operates by means of triplet energy transfer (EnT) catalysis, a versatile mode of reactivity in organic synthesis that is not accessible to biocatalysis at present. Installation of a genetically encoded photosensitizer into the beta-propeller scaffold of DA_20_00 converts a de novo Diels–Alderase into a photoenzyme for [2+2] cycloadditions (EnT1.0). Subsequent development and implementation of a platform for photoenzyme evolution afforded an efficient and enantioselective enzyme (EnT1.3, up to 99% enantiomeric excess (e.e.)) that can promote intramolecular and bimolecular cycloadditions, including transformations that have proved challenging to achieve selectively with small-molecule catalysts. EnT1.3 performs >300 turnovers and, in contrast to small-molecule photocatalysts, can operate effectively under aerobic conditions and at ambient temperatures. An X-ray crystal structure of an EnT1.3-product complex shows how multiple functional components work in synergy to promote efficient and selective photocatalysis. One limitation of biocatalysis is the lack of mirror-image enzymes for the formation of both enantiomeric products. Placing the genetically encoded BPA photosensitizer in an alternative position in the active site of the initial photoenzyme EnT1.0, gave rise to an enantiocomplimentary enzyme CEnT1.0. Here we develop the enantiocomplimentary enzyme using directed evolution to yield CEnT1.2 which promotes intramolecular [2+2] cycloadditions with an efficiency which rivals EnT1.3. This study opens up a wealth of new excited-state chemistry in protein active sites and establishes the framework for developing a new generation of enantioselective photocatalysts. |
| References: | [1] Trimble, J.S., Crawshaw, R., Hardy, F.J. et al. A designed photoenzyme for enantioselective [2+2] cycloadditions. Nature 611, 709-714 (2022). https://doi.org/10.1038/s41586-022-05335-3 |
| Figures: | 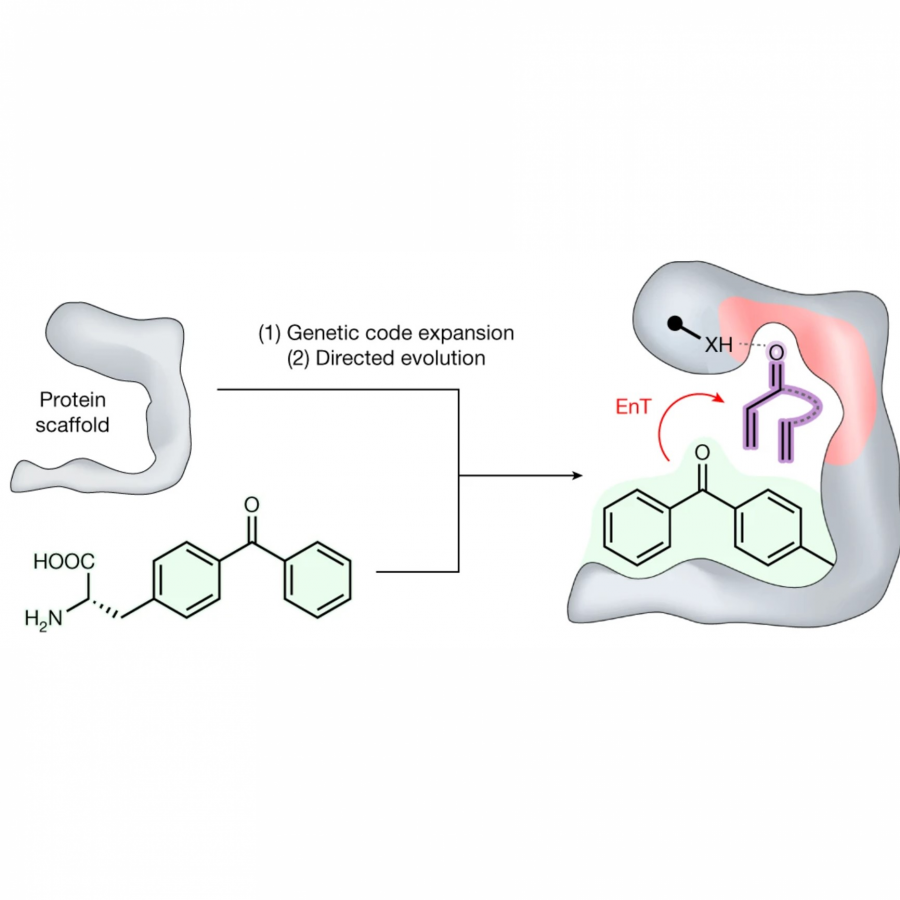 Our approach to photoenzyme development Selective installation of a genetically encoded photosensitizer into a protein scaffold and subsequent optimization of the modified protein by directed evolution. 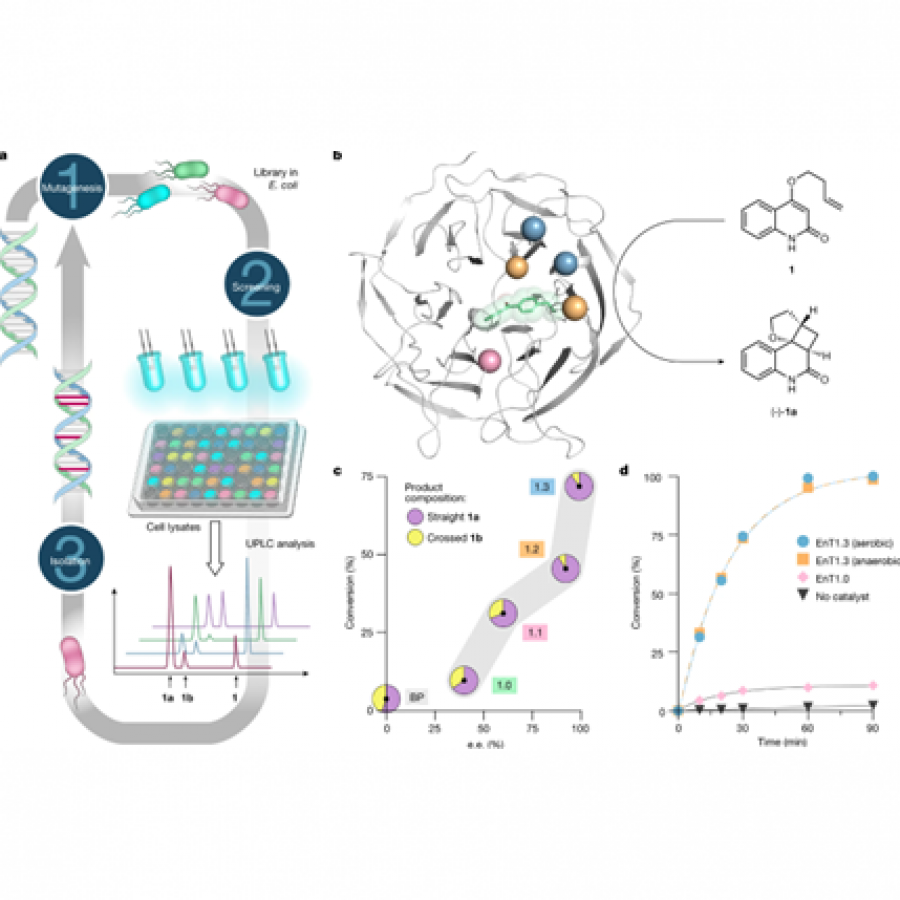 Directed evolution of an efficient and enantioselective photoenzyme. a, Schematic of the directed-evolution workflow for photoenzyme engineering. b, Mutational map of EnT1.3, highlighting the encoded photosensitizer BpA173 (green sticks and semitransparent CPK spheres) and residues installed during alanine scanning (pink s |
#1083 | COMPUTATIONAL INVESTIGATION OF HISTIDINE-ARGININE PAIRS IN ENZYME CATALYZED MORITA-BAYLIS-HILLMAN REACTIONS |
|
| Presenting author: | Ozge DIKMEN of YEDITEPE UNIVERSITY |
| Corresponding author: | Ozge DIKMEN of YEDITEPE UNIVERSITY |
| Other authors: | Melisa AKBULUT of YEDITEPE UNIVERSITY Sidal BULUT of YEDITEPE UNIVERSITY Nihan ÇELEBI of YEDITEPE UNIVERSITY DIKMEN Ozge of YEDITEPE UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Morita-Baylis-Hillman Reaction / Promiscuous Enzymes / Biocatalyst / Enzyme Design |
| Purpose: | The Morita−Baylis−Hillman (MBH) reaction is a C-C coupling reaction in which simple starting materials are converted into functionalized products in a catalytic process. It has numerous applications, especially in the synthesis of pharmaceutically important compounds. Despite the great synthetic potential, the practical application of the MBH reaction is rather limited due to low catalytic efficiencies. Use of biocatalysts could offer a promising solution to these challenges. Recent studies in the use of enzymes in organic synthesis have led to the exploration of promiscuous biocatalytic routes to the MBH reaction. Joshi et al. reported that lysozyme C, myoglobin and a couple of proteins containing a suitably positioned histidine and arginine pair in proximity catalyzed the MBH reaction with notable efficiencies.[1] 14 rounds of directed evolution on a de novo designed MBH enzyme based on a histidine nucleophile has introduced an arginine motif in the active site, suggesting the importance of arginine as an oxyanion hole motif.[2] In this study, the importance of histidine and arginine pairs for the catalytic activity was explored using quantum mechanical theozyme models.[3] Generated catalytic atom maps based on these theozymes were screened in the protein database to identify natural proteins that can present the catalytic amino acids in the three-dimensional arrangement required for optimal catalysis of the MBH reaction.[4] In light of the obtained results, new proteins with potential promiscuous activity towards the MBH reaction were proposed for experimental testing. |
| References: | [1] P. N. Joshi, L. Purushottam, N. K. Das, S. Mukherjee and V. Rai, RSC Adv., 2016, 6, 208-2011. [2] R. Crawshaw, A. E. Crossley, L. Johannissen, A. J. Burke, S. Hay, C. Levy, D. Baker, S. L. Lovelock and A. P. Green, Nat. Chem., 2022, 14, 313-320. [3] T. Ütnier, N. Çelebi-Ölçüm, Catal. Sci. Technol., 2023, 13, 329-341 [4] This research is funded by TÜBİTAK (project no 122Z506). |
#1084 | CO2 valorisation: Multienzymatic synthesis of lactic acid using CO2 |
|
| Presenting author: | Albert CARCELLER of UNIVERSITAT AUTONOMA DE BARCELONA |
| Other authors: | SADY ROBERTO RODRIGUEZ of UNIVERSITAT AUTONOMA DE BARCELONA David MUÑOZ of UNIVERSITAT AUTONOMA DE BARCELONA Oscar Enrique ROMERO of UNIVERSITAT AUTONOMA DE BARCELONA Marina GUILLEN of UNIVERSITAT AUTONOMA DE BARCELONA Gregorio ALVARO of UNIVERSITAT AUTONOMA DE BARCELONA Gloria CAMINAL of UNIVERSITAT AUTONOMA DE BARCELONA Gloria GONZALEZ of UNIVERSITAT AUTONOMA DE BARCELONA |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | multienzymatic system / cofactor regeneration / Carbon dioxide / pyruvate decarboxylase |
| Purpose: | Carbon dioxide has the highest residence time in the atmosphere, making it the most impactful greenhouse gas to the global warming. For that reason, efforts should be focused on reducing its emissions. The EU has set the objective to reduce CO2 emissions in the EU by 80% in 2050. Technological innovations are needed to reach this goal, leading to successful CCU (Carbon Capture and Utilization) technologies. The CO2 molecule is very stable, due to the highest valence state of carbon (+4), with a C=O bond breakage energy of 749 kJ mol-1. Hence, it requires high amounts of energy to transform it into added-value compounds. Biocatalysis presents a green approach for the development of CCU strategies, because the use of enzymes complies with 10 of the 12 principles of green chemistry. They can catalyze reactions under mild conditions of pressure, temperature and pH. A number of enzymatic systems for utilizing CO2 have already been reported in the literature. For example, formate dehydrogenase (FDH) catalyzes the transformation of CO2 into formic acid. But most of them are single-enzyme systems, which limits the variability of products that can be obtained. In addition, the majority require cofactors such as NADH/NAD+ for catalysis. Therefore, multi-enzymatic systems can tackle these limitations by widening the range of products obtained and allowing regeneration of cofactor in-situ. In this study, a multi-enzyme system for the synthesis of lactic acid using CO2 and ethanol, with a 100% atom economy, was studied. The system involves three enzymes: alcohol dehydrogenase which oxidizes ethanol into acetaldehyde consuming one NAD+ molecule, pyruvate decarboxylase which carboxylates acetaldehyde into pyruvic acid using a CO2 molecule, and lactate dehydrogenase which reduces pyruvic acid into lactic acid, using one NADH molecule, and regenerating the NAD+ needed for the first reaction. Firstly, each reaction was studied individually, revealing that PDC catalyzes an undesired secondary reaction which is highly favored. Therefore, two PDCs were studied to find which one produces less byproduct. Finally, by means of reaction medium engineering and enzyme ratio optimization, the lactic acid yield of the system could be increased, reaching 250 μM under the best conditions. So far, this is the highest concentration of lactic acid achieved using this multi-enzymatic system. |
| References: | [1] tong, x. biotech. and bioeng. 2011, 108, 465-469 |
| Figures: |  Multienzymatic synthesis of Lactic Acid ADH: Alcohol dehydrogenase
PDC: Pyruvate decarboxylase
LDH: Lactate dehydrogenase |
#1095 | Plastics to Surfactants |
|
| Presenting author: | Ross SMITHSON of UNIVERSITY OF MANCHESTER |
| Corresponding author: | Anthony GREEN of UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Directed Evolution / Up-cycling / / |
| Purpose: | The recent discovery of a hydrolytic enzyme, IsPETase, that can deconstruct poly(ethylene) terephthalate (PET), has sparked great interest in biocatalytic approaches to recycle plastics. Realisation of commercial utility will require the development of robust engineered enzymes that meet the demands of industrial processes. Recently, we have reported development and implementation of an automated, high-throughput directed evolution platform for engineering polymer degrading enzymes. Evaluation of >13,000 IsPETase variants, applying catalytic activity at elevated temperatures as a primary selection pressure, afforded a HotPETase variant with 21 mutations that has a melting temperature of 82.5°C and can therefore operate near or above the glass transition temperature of PET (60-70°C). HotPETase can depolymerise semi-crystalline PET more rapidly than previously reported PETases and can selectively deconstruct the PET component of a laminated packaging multi-material. This study established laboratory evolution as a platform to engineer useful plastic degrading enzymes to underpin biocatalytic plastic recycling processes, which we now want to apply to different plastics. We now aim to apply this high-throughput platform to alternative plastics and to up-cycle the degradation products to produce new anionic surfactants. |
#1097 | Engineering Glycerol Dehydratase Variants for Improved Resistance to Inactivation |
|
| Presenting author: | Abdul NASIR of SECOND AFFILIATED HOSPITAL OF ZHENGZHOU UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme engineering / Coenzyme B12 / Glycerol / Bioconversion |
| Purpose: | Glycerol is a byproduct in the production of biodiesels, and biological processes converting glycerol into chemicals have attracted much attention nowadays. Some of them are based on the dehydration reaction of glycerol into 3-hydroxypropanal catalyzed by glycerol dehydratase (GDHt), and the intermediate can be oxidized or reduced into 3-hydroxypropionic acid or 1,3-propanediol, respectively. These two products can be reacted further into useful chemicals. Even though GDHt has been used for developing many recombinant microorganisms to utilize glycerol, the enzyme has a critical limitation for wider applications. The cofactor coenzyme B12 is susceptible to chemical modification during the reaction or by oxygen molecules, which results in the inactivation of the enzyme. In this presentation, we will introduce our strategy to engineer GDHt variants more resistant to inactivation than the wild-type enzyme. Some of the designed variants have shown improved resistance to inactivation, and their characterization results will be described. Our engineered GDHt variants have the potential to overcome the limitation of the wild-type enzyme and accelerate the development of efficient and cost-effective industrial biological processes to convert glycerol into valuable biochemicals. |
#1103 | In Vitro one-opt production of 3-hydroxypropanal from C1 and C2 chemicals using two biocatalysts |
|
| Presenting author: | Min-Ju SEO of CHONNAM NATIONAL UNIVERSITY |
| Other authors: | Yeon-Ju JEONG of CHONNAM NATIONAL UNIVERSITY Soo-Jin YEOM of CHONNAM NATIONAL UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | deoxyribose-5-phosphate aldolase / methanol dehydrogenase / 3-hydroxypropanal / cascade enzymatic bioconversion |
| Purpose: | One- or two-carbon compounds are attractive substrates, which have been used for biotechnological production of value-added products such as 3-hydroxypropanal (3-HPA), 3-hydroxypropionic acid (3-HA), and 1,3-propanediol (3-PDO) that are used as starting materials to produce biocompatible plastic and polytrimethylene terephthalate. Methanol-derived formaldehyde (C1) and ethanol-derived acetaldehyde (C2) can be converted to 3-HPA (C3) via aldol condensation, which can also be utilized for 3-HA and 3-PDO synthesis. In this study, we optimized the reaction conditions for 3-HPA production from formaldehyde and acetaldehyde using deoxyribose-5-phosphate aldolase from Thermotoga maritima (DERATma), which showed the maximal activity at pH 7.0 and 40 °C. Under these optimized conditions, DERATma produced 7 mM 3-HPA from 25 mM formaldehyde and acetaldehyde for 60 min with a productivity of 520 mg/L/h. To investigate the one-pot production of 3-HPA from methanol and ethanol, we used methanol dehydrogenase from Lysinibacillus xylanilyticus (MDHLx), which converts methanol and ethanol to formaldehyde and acetaldehyde, respectively. One-pot cascade reaction with MDHLx and DERATma was performed under optimized reaction conditions to produce 3-HPA from alcohol-derived compounds (formaldehyde and acetaldehyde). To best of our knowledge, this is the first report on 3-HPA production from inexpensive alcohol substrates (methanol and ethanol). This study supported from National Research Foundation grant (NRF-2022R1C1C2003774, NRF-2022M3J5A1056169, NRF-2022M3J5A1085239; and Projects 1711195195 and RS-2023-00208002) funded by the Korean Ministry of Science and ICT. |
| Figures: |  Scheme of the synthetic pathway of 3-hydroxypropanal from methanol (C1) and ethanol (C2) using two biocatalysts 3-Hydroxypropanal can be produced from methanol-derived formaldehyde and ethanol-derived acataldehyde with DERA from Thermotoga maritima and MDH from Lysinibacillus xylanilyticus. |
#1104 | Discovery of polystyrene biodegradable bacteria from the soil and its application for the polystyrene biodecomposition |
|
| Presenting author: | Min-Ju SEO of CHONNAM NATIONAL UNIVERSITY |
| Other authors: | Hyun-Woo KIM of CHONNAM NATIONAL UNIVERSITY Soo-Jin YEOM of CHONNAM NATIONAL UNIVERSITY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Polystyrene / Biodegradation / Pseudomonas sp. / Synthetic plastic eating bacteria |
| Purpose: | Polystyrene (PS), the sixth type of plastics, is difficult to biodegrade due to its chemical structure, which has phenyl moieties attached to linear alkanes. We isolated a putative PS eating bacteria (Pseudomonas sp.) from the soil, which can biodegrade PS. To investigate biodegradation of PS, isolated Pseudomonas sp. was incubated in non-carbonaceous nutrient medium (M9 medium) with PS as the only carbon source. Growth rate of isolated Pseudomonas sp. increased with increasing the concentration of PS. Biodegradation of PS by isolated Pseudomonas sp. was conducted by fourier transform infrared spectroscopy (FT-IR), scanning electron microscope analysis (SEM), and water contact angle measurement (WCA) to confirm the PS chemical structure changes, biodegradation of PS-film, and increasing hydrophilicity of PS-film, respectively. These results provide significant insights into the discovery of a new type of Pseudomonas sp. for the biodegradation of PS, as well as its potential as PS eating bacteria. This study supported from National Research Foundation grant (NRF-2022R1C1C2003774, NRF-2022M3J5A1056169, NRF-2022M3J5A1085239; and Projects 1711195195 and RS-2023-00208002) funded by the Korean Ministry of Science and ICT. |
| Figures: |  Process of screening samples and Phylogenetic tree The screening process of PS-biodegrading microorganisms from soil (left) and Based on 16S rRNA gene sequences, showing bacterial populations present in Pseudomonas sp. JNU01 (rignt) |
#1107 | A microfluidic 3D-printed platform for resource efficient screening of immobilized unspecific peroxygenase variants |
|
| Presenting author: | Elena GKANTZOU of LEIBNIZ UNIVERSITY HANNOVER |
| Corresponding author: | Selin KARA of LEIBNIZ UNIVERSITY HANNOVER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | microfluidics / 3D printing / unspecific peroxygenases / continuous flow biocatalysis |
| Purpose: | Additive manufacturing, referring to tailor-made designs and fabrication techniques, has been claimed a game-changer in the construction of reactors and peripheral units in the field of flow chemistry[1]. 3D-printed modules have been elaborated in all stages of a continuous flow biocatalysis system[2], either (i) the reactor itself, (ii) the support material for biocatalyst confinement, or (iii) the peripheral accessories that can establish a highly controlled process[3]. Fungal unspecific peroxygenases (UPOs, EC 1.11.2.1) are heme-thiolate enzymes, displaying characteristic peroxidase activity and a unique peroxygenase activity[4]. UPOs are versatile biocatalysts with great importance in synthetic chemistry for an ensemble of highly selective C-H oxyfunctionalizations, while engineered UPO variants are currently under intensive study[5, 6]. In this concept, we are investigating the immobilization of UPOs on 3D-printed microfluidic reactors under the scope to facilitate screening processes of different enzyme variants. The microchannels are surface functionalized, and UPOs are confined in the reactor walls with a covalent immobilization method. Process parameters like (i) reactor geometry, (ii) flow rate, (iii) enzyme amount, and (iv) substrates’ concentration are investigated. The optimized configuration is elaborated for resource efficient screening of an engineered biocatalyst panel towards enzymes’ activity and operational stability. |
| References: | [1]De Santis, P., L.-E. Meyer and S. Kara, React. Chem. Eng., 2020, 5, 2155-2184 [2]Gomez de Santos, P., I. Mateljak, M. D. Hoang, S. J. Fleishman, F. Hollmann and M. Alcalde,J Am Chem Soc., 2023, 145, 3443-3453 [3]Hobisch, M., D. Holtmann, P. Gomez de Santos, M. Alcalde, F. Hollmann and S. Kara, Biotechnol Adv, 2021, 51, 107615 [4]Martin-Diaz, J., P. Molina-Espeja, M. Hofrichter, F. Hollmann and M. Alcalde, Biotechnol Bioeng, 2021, 118, 3002-3014 [5]Sans, V., Curr. Opin. Green Sustain. Chem., 2020, 25, 100367 [6]Zentel, K. M., M. Fassbender, W. Pauer and G. A. Luinstra, Adv. Chem. Eng., 2020, 56, 1, 97-137 |
| Figures: |  A microfluidic 3D-printed platform for efficient screening of immobilized UPO variants A 3D-printed microfluidic platform consists of different parts, for each step of the process. UPO enzyme is positioned in the main reaction chamber with a covalent immobilization procedure after surface functionalization of the microreactor walls. |
#1113 | Biocatalytic C–H Functionalization to Construct C–N Bonds via Nitrene Transfer |
|
| Presenting author: | Ziyang QIN of CALIFORNIA INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalytic C-H functionalization / protein engineering / propargyl and alpha-tertiary amines / N-heterocycles |
| Purpose: | Over the past decade, the Arnold laboratory has ventured beyond the native oxidation activities of cytochrome P450 monooxygenases to develop an array of non-natural functions based on transfer of nitrene-like intermediates, which paves the way for the efficient and selective biocatalytic construction of C–N bonds. To expand the repertoire of biocatalytic C–H amination via nitrene transfer, the group has pursued two main avenues: (1) introducing new nitrene precursors (nitrene donors) and (2) targeting various C–H bonds (nitrene acceptors). Within these two categories, we present the development of engineered cytochrome P411 enzymes (P450 monooxygenases with serine as the heme-ligating residue) for a diverse set of C–N bond-forming reactions. We will describe an efficient, selective, and scalable enzymatic platform for the enantioselective propargylic primary amination of alkynes using hydroxylamine derivatives as the nitrene precursor. Furthermore, we identified cytochrome P411 enzymes capable of catalyzing enantioenriched tertiary C–H primary amination, addressing a notorious challenge in chemical synthesis. Additionally, we developed a biocatalytic platform for enantioselective intramolecular C(sp3)–H amination to form chiral N-heterocycles using alkyl and aryl azides as nitrene precursors. These enzymatic aminations can be coupled with a P411-based carbene transferase or a tryptophan synthase to generate an α-amino lactone or a non-canonical amino acid, respectively, underscoring the power of new-to-nature biocatalysis in complexity-building chemical synthesis. |
| References: | [1] Qin, Z.-Y. †; Gao, S. †; Zou, Y.; Liu, Z.; Wang, J.; Houk, K. N.; Arnold, F. H. Biocatalytic Construction of Chiral Pyrrolidines and Indolines via Intramolecular C(sp3)-H Amination. Under Revision. [2] Liu, Z. †; Qin, Z.-Y. †; Zhu, L. D.; Athavale, S. V.; Sengupta, A.; Jia, Z.-J.; Garcia-Borràs, M.; Houk, K. N.; Arnold, F. H. An Enzymatic Platform for Primary Amination of 1-Aryl-2-alkyl Alkynes. J. Am. Chem. Soc. 2022, 144, 80- 85. |
| Figures: | 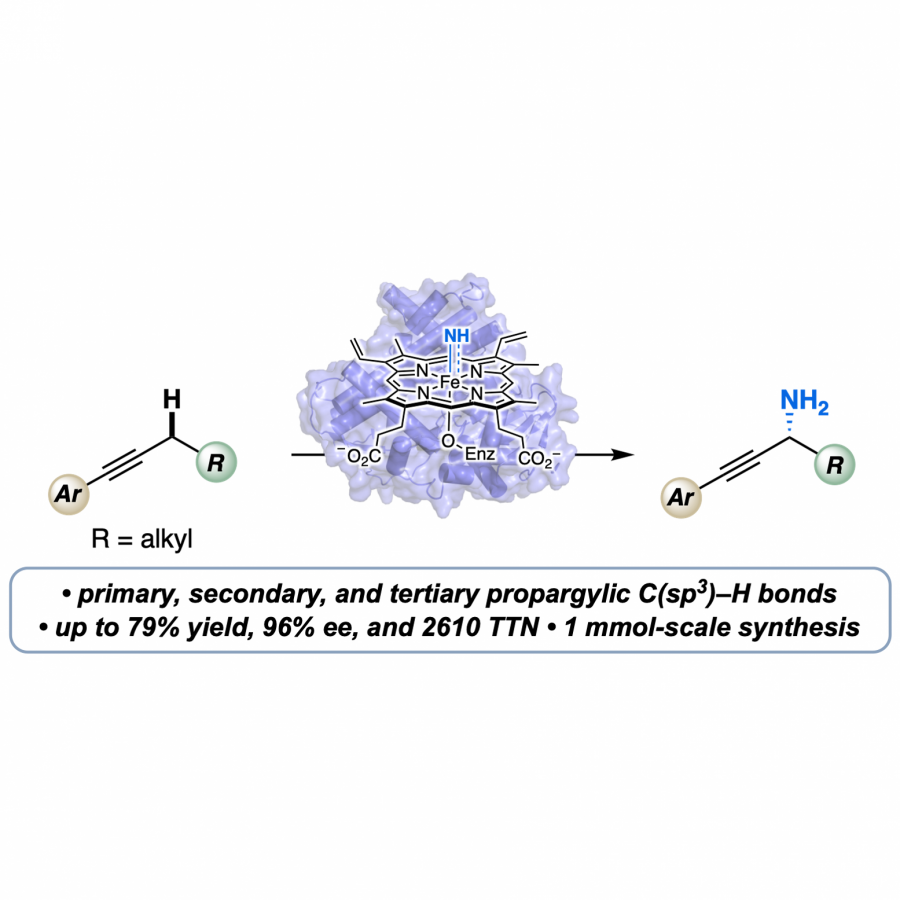 Enantioselective propargylic C-H amination An efficient, selective, and scalable enzymatic platform for the enantioselective propargylic primary amination of alkynes using hydroxylamine derivatives as the nitrene precursor. |
#1114 | A new binding mode expands the substrate scope of Aryl Malonate Decarboxylase (AMDase) |
|
| Presenting author: | Elske VAN DER POL of GRAZ UNIVERSITY OF TECHNOLOGY |
| Corresponding author: | Robert KOURIST of GRAZ UNIVERSITY OF TECHNOLOGY |
| Other authors: | Rolf BREINBAUER of INSTITUTE OF ORGANIC CHEMISTRY Thomas SCHLATZER of INSTITUTE OF ORGANIC CHEMISTRY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Mechanism / Probe / |
| Purpose: | Precise control of enantioselectivity is a critical feature of biocatalysts for many industrial applications. Bacterial aryl malonate (AMDase) is a cofactor-free enzyme that generates a wide spectrum of optically pure carboxylic acids, including several with anti-inflammatory activity such as flurbiprofen. AMDase cleaves the pro-R carboxylate of arylaliphatic malonic acids, followed by stereoselective protonation of a planar intermediate by a catalytic cysteine giving rise to aryl methyl and vinyl methyl carboxylic acids in high optically purity. Wildtype AMDase with C188 produces the pure (R)-products under inversion of the stereocenter. AMDase (V43I/G74C/A125P/V156L/M159L/C188G) (CGLIPL) has the catalytic cysteine allocated at the other side of the planar intermediate and gives rise to the (S)-products under retention of the configuration. The substrate scope of AMDase is limited to substrates bearing an alpha-substitutent not larger than a methyl group, presumably due to the steric hindrance in a small hydrophobic pocket in the active site of the decarboxylase. It was an unexpected finding that AMDase CGLIPL decarboxylated 2-methyl-2-vinyl malonic acid with low selectivity, giving rise to the final (S)-2-methyl-but-3-enoic acid with 66% ee [1]. As a possible explanation, an inverse binding mode resulting in the cleavage of the pro-S carboxylate presented itself, Scheme 1. We assumed that this binding mode should also allow the synthesis of carboxylic acids with larger alpha-substitutents, which would be a significant expansion of the substrate scope of the enzyme. Here we probe the activity of the enigmatic AMDase CGLIPL variant. Using a newly synthesized pro-R-13C-labelled vinyl methyl malonate, we identified the leaving group (pro-R (13C) or pro-S carboxylic acid). The analysis of the stereochemical pathways indicated that the protonation after decarboxylation of 2-methyl-2-vinyl malonate in two different binding modes proceeds exclusively under inversion of the stereocenter. Surprisingly, retention of the stereochemistry was not observed. We will show an analysis of the stereochemical pathways of AMDase and discuss mechanistic consequences. Furthermore, we will show how this novel binding mode unlocks the conversion of ethyl-substituted substrates with outstanding selectivity. |
| References: | [1] J. Enoki, C. Megge, D Tischler, K. Miyamoto, and R. Kourist, Chem. Eur. J. 2019, 25, 5071 - 5076 |
| Figures: | 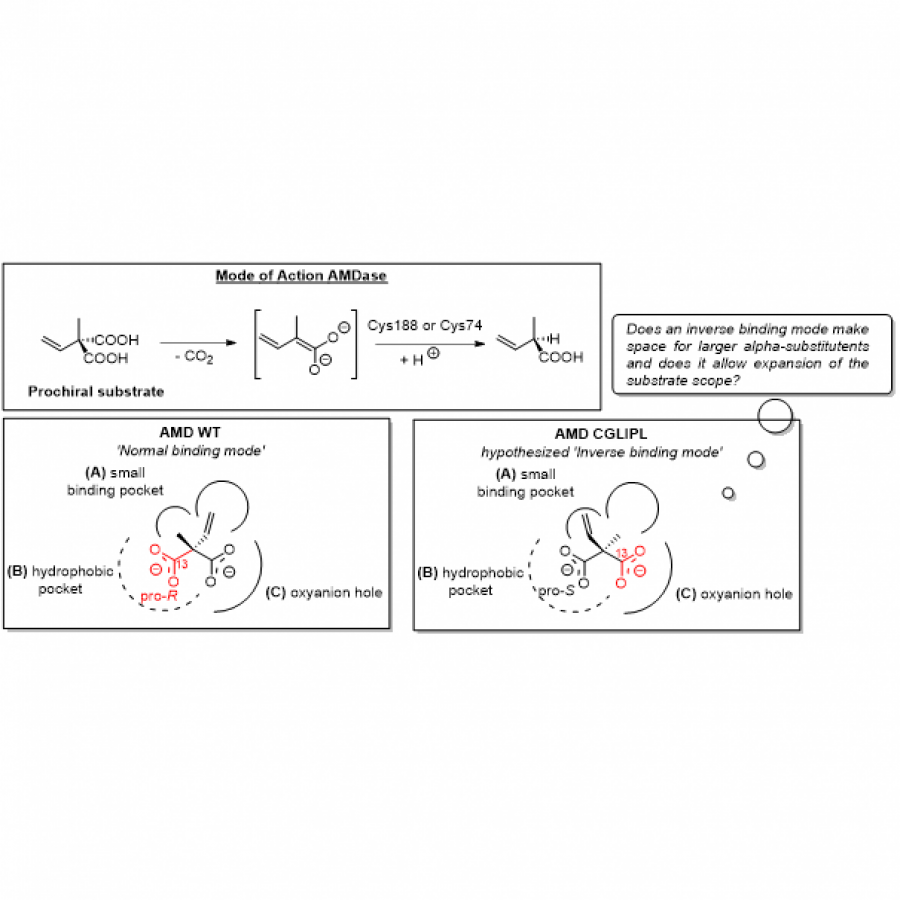 AMD binding modes Scheme 1. Mode of action of AMDase, and binding of 2-methyl-2-vinyl malonate in AMD WT and CGLIPL variant. |
#1115 | Crystal Structure and Expanded Substrate Spectrum of the α-keto acid C-Methyltransferases SgvM and MrsA |
|
| Presenting author: | Juliane BREILTGENS of UNIVERSITY FREIBURG |
| Corresponding author: | Michael MÜLLER of UNIVERSITY FREIBURG |
| Other authors: | Ziruo ZOU of UNIVERSITY FREIBURG Christina SOMMER-KAMANN of UNIVERSITY FREIBURG Florian KEMPER of UNIVERSITY FREIBURG Oliver EINSLE of UNIVERSITY FREIBURG Jennifer ANDEXER of UNIVERSITY FREIBURG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | C-methyltransferase / α-keto acid / crystal structure / site-directed mutagenesis |
| Purpose: | S-adenosylmethionine (SAM)-dependent methyltransferases (MTs) are involved in the C-methylation of a variety of natural products such as DNA, proteins, or small molecules.[1] Enzymatic C-methylation requires activation of the substrate’s carbon atom by an adjacent functional group to form a nucleophilic intermediate carbanion and to enable nucleophilic attack on the methyl residue of SAM. A few examples of C-MTs have been described that methylate enolizable β-branched α-keto acids, leading to precursors of non-proteinogenic amino acids.[2] The MTs SgvM from Streptomyces griseoviridis and MrsA from Pseudomonas syringae pv. syringae catalyze the methylation of the β-carbon atom of α-keto acids in the biosynthesis of the antibiotic natural products viridogrisein and 3-methylargenine, respectively.[3] MrsA shows high substrate selectivity for its native substrate 5-guanidino-2-oxovalerate, while other α-keto acids, such as the SgvM substrates 4-methyl-2-oxovalerate, 2-oxovalerate, and phenylpyruvate, were not accepted. Here, we report on the crystal structures of SgvM and MrsA in the apo form, in complex with their substrates, SAM, and methyladenosine, a degradation product of SAM, respectively. By investigating key substrate recognition residues in the active site of both enzymes and through site-directed mutagenesis, the substrate spectrum of MrsA was extended to accept the α-keto acid substrates of SgvM with uncharged and lipophilic β-residues. Our results showcase the possibility to transfer the substrate promiscuity of α-keto acid MTs from distinct biosynthetic pathways by rational design. |
| References: | [1] a) A.-W. Struck, M. L. Thompson, L. S. Wong, J. Micklefield, ChemBioChem 2012, 13, 2642; b) H. L. Schubert, R. M. Blumenthal, X. Cheng, Trends Biochem. Sci. 2003, 28, 329. [2] a) A. W. Schultz, D.-C. Oh, J. R. Carney, R. T. Williamson, D. W. Udwary, P. R. Jensen, S. J. Gould, W. Fenical, B. S. Moore, J. Am. Chem. Soc. 2008, 130, 4507; b) J. Feng, J. Wu, J. Gao, Z. Xia, Z. Deng, X. He, Appl. Environ. Microbiol. 2014, 80, 5021; c) C. Mahlert, F. Kopp, J. Thirlway, J. Micklefield, M. A. Marahiel, J. Am. Chem. Soc. 2007, 129, 12011. [3] a) S. D. Braun, J. Hofmann, A. Wensing, M. S. Ullrich, H. Weingart, B. Völksch, D. Spiteller, Appl. Environ. Microbiol. 2010, 76, 2500; b) Y. Xie, B. Wang, J. Liu, J. Zhou, J. Ma, H. Huang, J. Ju, ChemBioChem 2012, 13, 2745. |
| Figures: | 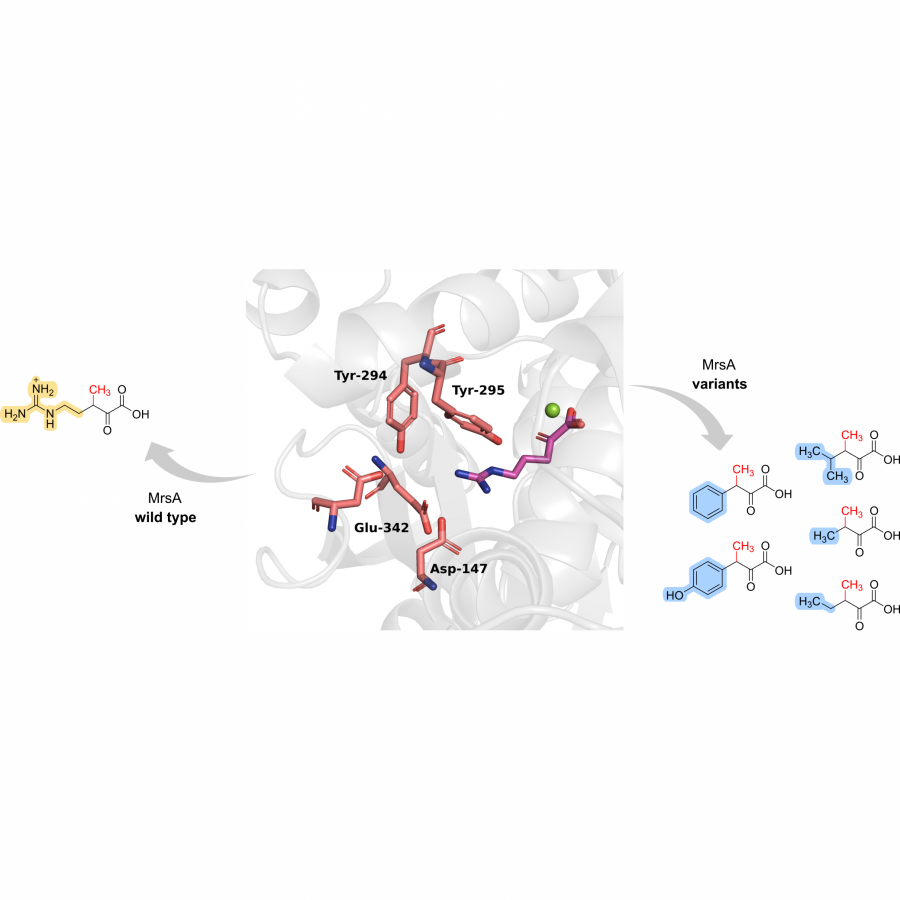 Expanded Substrate Spectrum of MrsA Active site of MrsA wild type in complex with its native substrate 5-guanidino-2-oxovalerate (dark grey) and Mg2+ ion (green). The residues of the mutation sites for MrsA variants are highlighted in red. |
#1117 | Exploiting ene-reductases promiscuous bioreduction of oximes to access tetrasubstituted pyrazines and aminoalcohols |
|
| Presenting author: | Francesco MASCIA of AUSTRIAN CENTRE OF INDUSTRIAL BIOTECHNOLOGY |
| Corresponding author: | Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Other authors: | Willem BREUKELAAR of UNIVERSITY OF GRAZ Stefan VELIKOGNE of UNIVERSITY OF GRAZ Nakia POLIDORI of UNIVERSITY OF GRAZ Amit SINGH of UNIVERSITY OF GRAZ Karl GRUBER of UNIVERSITY OF GRAZ Silvia GLUECK of AUSTRIAN CENTRE OF INDUSTRIAL BIOTECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ene-reductases / promiscuous activity / oxime reduction / pyrazines |
| Purpose: | Promiscuous enzymatic activities recently gained much attention for contributing to the expansion of the repertoire of biocatalytic transformations, including new-to-nature reactions [1]. Ene-reductases (EREDs) from old yellow enzyme family are well known for their capacity to reduce C=C bonds [2]. Recently, we identified a promiscuous activity of ene-reductases showing that these enzymes reduce oximes like α-oximo β-keto esters to their corresponding amines. In this reaction, α-oximo β-keto esters were reduced to the corresponding α-amino intermediate, which then spontaneously dimerized and oxidized, eventually leading to pyrazine products [3]. In this work, a library of eight α-oximo β-keto esters was tested with six different ene-reductases, showing that all the substrates could be converted to the corresponding amines, with good product formation and isolated yields. Furthermore, to prove the presence of a reactive amine intermediate, a cascade reaction was set up, where an ADH acting on the intermediate but not on the substrate was selected, which led to the formation of chiral amino-alcohols [4]. |
| References: | [1] S. Athavale et al., J. Am. Chem. Soc. 2022, 144(41), 19097-19105 [2] C. K. Winkler et al., Curr. Opin. Chem. Biol. 2018, 43, 97-105 [3] S. Velikogne et al., ACS Catal. 2020, 10(22), 13377-13382 [4] W. Breukelaar et al., ACS Catal. 2023, 13(4), 2610-2618 |
| Figures: | 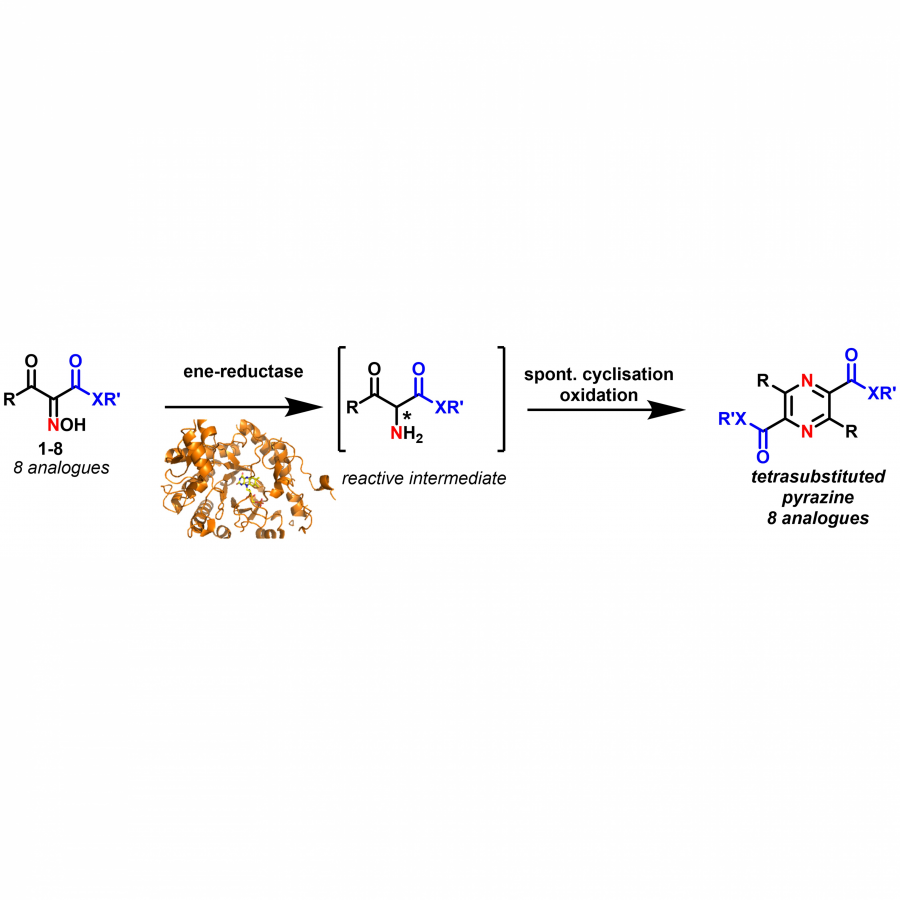 Enzymatic oxime reduction Reaction scheme of the ene-reductase catalysed oxime reduction to an amine intermediate, followed by the spontaneous oxidation and cyclisation leading to the tetrasubstituted pyrazines. |
#1119 | New features of the biocatalyst transketolase from Geobacillus stearothermophilus emerging from biochemical and structural analyses |
|
| Presenting author: | Camilla LEOGRANDE of GEORG AUGUST UNIVERSITÄT GÖTTINGEN |
| Other authors: | Fabian RABE VON PAPPENHEIM of GEORG AUGUST UNIVERSITÄT GÖTTINGEN Laurence HECQUET of UNIVERSITÉ CLERMONT AUVERGNE Wolf-Dieter FESSNER of TECHNISCHE UNIVERSITÄT DARMSTADT Kai TITTMANN of GEORG AUGUST UNIVERSITÄT GÖTTINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transketolase / thermostability / tetramerization / X-ray crystallography |
| Purpose: | Transketolases are enzymes with a high biocatalytic potential: they are able to transfer a chemical moiety from a ketose donor to an aldose acceptor thereby creating a new carbon-carbon bond. The transketolase from the thermophilic organism Geobacillus stearothermophilus (gstTK) has unique stability properties that make it particularly suitable for industrial applications [1]. Therefore, it has been elected to undergo extensive directed-evolution efforts for accepting non-natural substrates. Here, we report on the structural analysis of gstTK. We present the first experimental structure of gstTK solved by X-ray crystallography (PDB code 8CIP). The gstTK crystal was assigned to space group P1 with two homodimers in the asymmetric unit, a feature only shared with the sole other thermophilic TK released to date (ttTK from Thermus Thermophilus, PDB code 7WRR) and, interestingly, with the TK from Mycobacterium tubercolosis (mtTK, PDB code 3 RIM). While in vitro assays on mtTK relegated the tetramerization to a mere crystallization artifact, contrary evidence exists for ttTK. It was postulated that a higher oligomerization state could be a strategy adopted by the enzyme to cope with higher temperatures [2]. We here prove the existence of a gstTK tetramer in solution with several independent biochemical assays, using the well-studied mesophilic orthologue from Escherichia coli (ecTK) as comparison. Our support to the above-mentioned hypothesis could enable the engineering of new thermostable catalysts in the future. Acknowledgements: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 956631. |
| References: | [1] j. abdoul-zabar, i. sorel, v. helaine, f. charmantray, t. devamani, d. yi, v. de berardinis, d. louis, p. marliere, w.-d.fessner, l. hecquet, adv. synth. catal. 2012, 355(1), 116-128. [2] a. yoshihara, y. takamatsu, s. mochizuki, h. yoshida, r. masui, k. izumori, s. kamitori, appl. microbiol. biotechnol. 2023, 107(1), 233-245. |
| Figures: | 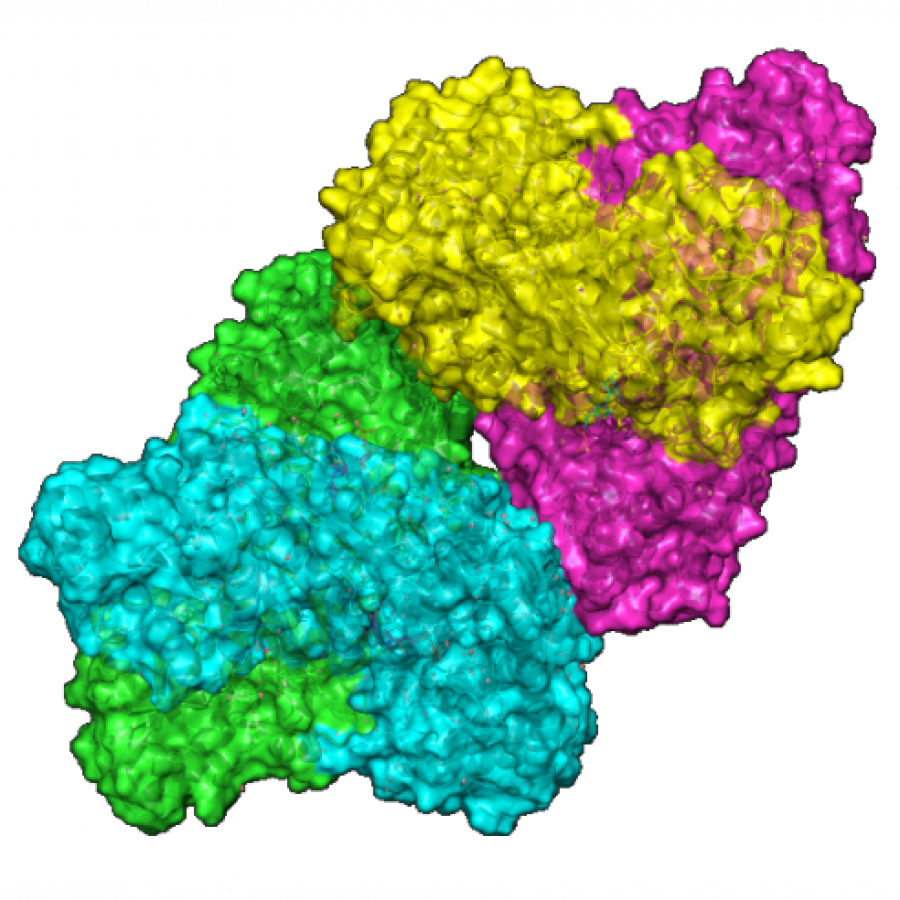 PDB 8CIP The gstTK tetramer as appears in the crystallographic asymmetric unit. |
#1121 | Disulfide engineering of tryptophan halogenases |
|
| Presenting author: | Caroline BESSE of BIELEFELD UNIVERSITY |
| Other authors: | Norbert SEWALD of BIELEFELD UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Disulfide Engineering / / Halogenase / |
| Purpose: | Flavin-dependent halogenases allow halogenation of electron-rich aromatic compounds under mild reaction conditions. Unlike chemical halogenation, flavin-dependent halogenases are highly regioselective even at electronically unfavoured positions. Flavin-dependent halogenases comprise several tryptophan halogenases that can halogenate positions 5, 6 or 7 of tryptophan. The resulting haloarenes are of interest to the pharmaceutical and agricultural industries and are also suitable for further functionalization, such as cross-coupling reactions. Although enzymatic halogenation was shown to provide gram amounts of halogenated tryptophan [1], improvement of the enzyme performance is still desirable. Previous reports showed that several tryptophan halogenases, like PyrH, RebH and PrnA tend to form dimers in solution and in crystal structure. The tryptophan-6-halogenase Thal, while forming dimers in all published crystal structures, was found to exist as a monomer in solution. ESI-MS analysis of Thal and thermostable Thal variants have shown that the thermostable Thal variants have a higher affinity for homodimer formation than the wild type.[2] Inspired by these results, the influence of dimerization on thermostability and activity was investigated using disulfide engineering. To generate a covalently bridged Thal dimer, two amino acids at the prospective dimer interface were mutated to cysteines to enable formation of disulfide bridges. Using size exclusion chromatography, SDS gel electrophoresis and mass spectrometric analysis under non-reducing conditions, it was demonstrated that the generated Thal variant exists as a disulfide-bridged dimer in solution. While the variant shows a significantly increased T50 value compared to the wildtype under non-reduced conditions, the T50 value under reduced conditions is decreased compared to the wild type. So, it could be shown that the dimerization is the driving force for the increased thermostability. The activity was not affected by the mutations and dimerization. To investigate whether a similar effect can be achieved with other halogenases, homologous mutations were introduced into the tryptophan-5-halogenase PyrH. Since these resulted in significantly increased thermostability, further mutants were generated that form covalent dimers and exhibit increased thermostability compared to the PyrH wildtype. The generated variants are a good starting point for further enzyme engineering campaigns to increase activity or expand the substrate spectrum. |
| References: | [1] M. Frese; N. Sewald. Angew. Chem. Int. Ed. 2015, 54, 298-301. [2] H. Minges; C. Schnepel; D. Böttcher; M. S. Weiß; J. Sproß; U. T. Bornscheuer; N. Sewald, ChemCatChem 2020, 12, 818. |
#1123 | Mono- and biphasic solvent systems for the enzymatic synthesis of tyrosol fructoside: Investigation of deep eutectic solvents and organic solvents |
|
| Presenting author: | Klaudia KARKESZOVÁ of DEPARTMENT OF CHEMICAL AND BIOCHEMICAL ENGINEERING, FACULTY OF CHEMICAL AND FOOD TECHNOLOGY, INSTITUTE OF CHEMICAL AND ENVIRONMENTAL ENGINEERING, SLOVAK UNIVERSITY OF TECHNOLOGY |
| Other authors: | Monika ANTOŠOVÁ of DEPARTMENT OF CHEMICAL AND BIOCHEMICAL ENGINEERING, FACULTY OF CHEMICAL AND FOOD TECHNOLOGY, INSTITUTE OF CHEMICAL AND ENVIRONMENTAL ENGINEERING, SLOVAK UNIVERSITY OF TECHNOLOGY Elena KARNIŠOVÁ POTOCKÁ of INSTITUTE OF CHEMISTRY, SLOVAK ACADEMY OF SCIENCES Vladimír MASTIHUBA of INSTITUTE OF CHEMISTRY, SLOVAK ACADEMY OF SCIENCES Milan POLAKOVIČ of DEPARTMENT OF CHEMICAL AND BIOCHEMICAL ENGINEERING, FACULTY OF CHEMICAL AND FOOD TECHNOLOGY, INSTITUTE OF CHEMICAL AND ENVIRONMENTAL ENGINEERING, SLOVAK UNIVERSITY OF TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | tyrosol fructoside / Saccharomyces cerevisiae β-fructofuranosidase / deep eutectic solvents / aqueous-organic solvent systems |
| Purpose: | Tyrosol and its glycosides protect cells from oxidative stress and have several health benefits. Herein, Saccharomyces cerevisiae β-fructofuranosidase catalysed synthesis of tyrosol fructoside (TyrFru) from sucrose and tyrosol is reported. The highest TyrFru yield of 20% was achieved under optimized conditions: 1.5 M sucrose, 10 g/L tyrosol, pH 6, and 40°C. Decreased product yield is caused by simultaneous fructosidase-specific reactions occurring in the reaction medium. Tyrosol, as the acceptor of fructosyl residues, competes with water in the hydrolysis reaction and even with sucrose in fructan synthesis. The addition of organic solvents to the reaction medium can reduce the side reactions dependent on water. Also, the solvents can cause conformational changes that affect the enzyme’s flexibility and change its substrate specificity. Hence, the effect of water-miscible and -immiscible organic solvents as well as of novel deep eutectic solvents (DESs) on the TyrFru biosynthesis was studied. Monophasic organic-water systems were constructed with nine organic solvents with log P values of -1.35 to 0.5. Effects of these polar solvents considering the initial reaction rates of all three reactions and product yield were investigated. An addition of even 5 % of co-solvent had an adverse effect on the product yield. All monitored solvents caused a decrease in the yield of TyrFru from 20 % obtained in the buffer to 15‒10 %. The measurements of the initial rates of concurrent reactions revealed that, contrary to expectations, the hydrolytic activity of the enzyme was promoted. At cosolvent concentration of 15%, β‒fructosidase was catalytically active only in DMSO, DMF, dioxane, and acetone, and the formation of TyrFru was even lower. However, the hydrolytic activity of the enzyme in 15 % (v/v) dioxane was still 1.38 times higher than in the cosolvent-free system. The addition of DESs was applied in the concentration range from 5 to 70% (v/v) to investigate the possibility of shifting the reaction equilibrium towards fructoside synthesis. The effect of choline chloride- (ChCl), choline acetate- (ChAc), and betaine-based DESs on the activity and TyrFru synthesis was studied (Karkeszová et al.2023). None of the twelve DESs investigated increased the enzyme activity. As the β‒fructosidase activity may not always correlate with its efficiency in tyrosol transfructosylation, five DESs were selected as the reaction medium for TyrFru synthesis. Among them, four DESs were selected because of the retention of medium and high enzyme activity and ChAc: Urea as the “worst-performing” solvent. The transfructosylation of tyrosol was monitored at three DES concentrations from 10% (v/v) to 40% (v/v); it decreased with the increasing concentration of DESs and completely stopped in the presence of 40% DESs. Surprisingly, among the tested DESs, ChAc: Urea had the best properties for TyrFru production. However, strong stabilizing effect of DES was confirmed at lower concentrations (Holla et al. 2021), suggesting that the loss of activity is due to the catalyst interference and not because of enzyme denaturation. However, the destabilizing effect of DES is more pronounced than the stabilizing effect with their increasing concentration. TyrFru synthesis in buffer/organic (70:30, v/v) biphasic systems was investigated. Tyrosol was more soluble in the organic phase formed by solvents with log P from 0.73 to 1.78. Due to the unavailability of the substrate for the enzyme, TyrFru production was low. In solvents with log P above 2.1, most of the tyrosol was in the aqueous phase and therefore the course of transfructosylation was very similar to that in water. The formed TyrFru was not soluble in any of the organic phases and therefore no shift in the reaction equilibrium was achieved by removing the product from the organic phase. Acknowledgments Grant of Slovak Research and Development Agency APVV- 18-0188. |
| References: | 1. Hollá, V.; Karkeszová, K.; Antošová, M.; Polakovič, M. Transglycosylation Properties of a Kluyveromyces Lactis Enzyme Preparation: Production of Tyrosol β-Fructoside Using Free and Immobilized Enzyme. Process Biochemistry 2021, 110, 168-175, doi:10.1016/j.procbio.2021.08.016. 2. Karkeszová, K.; Antošová, M.; Potocká, E.K.; Mastihuba, V.; Polakovič, M. Medium Engineering of Phenylethanoid Transfructosylation Catalysed by Yeast β-Fructofuranosidase. Bioprocess Biosyst Eng 2023, 46, 237-249, doi:10.1007/s00449-022-02828-3. |
#1132 | Unlocking the catalytic potential of an unusual thiamine diphosphate (ThDP)-dependent enzyme |
|
| Presenting author: | Lucrezia LANZA of UNIVERSITY OF FREIBURG |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Thiamine diphosphate-dependent enzymes / Carboligation / |
| Purpose: | Thiamine diphosphate (ThDP)-dependent enzymes are valuable biocatalysts for the synthesis of chiral building blocks needed in the pharmaceutical and chemical industries. Their substrate promiscuity, high stereo- and enantioselectivity, and ability to catalyse carbon–carbon bond formation, make them extremely attractive in this field. One of the peculiar features of these enzymes is that they have overall little sequence similarity but share a similar structural organization that favours the same ThDP cofactor binding and chemistry. There is one conserved motif that has been commonly identified among this diverse class of enzymes: the ThDP binding motif characterized by a -GDG-(X)26-28-NN- sequence [1]. This motif is involved in the correct positioning of the cofactors (ThDP and a divalent cation) in the active site and is therefore crucial for the catalytic activity of the enzymes [2]. Notably, some exceptions to this conserved motif exist in nature, but, to the best of our knowledge, have not been characterized yet. Here we present a novel ThDP-dependent enzyme with an unusual ThDP-binding motif. By combining in silico analysis, mutagenesis studies, and structural characterization, we investigate the mechanistic role of this uncommon motif. Furthermore, using biosynthetic gene cluster analysis we speculate on the physiological substrates of the enzyme and characterize it in terms of basic biochemical parameters, substrate promiscuity, and stereoselectivity. Finally, we exploit the enzyme for unusual product scopes with respect to biocatalytic applications. This project is supported by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie Grant Agreement no. 956631 — CC-TOP |
| References: | [1] Hawkins CF, Borges A, Perham RN. A common structural motif in thiamin pyrophosphate-binding enzymes. FEBS Lett. 1989 Sep 11;255(1):77-82. doi: 10.1016/0014-5793(89)81064-6. PMID: 2792374. [2] Candy JM, Duggleby RG. Investigation of the cofactor-binding site of Zymomonas mobilis pyruvate decarboxylase by site-directed mutagenesis. Biochem J. 1994 May 15;300 (Pt 1)(Pt 1):7-13. doi: 10.1042/bj3000007. PMID: 8198554; PMCID: PMC1138114. |
#1137 | Design and Optimisation of a De Novo Gold Artificial Metalloenzyme |
|
| Presenting author: | Elinor MORRIS of UNIVERSITY OF BASEL |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial Metalloenzyme / de novo design / gold catalysis / thermostable |
| Purpose: | The ability to design and produce metalloenzymes from scratch could enable chemists to engineer artificial metalloenzymes with a vastly superior biocatalytic toolbox than is currently accessible. Such an ability may allow for the creation of an ‘ideal’ enzyme for a given transformation, which could be expressed in high yields and with high (thermo)stabilities. The field of enzyme design has, however, lagged behind that of protein design in recent years, owing principally to the inherent computational challenges involved, as well as the many unknowns we still face with respect to how an enzyme’s structure relates to its function [1]. Herein, we present preliminary results showcasing a thermostable de novo tandem repeat scaffold [2], computationally designed to bind to an artificial NHC-Au cofactor. We demonstrate that this scaffold binds the cofactor with micromolar affinity and effects gold-catalyzed hydroamination with higher turnover numbers (TONs) than the cofactors alone. Moreover, we show that mutation of active site residues in the protein scaffold can improve the enzyme’s catalytic activity. |
| References: | [1] Lovelock, S. L.; Crawshaw, R.; Basler, S.; Levy, C.; Baker, D.; Hilvert, D.; Green, A. P., Nature 2022, 606, 49-58 [2] Doyle, L.; Hallinan, J.; Bolduc, J.; Parmeggiani, F.; Baker, D.; Stoddard, B. L.; Bradley., Nature 2015, 528, 585-588 |
| Figures: | 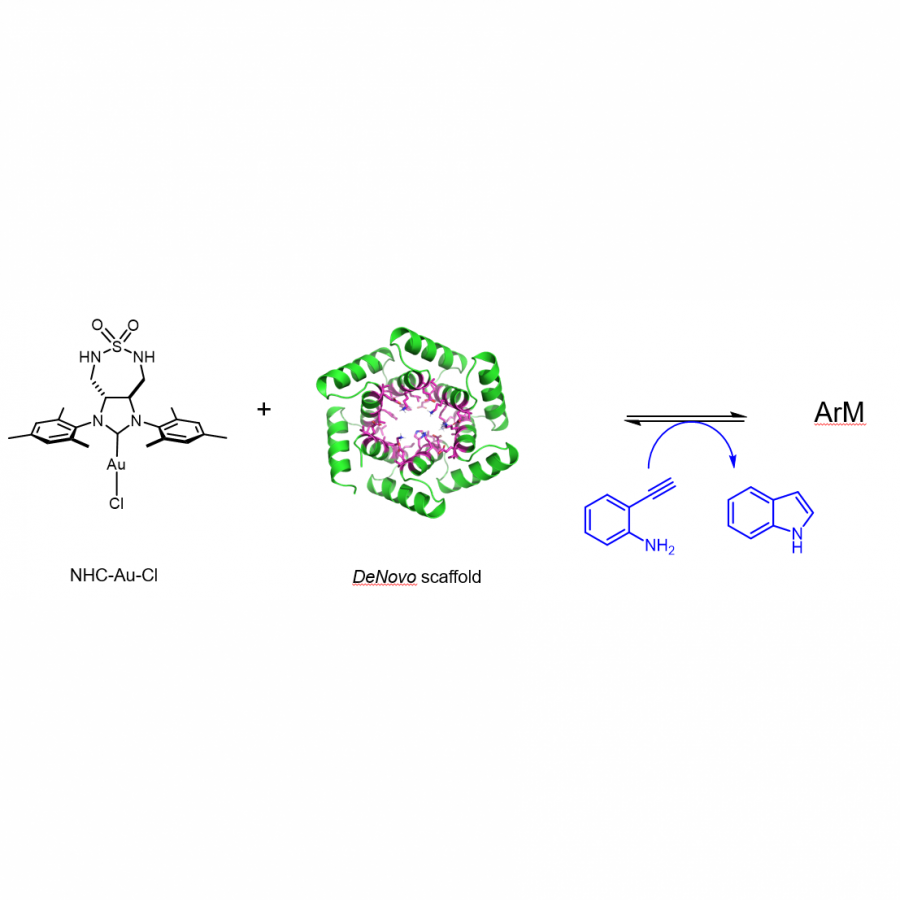 Artificial metalloenzyme (ArM) formation Incubation of the NHC-Au cofactor with the computationally designed protein leads to the formation of an artificial metalloenzyme (ArM) which can effect gold catalysis. For example, the hydroamination reaction presented here. |
#1138 | Characterization of a novel invertase from Trichoderma sp., a producer of a wide range of fructooligosaccharides |
|
| Presenting author: | Egle NARMONTAITE of CENTER OF MOLECULAR BIOLOGY SEVERO OCHOA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | fructooligosaccharides / transfructosylation / invertase / enzyme kinetics |
| Purpose: | Functional foods are known as foods that not only provide essential nutrients but also have additional functions such as health-promoting benefits and disease prevention. There is currently a growing biotechnological interest in the market for a new, safer functional foods with beneficial properties, where bioactive oligosaccharides are gaining relevance over compounds such as dietary fibers, peptides, or unsaturated fatty acids. Bioactive oligosaccharides are considered as prebiotics where fructooligosacharides (FOS), galacto-oligosaccharides (GOS) and glucooligosacharides (GlcOS) are the most common ones. They selectively stimulate the growth and activity of health-promoting lactobacilli and bifidobacter in the gut as well as inhibit the growth of pathogenic microorganisms, improves immune and cardiovascular systems, mineral absorption and even mental health [1]. FOS are basically oligomers of fructose units with a glucose moiety attached to the terminal fructose. A sucrose terminal group is formed as a result of this bond. Based on the linkage pattern between monosaccharides, FOS can be included into three main series: levan type, where fructose units are linked by β-(2→6) linkages (6F-FOS, e.g., 6-kestose), inulin-type with β-(2→1) bond (1F-FOS, e.g., 1-kestose), and neoseries where a β-(2→6) linkage connects fructose to the glucosyl moiety of sucrose (6G-FOS, e.g. neokestose). Invertases or β- fructofuranosidases (EC 3.2.1.26) are biotechnologically important enzymes that catalyze the release of β-fructose from the non-reducing termini of various β-D-fructofuranoside substrates. They belong to the GH32 glycosyl hydrolase family (CAZy; http://www.cazy.org), which also includes invertases, inulinases and fructosyltransferases. These enzymes may also catalyze the synthesis of short-chain FOS, in which some fructosyl moieties are linked to a sucrose unit [1,2]. Trichoderma is the most prevalent culturable genus of the soil fungi. Many species in this genus can be characterized as opportunistic avirulent plant symbionts. It is a filamentous soil fungus that functions as a biocontrol agent for a wide range of economically important aerial and soilborne plant pathogens. In this work we detected a sucrose hydrolyzing activity in a Trichoderma sp. and upon bioinformatical analysis, the enzyme responsible for this activity was identified. The GH32 family protein (possible invertase) TIGH32 was successfully heterologously expressed in Escherichia coli cultures, purified and characterized biochemically. By using different high-performance liquid chromatography (HPLC) techniques the enzyme’s potential to produce FOS threw transfructosylation reactions was analyzed and some of the products formed identified. A structural model of TIGH32 was generated using AlphaFold 2 to provide more information about its structure- function determinants. This study provides more insight into the enzymatic activity of the GH32 proteins and particular specificity of TIGH32 as a potential FOS producer. |
| References: | [1] Jimenez-Ortega, E., Narmontaite, E., Gonzalez-Perez, B., Plou, F.J., Fernandez-Lobato, M., Sanz-Aparicio, J., Int. J. Mol. Sci. 2022, 23, 14981. [2] Ramirez-Escudero, M., Gimeno-Perez, M., Gonzalez, B., Linde, D., Merdzo, Z., Fernandez-Lobato, M., Sanz-Aparicio, J. J. Biol. Chem. 2016, 291, 6843-6857. |
| Figures: |  Experimental design The work-flow of the experiments presented in this work. |
#1139 | Discovery of a novel ultra-thermostable Carbonic Anhydrase for enzymatic CO2 sequestration using high-throughput metagenomic analysis |
|
| Presenting author: | KONSTANTINOS RIGKOS of NATIONAL HELLENIC RESEARCH FOUNDATION (N.H.R.F) |
| Corresponding author: | DIMITRA ZARAFETA of NATIONAL HELLENIC RESEARCH FOUNDATION (N.H.R.F) |
| Other authors: | PAVLOS SARIDIS of NATIONAL HELLENIC RESEARCH FOUNDATION (N.H.R.F) GEORGIOS FILIS of NATIONAL HELLENIC RESEARCH FOUNDATION (N.H.R.F) DIMITRA ZARAFETA of NATIONAL HELLENIC RESEARCH FOUNDATION (N.H.R.F) GEORGIOS SKRETAS of BIOMEDICAL SCIENCES RESEARCH CENTRE “ALEXANDER, FLEMING “ |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme discovery / CO2 Capture / Carbonic Anhydrase / Bioinfomatic Analysis |
| Purpose: | Anthropogenic CO2 emissions have been dramatically increasing for the past 30 years, an outcome that has led to an enhanced greenhouse effect, causing the Earth's temperature to rise at an accelerated rate. This phenomenon, known as Global Warming, is expected to have devastating effects on humanity if not addressed. This is well reflected in the commitment the European Commission made in 2019 to achieve net-zero emissions by 2050. In this landscape, the need for novel, low cost and sustainable CO2 Capture and Storage Technologies (CCS) is urgent. Today, the go-to technology relies on the use of amines for the chemical separation of CO2 from flue gas. Unfortunately, amines have a negative environmental impact due to high toxicity and high-energy consumption related to their regeneration process. Consequently, more eco-friendly technologies to mitigate the negative impact of CO2 rising emissions are pursued by researchers worldwide. An alternative green technology for CO2 sequestration involves the use of carbonic anhydrase (CA), an enzyme that catalyzes the reversible hydration of CO2 to HCO3- and H+ ions. This catalytic activity is vital for many biological processes, including respiration and photosynthesis and because of this, CAs are found in all living organisms. Despite the fact that CAs are abundant in nature and can speed up CO2 absorption kinetics in aqueous media, the harsh conditions present in industrial setups (high temperatures, alkaline pH and high salinity) compromise the stability of common enzymes. A promising approach to overcome this issue is the identification of thermostable enzymes which belong to the protein repertoire of extremophilic organisms. A bottleneck to this strategy is the fact that the protein space of such organisms is not accessible through classic microbiology, as 99% of them cannot be cultured. Metagenomic analysis can overcome this limitation by giving access to this huge, unexplored protein space, and driving the discovery of extremozymes of interest, such as thermostable CAs. In the present study, the discovery and characterization of a novel carbonic anhydrase is presented. High-throughput bioinformatic analysis of large, metagenomic data sets found in NIH's Sequence Read Archive (SRA) and in the Joint Genome Institute (JGI) open-access databases, resulted in the identification of an ultra-thermostable CA that was produced recombinantly in E. coli and studied. The novel CA-KR1 carbonic anhydrase, shares low similarity to known enzymes and exhibits very high thermostability, which, to our knowledge ranks it amongst the most thermostable CAs reported to date. This trait renders CA-KR1 a very strong candidate biocatalyst for enzymatic industrial CO2 sequestration. |
| References: | [1] Ozensoy Guler O, Capasso C, Supuran CT. J Enzyme Inhib Med Chem. 2016, 31(5):689-94. [2] Angeli A, Supuran CT. J Enzyme Inhib Med Chem. 2023, 38(1):2166503. [3] Hanifa M, Agarwal R, Sharma U, Thapliyal PC, Singh LP. J CO2 Util. 2023, 67:102292. [4] Olabi AG, Wilberforce T, Sayed ET, Shehata N, Alami AH, Maghrabie HM, et al. Energies. 2022, 15(22):8639. [5] Dutcher B, Fan M, Russell AG. ACS Appl Mater Interfaces. 2015, 7(4):2137-48. [6] Anderson C, Harkin T, Ho M, Mumford K, Qader A, Stevens G, et al. Energy Procedia. 2013, 37:225-32. [7] Barzagli F, Giorgi C, Mani F, Peruzzini M. J CO2 Util. 2017, 22:346-54. [8] Talekar S, Jo BH, Dordick JS, Kim J. Curr Opin Biotechnol. 2022, 74:230-40. |
| Figures: |  SDS-PAGE & Protonography analysis of CA-KR1 protein. (A) Unboiled sample of purified CA-KR1 protein (Lane 2) and protein standard (Lane 1). (B) Protonography analysis of CA-KR1 (Lane 3). The color shift from blue to yellow indicates CA activity. 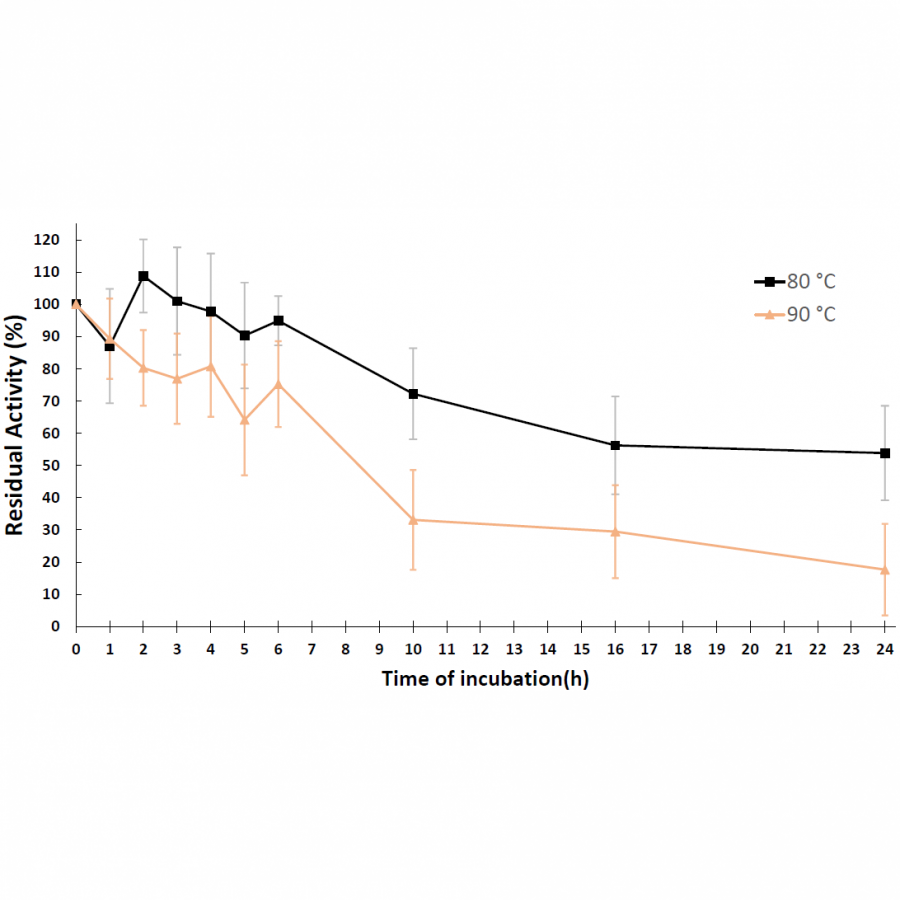 CA-KR1 Thermostability The residual activity of CA-KR1 was measured following incubation of the enzyme at 80oC and 90oC for up to 24 h. The reported values correspond to the mean value from five independent experiments performed at least in triplicate +/- SD. |
#1140 | Cell-Free Production of α-Ketoglutarate from D-Galacturonic Acid via a Multi-Enzymatic Cascade |
|
| Presenting author: | Luca SCHMERMUND of TECHNICAL UNIVERSITY OF MUNICH, TUM CAMPUS STRAUBING |
| Other authors: | Zahabiya MALUBHOY of TECHNICAL UNIVERSITY OF MUNICH, TUM CAMPUS STRAUBING Kathrin HÖRNSCHEMEYER of TECHNICAL UNIVERSITY OF MUNICH, TUM CAMPUS STRAUBING Volker SIEBER of TECHNICAL UNIVERSITY OF MUNICH, TUM CAMPUS STRAUBING |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymatic Cascade / Biomass / Dehydratases / Biocatlysis with Oxygen |
| Purpose: | Recently, D-glucuronate was converted to α-ketoglutarate (aKG) via a multi-enzymatic cascade using five enzymes, namely UDH, D-glucarate dehydratase (GlucD), KdgD, αKgsaDH and NOX. In this context, aKG represents an important chemical building block.[1] In this study, we wanted to use D-galacturonic acid as substrate for the enzymatic production of aKG (Scheme 1). D-galacturonic acid is the main component of pectin, which is present in most primary cell walls.[2] The utilization of biogenic raw materials like uronic acids for the production of base chemicals is of crucial importance to develop a bio-based economy. The conversion of D-galacturonate to aKG required the identification of a D-galactarate dehydratase (GalcD) to convert the intermediate galactarate to 5-keto-4-deoxyglucarate. Therefore, galactarate dehydratases from Actinobacillus succinogenes 130Z (As), Escherichia coli (Ec), Salmonella typhimurium (St) and Sodalis ligni (Sl) were selected via a data bank serach. The dehydratases were produced in E. coli with N-terminal His6-tag. All enzymes displayed activity toward D-galactarate. However, the EcGalcD and the AsGalcD were only active in the presence of 10-50 mM DTT and thus not suitable for the cascade. StGalcD and SlGalcD were active without the addition of a reducing agent and were applied in the cascade. With the StGalCD 20 mM (80%) aKG were obtained from 25 mM D-galacturonate. In the future, the efficiency of the cascade will be improved by combining process engineering and enzyme engineering approaches. Furthermore, the addition of a GlucD to the cascade may allow the simultaneous conversion of galacturonate and glucuronate to aKG in one cascade. |
| References: | [1] Beer, B.; Pick, A.; Sieber, V., Metab. Eng. 2017, 40, 5-13. [2] Zheng, L.; Xu, Y.; Li, Q.; Zhu, B., Bioresour. Bioprocess. 2021, 8, 79. |
| Figures: | 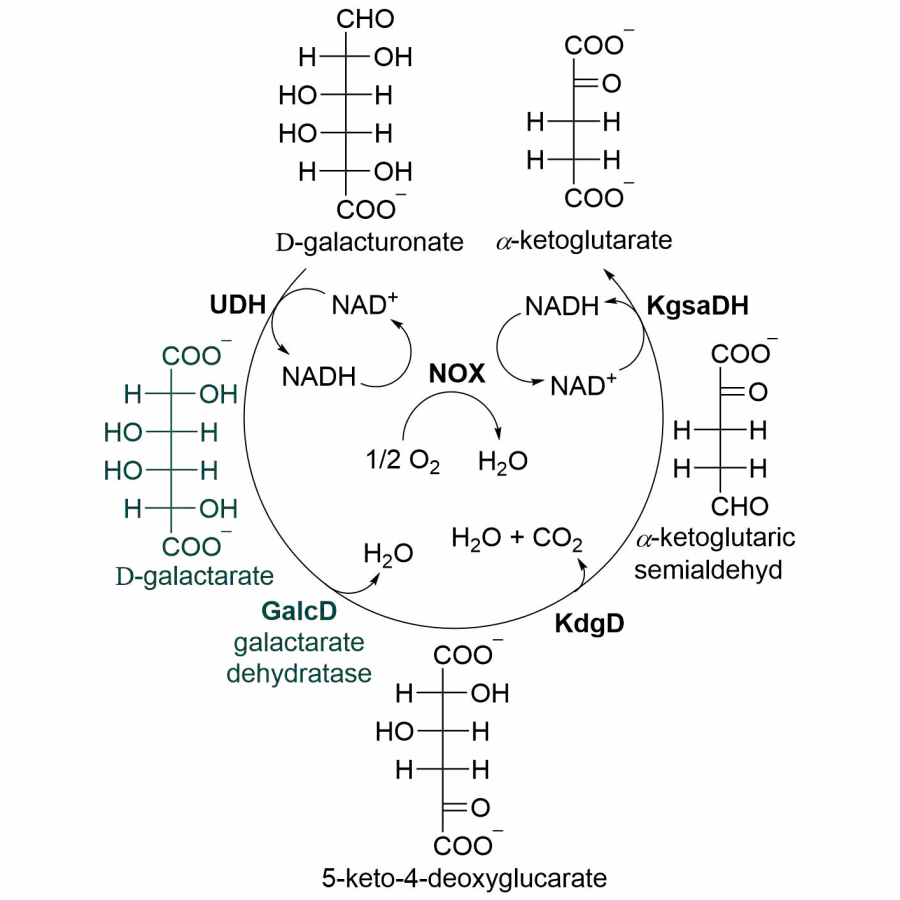 Scheme 1. Multi-enzymatic conversion of D-galacturonate to aKG. UDH: uronate dehydrogenase;
GalcD: galactarate dehydratase; KdgD: 5-keto-4-deoxyglucarate dehydratase; KgsaDH: a-ketoglutaric semialdehyde dehydrogenase; NOX: NADH oxidase.
|
#1144 | Enzymes immobilized on cellulose carriers: Renewable, biodegradable and economical |
|
| Presenting author: | Rob SCHOEVAART of CHIRALVISION BV |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme immobilization / renewable / biodegradable / cellulose |
| Purpose: | An increasing range of carriers with a wide choice of enzymes suiting both batch and continuous processes are commercially available. Macro- and gigaporous carriers suitable for any enzyme are on the market for large scale usage and guarantee minimized costs. As a result of these developments, the number of industrial applications is steadily growing and the ongoing progress will ensure continued success in meeting new challenges. Readily available renewable carriers are missing from this range of carriers, however. To make a 'green process greener' cellulose beads can be used as enzyme carriers to replace non-renewable polluting plastic beads currently used in most industrial biocatalytic processes. Cellulose is a natural biopolymer extracted from wood, a renewable feedstock that can be sourced sustainably. Cellulose is thermally and mechanically stable while at the same time being biodegradable in the presence of microbes (e.g. in wastewater treatment sludge). In collaboration with Naturbeads (Bath University) a new cellulose carrier was developed that can act as a replacement for standard acrylic beads. The cellulose beads have well defined properties such as uniform size, spherical shape and high porosity (>90% pore volume). A series of experiments was run to introduce a variety of functional groups including covalent and ionic binding groups. This functionalization allows for binding of enzymes via different binding modes ensuring high loading, good activity recovery and stable performance. Various enzymes were immobilized on the functionalized cellulose beads and subjected to biphasic process conditions. With a group of lipases hydrolysis tests were run at 40 ˚C in a Spinchem rotating bed reactor and for each formulation at least 10 recycles were performed. The best performing ones - among which lipase from Thermomyces lanuginosus – showed equal to even better performance and recyclability compared to the same amount of enzyme immobilized on a standard epoxide acrylic beads. This proofs that an enzyme immobilized on a cellulose bead can be a biodegradable and renewable alternative for plastic beads while at the same time having better economics. |
| Figures: | 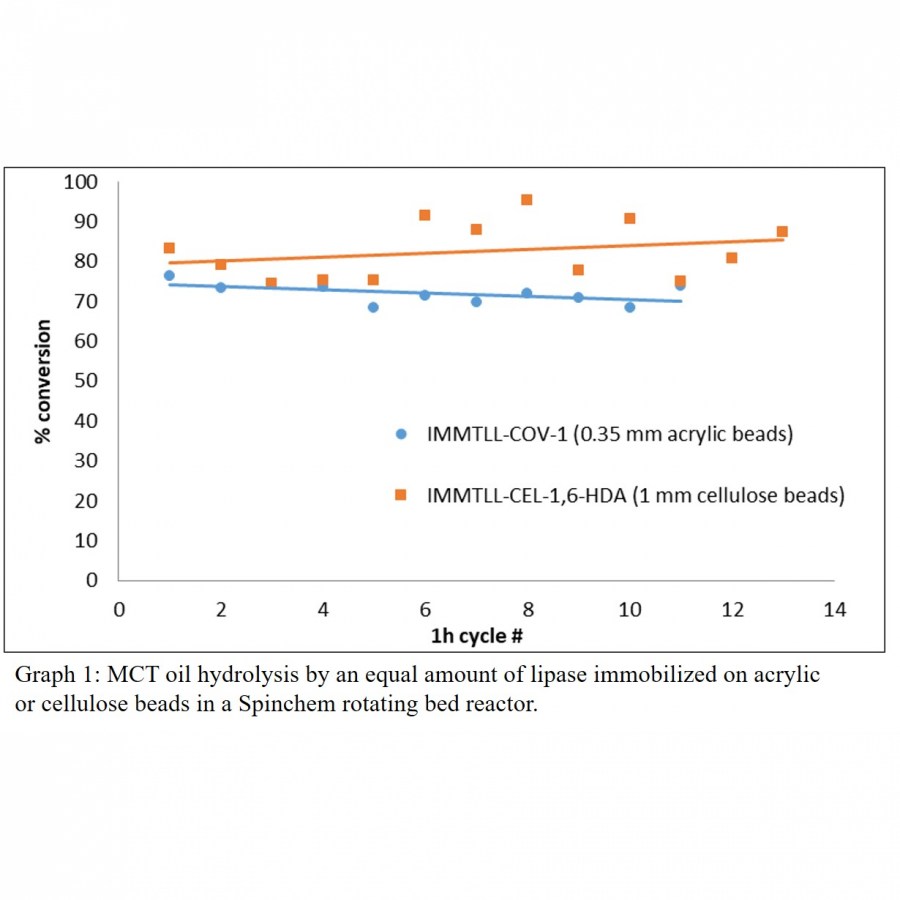 Graph 1 MCT oil hydrolysis by an equal amount of lipase immobilized on acrylic or celluose beads in a Spinchem rotating bed reactor. |
#1145 | Biocatalytical ethanolysis of edible oils with CaLA immobilized on acrylic beads and membranes |
|
| Presenting author: | Camiel DE RUITER of CHIRALVISION |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | immobilization / ethanolysis / CaLA / |
| Purpose: | In the process industry, downstream processes are the most energy and resource consuming steps in industrial operations. To contribute to Europe’s goal of a clean and liveable environment, new processes are required that have a very broadly applicable concept for an efficient integration of downstream operations in the overall process chain to reduce CAPEX and OPEX and therefore significantly enhance the competitiveness of the European process industry. The MACBETH consortium provides a breakthrough technology by combining catalytic synthesis with the corresponding separation units in a single, tailor-made, highly efficient catalytic membrane reactor (CMR). Within MACBETH for the first time 24 partners with manifold expertise and competencies in membrane technology are united to successfully transfer the technological concept to other sectors of the chemical industry. Based on a large variety of already established building blocks (such as biocatalysts, membranes, support materials and reactor concepts) a demo plant for bio-catalytical oil cleavage (BOC) is being developed, showing the commercial applicability of CMR in biotechnology for the first time. The BOC case will develop a CMR-based reactor combining enzyme-catalysed selective ethanolysis of oil fatty acids in an organic media system followed by an integrated membrane separation to isolate selected fatty acids as ethyl esters or as mono-, di- or triglycerides. In a 350 hour long run a column filled with lipase from Candida antarctica immobilized on acrylic bead (IMMCALA-COV-1) processed over 4 kg of oil per gram of immobilized enzyme in a continueous flow. The immobilized enzyme has been demonstrated to remain active for over 1000 hours under process conditions (40 ˚C). A demo plant is being set-up for further scale-up and even longer runs in combination with membrane separation of the reaction products. Enzymes have also been successfully immobilized directly onto the membrane to combine the enzymatic conversion with the separation step to form a catalytic membrane reactor for further efficiency improvement. |
| Figures: | 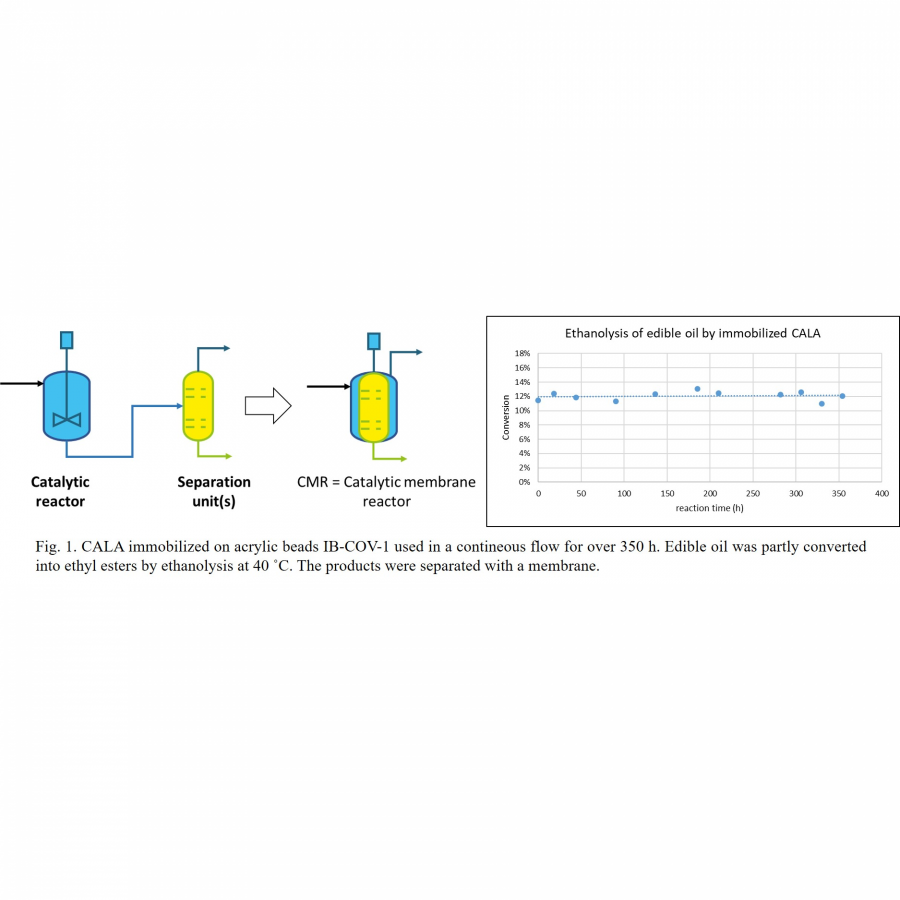 Fig 1 CALA immobilized on acrylic beads IB-COV-1 used in a contineous flow for over 350h. Edible oil was partly converted into ethyl esters by ethanolysis at 40 ˚C. The products were separated with a membrane. |
#1164 | Evaluation of the potential conversion of lignin into aromatic molecules by bacterial strains. |
|
| Presenting author: | Corinne IVALDI of UNIVERSITÉ DE REIMS, CHAIRE AFERE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Bioconversion / Bacteria / Lignin / |
| Purpose: | Corinne Ivaldi1, Brigitte Chabbert2, Roland Molinié3, David cronier2, David Mathiron4, Jean Xavier Fontaine3, Serge Pilard4, François Mesnard3, Harivony Rakotoarivonina1 1 Université de Reims Champagne-Ardenne, INRAE, FARE, AFERE, UMR A 614, Reims, France 2 Université de Reims Champagne-Ardenne, INRAE, FARE, UMR A 614, Reims, France 3 Université de Picardie Jules Verne, INRAE, BioEcoAgro, UMRT 1158, BIOPI, Amiens, France 4 Université de Picardie Jules Verne, Plateforme Analytique, Amiens, France Email: corinne.ivaldi@univ-reims.fr Faced with the scarcity of fossil resources and climatic change, it becomes essential to find environmental-friendly alternatives to develop sustainable products. Lignin is a renewable source of aromatic molecules. It is present in very large quantities throughout the world in plant biomasses but also as by-products of certain industries (paper industry, wood industry,...). Currently, lignin mainly is used to produce energy and some little valued in molecules of interest because of its complexity1. Lignin can be degraded by biological and enzymatic processes known to be respectful of the environment. The biocatalysts used during these processes are still not very efficient. In nature, several microorganisms are able to degrade lignin2. The use of bacteria can have several advantages: ease of implementation (culture, biomass production,...) ubiquitous in many environments (resistant to extreme conditions, metabolic capabilities, ...), available and efficient genetic tools (many bacteria can be genetically modified to obtain a desired function). However, most of the work in the field of bacterial conversion of lignin concerns model molecules whereas studies on lignins remain rare and do not necessarily reflect their ligninolytic capacities. Due to the complexity of lignins (structures and chemical composition), it is a need for efficient biological tools to valorize the aromatic moieties of this polymer. Our objective is to identify bacteria efficient in lignin conversion. Our strategy consists in selecting some "ligninolytic" bacteria identified in the literature belonging to various genus. We studied their capacity to use technical lignins by multiple approaches (growth, enzymes production, aromatic molecules and residue analysis). The results showed that the targeted bacteria behave differently in presence of lignin in terms of growth, production of enzymes and phenolic molecules. In conclusion, efficient use of lignins requires a suitable combination of biocatalysts/lignins. It is necessary to choose biocatalysts according to their metabolic capacities, their complementarity but also considering the structure and composition of the substrates to be valorized. This work was funded by the SFR Condorcet and the University of Reims Champagne-Ardennes. |
| References: | [1] J. Becker, Biotechnol. Advances 2019, 37, 107360. [2] F. Asina, Renew. Sustain. Energy Rev 2017, 77, 1179-1205. |
#1169 | Enzymatic depolymerization of polylactide into repolymerizable monomers using sustainable conditions. |
|
| Presenting author: | Alaric CATARD of LABORATORY OF REACTIONS AND CHEMICAL ENGINEERING - UNIVERSITÉ DE LORRAINE |
| Other authors: | Isabelle CHEVALOT of LABORATORY OF REACTIONS AND CHEMICAL ENGINEERING - UNIVERSITÉ DE LORRAINE Sandrine HOPPE of LABORATORY OF REACTIONS AND CHEMICAL ENGINEERING - UNIVERSITÉ DE LORRAINE |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Polylactide / Cyclo Depolymerization / Novozyme 435 / Green solvent |
| Purpose: | Bioplastics are often seen as a solution to answer the problems associated to petro-sourced plastics. One of the most famous is polylactide, also known as PLA, representing 24% of the global production of bio-based plastics (1). This is a bioplastic made from lactic acid (L-lactic acid mainly), produced by fermentation of renewable resources (e.g. corn, potato…) and is mainly produced by polymerization of L-lactide (i.e. cyclic dimer of L-lactic acid) by Ring Opening Polymerization (ROP). In addition to its bio-sourced nature, PLA has the advantage of having similar characteristics to some conventional plastics such as polystyrene (PS) or polyethylene terephthalate (PET) (2). Even if PLA is a bio-based thermoplastic, it can be at the origin of the same environmental pollution as conventional plastics, considering that it degrades hardly and slowly in nature. Moreover, the European Commission has stated its ambition to reach 100% recyclable packaging on the European market by 2030. Consequently, PLA recycling processes must be put in place to meet these different challenges. Concerning recycling of plastics, it is distinguished mainly two methods: mechanical recycling and chemical recycling. This work explored the way of chemical recycling of poly(L-lactic acid), by depolymerizing it into directly repolymerizable monomers (i.e. lactide/cyclic oligomers vs. lactic acid), which will be reused in the same (closed-loop) application enabling “infinite recycling” (3). It exists different ways to break down the PLA chains among which hydrolysis, alcoholysis and pyrolysis are highly studied in academic fields. Pyrolysis, based on the use of high temperatures (> 600 °C) makes the degradation of the polymer into easily repolymerizable products (e.g. cyclic oligomer, lactide…). However, this strategy is hard to implement at industrial scale and it causes side reactions (e.g. epimerization, acrylic acid generation…) (4). In order to use a lower temperature, from which the thermodynamic equilibrium is shifted towards the depolymerization side (known as ceiling temperature or Tc) and which limits the secondary reactions, solvents can be used (5). This strategy enables the recovery of useful repolymerizable cyclic oligomers and is often described as Ring Closing Polymerization (RCP) or cyclodepolymerization (CDP), which constitutes the opposite reaction of ROP (6). Cederholm et al. obtained high yields of lactide (> 95%) after short times (1-4h) using Sn(Oct)2 as a catalyst and different solvents (7). Takahashi et al achieved to produce cyclic oligomers via enzymatic depolymerization of PLLA into o-xylene (8). The present work aims to use renewable and sustainable biocatalyst (Novozyme 435®, an immobilized Lipase B from Candida antarctica) and green solvents (Dimethyl carbonate (DMC) and diethyl carbonate (DEC)) to enable the recovery of repolymerizable cyclic oligomers of lactid acid from PLA with various degrees of crystallinity. First, DMC and DEC have demonstrated a good ability to solubilize PLA reaching final concentrations of 120 g.L-1 in both solvents using a PLA with high crystallinity degree (χc = 49.66 ± 7,21 %) and high temperatures. Moreover, it has been demonstrated that Novozyme 435® (N435®) still maintains activity after different times (from 2 to 48 hours) at high temperatures (from 80 to 120 °C) in both solvents, making it suitable for the depolymerization of PLA at high temperature in solvent media. DMC and DEC, being organic carbonate solvents, their reactivity in presence of N435® at high temperatures have also been explored by Gas Chromatography (GC) analysis in order to check their stability. Finally, Size Exclusion Chromatography (SEC) analysis have been set up to evaluate the decrease in molar mass of the polymer. Future objectives will be to analyze low-weight oligomers produced (e.g. using 1H NMR) and investigate their repolymerizability by N435® in sustainable conditions. |
| References: | 1. Schneiderman DK, Hillmyer MA. 50th Anniversary Perspective : There Is a Great Future in Sustainable Polymers. Macromolecules. 23 mai 2017;50(10):3733‑49. 2. Haider TP, Völker C, Kramm J, Landfester K, Wurm FR. Plastics of the Future? The Impact of Biodegradable Polymers on the Environment and on Society. Angew Chem Int Ed. 2 janv 2019;58(1):50‑62. 3. Hatti-Kaul R, Nilsson LJ, Zhang B, Rehnberg N, Lundmark S. Designing Biobased Recyclable Polymers for Plastics. Trends in Biotechnology. janv 2020;38(1):50‑67. 4. Dogu O, Pelucchi M, Van de Vijver R, Van Steenberge PHM, D’hooge DR, Cuoci A, et al. The chemistry of chemical recycling of solid plastic waste via pyrolysis and gasification: State-of-the-art, challenges, and future directions. Progress in Energy and Combustion Science. mai 2021;84:100901. 5. Greer SC. Physical Chemistry of Equilibrium Polymerization. J Phys Chem B. 1 juill 1998;102(28):5413‑22. 6. Hodge P. Cyclodepolymerization as a method for the synthesis of macrocyclic oligomers. Reactive and Functional Polymers. juill 2014;80:21‑32. 7. Cederholm L, Wohlert J, Olsén P, Hakkarainen M, Odelius K. « Like Recycles Like »: Selective Ring-Closing Depolymerization of Poly(L-Lactic Acid) to L-Lactide. Angewandte Chemie International Edition. 2022;61(33):1‑9. 8. Takahashi Y, Okajima S, Toshima K, Matsumura S. Lipase-Catalyzed Transformation of Poly(lactic acid) into Cyclic Oligomers. Macromol Biosci. 15 mars 2004;4(3):346‑53. |
| Figures: | 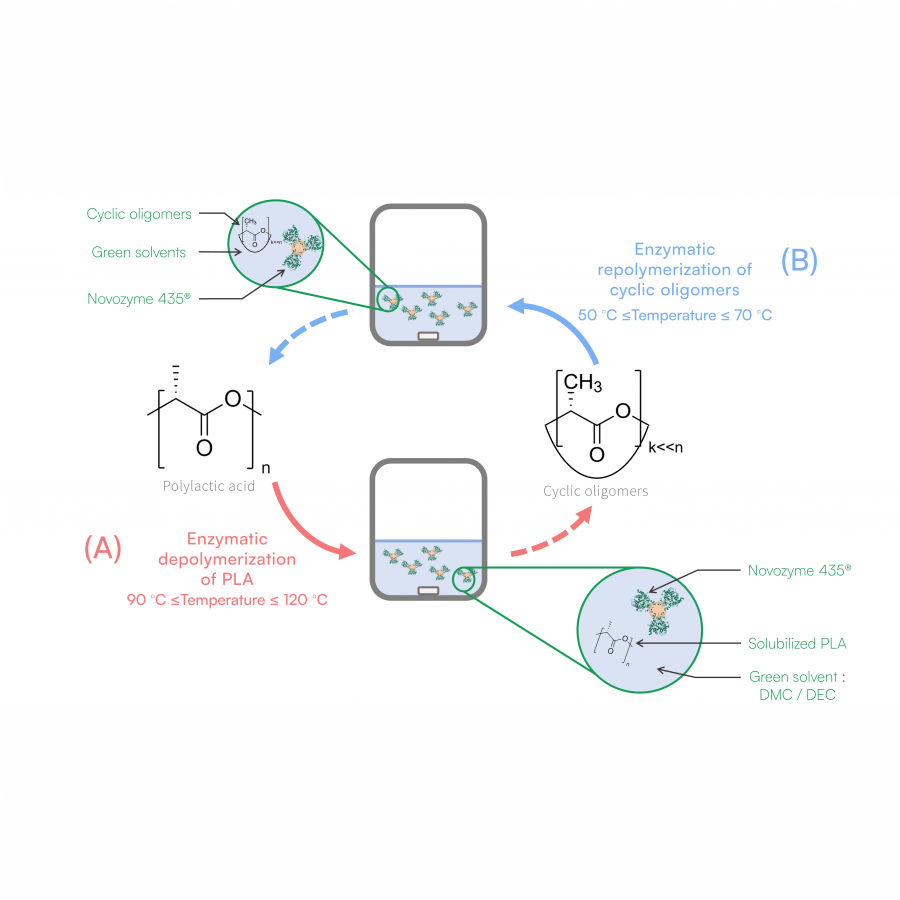 Representative schema of the enzymatic depolymerization of PLA into repolymerizable products and their enzymatic repolymerization (A) : Enzymatic depolymerization of PLA solubilized in green solvents (e.g. dimethyl carbonate or diethyl carbonate).
(B) : Enzymatic repolymerization of oligomers in green solvents |
#1170 | Synthesis of Deoxysugars from Aliphatic Ketoacids and Aldoses Catalysed by Thermostable Transketolase Variants from Geobacillus stearothermophilus |
|
| Presenting author: | Giuseppe ARBIA of UNIVERSITÉ CLERMONT AUVERGNE |
| Other authors: | Franck CHARMANTRAY of UNIVERSITÉ CLERMONT AUVERGNE Laurence HECQUET of UNIVERSITÉ CLERMONT AUVERGNE Aurélie LAGARDE of UNIVERSITÉ CLERMONT AUVERGNE Muriel JOLY of UNIVERSITÉ CLERMONT AUVERGNE Samantha GITTINGS of PROZOMIX LIMITED Kirsty M. GRAHAM of PROZOMIX LIMITED Nazim OCAL of UNIVERSITÉ CLERMONT AUVERGNE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / transketolase / stereoselectivity / D-amino acid oxidase |
| Purpose: | Transketolase (TK), catalyzes the synthesis of a wide range of ketoses by stereoselective C-C bond formation in the presence of thiamine diphosphate (ThDP) and Mg2+ as cofactors.7 Indeed, wild type TK transfers a ketol unit from 3-hydroxypyruvate (HPA), which is the most commonly used synthetic donor substrate, to an aldehyde as acceptor substrate and the product of the reaction is a (3S)-ketose or an analog depending on the acceptor substrate. The reaction is rendered irreversible by the release of carbon dioxide. Recent studies showed that aliphatic alpha-ketoacids can be recognized as donor substrates by specially designed TK variants.[1] Particularly, the thermostable TK from Geobacillus stearothermophilus (TKgst)[2] has been engineered to accept pyruvate, 2-oxobutyrate as donors in place of HPA (scheme 1).[3] We present a strategy for the enzymatic synthesis of 1-deoxy and 1,2 deoxyketoses from the aliphatic alpha-ketoacids, pyruvate and 2-oxobutyrate, as donors, and natural aldoses of variable chain length as acceptors, catalyzed by TKgst variants.[4] Analytical studies have been firstly carried out with the appropriate substrates allowing to select the best combinations of mutations. The preparative scale synthesis allowed to obtain 1-deoxy and 1,2 deoxy ketoses with good to excellent isolated yields (61%-86%). To optimize the strategy, and as a proof of principle, the alpha-ketoacids pyruvate and 2-oxobutyrate were generated in situ from the corresponding D-aminoacids D-alanine and D-homoalanine respectively, using a thermostable D-aminoacid oxidase DAAO4536 that was selected from a screening of 55 putative DAAOs provided by Prozomix Limited. Hence, a one-pot one step procedure was performed at 50 °C by coupling DAAO4536 and the best TKgst variant H102L/L118I/H474S in the presence of D-alanine or D-homoalanine as alpha-ketoacids precursors and D-erythrose as acceptor substrate. The corresponding 1-deoxy and 1,2-dideoxydeoxyketoses were isolated with good yields (64% and 72% respectively, out of two steps).[4] |
| References: | [1]. H. Yu, R. Icken Hernandez Lopez, D. Steadman, D. Mendez-Sanchez, S. Higson, A. Cazares-Korner, T. D. Sheppard, J. M. Ward, H. C. Hailes, P. A. Dalby, FEBS J. 2020, 9, 1758-1776. [2]. J. Abdoul Zabar, I. Sorel, V. Hélaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marlire, W.-D. Fessner, L. Hecquet, Adv. Synth. Catal. 2013, 355, 116-128. [3]. T. Saravanan, S. Junker, M. Kickstein, J. Hegen, S. Hein, M. K. Link, S. Witt, M. Lorillière, L. Hecquet, W.-D. Fessner, Angew. Chem. Int. Ed. 2017, 56, 5358-5362; b) H.Casajus, A. Lagarde, M. Leremboure, T. De Dios Miguel, L. Nauton, W.-D. Fessner, N. Duguet, F. Charmantray, L. Hecquet, ChemCatChem 2020, 12, 5772-5779. [4]. N. Ocal, G. Arbia, A. Lagarde, M. Joly, S. Gittings, K. M. Graham, F.Charmantray, L.Hecquet. Adv. Synth. Catal. 2023, 78-87 |
| Figures: | 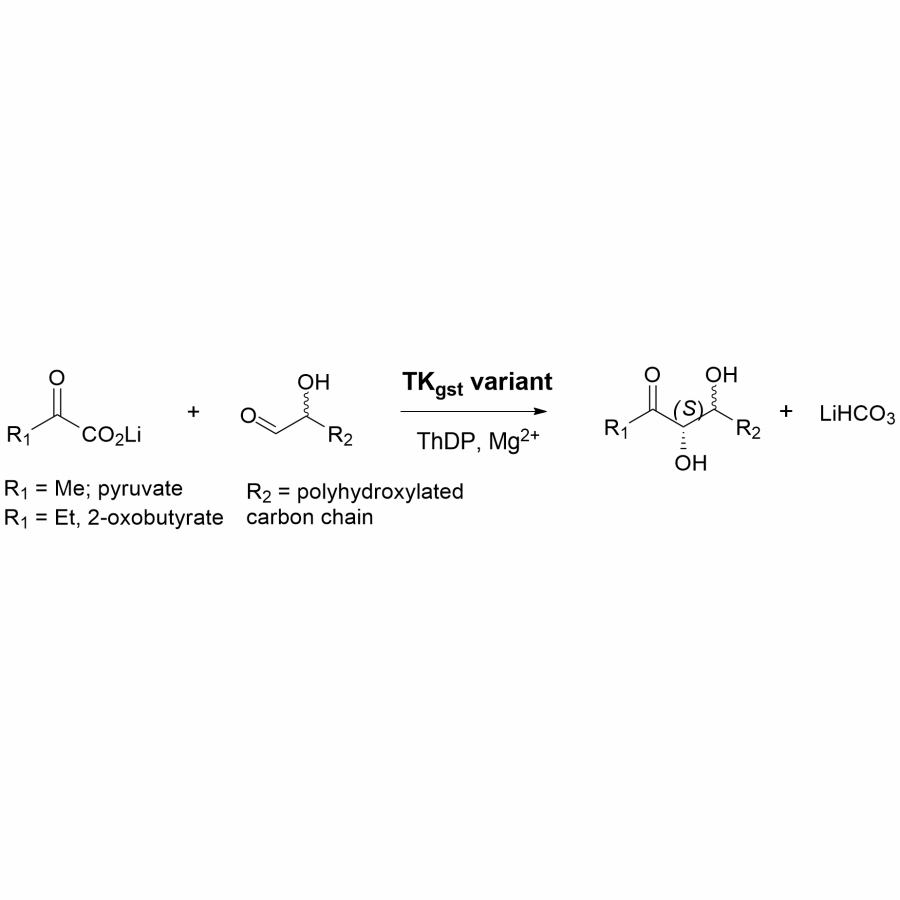 Transketolase reaction scheme 1 |
#1177 | 1-pot 2-step enzymatic synthesis for chiral diols - Enzyme engineering combined with reaction optimization and process development |
|
| Presenting author: | Torsten SEHL of FORSCHUNGSZENTRUM JÜLICH GMBH |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Chiral diols are valuable building blocks used in the polymer, fine chemical and pharmaceutical industries. Traditionally, these versatile compounds are produced by chemical syntheses such as chemical racemate cleavage, hydrogenation with heavy metal homogeneous catalysis, hydroxylation and condensation. Our enzymatic process allows the synthesis of chiral diols under mild reaction conditions, using non-toxic materials and ensuring high step and resource efficiency. The synthesis is performed with ketoreductases (KRED) and has the advantage of eliminating multistep workup and synthesis steps. While the production of (2R,4R)-pentane-2,4-diol served as a demonstrator, the strategy can be used for the production of many other diols. Suitable KREDs were selected and optimized in collaboration with Enzymaster using their in-silico computational engineering tool BioEngine®. Enzymes screening took place for (2R,4R)-pentane-2,4-diol on a mL laboratory scale followed by reaction condition optimization for positive screening hits. Subsequently, a process concept was established together with process engineers at RWTH Aachen University on a 200 mL scale and scaled up to the 10 L. With optimized KRED variants in combination with multi-parameter reaction engineering, we have succeeded in establishing a process for the production of (2R,4R)-pentane-2,4-diol. Compared to aqueous reactions, using a micro-aqueous reaction system (MARS) enables not only access to high product concentrations of >200 g/L, but also simplifies product work-up. Basic principles of the concept were subsequently used for the production of further diols. All diols can be produced in high chemical (>99%) and optical purities (>99% ee, de). Our enzymatic process for producting valuable, chiral diols combines simplicity in operation with reduced consumption of chemicals, energy, space and time, resulting in excellent step and resource efficiency. With this process, 12 valuable chiral diols can now be produced in industrially relevant titers including efficient downstream processing. |
#1180 | Digitalization of biocatalysis: the data exchange format EnzymeML |
|
| Presenting author: | Jürgen PLEISS of UNIVERSITY OF STUTTGART |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | research data management / FAIR data principles / enzyme kinetics / reproducibility |
| Purpose: | The design of biocatalytic reaction systems is highly complex due to the dependency of the estimated kinetic parameters on the enzyme, the reaction conditions, and the modelling method. Consequently, reproducing enzymatic experiments and reusing enzymatic data are challenging. To enable storage, retrieval, and exchange of enzymatic data such as the reaction conditions, the measured time courses of substrate and product, the selected kinetic model, and the estimated kinetic parameters, the XML–based markup language EnzymeML has been developed [1]. EnzymeML is based on SBML and the community Standards for Reporting Enzyme Data (STRENDA). The EnzymeML toolbox supports biocatalysis research by making enzymatic data findable, accessible, interoperable, and reusable [2]. An EnzymeML document contains information about reaction conditions and the measured time course of substrate or product concentrations (Fig. 1). It is generated from an EnzymeML spreadsheet or by the webtool BioCatHub. Kinetic modelling is performed by uploading EnzymeML documents to the modelling platforms COPASI or PySCeS. The estimated kinetic parameters are then added to the EnzymeML document. The EnzymeML document containing the experimental and modelling results is then uploaded to a Dataverse installation or to the reaction kinetics database SABIO-RK. The workflow of a project is encoded as Jupyter Notebook, which can be re-used, modified, or extended. EnzymeML serves as a seamless communication channel between experimental platforms, electronic lab notebooks, tools for modelling of enzyme kinetics, publication platforms, and enzymatic reaction databases (Fig. 2). The feasibility and usefulness of the EnzymeML toolbox was demonstrated in six scenarios, where data and metadata of different enzymatic reactions are collected, analysed, and uploaded to public data repositories for future re-use [3]. Because EnzymeML documents are structured and standardized, the experimental results encoded in an EnzymeML document are interoperable and reusable by other groups. Because an EnzymeML document is machine-readable, it can be used in an automated workflow for storage, visualization, data analysis, and re-analysis of previously published data, without limitations of the size of each dataset or the number of experiments. Thus, EnzymeML contributes to the digitalization of (bio)catalytic sciences. |
| References: | 1. Pleiss J, 2021. Standardized data, scalable documentation, sustainable storage - EnzymeML as a basis for FAIR data management in biocatalysis. ChemCatChem 13: 3909-3913; doi: 10.1002/cctc.202100822 2. Range J, Halupczok C, Lohmann J, Swainston N, Kettner C, Bergmann FT, Weidemann A, Wittig U, Schnell S, Pleiss J, 2022. EnzymeML - a data exchange format for biocatalysis and enzymology. FEBS J 289: 5864-5874; doi: 10.1111/febs.16318 3. Lauterbach S, Dienhart H, Range J, Malzacher S, Spöring JD, Rother D, Pinto MF, Martins P, Lagerman CE, Bommarius AS, Vang Høst A, Woodley JM, Ngubane S, Kudanga T, Bergmann FT, Rohwer JM, Iglezakis D, Weidemann A, Wittig U, Kettner C, Swainston N, Schnell S, Pleiss J, 2023. EnzymeML: seamless data flow and modeling of enzymatic data. Nat Methods; doi: 10.1038/s41592-022-01763-1 |
| Figures: | 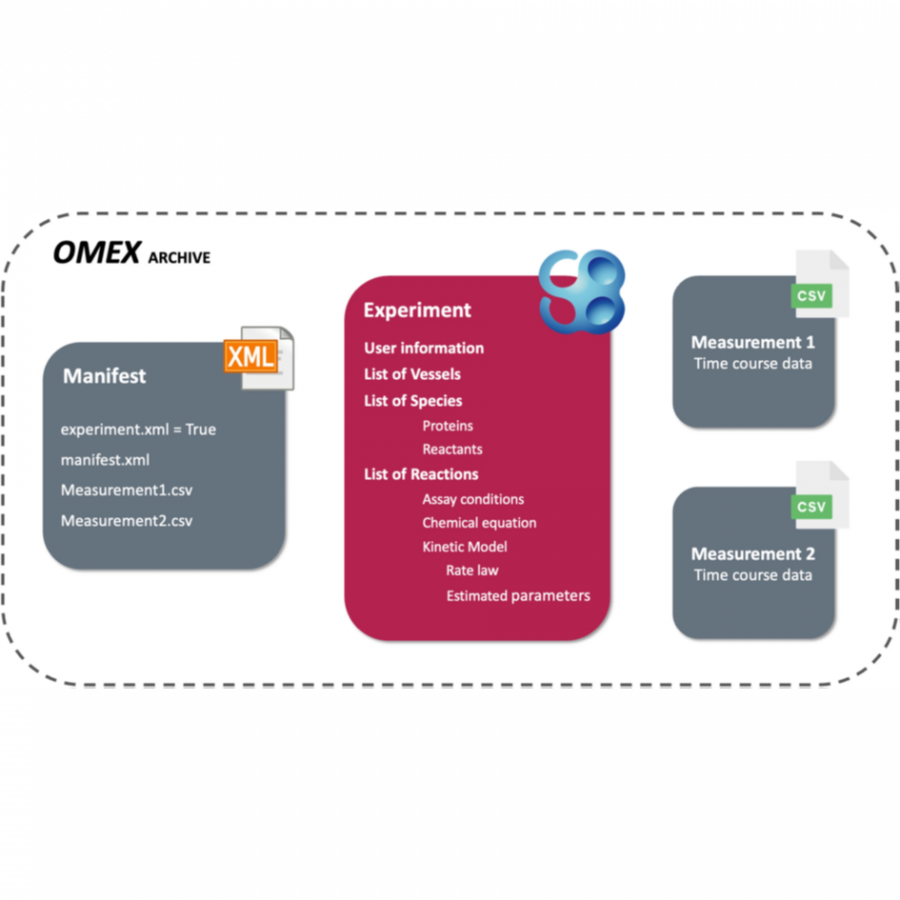 Structure of an EnzymeML document. An EnzymeML document is a ZIP container in OMEX format and contains the experiment file (SBML) and the measurement files (CSV). 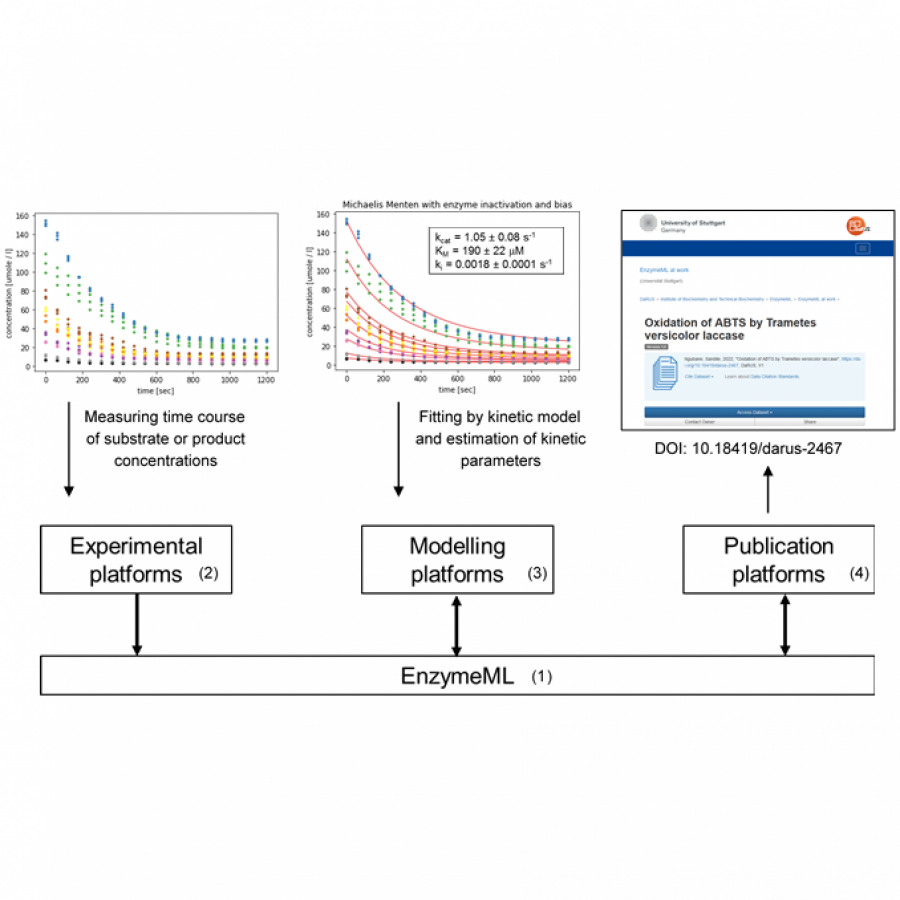 Seamless data transfer An EnzymeML document integrates (meta-)data from experiment and modelling), and serves as a communication channel between experimental, modelling, and publication platforms. |
#1183 | Efficient lipase-catalyzed esterification of carbohydrate polyols in reactive natural deep eutectic solvents |
|
| Presenting author: | Francisc PETER of UNIVERSITY POLITEHNICA TIMISOARA |
| Other authors: | Alina Ramona BUZATU of ” VICTOR BABES” UNIVERSITY OF MEDICINE ND PHARMACY Anamaria TODEA of UNIVERSITY POLITEHNICA TIMISOARA Diana Maria DREAVA of UNIVERSITY POLITEHNICA TIMISOARA Valentin BADEA of UNIVERSITY POLITEHNICA TIMISOARA Carmen Gabriela BOERIU of UNIVERSITY POLITEHNICA TIMISOARA Ioan BITCAN of UNIVERSITY POLITEHNICA TIMISOARA |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | reactive NADES / lipase / arabitol esters / green processes |
| Purpose: | Biocatalytic pathways for esterification of carbohydrates and carbohydrate polyols show the significant advantage of a single step operation, but also face the challenge of finding the appropriate solvent to enhance simultaneously the solubilization of sugars and the stability and regioselectivity of the biocatalyst in the reaction medium, while minimizing the environmental burden. Natural deep eutectic solvents (NADES) emerged recently as a novel class of green solvents and a prospective solution for most of the problems related to the reaction media in biocatalytic transformations, being nontoxic, biodegradable, and biocompatible. In this study, we present a new route for the enzymatic esterification of carbohydrate polyols using a reactive NADES (R-NADES) as reaction medium, acting both as a solvent and as a reagent pool. Binary hydrophilic R-NADES consisting of choline chloride (ChCl) as hydrogen bond acceptor (HBA) and sugar alcohols (i.e., D-sorbitol, xylitol, D-arabitol) as hydrogen bond donors (HBD) were prepared and characterized to determine the freezing temperature (Tf), the thermal stability and the viscosity-temperature profile. All polyol-based R-NADES were stable fluids at temperatures up to 300C and showed low viscosities between 40-80C, that is the optimal temperature range for lipase-catalyzed reactions. Polyol-based R-NADES were compatible reaction media for a number native and immobilized lipases. Lipase B from Candida antarctica immobilized on acrylic resins (LAR) showed significant esterification activity and high thermal stability in tested R-NADES, maintaining more than 90% of activity upon incubation at 70C for 72 hours. Moreover, LAR effectively catalyzed the synthesis of lauryl esters of carbohydrate polyols in the binary ChCl/polyol R-NADES tested and consequently, the synthesis of lauryl esters of D-arabitol, xylitol and D-sorbitol was successfully achieved. The design of experiments for D-arabitol esterification with lauric acid (LA) catalyzed by LAR revealed that enzyme load and temperature are decisive reaction parameters for conversion increase. Preparative reaction at optimal conditions achieved 80% conversion of lauric acid after 24 h. The major product was identified as 1,5-dilauryl-D-arabitol based on ESI-MS, FTIR, 1D- and 2D-NMR. The 1,5-DLDA product was isolated in 95 % yield. Minor amounts of monolauryl-D-arabitol were detected by RP-HPLC, showing an almost quantitative conversion into the diester. At similar conditions, 1,5-dilauryl-xylitol (1,5-DLX) and 1,6-dilauryl-D-sorbitol (1,6-DLS) were obtained, at a LA conversion of 56 mol% and 62 mol%, respectively. The results are unprecedented in literature reports and open the way to further develop green processes for the enzymatic synthesis of glycolipids. This work was supported by a grant of the Romanian Ministry of Education and Research, CCCDI-UEFISCDI, project number PN-III-P4-ID-PCE-2020-2177, within PNCDI III, contract number PCE 157/2021. |
#1184 | Exploring mass photometry for monitoring hydrodynamic properties of enzymes |
|
| Presenting author: | Saniye Gul KAYA of MOLECULAR ENZYMOLOGY, GRONINGEN BIOMOLECULAR SCIENCES AND BIOTECHNOLOGY INSTITUTE, UNIVERSITY OF GRONINGEN |
| Corresponding author: | Katarzyna M. TYCH of SINGLE MOLECULE BIOPHYSICS, GRONINGEN BIOMOLECULAR SCIENCES AND BIOTECHNOLOGY INSTITUTE, UNIVERSITY OF GRONINGEN |
| Other authors: | Yulia D. YANCHEVA of SINGLE MOLECULE BIOPHYSICS, GRONINGEN BIOMOLECULAR SCIENCES AND BIOTECHNOLOGY INSTITUTE, UNIVERSITY OF GRONINGEN Marco W. FRAAIJE of MOLECULAR ENZYMOLOGY, GRONINGEN BIOMOLECULAR SCIENCES AND BIOTECHNOLOGY INSTITUTE, UNIVERSITY OF GRONINGEN Matthias HEINEMANN of MOLECULAR SYSTEMS BIOLOGY, GRONINGEN BIOMOLECULAR SCIENCES AND BIOTECHNOLOGY INSTITUTE, UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enhanced enzyme diffusion / mass photometry / enzyme catalysis / |
| Purpose: | The growth and division of biological cells are crucial for life, yet the maximum rate of microbial cell growth is limited, and the reason for this is not fully understood [1]. Understanding this limit is important for biotechnological applications. Research suggests that cells, like machines, are unable to function beyond a critical rate of Gibbs energy dissipation, and this limit may be determined by the spatial displacement of certain enzymes during catalysis [2]. This displacement could lead to enhanced enzyme diffusion and cellular agitation, potentially disrupting biomolecular functions and limiting cell growth [3]. Enhanced enzyme diffusion (EED) is a debated phenomenon where some enzymes diffuse faster than expected from Brownian motion in the presence of their substrates and/or inhibitors [4]. Most experiments confirming EED have used fluorescence correlation spectroscopy (FCS), which requires fluorescent labelling, and is an ensemble averaging technique. Chen et al. [5] discuss an artefact in FCS caused by fluorescent labelling, which has been incorrectly interpreted as EED. Thus, there is a need for other suitable techniques to study the hydrodynamic properties of enzymes while being catalytically active. We explored the use of a relatively new technique called mass photometry to monitor the hydrodynamic properties of several enzymes. The technique only requires small samples (µL range), is non-invasive, is a label-free approach, while enabling measuring protein masses. Results obtained using this technique with several prototype enzymes will be presented. |
| References: | [1] Nielsen J, Keasling JD. Engineering Cellular Metabolism. Cell. 2016. doi:10.1016/j.cell.2016.02.004 [2] Niebel B, Leupold S, Heinemann M. An upper limit on Gibbs energy dissipation governs cellular metabolism. Nat Metab. 2019;1(1):125-132. doi:10.1038/s42255-018-0006-7 [3] Xu, M., Rogers, W. B., Ahmed, W. W., & Ross, J. L. (2020). Comparison of different approaches to single-molecule imaging of enhanced enzyme diffusion. arXiv preprint arXiv:2012.15424. [4] Hari S. Muddana et al. “Substrate catalysis enhances single-enzyme diffusion”. In: Journal of the American Chemical Society 132.7 (2010). issn: 00027863. doi: 10.1021/ja908773a. [5] Zhijie Chen et al. “Single-molecule diffusometry reveals no catalysis-induced diffusion enhancement of alkaline phosphatase as proposed by FCS experiments”. In: Proceedings of the National Academy of Sciences of the United States of America 117.35 (2020). issn: 10916490. doi: 10.1073/pnas.2006900117. |
#1188 | Directed evolution of an artificial alkane hydroxylase based on a human carbonic anhydrase protein |
|
| Presenting author: | Iori MORITA of UNIVERSITÄT BASEL |
| Corresponding author: | Thomas WARD of UNIVERSITÄT BASEL |
| Other authors: | Elias SALVISBERG of UNIVERSITÄT BASEL Adriana FARAONE of ICIQ Anastassia VOROBIEVA of VIB-VUB CENTER FOR STRUCTURAL BIOLOGY Bruno CORREIA of INSTITUTE OF BIOENGINEERING, EPFL |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzyme / peroxygenase / Fe-TAML / human carbonic anhydrase |
| Purpose: | Selective hydroxylation of alkanes enables a step-economical functionalization of inert C–H bonds. In this context, transition metal-catalyzed C-H functionalization has shown great potential, as well as complementary to enzymes [1]. However, chemo-, regio- and stereo-selective C-H hydroxylation remains among the most challenging reactions. In this context, we have previously reported an artificial alkane hydroxylase (ArAlHyd) taking advantage of the catalytic activity of Fe-TAML (tetraamide macrocyclic ligand) catalysts [2] and a protein scaffold providing an evolvable second coordination sphere [3]. Herein, we report our efforts to develop an ArAlHyd based on a thermostable mutant of human carbonic anhydrase (hCAIIts). We have designed a highly active Fe-TAML complex, equipped with a sulfonamide, thus ensuring its incorporation within hCAIIts, acting as a host protein. The structure of the cofactor was further optimized to ensure that the cofactor binds with hCAIIts firmly with sub-micromolar affinity. The hCAIIts scaffold was also optimized by insertion of a loop in the proximity of the cofactor. The optimized ArAlHyd showed improved hydroxylation activity than the free cofactor. We will present our efforts to optimize the activity by directed evolution further relying on cell-lysate assays. |
| References: | [1] Milan, M.; Bietti, M.; Costas, M. Chemical Communications 2018, 54, 9559-9570. [2] Collins, T. J.; Ryabov, A. D. Chemical Reviews 2017, 117, 9140-9162. [3] Serrano-Plana, J.; Rumo, C.; Rebelein, J. G.; Peterson, R. L.; Barnet, M.; Ward, T. R. Journal of the American Chemical Society 2020, 142, 10617-10623. |
| Figures: | 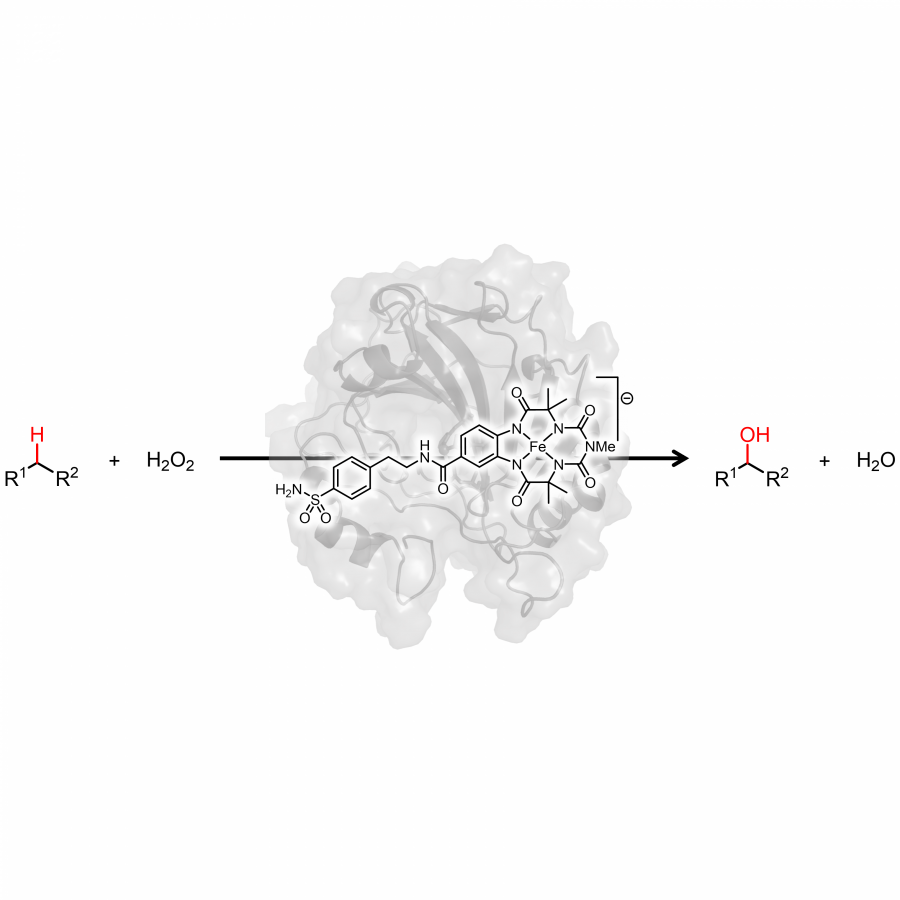 Figure 1 Hydroxylation of alkanes with H2O2 catalyzed by ArAlHyd |
#1198 | Structure-based incorporation of non-canonical amino acids toward the engineering of artificial metalloenzymes |
|
| Presenting author: | Emeline VERNHES of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) |
| Other authors: | Régis FAURÉ of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) Jérémy ESQUE of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) Gianluca CIOCI of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) Jean-Guy BERRIN of BIODIVERSITÉ ET BIOTECHNOLOGIE FONGIQUES Isabelle ANDRÉ of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) Sébastien NOUAILLE of TOULOUSE BIOTECHNOLOGY INSTITUTE (TBI) |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloprotein / non-canonical amino acid / protein engineering / crystal structure |
| Purpose: | Metals are present in almost half of all enzymes to fulfill structural and/or catalytic functions. Metalloenzymes have been successfully engineered for biocatalysis, often by modifying the substrate or metal specificity of a natural metalloenzyme [1]. An alternative to these enzymes consists in the insertion of a metal complex into a versatile protein scaffold able to accommodate various substrates and/or reactions [2]. In this study, the goal is to preserve the natural enzyme’s substrate specificity while adapting new-to-nature metal-based reactivity through active site remodelling. However, designing a metal-binding site is not trivial as it often requires a proper positioning of several amino acid residues (usually a histidine brace motif, plus an additional residue) to ensure an efficient coordination of both the metal ion and the ad hoc substrate. To circumvent this obstacle, bipyridyl-alanine (BpyA), a metal-chelating non-canonical amino acid (ncAA), was used. NcAA incorporation can be achieved by reassigning a nonsense codon to a ncAA by the use of an orthogonal amino-acyl tRNA synthetase (aaRS)/tRNA pair [3]. Despite some achievements over the last 15 years, this approach still suffers from highly variable and often low incorporation efficiencies, which can be deterrent. Based on the historical and widely used pEVOL incorporation system [4], we developed the pINS systems by altering the tRNA and aaRS expression levels. Several amino acid positions identified by molecular modelling were selected to incorporate BpyA in the vicinity of the catalytic site of our model enzyme. Using the pINS system, both BpyA incorporation efficiency and protein production were improved with success, reaching production levels comparable to the wild-type enzyme. For each targeted position, the proper incorporation of BpyA was confirmed by mass spectrometry and X-ray crystallography. Interestingly, the reorientation of a neighboring amino acid side chain was observed to obtain a complete metal ion coordination in the absence of a bound substrate. Moreover, the overall 3D structure and thermal stability of the mutants were found to be poorly affected by the introduction of BpyA. Future experiments will investigate the substrate-binding properties of the BpyA mutants and their catalytic potential. Acknowledgments: this work was performed in the framework of the Carnot 3BCAR project NeoZYM (2021-2023). |
| References: | [1] A. S. Klein, C. Zeymer, Protein Eng Des Sel 2021, 34, gzab003. [2] A. G. Jarvis, Curr Opin Chem Biol 2020, 58, 63-71. [3] S. Smolskaya, Y. A. Andreev, Biomolecules 2019, 9, 255. [4] T. S. Young, I. Ahmad, J. A. Yin, P. G. Schultz, J Mol Biol 2010, 395, 361-374. |
#1201 | A Design and Evolution of Artificial Radical Cyclase Enables Bicyclic Terpenoid Scaffold Construction via M-HAT reaction |
|
| Presenting author: | Dongping CHEN of UNIVERSITY OF BASEL |
| Corresponding author: | Thomas WARD of UNIVERSITY OF BASEL |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzyme / dual anchoring / transfer hydrogenation / directed evolution |
| Purpose: | Artificial metalloenzymes result from anchoring a metal cofactor within a host protein. Such hybrid catalysts combine the selectivity and specificity of enzymes with the versatility of (abiotic) transition metals to catalyze new-to-Nature reactions in an evolvable scaffold. To improve the localization of an arylsulfonamide-bearing iridium-pianostool catalyst within human carbonic anhydrase II (hCAII) for the enantioselective reduction of prochiral imines, we introduced a covalent linkage between the host and the guest. Herein we show that a judiciously positioned cysteine residue reacts with a p-nitropicolinamide ligand bound to iridium to afford an additional sulfonamide covalent-linkage. Three rounds of directed evolution, performed on the dually-anchored cofactor, led to improved activity and selectivity for the enantioselective reduction of harmaline (up to 97 % ee (R) and > 350 turnovers on a preparative scale). To evaluate the substrate scope, the best hits of each generation were tested with eight substrates. X-ray analysis, carried out at various stages of the evolutionary trajectory, was used to scrutinize i) the nature of the covalent linkage between the cofactor and the host as well as ii) the remodeling of the substrate-binding pocket. |
| References: | [1] Stein, A.; Chen, D.; Igareta, N. V.; Cotelle, Y.; Rebelein, J. G.; Ward, T. R. ACS Central Science 2021, 7 (11), 1874-1884. [2] Rebelein, J. G.; Cotelle, Y.; Garabedian, B.; Ward, T. R. ACS Catalysis 2019, 9 (5), 4173-4178. [3] Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M. M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J. C.; Ward, T. R. Chemical Reviews 2018, 118 (1), 142-231 |
| Figures: | 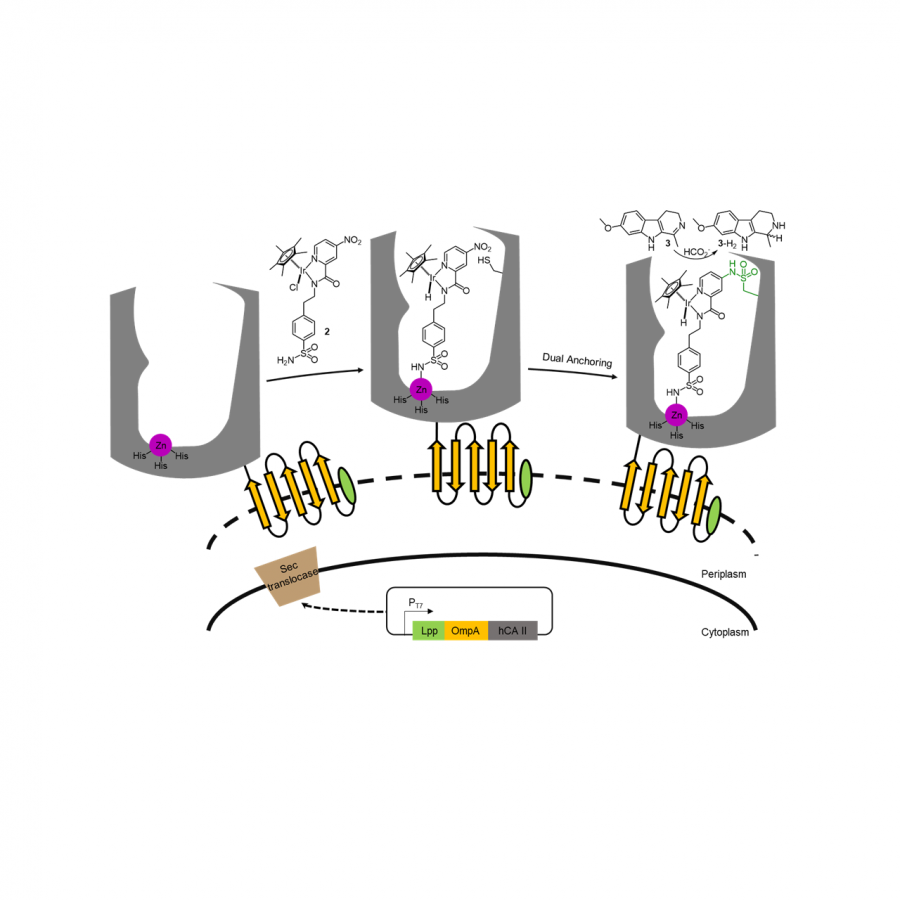 In cellulo assembly of artificial transfer hydrogenases (ATHases). HCAII is displayed on the cell surface of E. coli by fusion to a truncated lipoprotein and the outer membrane protein A (Lpp-OmpA). Following incubation with the sulfonamide-bearing IrCp* piano-stool complex 2, the resulting ArM catalyzes the enantioselec 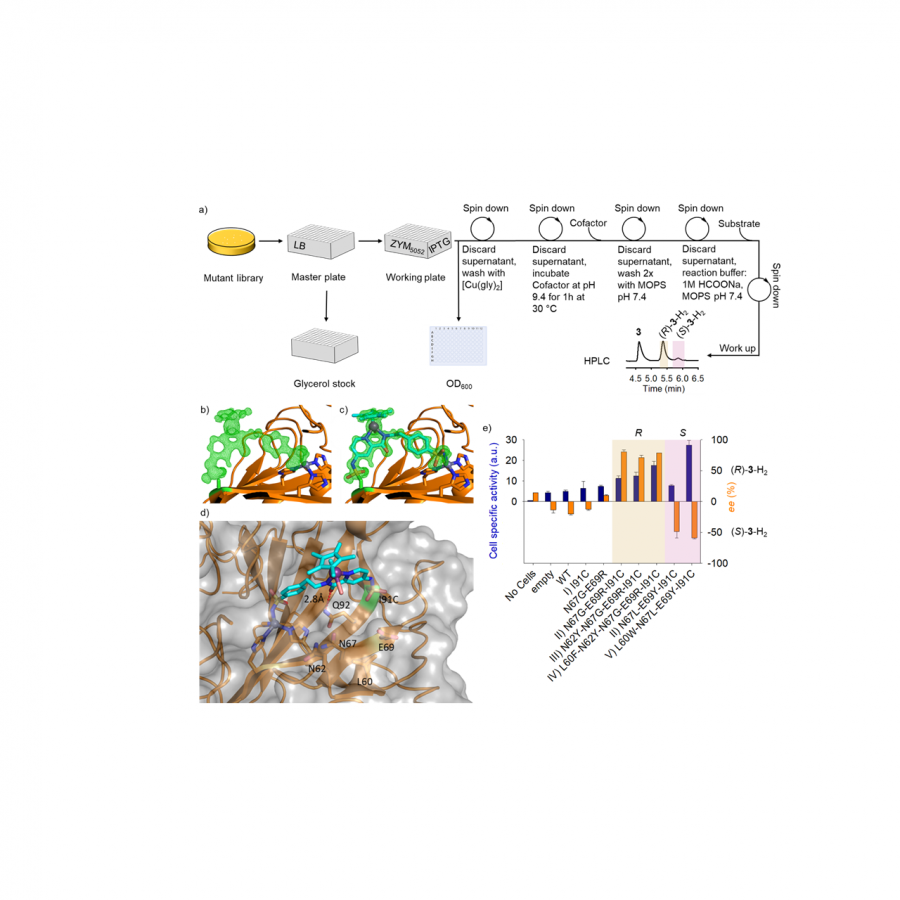 Directed evolution of ArMs resulting from the dual anchoring of cofactor 2 a) A streamlined screening workflow was developed that enables expression of hCAII mutants and subsequent evaluation of their activity and selectivity in a 96 well-plate format. See SI for a detailed description of the workflow. b-d) X-ray characterizatio |
#1204 | Substrate scope and selectivity of hydroxysteroid dehydrogenases in the biocatalyzed reduction of 1,2-diketones |
|
| Presenting author: | Daniela MONTI of SCITEC - CNR |
| Other authors: | Chiara TOGNOLI of UNIVERSITÀ DEGLI STUDI DI MILANO Susanna BERTULETTI of SCITEC - CNR Erica Elisa FERRANDI of SCITEC - CNR Marta VANONI of SCITEC - CNR Sergio RIVA of SCITEC - CNR Ivan BASSANINI of SCITEC - CNR |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | hydroxysteroid dehydrogenases / selectivity / substrate promiscuity / 1,2-diketone reduction |
| Purpose: | The biocatalytic reduction of 1,2-diketones to give chiral α-hydroxy ketones and/or vicinal diols has been previously studied using different alcohol dehydrogenases (ADHs) from various sources [1]. In our recent works, we have investigated the substrate promiscuity of a specific subfamily of ADHs, i.e., hydroxysteroid dehydrogenases (HSDHs), NAD(P)H-dependent oxidoreductases acting on their natural substrates, such as neutral steroids, bile acids and other steroid derivatives, with exquisite stereo- and regioselectivity [2]. These studies showed that selected HSDHs from our in-house collection, including novel biocatalysts from extreme environments identified by (meta)genome mining, can accept a wide range of different substrates, for example α-keto esters, tetralones and the racemic synthon Wieland-Miescher ketone [3]. In this work, we have extended our investigation of HSDHs promiscuity to a set of representative 1,2-diketones, i.e., the symmetric aliphatic/aromatic diketones 1 and 2 and the asymmetric synthon 1-phenyl-1,2-propanedione 7 (Scheme 1). The reactions were set up in the presence of a B. megaterium glucose dehydrogenase (BmGDH)-glucose system for NAD(P)H cofactor regeneration and analyzed at scheduled times to assess both substrate conversion and selectivity. Additionally, docking studies were carried out to get a deeper insight in the stereochemistry of 1,2-diketones reduction catalyzed by selected HSDHs. |
| References: | [1] Zhou, J.; Xu, G.; Ni, Y. ACS Catal. 2020, 10, 10954-10966. [2] Ferrandi, E. E. et al., Eur. J. Org. Chem. 2020, 2020 (29), 4463-4473. [3] a) Bertuletti, S. et al., Adv. Synth. Catal. 2020, 362 (12), 2474-2485; b) Bertuletti, S. et al., Eur. J. Org. Chem 2021, 2021 (29), 3992-3998; c) Nasti, R. et al., ChemBioChem 2022, 23 (8), e202200105; d) Ferrandi, E. E. et al., Int. J. Mol. Sci. 2022, 23 (20), 12153. |
| Figures: | 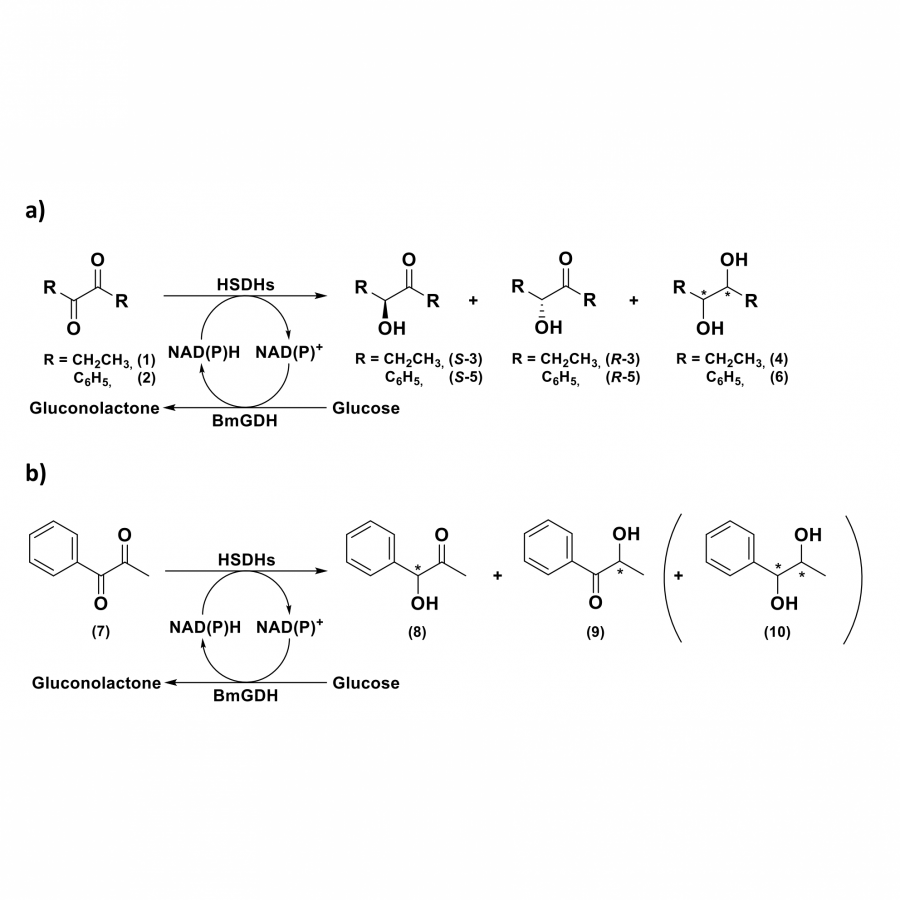 Scheme 1 HSDHs-catalyzed reduction of either symmetric (a) or asymmetric (b) 1,2-ketones. |
#1205 | Unlocking distal mutations in an artificial enzyme |
|
| Presenting author: | Fabrizio CASILLI of UNIVERSITY OF GRONINGEN |
| Other authors: | Gerard ROELFES of UNIVERSITY OF GRONINGEN |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | protein engineering / allostery / multi-sequence alignment / distal mutations |
| Purpose: | In Nature, enzymes have evolved to fulfill their specific biological function by fine-tuning not only the active site, but also the whole structure ensemble. This can be observed in the generation of allostery in many biocatalysts. However, the design of novel artificial enzymes is usually limited to only optimizing a rudimentary active site, which is fine-tuned to properly accommodate new chemistries. The importance of long-distance interactions is usually overlooked due to the inherent difficulty of sampling the complex protein sequence space. Recently, an artificial enzyme comprising a non-canonical para-aminophenylalanine (pAF) buried in the Lactococcal multidrug resistance regulatory protein (LmrR) pocket (LmrR_pAF) was found to catalyze a model hydrazone formation reaction via an iminium ion intermediate [1]. A previous directed evolution campaign yielded the triple mutant LmrR_pAF_RMH, which showed 57-fold higher catalytic efficiency (kcat/KM) than the initial template [2]. The evolution campaign was focused on residues surrounding the catalytic aniline side chain; favorable interactions in the outer sphere were not investigated. A small library of point mutations scattered outside the artificial active site was built based on information from a combination of computational tools and multiple sequence alignment (MSA). The convenient colored read-out provided by this reaction allowed an easy screening of the activity profile in 96-well format. This led to the identification of two mutations positioned at more than 11 Å of distance (Cα-Cα) from the catalytic ncAA, both showing 75% improvement from the parent enzyme. A closer investigation on the interactions underpinning the positions identified may provide a clearer understanding of the allosteric interaction network within LmrR. |
| References: | [1] Drienovska, I., Mayer, C., Dulson, C., and Roelfes, G., nat. chem. 2018, 10(9), 946-952 [2] Mayer, C., Dulson, C., Reddem, E., Thunnissen, A.-M. W. H., and, Roelfes, G., angew. chemie int. ed. 2019, 58(7), 2083-2087 |
#1206 | ENANTIOSELECTIVE SYNTHESIS OF BETA-HYDROXYSULFIDES USING KETOREDUCTASES |
|
| Presenting author: | Ariane MATTANA of UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Daniele CASTAGNOLO of UNIVERSITY COLLEGE LONDON |
| Other authors: | Fei ZHAO of UNIVERSITY COLLEGE LONDON James FINNIGAN of PROZOMIX Simon CHARNOCK of PROZOMIX |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Chiral sulfur compounds are an important class of chemicals which find applications across multiple industries. Enantiomerically pure sulfur compounds are key structural motifs in organic chemistry, in pharmaceutical ingredients as well as in natural compounds responsible for the aroma and flavours of many foods and beverages. Due to the importance of sulfur motifs in organic chemistry and in the food and pharmaceutical industries, there is a lot of focus on efficient approaches to their synthesis [1]. The number of chemical reactions catalysed by enzymes has increased exponentially over the last century [2]. Biocatalysis offers sustainable and cost-effective methods for the synthesis of chemicals and drugs and provides advantages over traditional chemo-catalytic methods. We recently reported the synthesis of enantiomerically pure chiral beta-hydroxysulfoxide compounds bearing a stereocentre at the C-O bond using ketoreductase enzymes [3-4]. Moreover, we have also reported the enantioselective synthesis of beta-hydroxysulfoxides bearing a stereocentre at the C-S bond [3]. Namely, two ketoreductases catalysed the enantioselective synthesis of the latter compounds with opposite enantioselectivites by dynamic kinetic resolution (DKR) of racemic alpha-thioaldehydes [3]. |
| References: | [1] M. B. Marakalala, E. M. Mmutlane and H. H. Kinfe. 2018. Beilstein J. Org. Chem. 14, 1668-1692. [2] M. Hall. 2021. RSC Chem. Biol. 2, 958-989. [3] F. Zhao, K. Lauder, S. Liu, J. D. Finnigan, S. B. R. Charnock, S. J. Charnock, D. Castagnolo. 2022. Angew. Chem., Int. Ed. 61, 31. [4] K. Lauder, A. Toscani, Y. Qi, J. Lim, S.J. Charnock, K. Korah, D. Castagnolo, 2018 Angew. Chem., Int. Ed. 57, 5803-5807. |
#1210 | a high-throughput screening assay for alkane halogenases |
|
| Presenting author: | Hannes MEINERT of UNIVERSITY OF GREIFSWALD |
| Corresponding author: | Uwe T. BORNSCHEUER of UNIVERSITY OF GREIFSWALD |
| Other authors: | Christoffel P. S. BADENHORST of UNIVERSITY OF GREIFSWALD Hannah MEIER of UNIVERSITY OF GREIFSWALD |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alkane Halogenase / HaloTag / FRET / HTS Assay |
| Purpose: | Alkane halogenases are an interesting class of enzymes due to their importance in late-stage modification of organic molecules. However, a huge disadvantage is their limited substrate scope.[1] We envisioned a concept for a high-throughput screening assay that could have the potential to drastically increase their substrate scope. The assay is based on the utilization of catalytically inactive haloalkane dehalogenases which bind their substrates irreversibly.[2] By utilizing these so called HaloTag proteins[3] in combination with a non-canonical fluorescent amino acid (flAA) and a fluorescent ligand a FRET effect can be employed to screen for halogenases that produce the desired haloalkane. In this context a positive signal would show the fluorescence of the flAA and a negative signal would be caused by the quenching of this fluorescence through the ligand which is added in a second incubation step (see Figure). At this stage of the project a suitable combination of amino acid and fluorescent ligand that quenches the flAA through FRET were identified. Currently, a position in the catalytically inactive haloalkane dehalogenase for the introduction of the fluorescent amino acid is searched for. The incorporation at this position should not impair ligand binding but should also be as close as possible to the active site that optimal quenching upon ligand binding occurs. |
| References: | [1] Romero et al., Angew. Chem. Int. Ed., 2021, 60, 16824-16855 [2] Pries et al., J. Biol. Chem., 1995, 270, 10405-10411 [3] Marques et al., JACS Au, 2022, 2, 1324-1337 |
| Figures: | 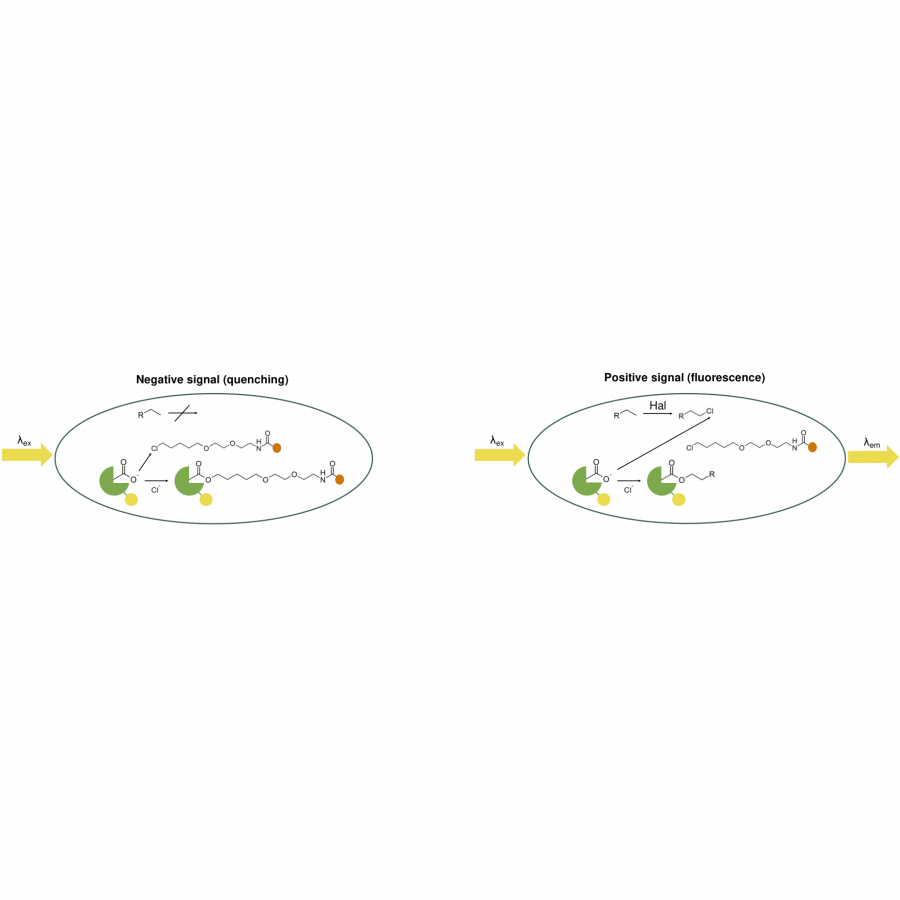 Assay Concept The negative signal (left) results when no haloalkane is formed due to the absence of an active halogenase resulting in the quenching of the flAA
(yellow) of the HaloTag (green) through the fluorescent ligand (orange). A positive signal (right) results fr |
#1219 | Environmentally friendly detoxification of rifampicin by bacterial CotA-laccase and hydrogen peroxide |
|
| Presenting author: | Paulo DURÃO of INSTITUTO DE TECNOLOGIA QUÍMICA E BIOLÓGICA |
| Other authors: | Patrícia BORGES of INSTITUTO DE TECNOLOGIA QUÍMICA E BIOLÓGICA Peter KIS of INSTITUTO DE TECNOLOGIA QUÍMICA E BIOLÓGICA Ivo CHELO of CE3C – CENTRE FOR ECOLOGY, EVOLUTION AND ENVIRONMENTAL CHANGES Rita VENTURA of INSTITUTO DE TECNOLOGIA QUÍMICA E BIOLÓGICA Lígia MARTINS of INSTITUTO DE TECNOLOGIA QUÍMICA E BIOLÓGICA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | antibiotic resistance / rifampicin / enzymatic degradation / environmentally-friendly |
| Purpose: | Antibiotics are a novel pollutant accumulating in our rivers and wastewaters ultimately leading to bacterial antibiotic resistance, a worldwide problem to which there is no current solution. We have developed a novel and environmentally friendly two-step process to transform the antibiotic rifampicin (RIF), a first-line antibiotic against tuberculosis, into a mix of non-antimicrobial compounds. The process involves a full oxidation of RIF through a reaction carried over by bacterial CotA-laccase enabling a subsequent bleaching step carried over by hydrogen peroxide into a mix of colourless final products. We have refined X-ray crystal structures of CotA-laccase soaked with rifampicin showing the antibiotic bound to the enzyme contributing to the elucidation of the molecular mechanism. Growth curves of a susceptible E. coli K12 MG1655 and four RIF resistant strains in the presence of 100 µg/mL of the final degradation products and measurements of the minimum inhibitory concentration (MIC) for the same strains show that final products are no longer an anti-microbial. Competitive fitness assays between susceptible and RIF resistant bacteria show that the final products do not exert selection pressure in favour of the resistant strains. Moreover, bioassays with the model organism C. elegans suggest that final products are not toxic to eukaryotic organisms. Overall, our results show that we have developed a robust and environmentally friendly process to effectively remediate rifampicin from antibiotic contaminated environments. |
| References: | [1] Durao, P., Balbontin, R., Gordo, I., 2018. Trends Microbiol. 26, 677-691. [2] Gullberg, E., Cao, S., Berg, O.G., Ilbäck, C., Sandegren, L., Hughes, D., Andersson, D.I., 2011. PLoS Pathog. 7, e1002158. [3] Bilal, M., Ashraf, S.S., Barceló, D., Iqbal, H.M.N., 2019. Sci. Total Environ. 691, 1190-1211. [4] Martins, L.O., Durao, P., Brissos, V., Lindley, P.F., 2015. Cell. Mol. Life Sci. 72, 911-922. |
| Figures: | 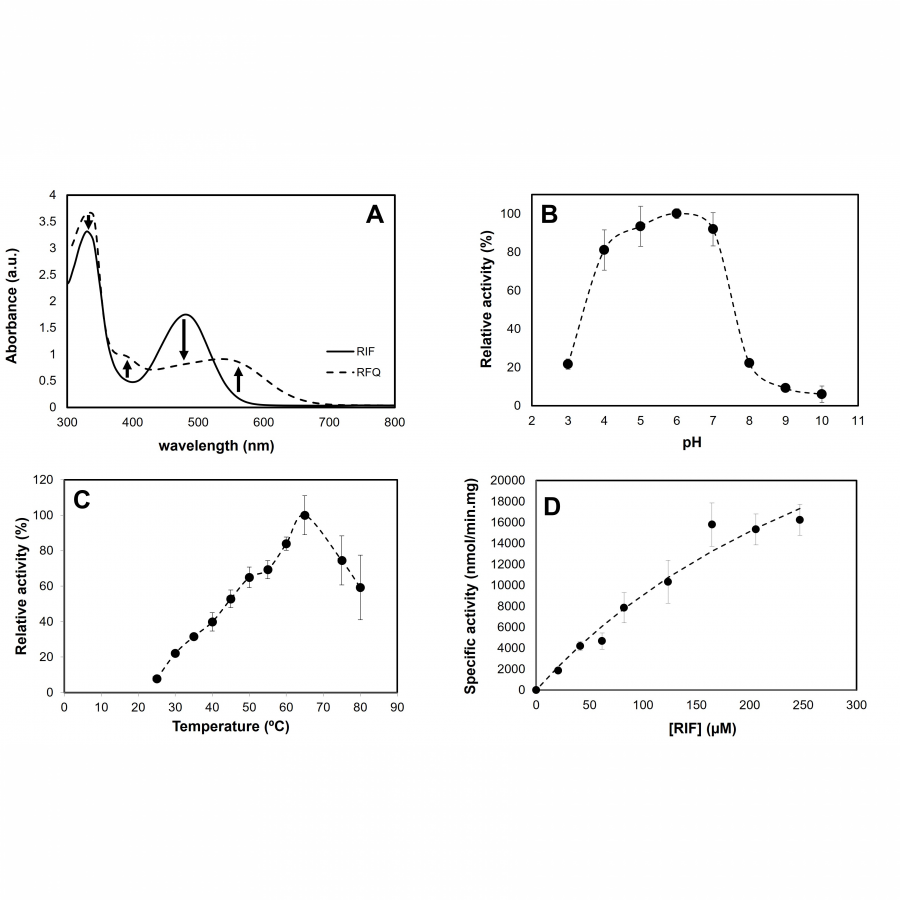 CotA oxidation of rifampicin A) Absorbance spectra of the of rifampicin (full line) and the product of the enzymatic oxidation by CotA laccase (dashed line). B) pH profile C) Optimal temperature D) Michaelis-Menten fit of the reaction. 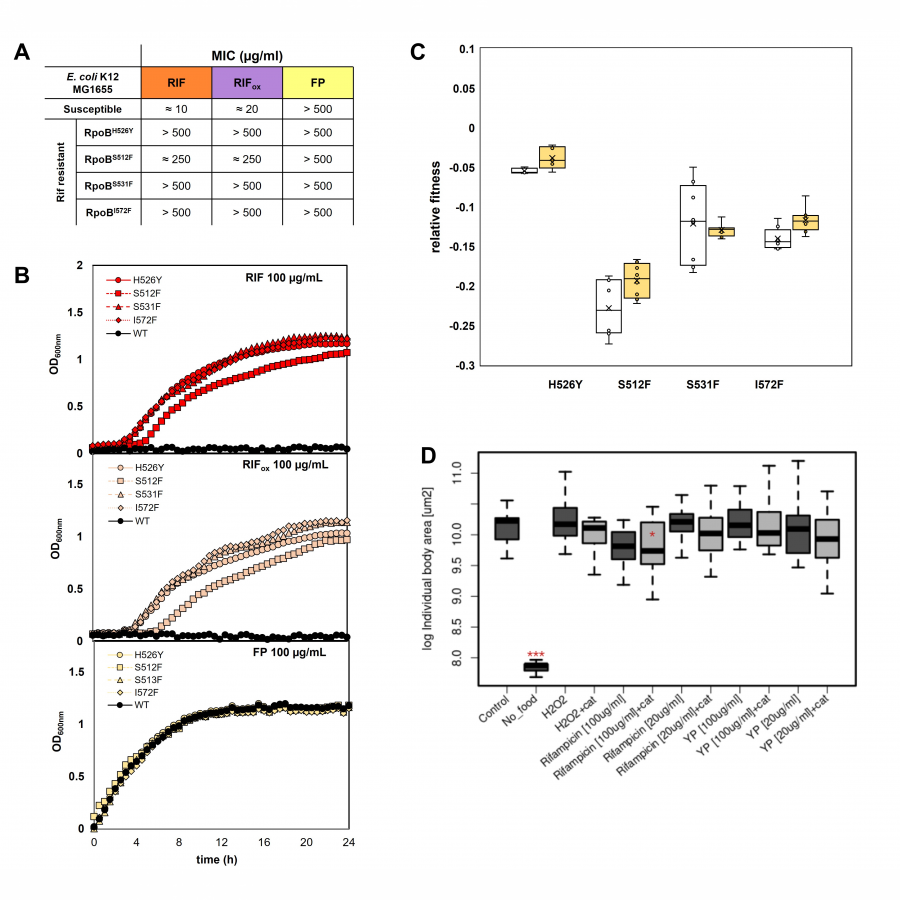 Assessment of the antimicrobial and toxicity properties of the degradation products MICs (A), growth curves (B) and competitive assays (C) were performed in the presence of degradation compounds using susceptible and RIF resistant E. coli MG1655 strains. D) Toxicity of final products in a bioassay with C. elegans was also assessed. |
#1220 | Side to side comparison of a novel wild-type polyesterase from Deinococcus maricopensis with LCC-ICCG indicates promising degradation of semi-crystalline post-consumer PET |
|
| Presenting author: | Konstantinos MAKRYNIOTIS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Corresponding author: | Evangelos TOPAKAS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | Efstratios NIKOLAIVITS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS Christina GKOUNTELA of NATIONAL TECHNICAL UNIVERSITY OF ATHENS Stamatina VOUYIOUKA of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | PET degradation / PETase / novel polyesterase / Deinococcus maricopensis |
| Purpose: | Synthetic plastics have become an integral part of modern life due to their versatile applications. However, their increased usage has resulted in a corresponding rise in waste generation, as their durability results in accumulation. Globally, according to recent studies, plastic waste primarily consists of polyesters, following the polyolefins, with PET being the most produced polyester resin in 2021 [1,2]. The majority of PET waste ends up in landfills, ultimately making its way into terrestrial and marine environments, as it presents low susceptibility in biodegradation because of the presence of aromatic components in its backbone [3–6]. Aiming to the hydrolysis of PET ester bonds, resulting to its degradation, recent advancements have led to the discovery of novel PET-degrading enzymes, such as the metagenome-derived LC-Cutinase (LCC) and its thermostable variant, LCC-ICCG, which demonstrates remarkable efficiency in depolymerizing amorphized PET waste [7,8]. As a result, LCC-ICCG is the only enzyme industrialized for PET biorecycling [9]. Concerning high crystallinity PET, it seems that LCC-ICCG presents room for improvement, as it follows the main trend of most well-known PETases whose effectiveness in PET degradation is significantly limited at increased crystallinity grades [10,11]. Hereby, we report the discovery of DmPETase, a novel wild-type polyesterase from the bacterium Deinococcus maricopensis, which depolymerase activity was tested on a broad range of synthetic materials, including PET and PU, as well as biodegradable polymers. Emphasizing on PET, our research focused on the effect of crystallinity on its degradation. Enzymatic reactions, with DmPETase and LCC ICCG, were conducted on virgin, amorphous, and semi-crystalline PET powder, as well as two types of post-consumer water bottles in their original and powdered form. DmPETase's unique characteristics form its own sub-branch on a phylogenetic tree of characterized PET-degrading enzymes, while it displays a dissimilar electrostatic surface to well-known benchmark PETases (Figure 1). Its biochemical characterization depicted a thermostable cutinase-like enzyme with the ability to degrade various synthetic polymers, mainly PCL and PBS. Notably, DmPETase was proved capable of degrading crystalline compartments of PET bottles, as well as semi-crystalline PET powder at the same level as LCC-ICCG , while getting less affected by PET crystallinity grade (Figure 2). These novel findings demonstrate the potential of DmPETase as a promising enzymatic platform for protein engineering, specifically aiming in enhancing PET degradation rate through modifications to its active site, given the enzyme's natural high affinity to semi-crystalline PET. |
| References: | [1] R. Geyer, Sci. Adv. 3 (2017) e1700782 [2] PlasticEurope: Plastics - The Facts 2022, Plastics - the Facts 2022, (2022). [3] L. Yang, Sci. Total Environ. 780 (2021) 146546 [4] P.E. Redondo-Hasselerharm, Sci. Adv. 6 (2020) eaay4054 [5] K. Cverenkárová, Life. 11 (2021) 1349 [6] Y.H.V. Soong, Bioengineering. 9 (2022) 98. [7] V. Tournier, Nature. 580 (2020) 216-219 [8] S. Sulaiman, Appl. Environ. Microbiol. 78 (2012) 1556-1562. [9] S. Thiyagarajan, RSC Adv. 12 (2022) 947-970 [10] V. Tournier, Chem. Rev. (2023) [11] Z. Ding, J. Hazard. Mater. 453 (2023) 131386 |
| Figures: |  (A) Superimposition of DmPETase (green) with LCC-ICCG (blue-PDB ID 7VVE) and IsPETase (pink- PDB ID 6EQH), highlighting important residues participating in structural features (cysteines for disulfide bonds) or catalytic and substrate binding. Ligand 2-hy 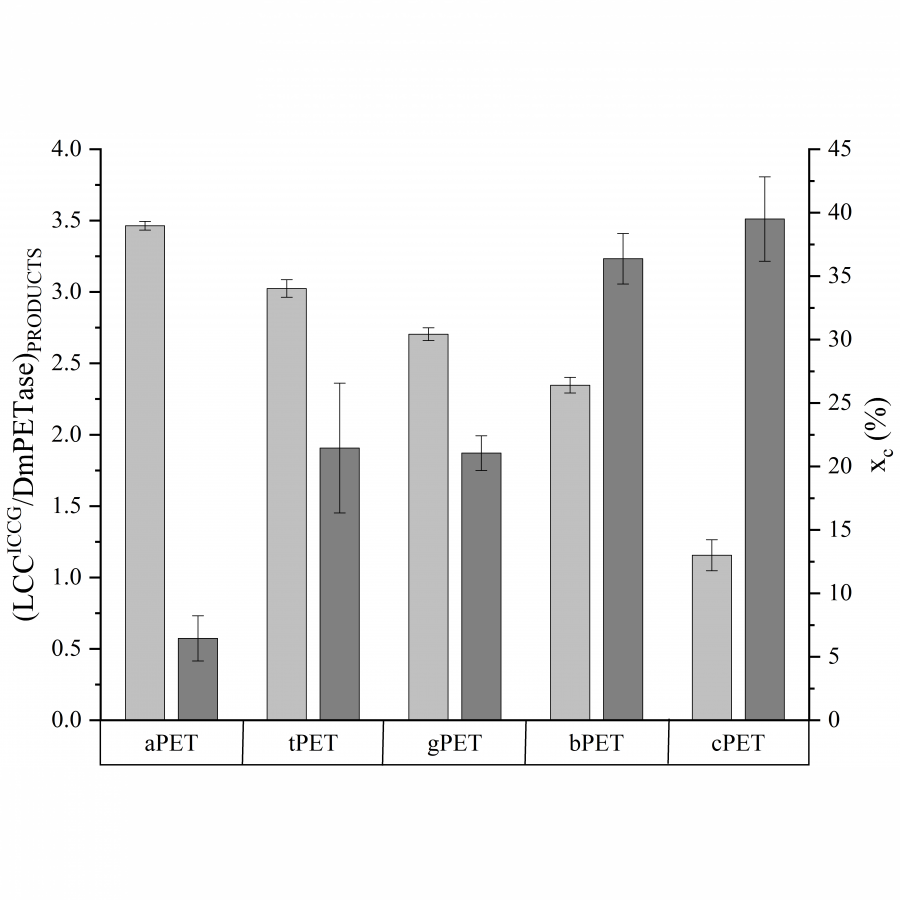 Ratio of hydrolysis products released by LCC-ICCG to DmPETase (light grey) of PET powders of variable crystallinities (dark grey). PET powders are: aPET- amorphous PET of Xc 5 %, tPET-transparent PET bottle of Xc 21 %, gPET-green PET bottle of Xc 21 %, cP |
#1222 | Access to thermostable enzymes and their application in flow biocatalysis |
|
| Presenting author: | Marius STOECKLE of KIT / IBG-1 |
| Other authors: | Martin PENG of KIT / IBG-1 Christof NIEMEYER of KIT / IBG-1 Kersten RABE of KIT / IBG-1 |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | thermostability / flow biocatalysis / protein-engineering / protein immobilization |
| Purpose: | The immobilization of biocatalysts in a continuous fluidic setup is one way to achieve compartmentalization and thus precise control over artificial reaction cascades for synthetic chemistry. We recently demonstrated the encapsulation of unmodified thermostable enzymes in a 3D printed, agarose-based thermoreversible hydrogel to enable multi-step sequential biotransformations.[1] To test the feasibility of the encapsulation strategy, we used a naturally thermostable alcohol dehydrogenase (ADH) as well as a ketoisovalerate decarboxylase (KIVD) from a mesophile organism as exemplary biocatalysts. KIVD was thermostabilized by different computational or evolutionary methods to increase the T50 value by up to 9°C.[1,2] After the successful proof-of-concept study, we further expanded the scope of this system by integrating phenacrylate decarboxylases (PAD) into this microfluidic system.[3] As an alternative for the hydrogel based immobilization strategy, thermostable enzymes can be covalently attached onto beads in a packed-bed reactor. In this context thermostable enzymes offer improved process stability and we selected a benzaldehyde lyase (BAL) as an example, since only one enzyme had been biochemically characterized before, which was rather instable.[4] To this end, we employed a computational prediction tool[5] for the identification of a novel thermostable benzaldehyde lyase and employed the enzyme for the continuous production of α-hydroxy-ketones. A homology-model based approach was used to create enzyme variants with altered substrate scope, which also showed further increased thermal stability. |
| References: | [1] M. Maier, et al. Angew. Chem. Int. Ed. 2018, 57, 5539. [2] M. Peng, et al. Biol. Chem. 2019, 400, 1519. [3] M. Peng, et al. Chem. - Eur. J. 2019, 25, 15998. [4] M. Peng, et al. ChemBioChem 2022, 23, e202100468. [5] G. Li, et al. ACS Synth. Biol. 2019, 8, 1411. |
| Figures: | 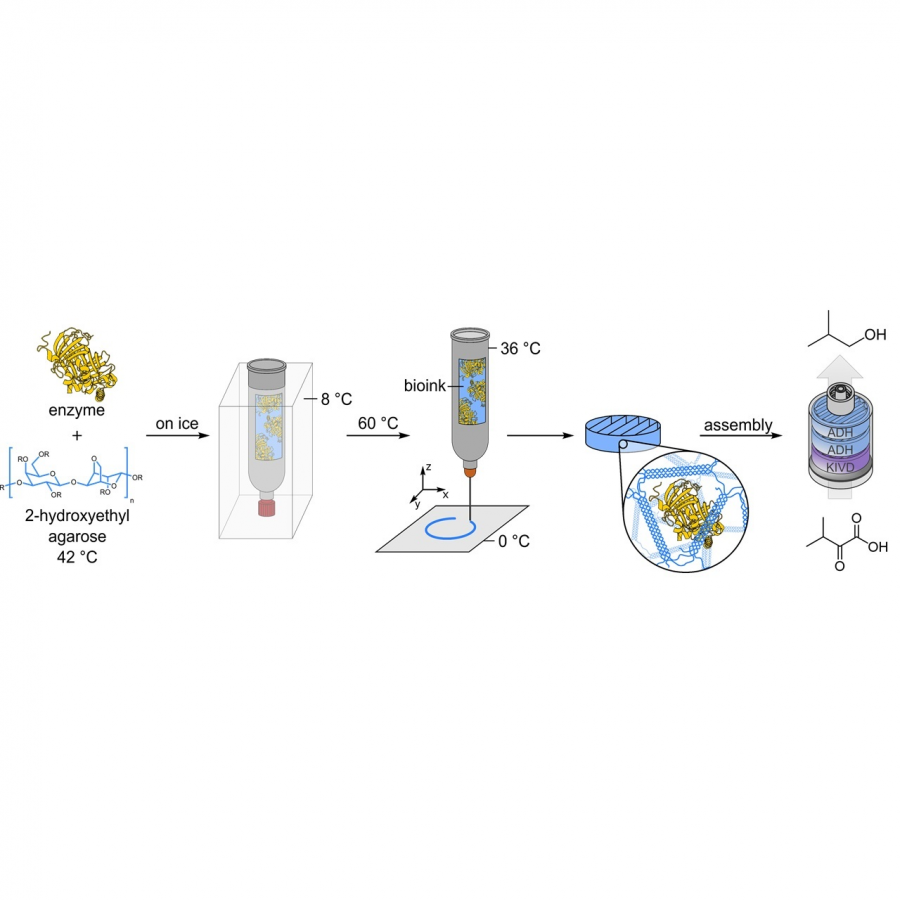 3D-Printed flow reactor modules Improved KIVD in an exclusively enzymatic conversion of α-ketoisovalerate into isobutanol together with a naturally thermostable alcohol dehydrogenase. |
#1224 | Terpene Factory |
|
| Presenting author: | Katia DUQUESNE of AIX-MARSEILLE UNIVERSITÉ |
| Other authors: | Létitia LEYDET of AIX-MARSEILLE UNIVERSITÉ Agnès AMOURIC of AIX-MARSEILLE UNIVERSITÉ Gilles IACAZIO of AIX-MARSEILLE UNIVERSITÉ |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | terpene mini-path / biocatalysis / synthetic biology / |
| Purpose: | Terpenoid are one of the most represented families of natural molecules on Earth. To date, more than 100,000 compounds [1] whose structural, biological (antibiotic, anticancer, anti-inflammatory, etc.) and physicochemical (cleaner, flavor, dye, etc.) properties hold the attention of the scientific community [2-3]. However, their access is limited because of the low available quantity by extraction from natural sources; an often expensive and laborious chemical synthesis; and long biosynthetic pathways. We have developed a new in vitro production pathway[4]: two enzymes making it possible to obtain diphosphates (DMAPP and IPP the universal precursors of terpenes), a prenyl transferase and finally the enzyme allowing cyclization. This synthesis is carried out from two alcohols with five carbon atoms in order to give different terpene compounds. By combining bioinformatics, biochemical and molecular biology approaches, we have developed this approach in vivo, by joining together two plasmids comprising the four genes necessary for this terpene pathway. Playing on the gene position, but also on the different regulatory elements (plasmid copy number, operonic organization,...), the quantity of terpenes obtained can vary: it is a question of optimizing the experimental conditions, from genetic construction to bioconversion. As a proof of concept we chose a fungal sesquiterpene synthase that produces d-cadinol (an anti-microbial, anti-fungal and anti-inflammatory compound)[5]. In the future the objective is to apply these approaches to all terpenoids: monoterpenes, triterpenes, diterpenes and teraterpenes. We propose a new biosynthetic tool to not only optimize and facilitate access to terpenes, but also to explore the biodiversity and characterize new terpene synthases. |
| References: | [1] Xiaoqiang Ma, Hong Liang, Qiuchi Pan, Kristala L. J. Prather, Anthony J. Sinskey, Gregory Stephanopoulos, and Kang Zhou. Optimization of the Isopentenol Utilization Pathway for Isoprenoid Synthesis in Escherichia coli. J. Agric. Food Chem. (2022) 70, 3512-3520. [2] Bohlmann J, Keeling CI. Terpenoid biomaterials. Plant J. (2008) 54(4):656-69. [3] Paramasivan K, Mutturi S. Progress in Terpene Synthesis Strategies through Engineering of Saccharomyces cerevisiae. Crit. Rev. Biotechnol. (2017) 37(8):974-989. [4] Couillaud J, Rico J, Rubini A, Hamrouni T, Courvoisier-Dezord E, Petit JL, Mariage A, Darii E, Duquesne K, de Berardinis V, Iacazio G. Simplified in Vitro and in Vivo Bioaccess to Prenylated Compounds. ACS Omega. (2019) 4(4), 7838-7849. [5] Ringel M, Dimos N, Himpich S, Haack M, Huber C, Eisenreich W, Schenk G, Loll B and Brück T. Biotechnological potential and initial characterization of two novel sesquiterpene synthases from Basidiomycota Coniophora puteana for heterologous production of δ-cadinol. Microbial Cell Factories (2022) 21 (1):64. |
| Figures: | 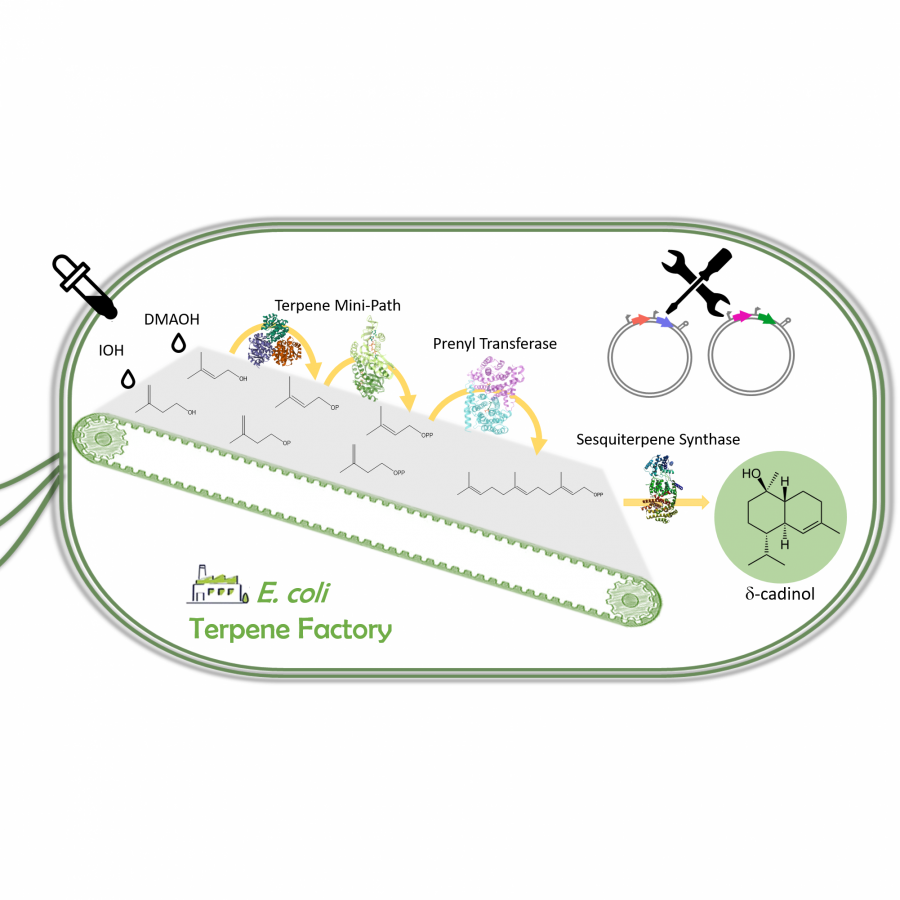 E. coli Terpene Factory Engineering Escherichia coli for in vivo d-cadinol production |
#1225 | Recycling Non-natural Cofactors for Selective Alkylation by Methyltransferases |
|
| Presenting author: | Aster VAN NOORD of TU DELFT |
| Other authors: | Lur ALONSO COTCHICO of ZYMVOL BIOMODELING Christian M. HECKMANN of TU DELFT Caroline E. PAUL of TU DELFT |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | alkylation / methyltransferases / SAM cofactor / |
| Purpose: | Carbon-carbon (C-C) bonds form the backbone of almost all organic molecules used in our society. The synthesis of those bonds, especially when introducing chiral centres, is often associated with non-selective and wasteful chemical processes. Enzymes are the key to alternative synthetic routes, as they catalyze reactions under mild conditions and with high selectivity. Carbon methyltransferases (cMTases) catalyze C-C bond formation via a methyl transfer, using the cofactor S-adenosyl methionine (SAM) as alkyl donor. Besides methyl groups, larger alkyl groups are also accepted by some cMTases [1]. The challenge for these alkylation reactions is their cofactor requirement. Non-natural SAM analogues (SAR) are needed to supply the cMTase with the desired alkyl group. The difficult chemical synthesis of SARs combined with their stoichiometric use, limits the application of cMTases. Previous research indicated that halide methyltransferases (HMTs) can alkylate the cofactor S-adenosylhomocysteine (SAH) using small alkyl iodides as alkyl donor [2-4]. In this research, we explore the capacity of HMTs to alkylate the SAH cofactor with larger alkyl substituents to form SARs (Figure 1). We used a computational approach to design HMT mutants that could accept larger alkyl substituents. Experimental results on the screening of designed mutants will be discussed. The SAR recycling system can subsequently be combined with cMTases to selectively introduce alkyl substituents to targeted substrates. |
| References: | [1] H. Stecher, M. Tengg, B.J. Ueberbacher, P. Remler, H. Schwab, H. Griengl, M. Gruber-Khadjawi, Angew. Chem. Int. Ed., 2009, 48, 9546-9548. [2] K.H. Schulke, F. Ospina, K. Hornschemeyer, S. Gergel, S.C. Hammer, ChemBioChem, 2021, 23, e202100632. [3] Q. Tang, C.W. Grathwol, A.S. Aslan-Uzel, S. Wu, A. Link, I.V. Pavlidis, C.P.S. Badenhorst, U.T. Bornscheuer, Angew. Chem. Int. Ed, 2021, 60, 1524-1527. [4] C. Liao, F.P. Seebeck, Nat. Catal., 2019, 2, 696-701. |
| Figures: |  Figure 1. Targeted selective C-C bond formation by cMTases via the transfer of large alkyl groups. This reaction is enabled by the formation of the SAR cofactor by an HMT using alkyl iodides as alkyl donor. |
#1226 | H2 and CO2 fueled reduction of carboxylic acids in Cupriavidus necator |
|
| Presenting author: | Marianna KARAVA of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, GRAZ UNIVERSITY OF TECHNOLOGY |
| Other authors: | Sara XHEMALAJ of PHARMACEUTICAL BIOTECHNOLOGY, BIBERACH UNIVERSITY OF APPLIED SCIENCES Vera LAMBAUER of INSTITUTE OF BIOTECHNOLOGY AND BIOCHEMICAL ENGINEERING, GRAZ UNIVERSITY OF TECHNOLOGY-AUSTRIAN CENTER OF INDUSTRIAL BIOTECHNOLOGY (ACIB) Regina KRATZER of INSTITUTE OF BIOTECHNOLOGY AND BIOCHEMICAL ENGINEERING, GRAZ UNIVERSITY OF TECHNOLOGY Margit WINKLER of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, GRAZ UNIVERSITY OF TECHNOLOGY - AUSTRIAN CENTER OF INDUSTRIAL BIOTECHNOLOGY (ACIB) Robert KOURIST of INSTITUTE OF MOLECULAR BIOTECHNOLOGY, GRAZ UNIVERSITY OF TECHNOLOGY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | autotrophic organism / carboxylic acid reductases / whole cell biotransformations / |
| Purpose: | The “Knallgas” bacterium Cupriavidus necator is an attractive bacterial host for sustainable synthesis of fine chemicals, due to its ability to utilize molecular H2 as electron donor, O2 as electron acceptor and CO2 as carbon source. In this work we demonstrate the development of a microbial platform for the reduction of carboxylic acids to aldehydes and alcohols with a wide range of applications in cosmetic and food industry [1]. To this end we have established an in vivo enzymatic cascade consisting of a carboxylic acid reductase (CAR) and C. necator endogenous alcohol dehydrogenases (ADHs) [1]. CARs catalyze the reduction of carboxylic acids to aldehydes while ADHs transform aldehydes to alcohols. NADPH and ATP cofactor regeneration necessary for the CAR activity is coupled to the autotrophic metabolism of C. necator powered by CO2 and H2 [2]. Heterologous expression of the CAR was performed both in wild type and in a mutant C. necator strain unable to produce poly(3-hydroxybutyrate). The generated strains were cultivated under heterotrophic and autotrophic conditions and tested in terms of activity. Our results highlight the potential of C. necator in chemical synthesis and pave the way for the sustainable synthesis of aldehydes and alcohols utilizing CO2 emissions and H2. |
| References: | [1] m. winkler, j. g. ling, ChemCatChem 2022, 14, e202200441. [2] l. assil-companioni, s. schmidt, p. heidinger, h. schwab, r. kourist, ChemSusChem 2019, 12, 2361-2365. |
| Figures: |  H2 and CO2 fueled reduction of carboxylic acids in Cupriavidus necator Figure 1. Production of C. necator cell biomass under autotrophic conditions. Cells are subsequently used for whole cell biotransformations of carboxylic acids. Cofactor recycling is coupled to the activity of the endogenous hydrogenases. |
#1228 | Unspecific Peroxygenase can be Tuned for Oxygenase or Halogenase Activity by Controlling the Reaction pH |
|
| Presenting author: | Verity BARBER of UNIVERSITY OF YORK |
| Other authors: | Alba DÍAZ-RODRÍGUEZ of GSK William UNSWORTH of UNIVERSITY OF YORK Gideon GROGAN of UNIVERSITY OF YORK |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Unspecific Peroxygenase / Halogenation / Oxygenation / Cascade |
| Purpose: | Unspecific peroxygenases (UPOs) have gained significant interest due to their ability to catalyse the selective oxygenation of a large range of organic substrates at the expense of only hydrogen peroxide.[1] UPOs are phylogenetically related to the heme-containing enzyme, chloroperoxidase (CPO).[2] CPO catalyses the oxidation of chloride and bromide to the corresponding hypohalous acids that can effect the halogenation of a number of organic substrates.[3,4] The structural similarity of UPOs to chloroperoxidase (CPO) means that UPOs can also catalyze halogenation reactions based upon the generation of hypohalous acids from halide and peroxide. Ullrich and Hofrichter demonstrated the halogenating capability of the UPO from Agrocybe aegerita (AaeUPO) with the bromination of phenol to from 2- and 4-bromophenol although the phenomenon was not extensively explored.[5] Whilst investigating the halogenating activity of AaeUPO we discovered that this enzyme can be tuned for oxygenase or halogenase activity by simply controlling the pH. Here we show that using simple aromatic compounds such as thymol at pHs of 3.0 and 6.0, either brominated or oxygenated products are evolved. In addition, we have developed a one-pot oxygenation-bromination cascade reaction using 4-ethylanisole, in which the pH is adjusted at the halfway stage, yielding the selectively brominated and oxygenated product 3’-bromo-4’-methoxyacetophenone. These results identify UPOs as an unusual example of a biocatalyst that is tunable for entirely different chemical reactions, dependent upon the reaction conditions. |
| References: | [1] Y. Wang, D. Lan, R. Durrani and F. Hollmann., Curr. Opin. Chem. Biol., 2017, 37, 1-9. [2] M. Hofrichter and R. Ullrich., Appl. Microbiol. Biotechnol., 2006, 71, 276-288. [3] L.P. Hager, D.R. Morris, F.S. Brown and H. Eberwein., J. Biol. Chem., 1966, 241, 1769-1777. [4] H. Yamada, N. Itoh and Y. Izumiy., J. Biol. Chem., 1985, 260, 11962-11969. [5] R. Ullrich and M. Hofichter., FEBS Lett., 2005, 579, 6247-6250. |
| Figures: | 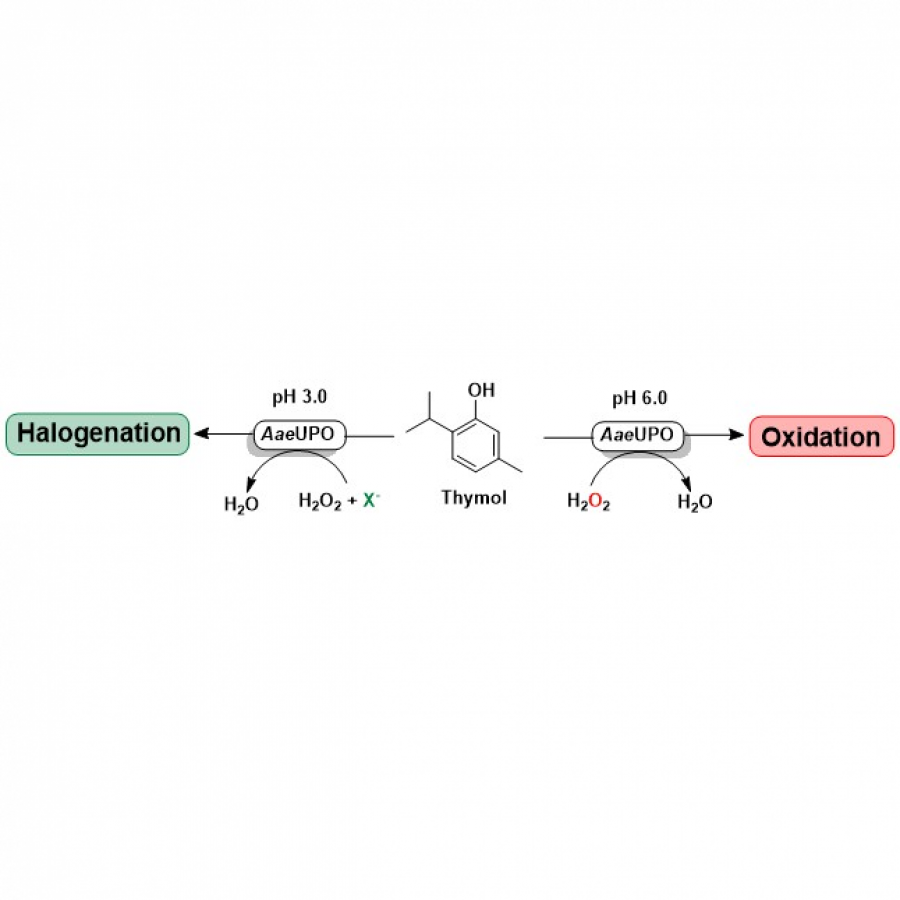 Scheme 1. By controlling the pH of the solution, the unspecific peroxygenase from Agrocybe aegerita can be tuned for either oxygenase or halogenase activity.
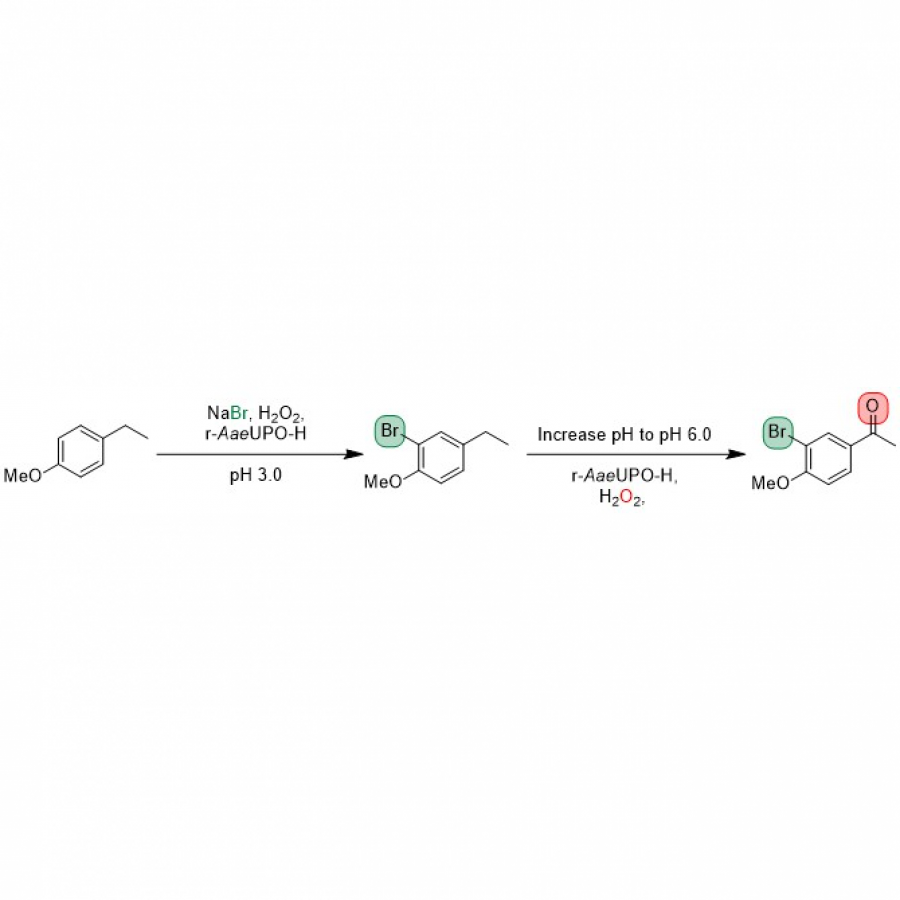 Scheme 2. Halogenation and oxygenation cascade catalysed by rAaeUPO-PaDa-I-H. |
#1229 | An Immobilised Silicon-Carbon Bond-Forming Enzyme for Anaerobic Flow Biocatalysis |
|
| Presenting author: | Esther MITTMANN of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Kersten S. RABE of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Other authors: | Sabrina GALLUS of KARLSRUHE INSTITUTE OF TECHNOLOGY Annika WEBER of KARLSRUHE INSTITUTE OF TECHNOLOGY Christof M. NIEMEYER of KARLSRUHE INSTITUTE OF TECHNOLOGY Kersten S. RABE of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flow Biocatalysis / Cytochromes / Immobilization / Organosilicons |
| Purpose: | The recent development of tailored cytochrome enzymes via directed evolution has enabled “new-to-nature” reactivities, such as the biocatalytic formation of carbon-silicon bonds using the cytochrome c from Rhodothermus marinus (CC).[1] The key to successful biocatalytic applications lies in the design of efficient overall processes in suitable reactor systems. Hence, enzyme immobilization techniques providing precise temporal and spatial reaction control in flow reactors are needed. To maximise the potential of this remarkable biocatalyst by increasing its turnover numbers (TON) and to enable its reusability in continuous processes, we recently reported the use of the SpyTag/SpyCatcher[2] bioconjugation system to immobilise this enzyme.[3] Our results showed that we were able to successfully modify the enzyme with a SpyTag (ST) without significantly impacting its catalytic activity. Even after immobilization on agarose microparticles, the enzyme retained 60% of its activity. We further developed analytical methods to quantify and compare TONs for the free and immobilized enzyme. By optimizing the reaction conditions and immobilizing CytC(TDE), we gained better insight into factors limiting TON and modulating the stereoselectivity of this interesting biocatalyst. Our investigations of the enzymatic activity of ST-CC revealed that using 25% acetonitrile as a co-solvent resulted in increased product formation. However, the yield of the reaction was restricted by the availability of the substrate dimethylphenylsilane and the reduced lifetime of the second substrate ethyl 2-diazopropanoate in aqueous solutions. To overcome these limitations, we utilized a continuously operated packed-bed reactor with inline mixing under anaerobic conditions, in which the substrates are supplied as a solution in pure acetonitrile. This minimized preincubation time with buffer and ensured optimal cosolvent concentration, resulting in a residence time of 2.6 min in the packed-bed reactor. When the immobilized enzyme was used in sequential batch or continuous flow to produce an organosilicon, we observed up to 6-fold TONs over a total period of 10 days compared to the free enzyme reaction, and thus much higher space-time-yields. However we observed a drop in stereoselectivity under these conditions. Further optimisation of the enzyme, the reaction conditions as well as the flow setup might enable improvements towards a more economically feasible application of this and similar heme-containing enzymes. |
| References: | [1] S. B. Kan, R. D. Lewis, K. Chen, F. H. Arnold (2016) "Directed evolution of cytochrome c for carbon-silicon bond formation: Bringing silicon to life", Science, 354, 1048-1051, DOI: 10.1126/science.aah6219. [2] B. Zakeri, J. O. Fierer, E. Celik, E. C. Chittock, U. Schwarz-Linek, V. T. Moy, M. Howarth (2012) "Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin", P Natl Acad Sci USA, 109, E690-E697, DOI: 10.1073/pnas.1115485109. [3] S. Gallus, E. Mittmann, A. J. Weber, M. Peng, C. M. Niemeyer, K. S. Rabe "An Immobilised Silicon-Carbon Bond-Forming Enzyme for Anaerobic Flow Biocatalysis", ChemCatChem, e202300061, DOI: 10.1002/cctc.202300061. |
| Figures: | 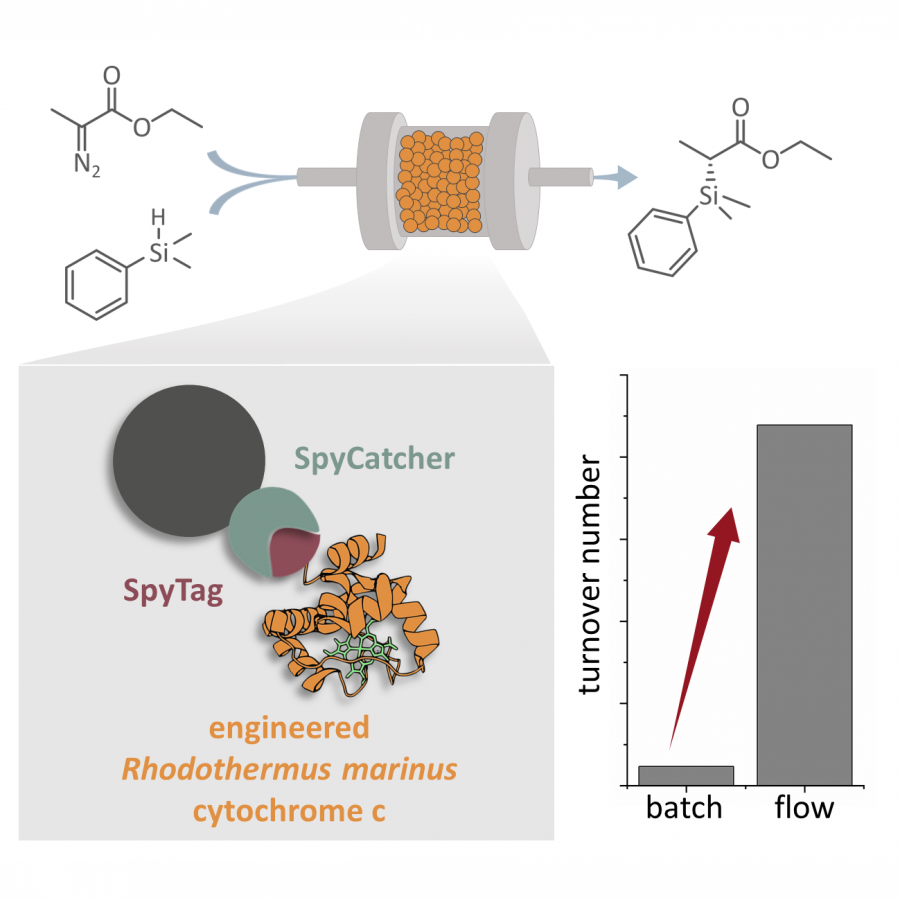 Flow biocatalysis for carbon-silicon bond formation A genetically engineered cytochrome c was immobilised with the SpyTag/SpyCatcher system and used in an anaerobic flow reactor, enabling it to produce organosilicons with up to 6-fold higher TON compared to the free enzyme in conventional batch experiments |
#1236 | CONTINUOUS BIOCATALYSED REDOX REACTIONS FOR THE ECOFRIENDLY SYNTHESIS OF PHARMACEUTICALLY RELEVANT COMPOUNDS |
|
| Presenting author: | Francesca ANNUNZIATA of UNIVERSITY OF MILAN |
| Corresponding author: | Lucia TAMBORINI of UNIVERSITY OF MILAN |
| Other authors: | Martina Letizia CONTENTE of UNIVERSITY OF MILAN Raffaella GANDOLFI of UNIVERSITY OF MILAN Cecilia PINNA of UNIVERSITY OF MILAN Sabrina DALLAVALLE of UNIVERSITY OF MILAN Andrea PINTO of UNIVERSITY OF MILAN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flow chemistry / Deep eutectic solvents / Redox reactions / Green ehemistry |
| Purpose: | Biocatalysis and flow chemistry are ideal partners for accessing novel chemical spaces and define sustainable and efficient synthetic tools with high level of intensification [1]. Biocatalytic redox reactions are very attractive since they often occur with high stereo- and regio-selectivity under mild and environmentally friendly conditions. The aim of our work was to develop greener and scalable routes for the synthesis of high value chemicals, using both whole cells and isolated enzymes. Moreover, new solvent systems such as Natural Deep Eutectic Solvent (NADES) have been investigated as green co-solvents in enzymatic transformations. The processes have been designed integrating in-line work ups and purification procedures to reduce manual handling, downstream processes and costs. For this purpose, meso reactors were employed in order to demonstrate that with a biocatalytic approach was also possible to obtain, in some cases, gram amounts of pure final compound improving the total productivity of the system. Two studies will be described: i) Enantioselective reduction of β-ketonitriles Immobilised whole cells of Rhodotorula rubra MIM 147, a widespred yeast genus, have been exploited in a fully automated system for the enantioselective synthesis of β-hydroxynitriles, starting from β-ketonitriles (Scheme 1) [2]. We selected a natural deep eutectic solvent made by choline chloride/glucose with a dual purpose as a co-solvent and as an internal source of glucose, fundamental for the cofactor regeneration. With the aim of developing a fully automated procedure for the obtainment of a key building block for the synthesis of the antidepressant drug duloxetine, i.e., (S)-3-hydroxy-3-(thiophen-2-yl)propanenitrile, an in-line purification procedure has been developed (scheme 1, compound 2f). An in-line extraction of the desired product was performed adding a flow stream of ethyl acetate followed by a liquid/liquid separation. The organic stream with any unreacted substrate and the desired product was further purified by flowing it through a column packed with polymer supported benzylamine (PS-BZA), allowing the recovery of the pure final product 2f after simple solvent evaporation. The optimised protocol allowed the isolation of the desired product in 80 minutes of residence time with >90% conversion and >99% e.e. and resulted to be versatile for the enantioselective reduction of different β-ketonitriles [3]. ii) Oxidation of tyrosol to hydroxytyrosol Tyrosol (Ty) and hydroxytyrosol (HTy) are valuable dietary phenolic compounds present in olive oil and wine and possess a range of biological effects [4]. The availability of gram amounts of pure Ty, HTy, their metabolites and derivatives are highly appealing for a deep biological evaluation. Furthermore, it must be considered that the price of HTy compared to the cost of Ty is more than 1000 higher. In this context, we performed the oxidation of easily accessible Ty using a free tyrosinase from Agaricus bisporus in presence of oxygen and ascorbic acid. The aqueous flow stream was then extracted in-line with ethyl acetate and the aqueous layer, containing the biocatalyst and the excess of ascorbic acid, was recirculated to improve the overall efficiency. An in-line purification for the collection of pure HTy was also designed (Scheme 2). Then, a bioreactor packed with an immobilised acyltransferase from Mycobacterium smegmatis (imm-MsAcT) was used to produce more lipophilic acetate derivatives [5]. With this modular set up, HTy was obtained with an isolated yield of 75%, whereas the acetate metabolites showed yields up to 80% in 10 minutes [6]. |
| References: | [1] L. Tamborini.; P. Fernandes; F. Paradisi; F. Molinari, Trends Biotechnol., 2018, 36, 73-8 [2] G. Facchetti; R. Gandolfi.; M. Fusè; D. Zerla; E. Cesarotti; M. Pellizzoni; I. Rimoldi, New J. Chem., 2015, 39, 3792 - 3800 [3] F. Annunziata, A. Guaglio, P. Conti, L. Tamborini, R. Gandolfi, Green Chem, 2022, 24, 950-965 [4] A- Karkovíc Markovíc; J. Toríc; M. Barbaríc; C. Jakobušíc Brala, Molecules, 2019, 24, 2001 [5] M.L Contente; S. Farris; L. Tamborini; F. Molinari; F. Paradisi, Green Chem. 2019, 21, 3263-3266 [6] F. Annunziata, M.L. Contente, C. Pinna, L. Tamborini, A. Pinto, Antioxidants, 2021, 10, 1142 |
| Figures: | 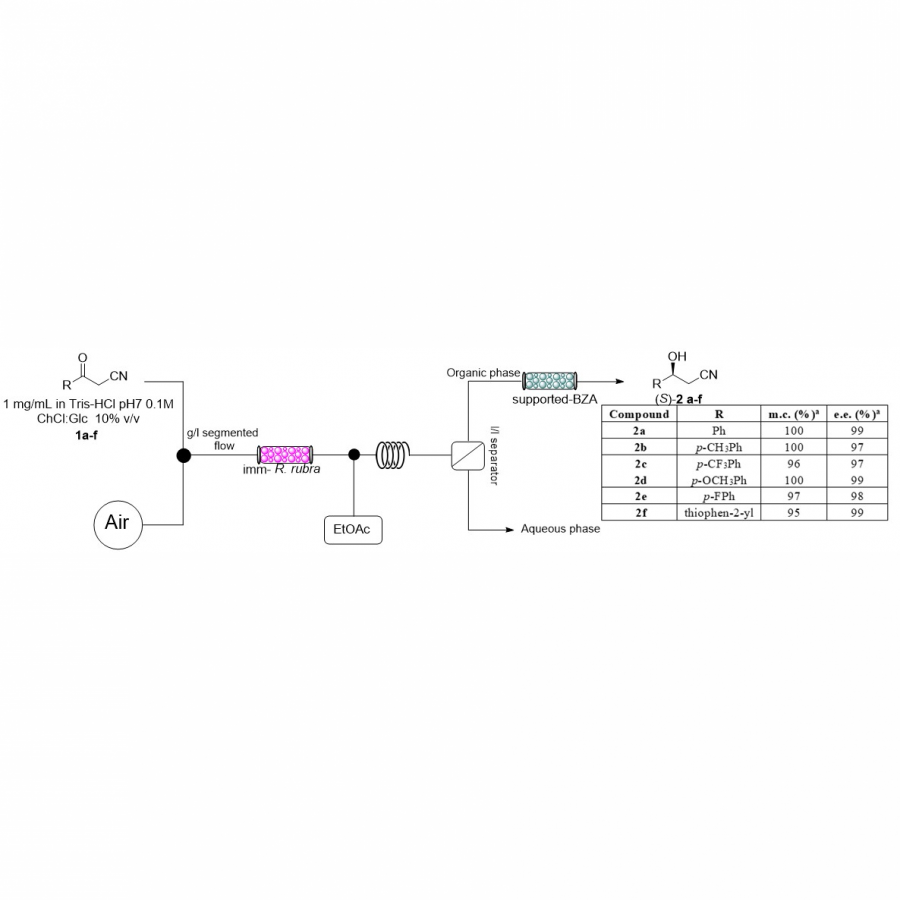 Scheme 1 Representation of the reactor configuration for the biocatalysed enantioselective reduction of different β-ketonitriles 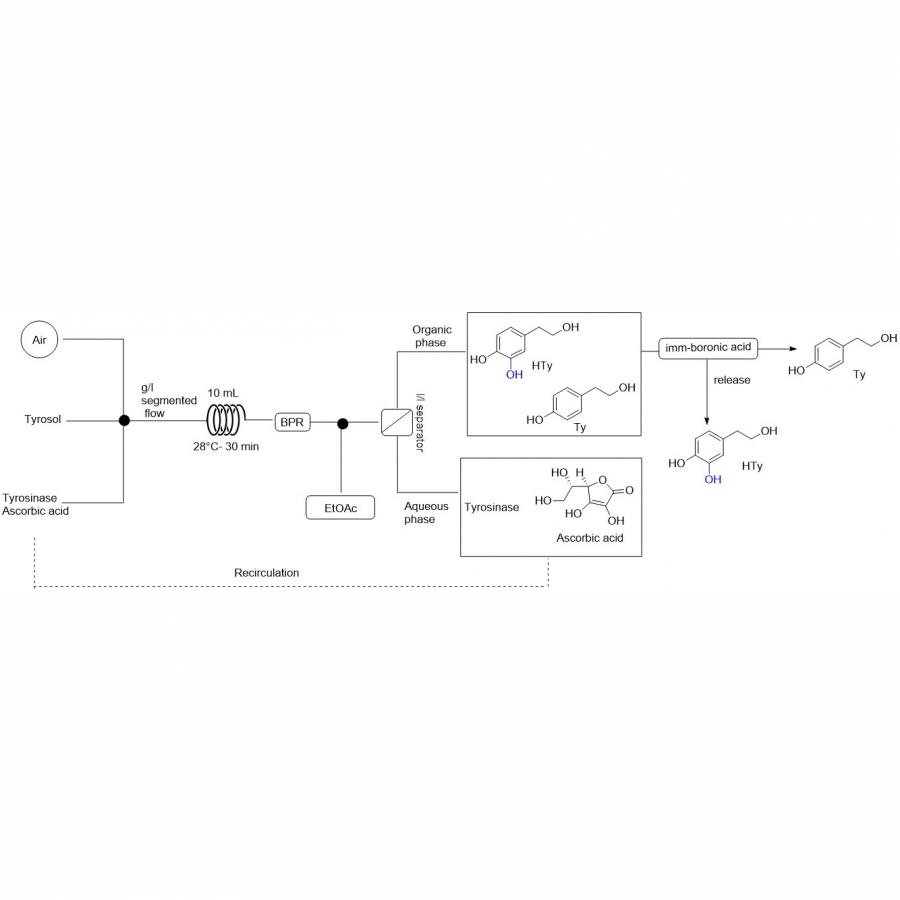 Scheme 2 Optimised flow reactor configuration for the bio-oxidation of Ty to HTy and in-line purification. The solutions of Ty and tyrosinase with ascorbic acid were prepared in sodium phosphate buffer 0.1 M, pH 7.0. BPR: 40 psi |
#1237 | Mapping redox enzymes’ active sites for coenzyme versatility |
|
| Presenting author: | Alice GUARNERI of LABORATORY OF ORGANIC CHEMISTRY, WAGENINGEN UNIVERSITY |
| Corresponding author: | Caroline E. PAUL of BIOCATALYSIS, DEPARTMENT OF BIOTECHNOLOGY, DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Georg STEINKELLNER of INNOPHORE GMBH Christian GRUBER of INNOPHORE GMBH Maurice C.R. FRANSSEN of LABORATORY OF ORGANIC CHEMISTRY, WAGENINGEN UNIVERSITY Willem J. H. VAN BERKEL of LABORATORY OF FOOD CHEMISTRY, WAGENINGEN UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / synthetic coenzymes / oxidoreductases / bioinformatics |
| Purpose: | Synthetic nicotinamide coenzyme biomimetics (NCBs) are attractive alternatives to the natural pyridine dinucleotides NAD(H) and NADP(H) for applied biocatalysis, allowing access to more cost-effective processes for enzyme-catalyzed redox reactions.[1] Their use has been hampered by the fact that only a limited number of oxidoreductases are known to accept them as hydride donors or acceptors during catalysis. To overcome the lack of coenzyme versatility, we developed a bioinformatics approach to create a portfolio of novel NCB-accepting enzymes. To do so, we computationally mapped the coenzyme-binding sites of biocatalysts active with synthetic coenzymes and searched the Protein Data Bank for enzymes presenting an active site with comparable characteristics. Several oxidoreductase candidates were identified and four of them were tested with NCBs, resulting in activity for three enzymes: actinorhodin ketoreductase from Streptomyces coelicor, α-ketoglutarate semialdehyde dehydrogenase from Azospirillum brasilense and isopiperitenone reductase from Mentha piperita. In conclusion, the designed computational approach was able to correctly predict coenzyme versatility. This novel in silico method has the potential to be used as platform for non-natural coenzyme engineering designs in the future. The results of the biocatalytic conversions, the relative efficiency of synthetic coenzymes compared to the natural NAD(P), and the implications for applied biocatalysis are discussed. |
| References: | [1] Guarneri, A. et al., Curr. Opin. Biotechnol. 2019, 60: 63-71 |
| Figures: | 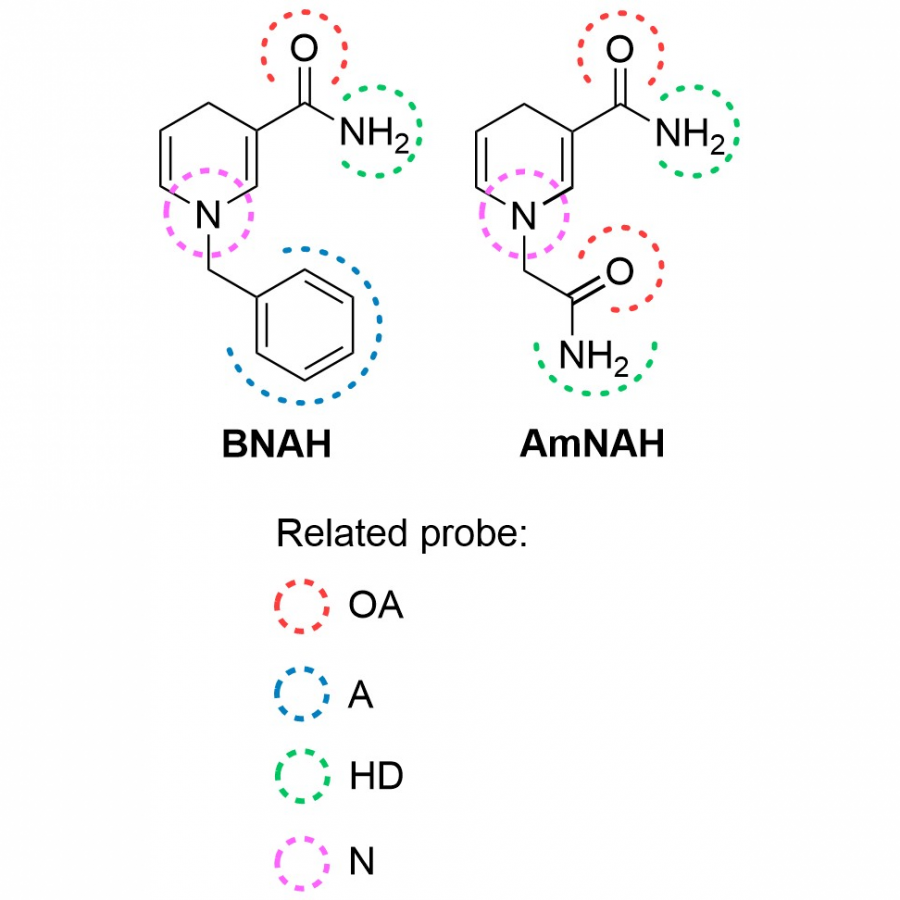 NCBs used in this work Chemical structures of two NCBs (BNAH, AmNAH) and corresponding chemical features (OA: oxygen as H-bond acceptor; A: aromatic functional group; HD: H-bond donor; N: non-hydrogen bonding nitrogen) 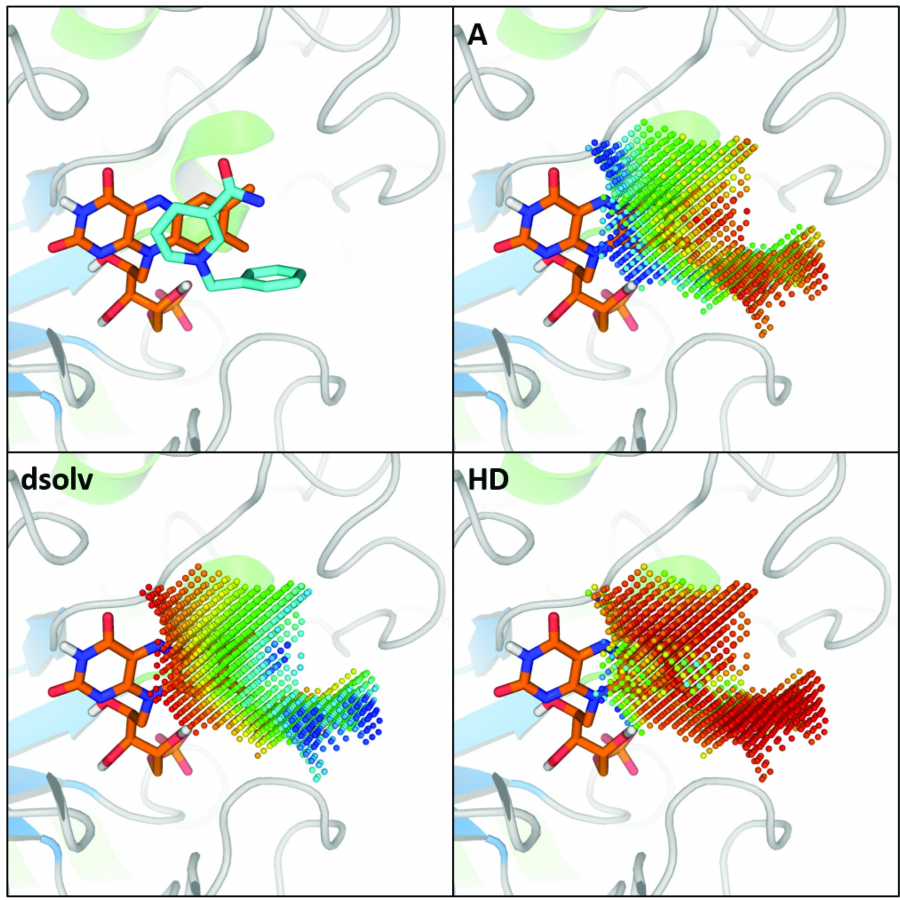 Exemplary characterization of the BNAH-binding pocket of the Old Yellow Enzyme 1 from Saccharomyces pastorianus NCB´s docking pose in the upper left; the response of its binding cavity to different chemical (A, HD) and structural (dsolv: desolvation) probes are shown as 3D-point clouds: blue (negative) to red (positive response the physicochemical functionality) |
#1238 | Enzymatic synthesis of novel amino acid-based biosurfactants from microbial oil |
|
| Presenting author: | CHEVALOT Isabelle of LRGP-UL |
| Corresponding author: | Isabelle CHEVALOT of UNIVERSITÉ DE LORRAINE, CNRS, LRGP |
| Other authors: | Dimitris KARAYANNIS of DEPARTMENT OF FOOD SCIENCE AND HUMAN NUTRITION, AGRICULTURAL UNIVERSITY OF ATHENS Emma BOULANGER of UNIVERSITÉ DE LORRAINE, CNRS, LRGP Seraphim PAPANIKOLAOU of DEPARTMENT OF FOOD SCIENCE AND HUMAN NUTRITION, AGRICULTURAL UNIVERSITY OF ATHENS Isabelle CHEVALOT of UNIVERSITÉ DE LORRAINE, CNRS, LRGP |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biosurfactants / microbial lipids / aminoacylases / N-acylation |
| Purpose: | Biosurfactants, a class of green and sustainable surfactants are naturally synthetized from microorganisms (bacteria, fungi, yeast and microalgae) or they can be generated from the association of a polar amino acid (hydrophilic moiety) and a non- polar long-chain compound (hydrophobic moiety) from renewable sources. To date, amino acid-based surfactants are industrially produced via a chemical route, called the Schotten- Baumann reaction, which are being related with major environmental drawback (e.g., salted wastes, acyl chlorides, organic solvents etc). These amino acid-based surfactants have low degree of toxicity, low haemolytic activity and are easily biodegradable. They are widely applied in the pharmaceutical, food and cosmetic industries due to their excellent emulsifying and antimicrobial activities. The present work is divided into two main objectives, in order biosurfactants be synthetised. The first was the production of microbial lipids (MLs) from oleaginous yeast cultivated on biodiesel-derived glycerol and the second was the enzymatic synthesis of high-value biosurfactant-type molecules, with MLs implicated as acyl donors (ADs). These novel biosurfactants were produced through the enzymatic N-acylation of lysine from MLs, catalyzed by aminoacylases produced from Streptomyces ambofaciens ATCC 23877 under aqueous conditions [1] and by Candida antarctica lipase B (CALB) under non-aqueous conditions [2]. The N-acylation reaction was evaluated and characterized based on regioselectivity, productivity and the substrate specificity of the enzyme towards the major fatty acids (FAs) found in MLs (e.g., linoleic acid). The oleaginous yeast Cryptococcus curvatus ATCC 20509 accumulated lipids up to 50% w/w in dry cell weight (DCW) (MLsmax = 6.1 g/L) after the optimization of the bioprocess according to the nature of the nitrogen source and the initial concentration of glycerol (Glyc0) employed in the medium. Aminoacylases revealed excellent activity towards the synthesis of acyl-lysine only when free fatty acids (FAs) were used, and the rare regioselectivity in the α-amino group, which has a great impact on the preservation of the functional side chains of any amino acids or peptides. CALB was able to catalyse the N- acylation of lysine by direct or trans-esterification. Unlike aminoacylases, the regioselectivity of CALB was oriented towards the ε-position of lysine. The substrate specificity of both enzymes was studied, and a new parameter was defined, viz. Specificity factor (Sf), which expresses the relative substrate specificity of an enzyme towards a FA present in a FA mixture (oil). HPLC-MS2 analysis revealed that aminoacylases have Sf in favor of palmitic acid, while CALB have Sf towards linoleic acid. In addition, a successful di-acylation of lysine exploiting the different regioselectivities of both enzymes is presented. To the best of our knowledge, this is the first time that a microbial oil has been successfully used as AD for biosurfactant synthesis [3] and that double N-acylation of lysine in the α and ε positions using two enzymes has been reported. This bio-refinery approach illustrates the concept of a state-of-the-art combination of biocatalysis and yeast culture technology. |
| References: | 1. Bourkaib, M.C.; Delaunay, S.; Framboisier, X.; Hôtel, L.; Aigle, B.; Humeau, C.; Guiavarc'h, Y.; Chevalot, I. N-acylation of L- amino acids in aqueous media: Evaluation of the catalytic performances of Streptomyces ambofaciens aminoacylases. Enzyme Microb. Technol. 2020, 137, 109536. 2. Dettori, L.; Jelsch, C.; Guiavarc’h, Y.; Delaunay, S.; Framboisier, X.; Chevalot, I.; Humeau, C. Molecular rules for selectivity in lipase-catalysed acylation of lysine. Process Biochem. 2018, 74, 50-60. 3. Karayannis, D.; Papanikolaou, S.; Vatistas, C.; Paris, C.; Chevalot, I. Yeast Lipid Produced through Glycerol Conversions and Its Use for Enzymatic Synthesis of Amino Acid-Based Biosurfactants. Int. J. Mol. Sci. 2023, 24, 714. |
| Figures: | 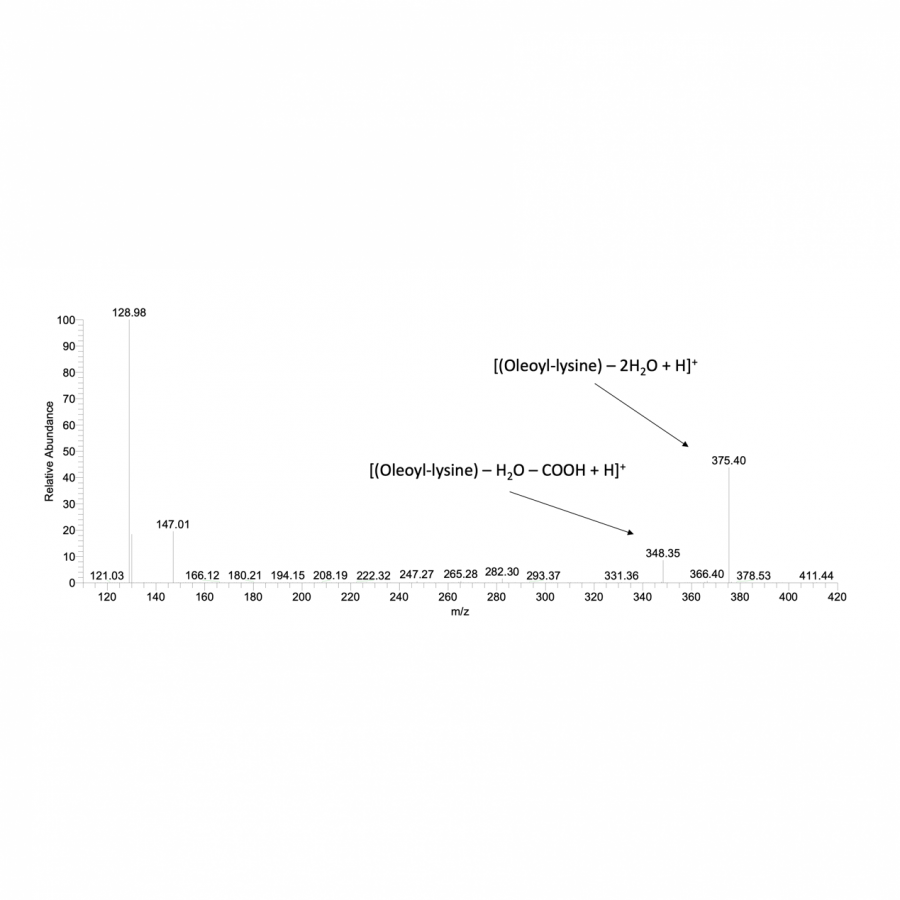 HPLC-MS2 spectrum of the product Fragmentation of the alpha-oleoyl-lysine, parent ion (m/z = 410), synthesised by aminoacylases. 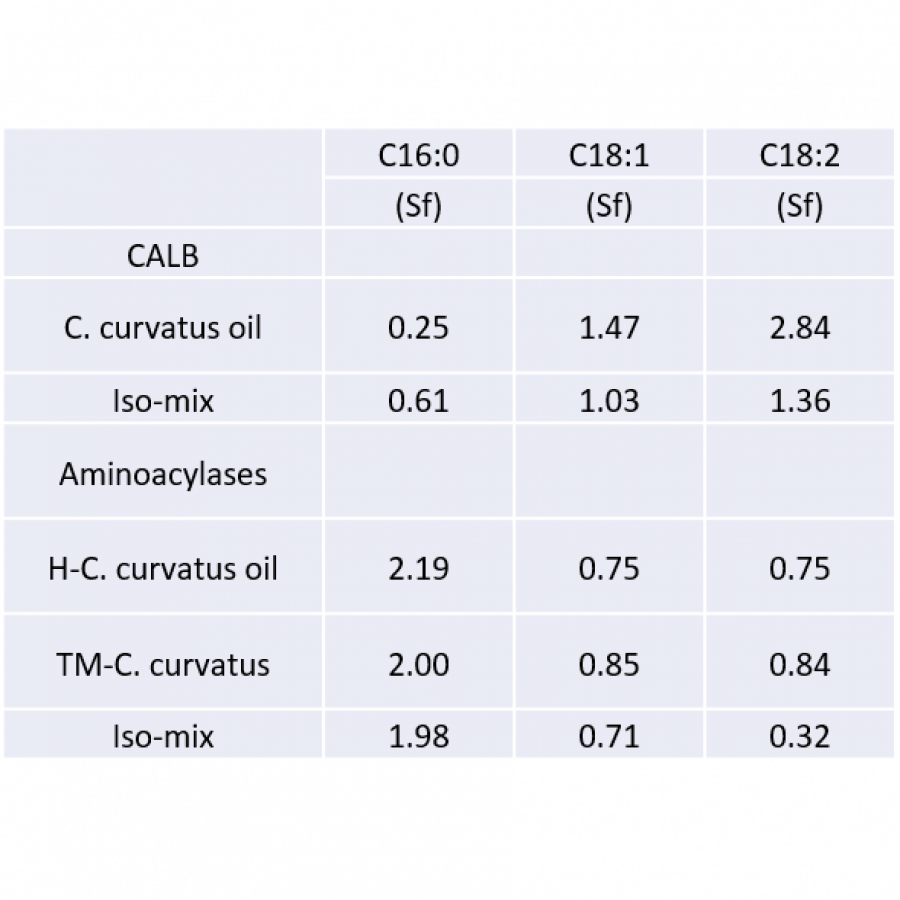 Specificity factor of CALB and aminoacylases towards the 3 major fatty acids of oils and tailor-made solutions. H-C. curvatus oil: Hydrolysed Cryptococcus curvatus oil; TM-C. curvatus: Tailor-made solution of the major fatty acids of Cryptococcus curvatus oil; Iso-mix: Tailor made solution containing 1/3 palmitic acid, 1/3 oleic acid and 1/3 palmitic acid. |
#1244 | Bayesian Process Optimization of an In Vitro ATP Producing and Regenerating Enzyme Cascade |
|
| Presenting author: | Regine SIEDENTOP of TU DORTMUND |
| Other authors: | Maximilian SISKA of FORSCHUNGSZENTRUM JÜLICH Niklas MÖLLER of TU DORTMUND Hannah LANZRATH of FORSCHUNGSZENTRUM JÜLICH Eric VON LIERES of FORSCHUNGSZENTRUM JÜLICH Stephan LÜTZ of TU DORTMUND Katrin ROSENTHAL of CONSTRUCTOR UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme cascade / ATP regeneration / Bayesian optimization / Gaussian process regression |
| Purpose: | In vitro enzyme cascades are an emerging alternative for traditional syntheses, which combine the benefits of biocatalysis and one-pot multi-step reactions. However, including a higher number of enzymes increases the complexity of the system and optimization is often necessary to achieve the desired cascade performance. Challenges are the high number of influencing parameters and the mutual interactions between the cascade’s components [1]. Bayesian optimization offers a statistical, data-driven optimization method with which the mentioned challenges can be addressed, while the number of experiments remains relatively low, compared to conventional approaches such as factorial experimental designs. It allows for a goal-oriented optimization for multi-parametric and multi-objective systems such as enzyme cascades with the simultaneous adjustment of multiple parameters and without relying on mechanistic understanding [2]. Bayesian optimization is already used in various fields, but in this work it was applied to an enzyme cascade, to the best of our knowledge, for the first time [3]. Here, Bayesian optimization was applied to an ATP-producing and -regenerating system. ATP is an important and energy-rich cofactor and many enzymes are dependent on it. In in vitro enzyme cascades, it can play a crucial role for the performance of the system and the in situ production and regeneration of the nucleotide by enzymes such as polyphosphate kinases (PPKs) can be beneficial. We implemented two PPKs for ATP production and regeneration for the ATP-dependent enzyme mevalonate kinase (MVK) that catalyzes the phosphorylation of mevalonate (MVA) to phosphomevalonate (MVAP). PPK and substrate concentrations were optimized for an increased specific activity of MVK [3]. With a total of 16 experiments, the initial concentrations of the three components were iteratively optimized, performing three experiments per round. We discovered, that the AMP concentration has a low impact and the PPK concentrations have a major influence on the reaction of MVK. The specific activity and the product concentration were increased at the same time during the optimization process. With a reference experiment with stoichiometric ATP amounts, a specific activity of 8.8 U mg-1 was achieved, which was even exceeded by the reaction with regenerated ATP with 10.2 U mg-1 [3]. With Bayesian optimization, the ATP regeneration by PPKs was successfully optimized for an increased activity of MVK. |
| References: | [1] R. Siedentop; C. Claassen; D. Rother; S. Luetz; K. Rosenthal, Catalysts 2021, 11, 1183. [2] B.J. Shields; J. Stevens; J. Li; M. Parasram; F. Damani; J.I.M. Alvarado; J.M. Janey; R.P. Adams; A.G. Doyle, Nature 2021, 590, 89-96. [3] R. Siedentop; M. Siska; N. Moeller; H. Lanzrath; E. von Lieres; S. Luetz; K. Rosenthal, Catalysts 2023, 13, 468. |
#1245 | Accelerating The Adoption of Biocatalysis In the Pharmaceutical Industry |
|
| Presenting author: | Alba DIAZ-RODRIGUEZ of GSK |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | The use of biocatalysis in the production of active pharmaceutical ingredients has seen a marked increase over the past decade. The biocatalytic toolbox available to chemists has continued to expand and new enzymes collections are commercially available. Despite this, the adoption of biocatalysis in early discovery chemistry or pharmaceutical manufacturing routes has a number of specific challenges. In this poster we will explain our approach to using biocatalysis and new technologies in early discovery but also later in development. We will be highlighting a few recent capabilities and enzyme classes we have worked on as well as recent internal and external examples. |
#1250 | Hybrid Organometallic and Enzymatic Tandem Catalysis for Oxyfunctionalisation Reactions |
|
| Presenting author: | DOMESTICI Chiara of University of Perugia/Delft University of Technolo |
| Corresponding author: | Chiara DOMESTICI of UNIVERSITY OF PERUGIA |
| Other authors: | Yinqi WU of DELFT UNIVERSITY OF TECHNOLOGY THOMAS HILBERATH of DELFT UNIVERSITY OF TECHNOLOGY Frank HOLLMANN of DELFT UNIVERSITY OF TECHNOLOGY Alceo MACCHIONI of UNIVERSITY OF PERUGIA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chemoenzymatic tandem / iridium FMN reduction / peroxygenases / oxyfunctionalisation |
| Purpose: | The biocatalytic insertion of oxygen into non-activated organic molecules is attracting an increasing interest in pharmaceutical and industrial chemistry.[1] Particularly, enzyme-mediated oxyfunctionalisation reactions usually reach high regio- and enantioselectivity representing alternative routes to the challenging conventional chemical methods. Although heme-containing monooxygenases (P450s) are widely used in this context, their reaction mechanisms via reductive activation of the molecular oxygen[2] poses several difficulties such as dependence on costly nicotinamide cofactors, intricate electron transport chains as well as the so-called Oxygen Dilemma.[3] This ampers the large-scale usage of P450s. Beside P450s, peroxygenases (UPOs) are receiving increased attention as selective oxyfuctionalisation catalysts as a consequence of the simpler reaction mechanism that enables the access to the same wide array of reactions and products, as they are considered the Swiss Army Knife of the oxyfunctionalisation chemistry.[4] However, the H2O2-dependence of UPOs also bears the challenges of irreversible oxidative enzymatic inactivation.[5] Thus, continuous low-level supply or in situ generation of hydrogen peroxide (H2O2) is essential for the stability of peroxygenases. A class of organometallic compounds potentially exploitable for H2O2 supply from formate, molecular oxygen and FMN as cocatalyst is constituted by Cp* iridium pyridincarboxyamidate complexes. They are extremely stable and water-soluble, and already used successfully as catalysts for hydrogenation reactions,[6] formic acid dehydrogenation,[7] water oxidation,[8] reductive amination of ketons[9] and hydrogen peroxide generation.[10] In this contribution, we report that the organometallic complex [Cp*Ir(H-Pica)NO3] {pica = picolinamidate =κ2-pyridine-2-carboxamide ion (–1)} 1 (Scheme 1) is indeed a good catalyst for the H2O2-regeneration using simple flavin mononucleotide as co-catalyst and formate as hydride source. Furthermore, we show that 1 can successfully cooperate with rAaeUPO, which takes advantage by the in situ produced H2O2. Thus, we disclosed for the first time the chemoenzymatic tandem combining 1 and rAaeUPO to catalyse oxyfunctionalisation reactions. The feasibility of the proposed hybrid system is demonstrated by the wide portfolio of the reactions it can perform, including aromatic and (cyclo)aliphatic hydroxylation, epoxidation and sulfoxidation. |
| References: | (1) J. Dong, E. Fern?ndez-Fueyo, F. Hollmann, C. Paul, M. Pesic, S. Schmidt, Y. Wang, S. Younes, W. Zhang, Angew. Chem., 2018, 130, 9380-9404, Angew. Chem. Int. Ed. 2018, 57, 9238-9261. (2) V. B. Urlacher, M. Girhard, Trends Biotechnol. 2019, 37, 882-897. (3) D. Holtmann, F. Hollmann, ChemBioChem 2016, 17, 1391-1398. (4) M. Hobisch, D. Holtmann, P. G. de Santos, M. Alcalde, F. Hollmann, S. Kara, Biotechnol. Adv. 2021, 51, 107615. (5) B. Valderrama, M. Ayala, R. Vazquez-Duhalt, Chem. Biol. 2002, 9, 555-565. (6) a) R. Kanega, N. Onishi, S. Tanaka, H. Kishimoto, Y. Himeda, J. Am. Chem. Soc. 2021, 143, 1570-1576; b) L. Tensi, A. V. Yakimov, C. Trotta, C. Domestici, J. De Jesus Silva, S. R. Docherty, C. Zuccaccia, C. Cop?ret, A. Macchioni, Inorg. Chem. 2022, 61, 10575-10586. (7) G. Menendez Rodriguez, C. Domestici, A. Bucci, M. Valentini, C. Zuccaccia, A. Macchioni, Eur. J. Inorg. Chem. 2018, 2018, 2247-2250. (8) A. Bucci, S. Dunn, G. Bellachioma, G. Menendez Rodriguez, C. Zuccaccia, C. Nervi, A. Macchioni, ACS Catal. 2017, 11, 7788-7796. (9) K. Tanaka, T. Miki, K. Murata, A. Yamaguchi, Y. Kayaki, S. Kuwata, T. Ikariya, M. Watanabe, J. Org. Chem. 2019, 84, 10962-10977. (10) H. T. H. Nguyen, L. H. Do, Chem. Commun. 2020, 56, 13381-13384. |
| Figures: | 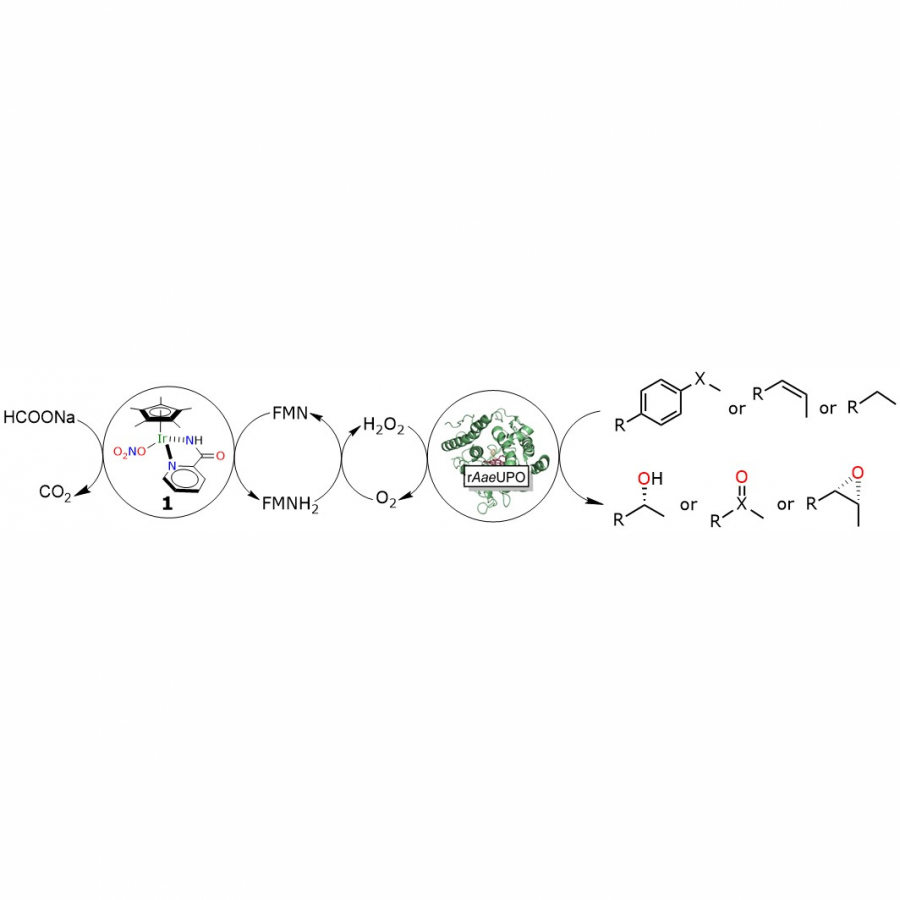 Scheme 1 Scheme of the chemoenzymatic tandem reaction
combining 1 with rAaeUPO to catalyse
oxyfunctionalisation reactions. |
#1258 | Biocatalytic oxidative cleavage of alkenes using novel metal-dependent aromatic dioxygenases |
|
| Presenting author: | Astrid SCHIEFER of TECHNISCHE UNIVERSITÄT WIEN |
| Corresponding author: | Florian RUDROFF of TECHNISCHE UNIVERSITÄT WIEN |
| Other authors: | Lukas SCHOBER of TECHNISCHE UNIVERSITÄT GRAZ Margit WINKLER of TECHNISCHE UNIVERSITÄT GRAZ |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ABAO assay / aromatic dioxygenases / fluorometric/photometric aldehyde detection / oxidative carbon-carbon double bond cleavage |
| Purpose: | Oxidative cleavage of alkenes can be used to obtain carbonyl compounds, valuable building blocks in various areas, including food, flavor, and the pharmaceutical industry. (1) While this can be done via chemical-synthetic approaches, like ozonolysis (2), we are interested in milder and safer enzymatic approaches using novel metal-dependent aromatic dioxygenases (ADOs). In the presence of oxygen, these enzymes enable the oxidative cleavage of substrates such as isoeugenol 1a to the corresponding aldehydes (Figure 1). In order to screen numerous substrates in a short time period, we are developing a pooling approach using the reported ABAO-assay (3), a photometric/ fluorometric assay for the detection of the produced aldehydes. The assay is based on the rapid reaction of aldehydes with 2-aminobenzamidoxime (ABAO), forming quinazolines 5 that exhibit UV absorption and fluorescence properties. Using this assay, we want to quickly analyze biotransformations with several pooled substrates by detecting positive/negative photometric/fluorometric responses. In the case of a positive response, GC/HPLC is used to identify the formed aldehydes/the accepted substrate. So far, substrates such as isoeugenol 1a, hydroxyanethole 1b , and 4-vinylguaiacol 1c were found to be successfully converted to the respective aldehydes in a whole-cell system reaction. Among the found VsADO from Valsa sordida, the PaADO from Podospora anserina, the TsADO from Talaromyces stipitatus, and the CpADO from Coniochaeta pulveracea, especially the MapADO from Moesziomyces aphidis showed promising properties and converted 10 mM isoeugenol to vanillin in a whole cell approach within 1 h. This project receives funding from FWF (T163034-2008). |
| References: | (1) Kazimirova, V.; Rebros, M. Production of Aldehydes by Biocatalysis. Int. J. Mol. Sci. 2021, 22 (9), 4949. (2) Fisher, T. J.; Dussault, P. H. Alkene ozonolysis. Tetrahedron 2017, 73 (30), 4233. (3) Ressmann, A. K.; Schwendenwein, D.; Leonhartsberger, S.; Mihovilovic, M. D.; Bornscheuer, U. T.; Winkler, M.; Rudroff, F. Substrate-Independent High-Throughput Assay for the Quantification of Aldehydes. 2019, 361 (11), 2538. |
| Figures: | 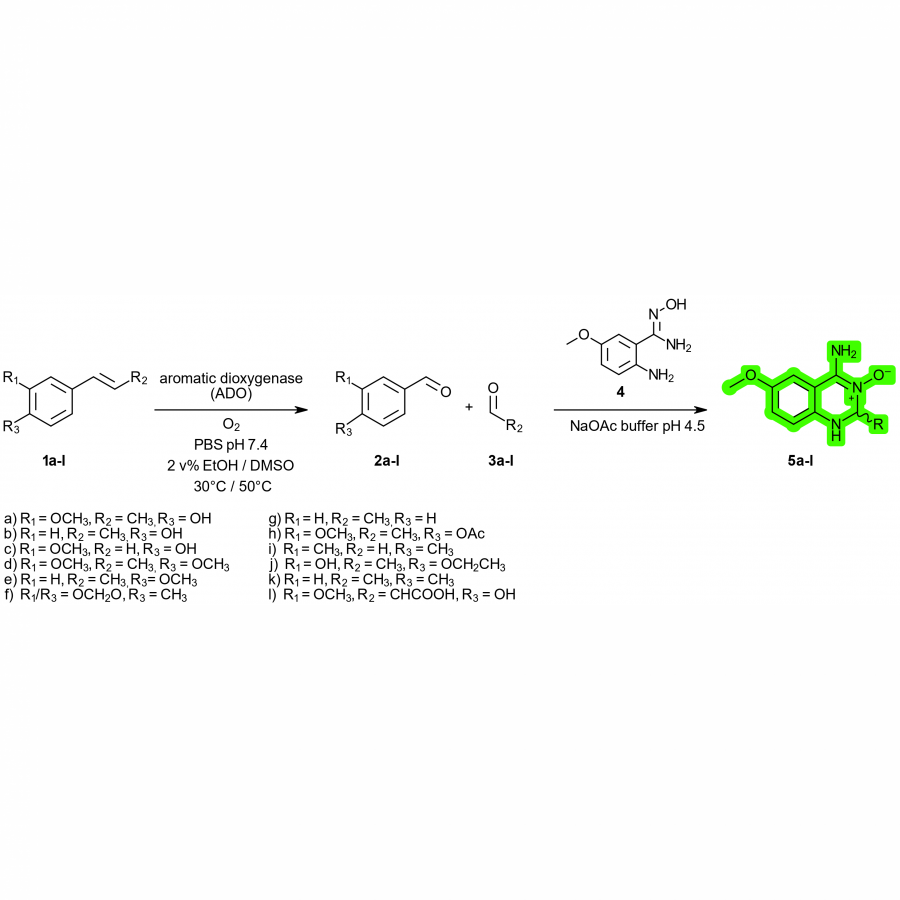 Overview of the enzymatic oxidative cleavage reaction and the detection strategy. Oxidative cleavage of 1 by aromatic dioxygenases to the corresponding aldehydes (2, 3). Detection of the formed aldehydes via ABAO-assay using 2-aminobenzamidoxime derivate 4. The resulting quinazolines 5 exhibit UV absorption and fluorescence properties. |
#1260 | Biocatalytic route for castor oil and furan derivatives valorisation |
|
| Presenting author: | Ioan BITCAN of POLITEHNICA UNIVERSITY TIMISOARA |
| Other authors: | Anamaria TODEA of POLITEHNICA UNIVERSITY TIMISOARA Diana DREAVĂ of POLITEHNICA UNIVERSITY TIMISOARA Iulia PĂUȘESCU of POLITEHNICA UNIVERSITY TIMISOARA Francisc PETER of POLITEHNICA UNIVERSITY TIMISOARA Lajos NAGY of UNIVERSITY OF DEBRECEN Sándor KÉKI of UNIVERSITY OF DEBRECEN |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ricinoleic acid / 5-hydroxymethyl-2-furancarboxylic / enzymatic polymerization / biocatalytic one-pot synthesis |
| Purpose: | The utilization of vegetable oils and their derivatives as raw materials came in the spotlight of green chemistry in the past decades. Their universal availability, low price, and biodegradability place oil derivatives as important biobased sources for polymer synthesis, as well [1]. Castor oil is one of the most studied natural oils due to its high content of ricinoleic acid, a natural hydroxy acid (> 80%). The presence of the double bond in the ricinoleic acid molecule allows additional functionalization by grafting reactions. Copolymers of hydroxy acids have been already synthesized by biocatalysis and have promising applications in several fields [2]. 5-Hydroxymethylfurfural (HMF), a biobased platform chemical obtained during biomass pretreatment allows a wide range of chemical modifications, leading to important value-added products. Recently, it has been quantitatively oxidized to 5-hydroxymethyl-2-furoic acid (5OH2FA) by Gluconobacter oxydans DSM 50049 cells [3]. A combination of vegetable oil and furan derivatives represents a sustainable synthetic way to materials with original properties obtained through simple, green, and efficient processes. In this study, the polymerization of ricinoleic acid and 5OH2FA was investigated by a green biocatalytic pathway. The reactions were carried out using commercially available native and immobilized lipases as catalysts in organic solvents, ionic liquids, and solvent-free systems, at various molar ratios and temperatures up to 80°C. The lipase from Pseudomonas stutzeri showed the highest catalytic efficiency at 50°C in t-BuOH. The insertion of 5OH2FA units into the ricinoleic acid estolide backbone was demonstrated by MALDI-TOF MS and 2D NMR analysis. The thermal properties of the oligoester products were evaluated by TG and DSC. The organogelator ability of the resulted oligoesters in the absence or presence of other components was tested by using several organic solvents with different log P values, at different temperatures. The morphological, and rheological properties of the resulted organogels were evaluated. This work was supported by a grant of the Romanian Ministry of Education and Research, CNCS - UEFISCDI, project number PN-III-P1-1.1-TE-2019-1573, within PNCDI III, contract number TE 101 and partially financially supported by the Project “Network of excellence in applied research and innovation for doctoral and postdoctoral programs / InoHubDoc”, project co-funded by the European Social Fund financing agreement no. POCU/993/6/13/153437 |
| References: | [1] A. Todea, I. Bîtcan, D. Aparaschivei, I. Păușescu, V. Badea, F. Peter, V. Gherman, G. Rusu, L. Nagy, S. Kéki, Polymers, 2019, 11, 9. [2] A. Todea, D. Aparaschivei, V. Badea, C.G. Boeriu, F. Peter, Biotechnol. J., 2018, 13, 6. [3] M. Sayed, S.-H. Pyo, N. Rehnberg, R. Hatti-Kaul, ACS Sustainable Chem. Eng., 2019, 7, 4, 4406. |
#1262 | Computational and enzymatic experimental strategies for polymers circularity |
|
| Presenting author: | Anamaria TODEA of UNIVERSITY OF TRIESTE |
| Other authors: | Demi VATTOVAZ of UNIVERSITY OF TRIESTE Sara FORTUNA of UNIVERSITY OF TRIESTE Danilo DI STEFANO of ESTECO SPA Monia RENZI of 3DEPARTMENT OF LIFE SCIENCES, UNIVERSITY OF TRIESTE Lucia GARDOSSI of UNIVERSITY OF TRIESTE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | rational design / degradation / lipases / cutinases |
| Purpose: | The interest in polymers derived from renewable sources has amplified as demonstrated by the large number of recent patents and publications. The material and polymer sectors are facing the challenge of integrating the sustainability of both processes and products, including their management after disposal [1,2]. Biocatalysis can boost such innovation, leveraging on enzymes that overcome the limitations of conventional chemical strategies by catalyzing, under highly selective and mild conditions, the targeted modification, synthesis or degradation of polymers and, most importantly, biobased polymers . Hydrolases, such as lipases and cutinases, were successfully used for in vitro polycondensation of bio-based diacids and polyols, leading to biodegradable polyesters with controlled structures. In parallel the capacity of several cutinases and lipases to degrade polyesters was evaluated by several groups [3,4]. The possibility to correlate structural features of a polymer with the catalytic properties of an enzyme would allow the rational design of environmentally safe new tailor-made biodegradable polymers. The RenEcoPol project aim was to develop alternative routes for recyclable polyester synthesis based on biobased building blocks using green processes such as biocatalysis. Within RenEcoPol a computational procedure able to analyze and evaluate the ability of different hydrolases to interact and to accept short chain substrates either for synthetic or degradative processes. In this respect specific modelling software's for proteins and protein-ligands interactions such as molecular dynamics and docking were selected and integrated into an automatic workflow. A series of bio-based monomers and enzymes were screened up to now and the computational studies results were correlated with some available experimental data and the results indicate that ability of enzymes to hydrolyze or synthesize polyesters can rationally selected by integrating different computational tools. The pipeline, here implemented in modeFRONTIER software allows to select, from a pool of enzymatic structures, the optimum biocatalyst for catalyzing the synthesis and/or the hydrolysis of polyesters. The new biobased polyesters will be characterized in detail by several analytical techniques for structure confirmation and assessment of the physico-chemical properties. In the third step the biodegradability and ecotoxicity in different natural and synthetic conditions of the synthesized materials was evaluated and in the last step a strategy for the recoverment of the components was demonstrated as proof of concept. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 101029444. |
| References: | [1] Pellis A., Green Chem., 2017, 19, 490-502. DOI: 10.1039/c6gc02142e [2]. Guarneri A, Adv Synth Catal 2019;361:2559-73. [3]. A Todea Processes, 2021, 9 (4), 646, 1-26. [4].Todea A. et al, ChemSusChem, 2022, 15 (9), e202102657. [5]. S. Fortuna, Catalysts, 2021, 11, 7, 748. [6] Zappaterra, F. et al Polymers 2023, 15, 1536 |
| Figures: | 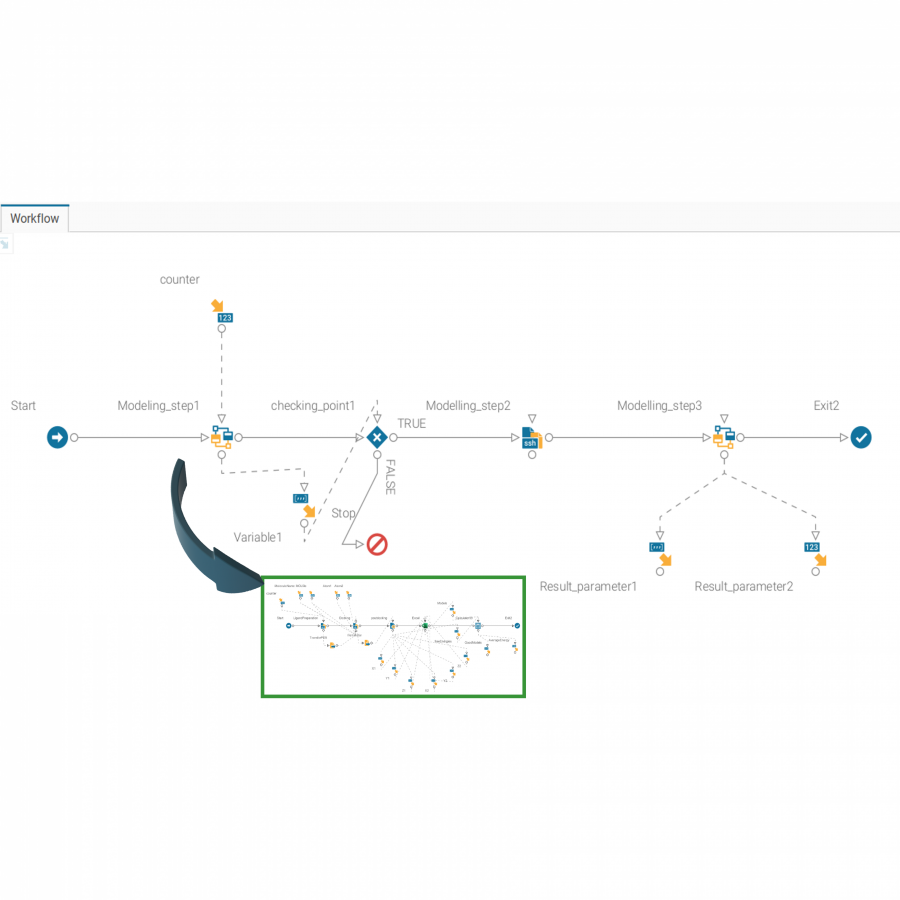 Figure 1 Workflow used for the automatic in-silico screening of hydrolases including docking and molecular dynamics steps 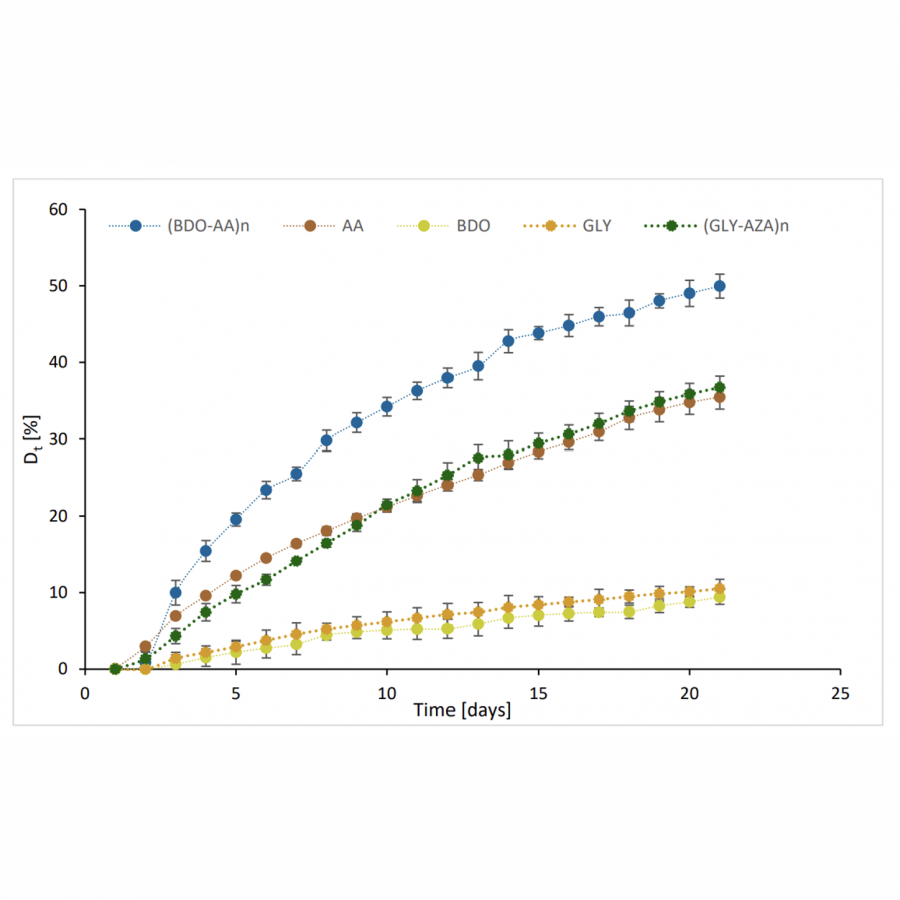 Figure 2 Degree of degradation of the oligoesters after 21 days of incubation in a marine environment; the data were normalized by subtracting the values of the control samples |
#1268 | Investigation into PHA synthase stereoselectivity for the production of PHB with improved material properties |
|
| Presenting author: | Marcel MAYER of TECHNICAL UNIVERSITY OF MUNICH |
| Corresponding author: | Volker SIEBER of TECHNICAL UNIVERSITY OF MUNICH |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Bioplastics / Sustainability / Polyhydroxybutyrate / Stereoselectivity |
| Purpose: | The transition to a sustainable future requires decarbonization in all sectors. This comprises the plastics industry that heavily relies on petroleum-based resources. An alternative to petroleum-based plastics is poly-3-hydroxybutyric acid (PHB), which is obtained by microorganisms via fermentation of renewable feedstock. However, the major two challenges in the production of PHB are its high manufacturing cost and inferior material properties compared to similar but petroleum-based plastics such as polyethylene [1]. As a highly crystalline material, PHB is brittle and breaks easily, limiting its range of applications. Additionally, a small window between melting and degradation temperature makes processing of PHB without thermal degradation challenging. It is commonly believed that PHB is an isotactic polymer made exclusively from (R)-3-hydroxybutyric acid (HBA) repeating units, which contributes to its poor material properties. However, it was shown that as the content of (S)-HBA increases, brittleness decreases and the processing window between melting and degradation temperature widens [2]. Introducing (S)-HBA into PHB could therefore be an unexplored approach to enhance its material properties. Despite that, no investigation into the stereoselectivity of the PHB-producing enzymes, poly-3-hydroxyalkanoate (PHA) synthases, has been performed yet. In our study, we investigated stereoselectivity of various wildtype PHA synthases by producing PHB from racemic substrate in vitro. From structural comparison of PHA synthases, we hypothesized that hydrophobicity of the active site pocket played a role in stereorecognition. Increased hydrophobicity could result in PHB with higher contents of (S)-HBA. Furthermore, size of the lower part of the active site pocket might also influence stereoselectivity by providing a larger volume for the recognition of unnatural substrates. This study could help to aid further work into the stereoselectivity of PHA synthases with a final goal to produce PHB with enhanced material properties. |
| References: | [1] Turco, R., Front. Bioeng. Biotechnol, 2020, 619266. [2] Haslboeck, M., Macromolecules 2019, 14, 5407-5418. |
#1269 | Biocatalysis for the production of renewable chemicals |
|
| Presenting author: | Max LUBBERINK of WAGENINGEN UNIVERSITY AND RESEARCH |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | pichia / PET / Fatty acids / |
| Purpose: | Wageningen Food & Biobased Research (WFBR) is a research institute that conducts contract research specializing in sustainable innovations in food and biobased products. The Biobased Products division focuses on the valorization and upcycling of biomass or industrial waste streams using novel, sustainable methods. Enzymatic methods are applied in a range of projects within this domain for their high chemical selectivity and mild reaction conditions. Activities involving biocatalysis include the production of enzymes in bacterial and fungal hosts, development of biocatalysts through engineering or immobilization, and development and optimization of biocatalytic processes. Within the WFBR institute, there is a close collaboration between different expertises, like biorefinery, chemical synthesis, fermentation technology, materials science and downstream processing. The combination of these research fields allows for the development of highly streamlined and optimized processes. In this poster, various topics of research in biocatalysis within WFBR will be presented, including the production of enzymes in Pichia pastoris, the enzymatic breakdown of plastics, the enzymatic synthesis of selectively functionalised fatty acids, and the enzymatic conversion of waste-CO2 to chemicals. |
#1272 | Thermomyces lanuginosus lipase applied in licuri oil biotransformation: an experimental and computational study. |
|
| Presenting author: | César ALMEIDA RODRIGUES of TIRADENTES UNIVERSITY |
| Other authors: | Jefferson BARROS DOS SANTOS of TIRADENTES UNIVERSITY Milson BARBOSA of INSTITUTO FEDERAL DA PARAÍBA (IFPB) Milena CHAGAS LISBOA of UNIVERSITÉ CATHOLIQUE DE LOUVAIN, UCLOUVAIN, INSTITUT DE LA MATIÈRE CONDENSÉE ET DES NANOSCIENCES Matheus PEREIRA of UNIVERSITY OF COIMBRA, CIEPQPF, DEPARTMENT OF CHEMICAL ENGINEERING Álvaro LIMA of TIRADENTES UNIVERSITY Cleide SOARES of TIRADENTES UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Lipase / Licuri oil / Hydrolysis / Molecular docking |
| Purpose: | Licuri almond (Syagrus coronata (Mart.) Becc.) has a high lipid content (around 49% w/w) being abundant in saturated fatty acids, particularly lauric acid, which comprises 42% (w/w) of the total fatty acid composition (LA SALLES et al., 2010). Fatty acids are the building blocks to produce higher-valued products in the cosmetic, pharmaceutical, biofuel industries through the catalyst appropriated selection and application. Among several catalytic processes, enzymatic-based methods offer promising advantages for developing sustainable and cost-effective vegetable oils hydrolysis (ALVES et al., 2019). However, the process efficiency is directly influenced by the lipase selectivity to catalyze the fatty acids release from the triglycerides in the oil. This phenomenon, which is not yet fully understood, remains a challenge for biocatalysis research. Thus, molecular docking analysis is a simple and fast approach to elucidate the molecular interaction mechanism between lipase and substrates, providing useful information on how different substrates can bind to the enzyme surface. So, the present study aims to evaluate the efficiency of Thermomyces lanuginosus lipase in the licuri oil biotransformation into free fatty acids (FFA) and identify the molecular mechanism that promote the enzymatic hydrolysis. Firstly, the oil was extracted from licuri fruit almonds using n-hexane as solvent through Soxhlet method (LA SALLES et al., 2010). The enzymatic hydrolysis was carried out in tank reactors at 45 °C, in the ratio of 25% oil and 75% water (v/v), under mechanical agitation at 1000 rpm, for a maximum period of 120 min. Commercial lipase from Thermomyces lanuginosus (TLL) was applied as a biocatalyst in the reaction with a ratio of 20 mgprotein/50 gsubstrate. Then, molecular docking was carried out to investigate interactions among lipases and triglycerides in Licuri oil (Caprilyl–dilauryl–glycerol (CyLaLa), capryl–dilauryl–glycerol (CaLaLa), capryl–lauryl–myristoyl–glycerol (CaLaM), and dilauryl–myristoyl–glycerol (LaLaM), using the AutoDock Vina v.1.1.2 software (TROTT; OLSON, 2010). FTIR analyzed the reaction product to confirm the occurrence of oil biotransformation. Until 120 min, TLL promoted a conversion and productivity of around 54% and 84,000 µmol/h·g, respectively. According to molecular docking results (Fig: 1A-B), TLL interacts with 3 of the 4 major triglycerides in licuri oil, through the amino acid His258, which belongs to TLL catalytic triad (Ser146, Asp201, and His258). The lipase highest catalytic performance is correlated to lower affinity energy in the following order: CaLaLa (-5.3 Kcal.mol-1)CaLaM (-5.2 Kcal.mol-1) CyLaLa (-5.0 Kcal.mol-1). LaLaM showed the highest affinity energy and did not interact with any amino acids present in TLL active site. Finally, the FTIR analyses confirmed the licuri oil biotransformation into FFA, by modifying the 1744 cm-1 band characteristic of the C=O ester bonds present in triglycerides, to the 1708 cm-1 band (Fig 1C) C=O of carboxylic acids, which are more intense due to oil biotransformation (CRUZ et al., 2019; ROHMAN; MAN, 2010). In this way, it was possible to understand better the TLL mechanism to catalyze the hydrolysis of licuri oil and create a new segment for licuri oil production chain by introducing its FFA concentrates as a new product for the industrial sector. Acknowledgments The work was funded by the Fundação de Pesquisa e Inovação Tecnológica do Estado de Sergipe -FAPITEC/SE (PROCESSO Nº: 01302440586/PDPG-SEMIARIDO1932116P), Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq da Chamada MCTIC/CNPq Nº 28/ 2018 Process:429557/2018-3), and in part by the Coordenação de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES) - financial code 001. And CIEPQPF is supported by the FCT through the projects UIDB/EQU/00102/2020 and UIDP/EQU/00102/2020. |
| References: | BARBOSA, M. S. et al. Optimization of the enzymatic hydrolysis of Moringa oleifera Lam oil using molecular docking analysis for fatty acid specificity. Biotechnology and Applied Biochemistry, v. 66, n. 5, p. 823-832, 2019. CASAS-GODOY, L. et al. Lipases: An Overview. Lipases and Phospholipases, v. 861, p. 3-30, 2012. CRUZ, M. et al. Monitoring Enzymatic Hydroesterification of Low-Cost Feedstocks by Fourier Transform InfraRed Spectroscopy. Catalysts, v. 9, p. 535, 2019. LA SALLES, K. T. DA S. DE et al. Characterization of Syagrus coronata (Mart.) Becc. oil and properties of methyl esters for use as biodiesel. Industrial Crops and Products, v. 32, n. 3, p. 518-521, 2010. ROHMAN, A.; MAN, Y. B. C. Fourier transform infrared (FTIR) spectroscopy for analysis of extra virgin olive oil adulterated with palm oil. Food Research International, v. 43, n. 3, p. 886-892, 2010. SEGALL, S. D. et al. Ouricuri (Syagrus coronata) Triacylglycerol Analysis Using HPLC and Positive Ion Electrospray Tandem MS. JAOCS, Journal of the American Oil Chemists’ Society, v. 81, n. 2, p. 143-149, 2004. TROTT, O.; OLSON, A. J. Software News and Update AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading OLEG. Journal of computational chemistry, v. 31, p. 455-461, 2010. |
| Figures: | 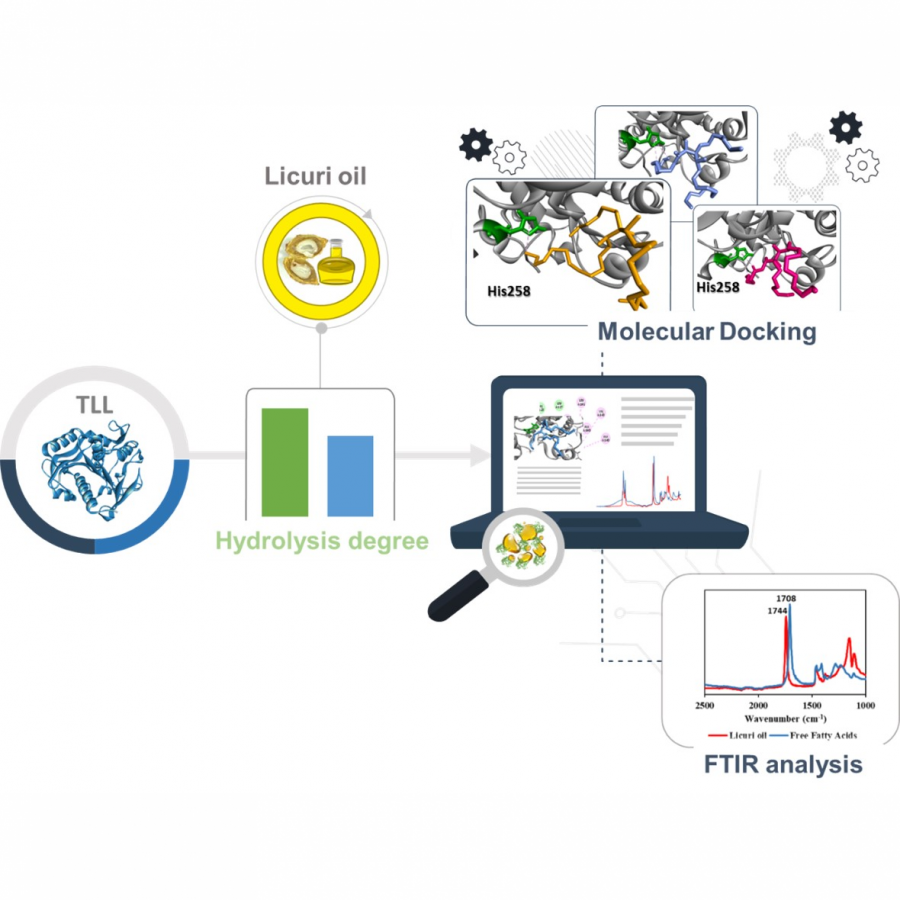 Graphical abstract Licuri oil biotransformation catalyzed per TLL and identified through experimental and computational analyses. |
#1276 | One-step production and immobilization approach for unspecific peroxygenases |
|
| Presenting author: | Niklas TEETZ of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN |
| Other authors: | Selina LANG of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN Dirk HOLTMANN of KARLSRUHE INSTITUTE OF TECHNOLOGY (KIT) |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | unspecific peroxygenases / immobilization / cell surface display / |
| Purpose: | One-step production and immobilization approach for unspecific peroxygenases Niklas Teetz, Department of Bioprocess Intensification, THM Gießen, Department of Technical Biology, Karlsruhe Institute of Technology, Karlsruhe; Selina Lang, Department of Bioprocess Intensification, THM Gießen; Dirk Holtmann, Department of Bioprocess Intensification, THM Gießen, Department of Technical Biology, Karlsruhe Institute of Technology, Karlsruhe Unspecific peroxygenases (UPOs) are regarded as a “dream catalyst” [1] for a variety of selective oxyfunctionalization reactions like hydroxylations, epoxidations and oxygenations [2]. The catalyzed reactions are similar to those of the well-known P450 monooxygenases [3] but UPOs offer independence from reduced nicotinamide cofactors and electron transport chains by using the easily produced hydrogen peroxide as only cofactor [1]. Here, we present the display of the model UPO rAaeUPO (PaDaI) on the cell surface of the commonly used heterologous production host Pichia pastoris (Komagataella phaffii) as a 1-step production and immobilization approach. Multiple genes were cloned, combining the coding sequence for PaDaI with genes or part of genes coding for cell wall proteins from Saccharomyces cerevisiae. The genes were transformed into P. pastoris via an integration vector for production of the fusion proteins and subsequent comparison of the different systems among each other and with secreted, free PaDaI. All used systems yielded active UPOs, immobilized on the cell surface. Only minimal to no enzyme activity was detected in the supernatant, indicating a near complete surface display of the produced enzymes. One system in particular, a C-terminal fusion of PaDaI and SAG1, which is a glycoprotein involved in cell-cell contact during yeast mating, yielded identical ABTS activity per volume culture broth to the secreted PaDaI with ~90 % of the activity being in the cell pellet. The presented cell surface system of UPOs offers even easier downstream processing than systems with secreted UPOs do and already includes immobilization on a cheap, easily retainable and replacable matrix, that is the cells of the production host themselves. |
| References: | [1] Y. Wang et al, Current Opinion in Chemical Biology 2017, 37:1-9 [2] Hofrichter et al, Antioxidants 2022, 11, 163 [3] S. Bormann et al, Molecular Catalysis 2020, 492, 110999 |
| Figures: | 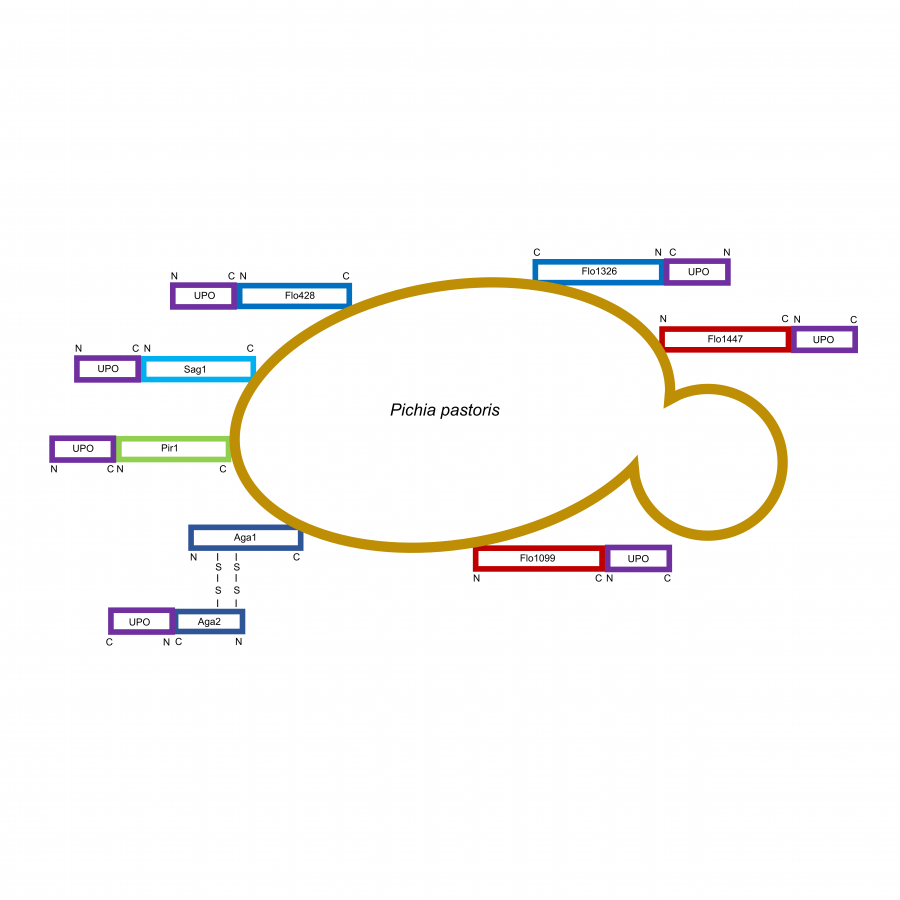 Scheme 1: Overview over the used cell surface display systems N: N-terminus
C: C-terminus
S--S: disulfide bond
SAG1: alpha-agglutinin
AGA1: anchoring subunit of a-agglutinin
AGA2: receptor binding subunit of a-agglutinin
PIR1: protein containing internal repeats
FLO: protein involved in flocculation 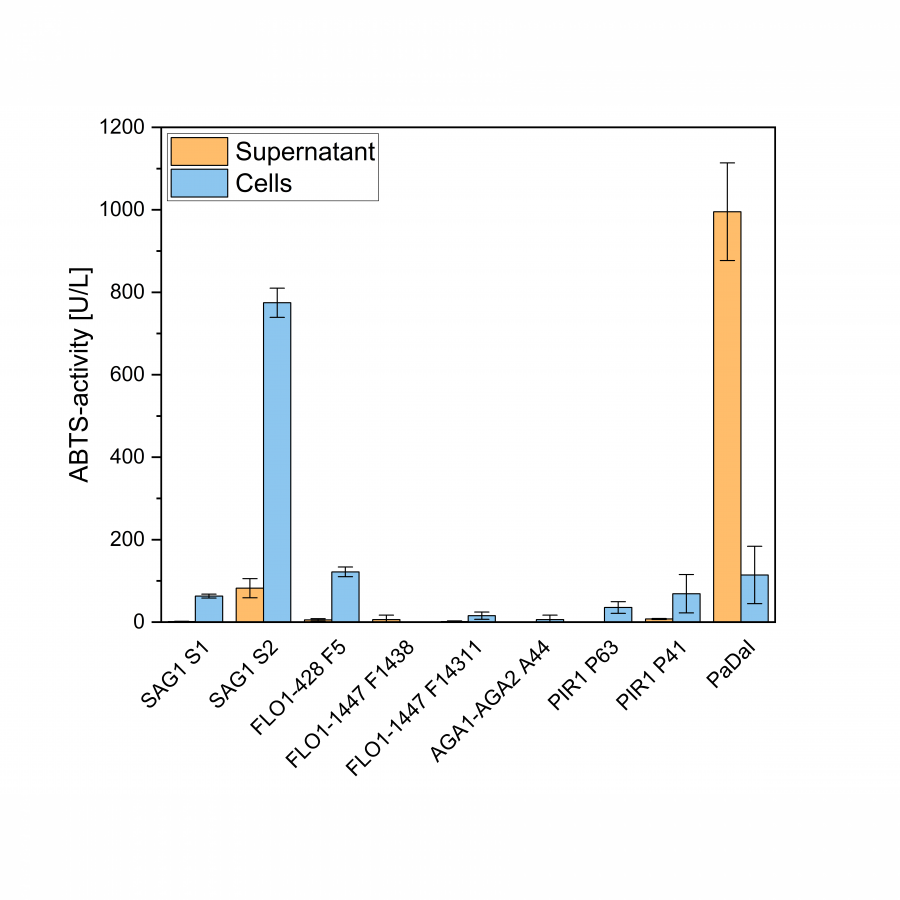 Comparison of ABTS-activity of different cell surface display systems and secreted PaDaI orange bars: activity in supernatant
blue bars: activity in cell pellet, resuspended in 100
mM KPi and normalized to initial culture volume |
#1277 | The Enzymatic Synthesis of S-Adenosyl-L-Homocysteine (SAH) and Its Derivatives for Enzyme-Catalyzed Methylation |
|
| Presenting author: | Xiaojin WEN of UNIVERSITY OF BASEL |
| Other authors: | Florian SEEBECK of UNIVERSITY OF BASEL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | S-Adenosyl-L-Homocysteine / Enzymatic Synthesis / / |
| Purpose: | S-Adenosyl-L-homocysteine (SAH) and related nucleosides have been found as key regulator compounds in many biological transmethylation systems and their potential in the pharmaceutical and chemotherapeutic fields has been suggested.[1] So far, for enzymatic methylation, SAH has been used as the substrate for one-step regeneration of S-adenosylmethionine (SAM), the key co-substrate of most methyltransferases (MTs) in nature.[2-4] Due to their natural promiscuity, or as a result of introduced mutations, many MTs can also transfer larger alkyl chains or aromatic fragments to SAH to form plenty of SAM analogues.[5-7] Therefore, SAH and its derivatives have recently attracted great interest. However, SAH is so expensive that low-cost methods for SAH synthesis are in high command.[8,9] Herein, we developed a three enzyme-catalyzed cascade for in vitro SAH biosynthesis from two low-priced starting materials D/L-homocysteine thiolactone and adenosine. This enzyme cascade could also be used for production of SAH derivatives from their corresponding adenosine analogues. |
| References: | [1] P. M. Ueland, Pharmacol. Rev. 1982, 34, 223-253; [2] C. Liao, F. P. Seebeck, Nat. Catal. 2019, 2, 696-701; [3] X. Wen, F. Leisinger, V. Leopold, F. P. Seebeck, Angew. Chem. Int. Ed. 2022, 61, e202208746; [4] F. Ospina, K. H. Schülke, J. Soler, A. Klein, B. Prosenc, M. Garcia-Borràs, S. C. Hammer, Angew. Chem. Int. Ed. 2022, 61, e202213056; [5] Q. Tang, I. V. Pavlidis, C. P. S. Badenhorst, U. T. Bornscheuer, ChemBioChem 2021, 22, 2584-2590; [6] Y. Motorin, J. Burhenne, R. Teimer, K. Koynov, S. Willnow, E. Weinhold, M. Helm, Nucleic Acids Res. 2011, 39, 1943-1952; [7] C. Dalhoff, G. Lukinavicius, S. Klimasăuskas, E. Weinhold, Nat. Chem. Biol. 2006, 2, 31-32; [8] G. de la Haba, G. L. Cantoni, J. Biol. Chem. 1959, 234, 603-608; [9] S. Shimizu, T. Ohshiro, S. Shiozaki, H. Yamada, J. Biotechnol. 1986, 4, 91-100. |
#1279 | Exploring the substrate scope of the Unspecific Peroxygenase from Daldinia caldariorum |
|
| Presenting author: | Angelique POTHUIZEN of DELFT UNIVERSITY OF TECHNOLOGY |
| Corresponding author: | Frank HOLLMANN of DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Rosalie WOUTERS of DELFT UNIVERSITY OF TECHNOLOGY Thomas HILBERATH of DELFT UNIVERSITY OF TECHNOLOGY Hugo BRASSELET of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Oxyfunctionalisation / Unespecific Peroxygenases / Biocatalysis / Daldinia caldariorum |
| Purpose: | Since their discovery in 2004, Unspecific Peroxygenases (UPOs) have gained attention because of their ability to selectively perform oxyfunctionalisation reactions on a broad range of substrates. From a chemical point of view, these reactions are very difficult to perform and often require harsh reaction conditions while still lacking selectivity. UPOs could provide an alternative to chemical synthesis of oxyfunctionalised hydrocarbons. Today, however, the number of UPOs available is rather limited. In this project, an UPO from Daldinia caldariorum (DcaUPO), as previously described by Linde et al. (2020) was heterologously expressed in E. coli and characterised. The main focus of this project was to explore the substrate scope of this UPO, therefore its activity was tested for a broad range of different substrates, including alcohols, aromatic hydrocarbons, polymer model compounds, sulphides and terpenes. Activity was observed towards multiple substrates from all substrate classes. Observed reaction types include alcohol oxidation, aromatic hydroxylation, aliphatic hydroxylation, N-dealkylation and sulfoxidation. In addition to exploring the substrate scope, a small effort was made into reaction optimisation for improving the stability of the enzyme. An excess of H2O2 can lead to heme degradation and inactivation of the enzyme, therefore a balance needs to be found between the supply of H2O2 to proceed with the reaction without inactivating the enzyme. To summarise, this poster shows the large potential of UPOs for synthesis of oxyfunctionalised compounds, but it also shows the work that still needs to be done before these enzymes are ready for large-scale applications. |
| References: | [1] Linde D, Olmedo A, GonzálezBenjumea A, Estévez M, Renau-Mínguez C, Carro J, Fernández-Fueyo E, Gutiérrez A,Martínez AT. 2020. Two new unspecific peroxygenases from heterologous expression of fungal genes in Escherichia coli. Appl Environ Microbiol 86:e02899-19. https://doi.org/10.1128/AEM.02899-19. |
#1281 | Molecular Engineering of Lytic Polysaccharide Monooxygenase TthLPMO9G: Rational Design of Mutations and evaluation of Substrate Recognition, Mode of Action, and Reducing Agent Specificity |
|
| Presenting author: | Koar CHOROZIAN of NATIONAL AND TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | Anthi KARNAOURI of AGRICULTURAL UNIVERSITY OF ATHENS Antonis KARANTONIS of NATIONAL AND TECHNICAL UNIVERSITY OF ATHENS Evangelos TOPAKAS of NATIONAL AND TECHNICAL UNIVERSITY OF ATHENS |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lytic polysaccharide monooxygenases / point mutations / oxidoreductases / cellulose |
| Purpose: | Lytic polysaccharide monooxygenases (LPMOs) are copper-dependent enzymes that play a crucial role in the oxidative degradation of recalcitrant carbohydrate polymers. Study of their activity provides insights into the biological mechanisms employed in nature for the polysaccharide degradation. Introducing point mutations is a common way to elucidate the catalytic activity of enzymes and shed light on the importance of targeted amino acids. In this study, three LPMOs from the thermophilic fungus Thermothelomyces thermophila, including the wild-type TthLPMO9G and two mutants generated following a rational mutation approach, H140A and S28A, were evaluated for their activity on cellulose, the specificity towards reducing agents with either O2 or H2O2 as co-substrate, as well as the stability against temperature and oxidative stress. Notably, the H140A mutant exhibited a weakened catalytic activity towards cellulose, although its stability and its monooxygenase and peroxygenase reactivity in the absence of substrate remained unaltered, which highlights the importance of His in this position for substrate recognition and catalysis. The S28A mutant exhibited a different pattern of soluble products released when acting on cellulose, which implies an altered mode of action compared to the wild-type enzyme when reduced by specific electron donors. The results showed that mutation in specific amino acids has a significant effect on the substrate recognition and activity of TthLPMO9G on cellulose, as well as the type of the produced oxidized oligosaccharides. |
| References: | [1] Stepnov, A.A., Eijsink, V.G.H. & Forsberg, Z. Enhanced in situ H2O2 production explains synergy between an LPMO with a cellulose-binding domain and a single-domain LPMO. Sci Rep 12, 6129 (2022). [2] Glyn R. Hemsworth; Revisiting the role of electron donors in lytic polysaccharide monooxygenase biochemistry. Essays Biochem 18 April 2023; 67 (3): 585-595. |
| Figures: | 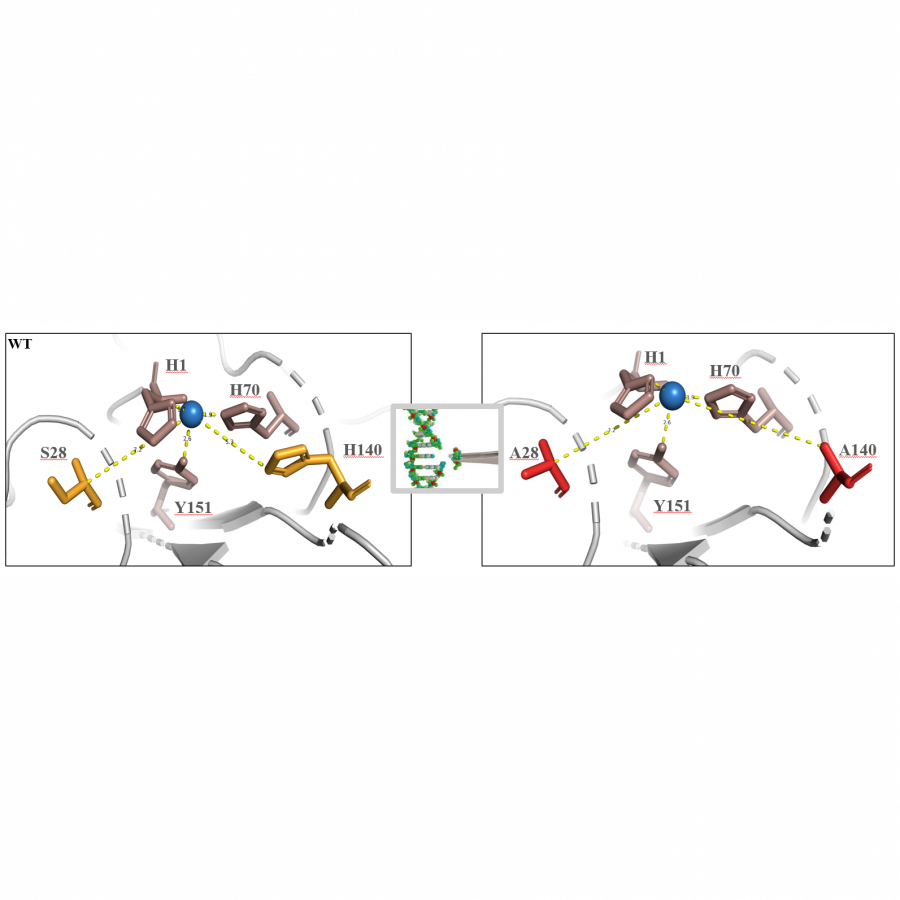 Comparison of wild-type TthLPMO9G and two point mutants. The wild-type TthLPMO9G protein is shown in gray and the amino acids of interest are shown in orange color, while the H140A and S28A mutants are shown in red, respectively. The H140A mutation was introduced at position 140, which is located next to the a |
#1282 | Regioselective oxidation of bulky substrates by expression of new unspecific peroxygenases (UPOs) with broader tunnels and automated in silico screening |
|
| Presenting author: | Bonnie WINTER of MARTIN-LUTHER-UNIVERSITÄT HALLE-WITTENBERG |
| Other authors: | Ahmed ABDELFATTAH of LEIBNIZ INSTITUTE FOR PLANT BIOCHEMISTRY Martin WEISSENBORN of MARTIN-LUTHER-UNIVERSITÄT HALLE-WITTENBERG Mehdi DAVARI of LEIBNIZ INSTITUTE FOR PLANT BIOCHEMISTRY |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | unspecific peroxygenase / computational / regioselectivity / oxidation |
| Purpose: | Unspecific peroxygenases (UPOs) are versatile biocatalysts capable of oxidizing various organic compounds. However, the small tunnels and binding pockets of currently expressed UPOs limit their use in converting bulkier substrates that are readily transformed by P450 monooxygenases. 1-3 Achieving selective conversions of bulky substrates typically involves medium to high-throughput screening, which can be a tedious and time-consuming laboratory process. This study aims to express new UPOs with broader tunnels and substrate pockets that can convert bulkier substrates as well as design an automated program for screening substrate/product libraries with a set of selected UPOs to predict regioselective oxidation products. To achieve this, we successfully expressed 9 out of 13 new UPOs using signal peptide shuffling4 in Saccharomyces cerevisiae and developed an automated in silico screening program based on docking and the concept of near-attack conformation states. Docking poses that meet the restrictions of the near-attack conformation state are assumed as productive and are further filtered for the desired regioselectivity. The method was first validated by a set of standard substrates. In the upcoming months, the program will be applied to a list of ~50 bulky substrates with industrial relevance to be oxidized selectively. Ten substrates that were predicted to be converted with high regioselectivity and which docking poses are in a productive conformation will be tested in the lab by a set of new UPOs. Our approach will enable efficient identification of the optimal UPO for each desired substrate-product pair, thus facilitating the development of more efficient and sustainable biocatalytic processes for a wide range of industrial applications. |
| References: | 1. Urlacher, V. B.; Girhard, M., Cytochrome P450 Monooxygenases in Biotechnology and Synthetic Biology. Trends in Biotechnology 2019, 37 (8), 882-897. 2. Grogan, G., Hemoprotein Catalyzed Oxygenations: P450s, UPOs, and Progress toward Scalable Reactions. JACS Au 2021, 1 (9), 1312-1329. 3. Münch, J.; Püllmann, P.; Zhang, W.; Weissenborn, M. J., Enzymatic Hydroxylations of sp3-Carbons. ACS Catalysis 2021, 11 (15), 9168-9203. 4. Pullmann, P.; Knorrscheidt, A.; Munch, J.; Palme, P. R.; Hoehenwarter, W.; Marillonnet, S.; Alcalde, M.; Westermann, B.; Weissenborn, M. J., A modular two yeast species secretion system for the production and preparative application of unspecific peroxygenases. Commun Biol 2021, 4 (1), 562. |
| Figures: |  Process of the project Starting with the expression of new UPOs with broader substrate tunnels, followed by the program development for automated docking and docking evaluation, which is applied to library of bulky substrates to finally test the best enzyme-substrate pairs. |
#1283 | Understanding Enzyme Mechanisms Under Low Moisture Conditions for Laundry Detergent Developments |
|
| Presenting author: | Ellie ASCHROFT of NORTHUMBRIA UNIVERSITY |
| Other authors: | Jose MUNOZ of NORTHUMBRIA UNIVERSITY GARY BLACK of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | glycoside hydrolases / dry environments / sustainable detergents / enzymology |
| Purpose: | It has long been understood that water is an essential requirement to maintain protein stability, mediate folding and aid catalysis. Although previous research has shown enzymes such as lyases or esterases can operate under low if not zero hydration, the overall understanding of how catalysis and folding occur with minimal water has not been thoroughly explored. In collaboration with Procter and Gamble, we aim to identify enzymes capable of degrading food stains on garments under low moisture conditions before washing. Therefore, reducing the need for long wash cycles at high temperatures whilst still maintaining effective stain removal. Successful stain removal from low moisture enzymes would not only be a break though, in the scientific laundry field, but the product developed using this technology will also aim to reduce carbon emissions from wash cycles and help save energy costs for consumers due to shorter washing time. To distinguish enzymes that can catalyse reactions with water absent, their folding and active site mechanisms will need to be understood. We are going to understand the mechanism of these enzymes, dissecting the structure/activity relationship under these challenged circumstances. We will explore biocatalysts from microorganisms commonly known to function in low or dry conditions such as enzymes from xerophilic bacteria. To fully understand this catalytic mechanism, we are using organic solvents as starting point to mimic the low water conditions and screen for the right enzymatic activities. In addition, the structure and residues specific to this hydrophobic activity are being investigated using protein engineering and proteomics to identify which of the enzymes are surface-placed proteins in their respective xerophilic bacteria. Finally, in this project we face the challenge to develop the correct enzymatic assay. For that, we have targeted starch stains as this glycan has been shown to be highly concentrated in laundry baskets. |
#1285 | Investigating the degradation pathways of fungal mannoproteins as carried out by different bacteria found in the human gut |
|
| Presenting author: | JONATHON WOODS of NORTHUMBRIA UNIVERSITY |
| Other authors: | Jose MUNOZ of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | gut microbiota bioprospecting / glycoside hydrolase / mannan / enzymology |
| Purpose: | As we transition to more sustainable food sources in order to reduce resource consumption and our ecological footprint, it is critical we investigate the effects new food sources have on the health of consumers. Changes in diet can particularly influence the structure and diversity of the human gut microbiome (HGM) which previous research has indicated contributes to overall host health, particularly of the gut and brain via the gut-brain axis [1]. Quorn is currently one of the leading brands in Europe promoting sustainably produced meat alternatives manufactured from the fermentation of the fungal species Fusarium venenatum (F. venenatum) [2]. Humans lack the capacity to fully digest F. venenatum due to not having the necessary enzymes to degrade the complex sugars (dietary fibres) that constitute its cell wall, instead, we rely solely on bacteria that make up the HGM. Different bacterial species endogenous to the gut utilise diverse dietary fibre through the process of fermentation by primary and secondary degraders. Cross-feeding interactions between different degraders are essential to ensuring the health of the HGM during dietary changes. To degrade a polysaccharide, bacteria rely on glycoside hydrolases (GHs) which cleave specific bonds in the structure of a polysaccharide through hydrolysis reactions. Bacteria native to the HGM group enzymes (GHs) specific to a single substrate in co-localised, co-regulated gene clusters known as polysaccharide utilisation loci (PUL) the transcription of which is initiated in the presence of their target polysaccharide [3]. This project aims to find out which known gut bacteria are capable of growth on mannoproteins (fibre) obtained from F. venenatum as the sole carbon source and the methods in which they do so. It is beneficial to determine which PULs and GHs the primary degrader species use in order to ascertain some of the structural elements of this novel fibre and the products of fermentation that may be made available to secondary degrader species. Mannoproteins are the fibre of interest as they form the outermost layer of the cell wall, so are responsible for coordinating many of F. venenatum’s extracellular interactions, especially during fermentation and processing to form Quorn’s products. Based on current research we expect PULs that degrade mannoproteins to include alpha-mannanases (e.g. GH76) as the mannoproteins structure is majoritively alpha-mannan bonds [4], once PULs containing alpha-mannanases are identified from known primary degrader species, other enzymes in the PULs may be investigated to determine what activity they may have on mannoproteins in order to build a comprehensive picture of its total degradation. The implications of this research will pave the way to an increased understanding of how the different enzymatic mechanisms deployed by bacteria native to the HGM maintain gut health in the presence of new food sources. [1] P. Kesika, N. Suganthy, B.S. Sivamaruthi, C. Chaiyasut, Life Sci, 2021, 264 [2] T. Finnigan, L. Needham, C. Abbott, Sustainable Protein Sources, 2016, 305-326 [3] P. Lapebie, V. Lombard, E. Drula, N. Terrapon, B. Henrissat, Nature Communications, 2019, 10 [4] I. Stalls, K. Sandra, S. Geysers, R. Contreras, J. Beeumen, M. Claeyssens, Glycobioogy, 2004, 14, 713-724 |
#1287 | Development of immobilized lipases for the production of 2nd generation biodiesel |
|
| Presenting author: | ANDRONIKI G. SPANOU of UNIVERSITY OF CRETE-DEPARTMENT OF CHEMISTRY |
| Corresponding author: | IOANNIS V. PAVLIDIS of UNIVERSITY OF CRETE-DEPARTMNET OF CHEMISTRY |
| Other authors: | CHRISTINA FIOTAKI of UNIVERSITY OF CRETE-DEPARTMENT OF CHEMISTRY ELENI THEODOSIOU of INSTITUTE OF APPLIED BIOSCIENCES-CENTRE FOR RESEARCH AND TECHNOLOGY HELLAS AGGELIKI ANDREADELLI of INSTITUTE OF APPLIED BIOSCIENCES-CENTRE FOR RESEARCH AND TECHNOLOGY HELLAS ANTONIOS M. MAKRIS of INSTITUTE OF APPLIED BIOSCIENCES-CENTRE FOR RESEARCH AND TECHNOLOGY HELLAS |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biodiesel / Lipases / Immobilization / Biocatalysis |
| Purpose: | The production of biofuels has gained significant attention in recent years due to the increasing demand for renewable and sustainable energy sources. Among them, biodiesel has emerged as a promising alternative to fossil fuels (Lv et al., 2021). Lipases, enzymes that catalyze the breakdown of fats and oils, have been widely used in the production of biodiesel. However, the high cost and limited stability of free lipases pose significant challenges in large-scale production (Martínez Gil et al., 2022). Immobilization has been explored as a solution to overcome these challenges (Arana-Peña et al., 2020). The objective of the present work is to develop robust biocatalysts for the production of 2nd generation biodiesel, utilizing industrial sunflower and acid oil. In this research, we investigated the potential of four biocatalysts for the production of 2nd generation biodiesel. These biocatalysts include Rhizopus oryzae lipase (Biolipasa-R, Biocon®-Spain), immobilized Candida antarctica lipase B (Novozym® 435), LIP2 lipase from Yarrowia lipolytica, and whole cells of Y. lipolytica after surface display of the LIPL2. Specifically, recombinant Y. lipolytica strains have been constructed using vectors bearing the strong promoters, i.e., Histone 3 and Exp1, and two LIP2 gene copies, in order to enhance the LIP2 secretion in the culture medium (Georgiadis et al., 2023). The presence of two LIP2 copies improved the specific activity of the enzyme approximately 55 times (418 U/mg) in comparison to the wild type strain (7.69 U/mg), making it an attractive candidate for biocatalyst development. In addition, Y. lipolytica cells were employed as whole-cell biocatalyst after surface engineering with the cell wall protein YlPIR1. Biodiesel production has been tested with this whole-cell biocatalyst and the results so far have shown up to 5 % conversion. Apart from the recombinant strains, biodiesel production has been tested with R. oryzae lipase and C. antarctica lipase B and the results have shown up to 100 % conversion. In this study, we compared the efficiency of these four biocatalysts in terms of biodiesel yield/conversion, reaction time, stability under different conditions, and immobilization efficiency. To attain the desired productivity, lipases were immobilized on Diatomaceous earth, an affordable and environmentally friendly silicon dioxide-based material, as well as on commercial supports like Purolite® methacrylate resins. Immobilization efficiency reached 100 % for Biolipasa-R and 81 % for the engineered secreted LIP2, using the commercial resins. The findings of this study aim to provide useful insights into the development of efficient and cost-effective biocatalysts for the production of biodiesel. This research has been co-financed by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH – CREATE – INNOVATE (project code:Τ2ΕDΚ-00573 ) |
| References: | [1] Liangliang, Lv., et al. Processes, 2021, 9(2), 1-10. [2] Martinez, Gil., et al. ACS Omega, 2022, 7(46), 41882-41904. [3] Arana-Pena, S., et al. Frontiers in Bioengineering and Biotechnology, 2020, 8. [4] Georgiadis, I., et al. Microorganisms, 2023, accepted. |
| Figures: |  Co-finance Co-finance |
#1288 | Highly Accurate Enzymatic Activity Predictions Based on a Machine Learning Algorithm Trained on a Transaminase Screening Data Set |
|
| Presenting author: | Oscar ALVIZO of CODEXIS |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Machine Learning / Enzyme Engineering / Transaminases / |
| Purpose: | Directed evolution is a well-established method for improving the performance of an enzyme catalyst. One of the challenges in the process is identifying a sequence that can serve as the starting sequence for the evolution campaign. Ideally, this sequence must be active on the target substrate and produce the desired product with the intended stereochemistry. The duration of an evolutionary campaign is highly dependent on the initial activity of the starting enzyme under the desired process conditions. The more active the starting sequence, the less rounds of evolution required to identify a highly performing variant that meets the required commercial criteria. Here we discuss a machine learning approach used to identify enzymes that can act on the target substrate. The ML model was trained on the activity of transaminases from multiple directed evolutionary lineages and dozens of substrates. The best results were observed when an ensemble of models was used for the predictions. The final optimized pipeline was shown to be 80-90% accurate. |
| References: | [1] Yang, K.K., Z. Wu, and F.H. Arnold, Nat Methods, 2019. 16(8): p. 687-694. [2] Singh, N., et al., Emerg Top Life Sci, 2021. 5(1): p. 113-125. [3] Wu, Z., et al., Proc Natl Acad Sci U S A, 2019. 116(18): p. 8852-8858. |
| Figures: | 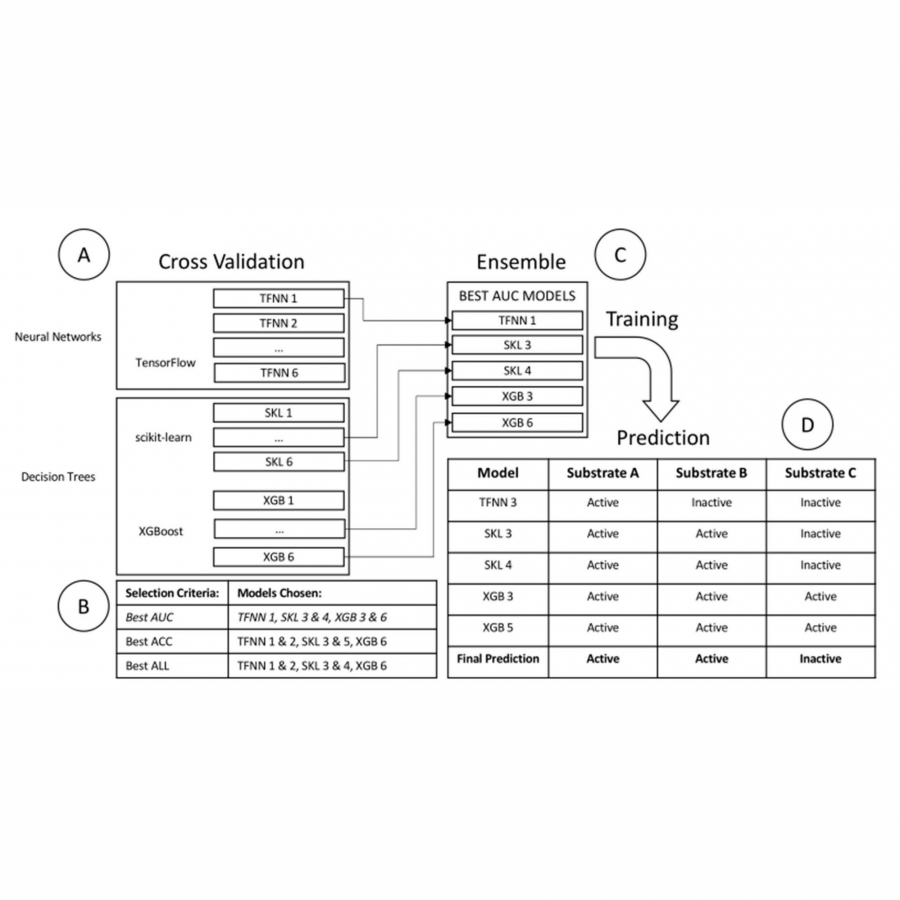 Architectural Flow Diagram A diagram depicting the steps used to generate the machine learning model for the prediction of active enzymes. |
#1289 | Lipase B from Candida antarctica in AOT-Water-Isooctane reverse micellar systems under high salt conditions to enhance butyl oleate synthesis. |
|
| Presenting author: | JOSÉ MARTÍN MÁRQUEZ VILLA of CIATEJ, A. C. |
| Corresponding author: | ROSA MARÍA CAMACHO-RUÍZ of CIATEJ, A. C. |
| Other authors: | JUAN CARLOS MATEOS-DÍAZ of CIATEJ, A. C. JORGE ALBERTO RODRÍGUEZ-GONZÁLEZ of CIATEJ, A. C. |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Reverse micelle / AOT / lipase-catalyzed esterification / halophilic enzymes |
| Purpose: | Reverse micelles act as a biocompatible shield to host biocatalysts in an aqueous medium against the aggressive organic solvent, where this nanoreactor delimits an inner water core and an outer organic solvent (Bhavya, Priyanka, & Rastogi, 2012; Mohd-Setapar, Mohamad-aziz, Harun, & Mohd-azizi, 2012; Pileni, 1993). Therefore, reverse micelles are particularly favorable systems for lipases, due to the enhanced selective solubility of the substrates by the intimate liquid-liquid contact they maintain with the organic solvent and the polar characteristics of the core that provide a physiological environment for lipases (Mohd-Setapar et al., 2012). In the present investigation, the synthesis of butyl oleate by Candida antarctica lipase B (CalB) under extreme halophilic conditions was investigated using the AOT/ Water/ Isooctane reverse micellar system. The impact of aqueous content (Wo=[H2O]/([Surfactant])) and NaCl variation on the enzymatic activity of CalB in the reaction of butyl oleate in reverse micelles was explored. The results indicated that, as a function of increasing NaCl, it is remarkable to achieve higher enzymatic activity parameters, such as 444.85 μmol/min at 5 M NaCl and Wo = 10, as the best esterification conditions at pH 7.2 and 30 °C. However, it was clear that the synthesis of butyl oleate by CalB lipase increased as a function of the reduction of the average reverse micelle size, where the reverse micelle sizes were determined by dynamic light scattering (DLS) and it was observed that the behavior of the reverse micelle size can be described as a function of water content and salt ion fluctuation. Consequently, the increase in butyl oleate synthesis demonstrated the potential of reverse micelles as systems that enhance mass transport phenomena in heterogeneous biocatalysis. Furthermore, reverse micelles are promising systems for the investigation of extreme halophilic lipases. |
| References: | 1. Bhavya, S. G., Priyanka, B. S., & Rastogi, N. K. (2012). Reverse Micelles-Mediated Transport of Lipase in Liquid Emulsion Membrane for Downstream Processing. Biotechnology Progress, 28(6), 1542-1550. https://doi.org/10.1002/btpr.1637 2. Mohd-Setapar, S. H., Mohamad-aziz, S. N., Harun, N. H., & Mohd-azizi, C. Y. (2012). Review on the extraction of biomolecules by biosurfactant reverse micelles. Procedia APCBEE, 3, 78-83. https://doi.org/10.1016/j.apcbee.2012.06.050 3. Pileni, M. P. (1993). Reverse micelles as microreactors. J. Phys. Chem., 97, 6961-6973. https://doi.org/10.1021/j100129a008 |
#1290 | Discovery of Bacterial Reductive Aminases via Enzyme Mining |
|
| Presenting author: | Ewald JONGKIND of DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Melis TEKE of DELFT UNIVERSITY OF TECHNOLOGY Marcel VAN DEN BROEK of DELFT UNIVERSITY OF TECHNOLOGY Caroline PAUL of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Reductive amination / Biocatalysis / / |
| Purpose: | The synthesis of chiral amines is highly valuable for the production of pharmaceutical drugs. Over the last decade, several enzyme classes have been employed to produce mainly chiral primary amines with high efficiency and enantioselectivity. Since 2017, Turner and co-workers have characterized several fungal reductive aminases (RedAms) that catalyze both imine formation and asymmetric reduction, resulting in secondary and tertiary amines.[1] Several bacterial imine reductases (IREDs) also displayed reductive aminase activity, albeit with different catalytic residues in their active site than those of RedAms.[2] Only one recent example of a bacterial RedAm sequence has been reported so far.[3] We envisioned to explore new RedAm sequences from bacterial organisms in genomic databases. After an initial BLAST search, we discarded all hits with a sequence identity of fifty percent or higher to exclude fungal RedAms. Using EnzymeMiner,[4] we selected seven hits from different organisms that contained the reported catalytic residues in their active site. We produced the new enzymes by recombinant expression in E. coli, and observed reductive amination activity towards different carbonyl substrate and amine donor combinations. We also applied an Hidden Markov-Model to search for hits in our in-house Rhodococcus database. We retrieved a sequence that gave a specific activity of 6 U/mg towards hexanal and allylamine, and in contrast to other RedAms, showed higher activity at pH 7.0 than at 9.0. Further screening of different carbonyl substrates and amine donors is ongoing. |
| References: | [1] G. A. Aleku, S. P. France, H. Man, J. Mangas-Sanchez, S. L. Montgomery, M. Sharma, F. Leipold, S. Hussain, G. Grogan, N. J. Turner, Nat. Catal. 2017, 9, 961-969. [2] S. L. Montgomery, A. Pushpanath, R. S. Heath, J. R. Marshall, U. Klemstein, J. L. Galman, D. Woodlock, S. Bisagni, C. J. Taylor, J. Mangas-Sanchez, J. I. Ramsden, B. Dominguez, N. J. Turner, Sci. Adv. 2020, 6, eaay9320. [3] K. Zhang, Y. He, J. Zhu, Q. Zhang, L. Tang, L. Cui, Y. Feng, Front Bioeng Biotechnol 2021, 9, 798147. [4] J. Hon, S. Borko, J. Stourac, Z. Prokop, J. Zendulka, D. Bednar, T. Martinek, J. Damborsky, Nucleic Acids Res. 2020, 48, W104-W109. |
| Figures: | 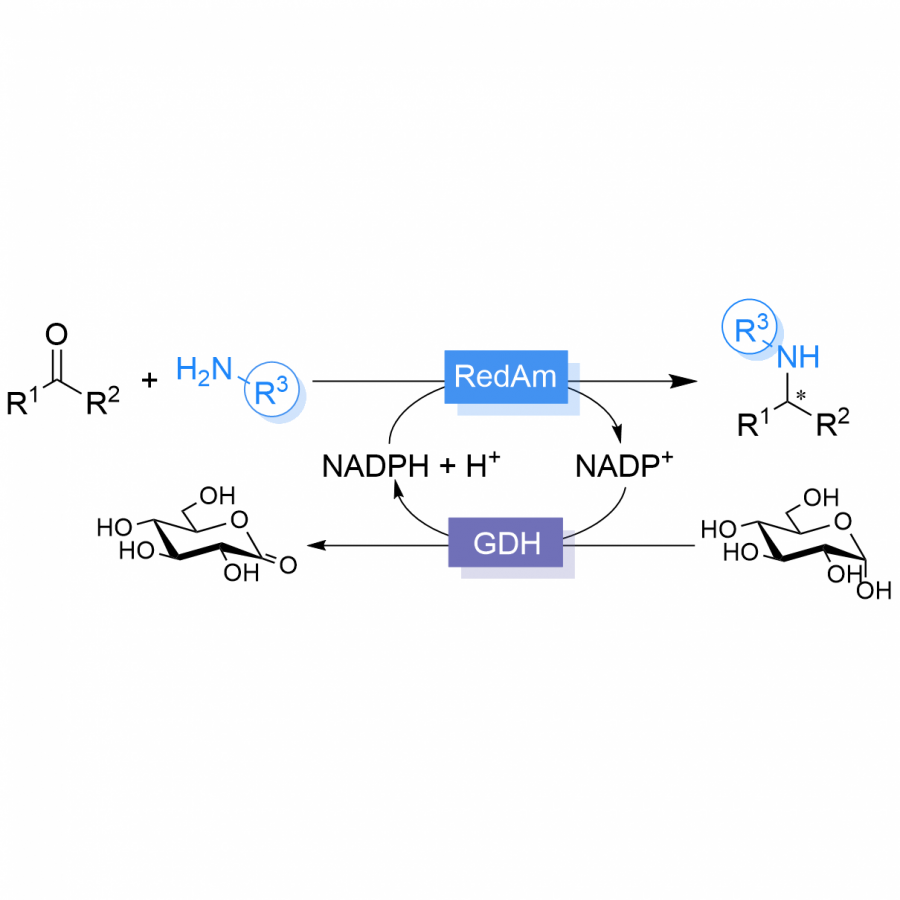 Reductive amination of ketones and aldehydes catalysed by potential bacterial reductive aminases, using a glucose dehydrogenase for cofactor recycling. . |
#1291 | Enzymatic modifications of sophorolipids: From biorefineries to valorization. |
|
| Presenting author: | Lorena MATEŠA of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB |
| Other authors: | Martina SUDAR of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB Pablo DOMÍNGUEZ DE MARÍA of SUSTAINABLE MOMENTUM SL. Zvjezdana FINDRIK BLAŽEVIĆ of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sophorolipids / biorefineries / reaction optimization / mathematical modeling |
| Purpose: | Sophorolipids (SLs) are a broadly known family of biosurfactants, composed of a sophorose head glycosidicaly bonded to the carbon of the long hydroxyl fatty acid [1]. Sophorolipids are typically produced by non-pathogenic yeasts, with main microorganism used for cultivation being Candida bombicola. Sophorolipids obtained in this manner are a mixture of structurally related molecules [2]. Different structures of sophorolipid molecules result in differences in their biological and physicochemical properties and, consequently, in several fields of application [3]. Sophorolipid molecules obtained by fermentation have industrial applications on their own, but some structural variations are desirable to further broaden their scope [4]. Enzymatic modifications offer the possibility to synthesize new derivatives by inserting other functional groups, which cannot be obtained via the fermentation process. Exposure of sophorolipids to enzymes could provide means towards the defined modification of sophorolipids [5]. Many examples are given on alteration of the fatty acid carboxylic end and the sophorose acetylation pattern, although examples of modifications of the sugar part of the sophorolipid are limited [6]. In this work, the main promising working lines for sophorolipid valorization are described, to establish a rational strategy to afford novel sophorolipid compounds with different applications and properties. Moreover, strategies for the synthetic improvement of sophorolipid derivatives, e.g., biorefinery-like cascading, use of biobased solvents, and reaction optimization using mathematical modelling, will be discussed. Acknowledgement: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No 101000560. |
| References: | [1] Ribeiro, I. A., J. Chromatogr. B 2012, 899, 72-80. [2] Saerens, K. M. J., Biotechnol. Bioeng. 2011, 108, 2923-2931. [3] Ma, X., Appl. Microbiol. Biotechnol. 2011, 91, 1623-1632. [4] Saerens, K. M. J.,. FEMS Yeast Res. 2015, 15. [5] Otto, R. T., Appl. Microbiol. Biotechnol. 1999, 52, 495-501. [6] Van Bogaert, I. N. A., Appl. Microbiol. Biotechnol. 2007, 76, 23-34. |
#1292 | Unraveling the benzylsuccinate synthase catalytic properties: modeling vs the experimental approach |
|
| Presenting author: | Gabriela OLEKSY of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHE |
| Other authors: | Anna SEKUŁA of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHE Kai KRÄMER of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Ivana ALEKSIĆ of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHE Maciej SZALENIEC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHE Johann HEIDER of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Antonio PIERIK of FACULTY OF CHEMISTRY, TECHNICAL UNIVERSITY OF KAISERSLAUTERN, GERMANY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | anerobic enzymes / biodegradation / EPR / |
| Purpose: | Benzylsuccinate synthase (BSS) is a model Fumarate Adding Enzyme (FAE), which belongs to the glycyl radical enzyme (GRE) family. The presence of a glycyl radical in the active centres of FAE makes those enzymes extremely sensitive to oxygen exposure. The role of FAEs in the environment is the initiation of anaerobic degradation pathways of aliphatic and aromatic hydrocarbons. BSS is produced by various anaerobic microorganisms, such as Thauera aromatica K172 and catalyzes the stereospecific addition of toluene to fumarate yielding (R)-benzylsuccinate. Due to the severe susceptibility of BSS to inactivation, the best strategy to study its catalytic mechanism is a combination of experimental and in silico techniques. The presented research focuses on the investigation of the catalytic properties of BSS produced natively by toluene-grown T. aromatica K172 and a recombinant BSS produced in the related species Aromatoleum evansii, which does not degrade toluene. The goal of this experiment was to verify the probable mechanism for the formation of an intermediate radical product with the inhibitors benzyl alcohol/benzaldehyde and fumarate. We followed changes in the glycyl-radical EPR signal in the presence of two inhibitors i.e., benzyl alcohol (B-OL) and benzaldehyde (B-ADH). B-OL and B-ADH inhibition kinetics were determined with T. aromatica K172 cell extracts under a strictly anaerobic environment at a pH of 7.8, at constant substrate concentrations (5 mM fumarate and 2 mM toluene) and inhibitors added to 5-100 µM. Benzylsuccinate production was quantified by the LC-DAD method, while product identification was conducted by LC-MS/MS. Interpretation of the observed EPR signals was supported by the simulation of spectra using the EasySpin program. The experimental spectra obtained with unlabeled and isotopically labelled (2H and 13C) B-OL and fumarate were analyzed. Isotropy and anisotropy of the hyperfine coupling constants (A) were tested, as well as a varying range of A, g value and linewidth parameters. Inhibition tests showed that adding 5 µM B-OL results in a 70% decrease of BSS activity and 100 µM led to almost complete inhibition. The B-ADH was a weaker BSS inhibitor caucusing a 50% and 95% decrease in activity, respectively, at 5 and 100 µM. The modelling result suggests the formation of a resonance-stabilized dehydrated allylic radical intermediate. The substrate preference of BSS for cresols and xylenes was investigated with cell extracts of recombinant WT and Y193S and Y193F mutants of BSS from A. evansii. The MS/MS analysis confirmed the activity of Y193F mutants with cresols and xylenes. A substrate preference shift from m-xylene to o-xylene was observed for the Y193F mutant. Acknowledgement. The authors acknowledge support of DFG/NCN (2018/31/F/NZ1/01856 and He2190/13-1), PL-Grid infrastructure, and Polish NAWA Programme STER-Internationalisation of doctoral schools (PPI/STE/2020/1/00020) |
| References: | 1. von Netzer F., Pilloni G., Kleindienst S., Krüger M., Knittel K., Gründger F., Lueders T. 2013. Enhanced gene detection assays for fumarate-adding enzymes allow uncovering of anaerobic hydrocarbon degraders in terrestrial and marine systems. Appl Environ Microbiol. 79(2):543-52 2. Heider J., Szaleniec M., Martins B., M., Seyhan D., Buckel W., Golding B., T. 2016. Structure and Function of Benzylsuccinate Synthase and Related Fumarate-Adding Glycyl Radical Enzymes. J Mol Microbiol Biotechnol. 26:29-44 |
#1293 | Biosynthesis of Furfurylamines in Batch and Continuous Flow by Immobilised Amine Transaminases |
|
| Presenting author: | Luisa MERZ of KTH ROYAL INSTITUTE OF TECHNOLOGY / VIAZYM B.V. |
| Other authors: | Tobias HEINKS of BIELEFELD UNIVERSITY Jan LIEDTKE of BIELEFELD UNIVERSITY Matthias HÖHNE of TECHNICAL UNIVERSITY BERLIN Luuk VAN LANGEN of VIAZYM B.V. Uwe BORNSCHEUER of UNIVERSITY OF GREIFSWALD Gabriele FISCHER VON MOLLARD of BIELEFELD UNIVERSITY Per BERGLUND of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Immobilization / Transaminase / Flow / HMF |
| Purpose: | Amines are important building blocks in the pharmaceutical industry, and their production through sustainable methods is becoming increasingly desirable. Biocatalytic syntheses and chemicals derived from renewable resources are being explored to achieve sustainable production of these amines. Furfurylamines, especially 5-(hydroxymethyl)furfurylamine (HMFA) and 2,5-di(aminomethyl)furan (DAF) derived from biomass, are gaining attention due to their renewable nature and functionality. We expanded previous work on the production of HMFA and DAF through the use of immobilised transaminsases in batch and flow synthesis.[1] We identified four different amine transaminases (ATAs) that can catalyse the reductive amination of 5-(hydroxymethyl)furfural (HMF) and 2,5-diformylfuran (DFF). To immobilise these ATAs, we used glutaraldehyde-functionalised amine beads and site-selective binding.[2] All four immobilised ATAs were successfully reused in five repetitive cycles of reductive amination of HMF with alanine and isopropylamine as a co-substrate. The transaminase from Silicibacter pomeroyi, ATA-Spo, worked especially well in batch reactions, yielding a conversion rate of 87% for both HMFA and DAF when alanine was used as the amine donor, and a conversion rate of 99% and 98% for HMFA and DAF, respectively, when isopropylamine was used. We further advanced the process by applying the immobilised enzymes in continuous flow using alanine and isopropylamine for the amination of HMF. ATA-Spo achieved high conversion rates of 48% and 41% after 12 days with alanine and isopropylamine, respectively.[3] Overall, this study highlights the potential of using renewable materials like furfurals and biocatalytic synthesis methods for the sustainable production of amines, specifically HMFA and DAF, using ATAs. The findings of this study could have implications in the pharmaceutical industry and other related fields. |
| References: | [1] Dunbabin, A., Subrizi, F., Ward, J. M., Sheppard, T. D. & Hailes, H. C. Furfurylamines from biomass: transaminase catalysed upgrading of furfurals. Green Chem. 19, 397-404 (2017). [2] Heinks, T., Montua, N., Teune, M., Liedtke, J., Höhne, M., Bornscheuer, U. T. & Fischer von Mollard, G. Comparison of Four Immobilization Methods for Different Transaminases. Catalysts 13, 1-17 (2023). [3] Heinks, T., Merz, L. M., Liedtke, J., Höhne, M., van Langen, L. M., Bornscheuer, U. T., Fischer von Mollard, G. & Berglund, P. Biosynthesis of Furfurylamines in Batch and Continuous Flow by Immobilized Amine Transaminases. Catalysts (2023) (accepted). |
| Figures: | 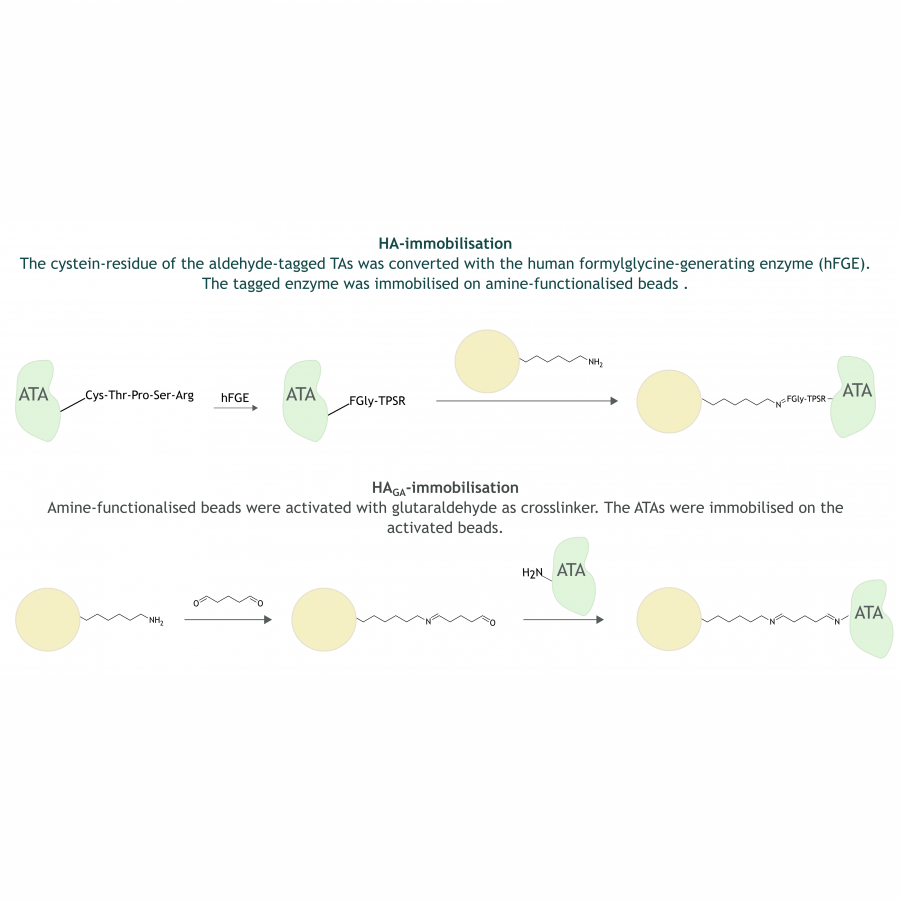 HA and HAGA Immobilisation Depicted are the two immobilisation methods applied in this work. 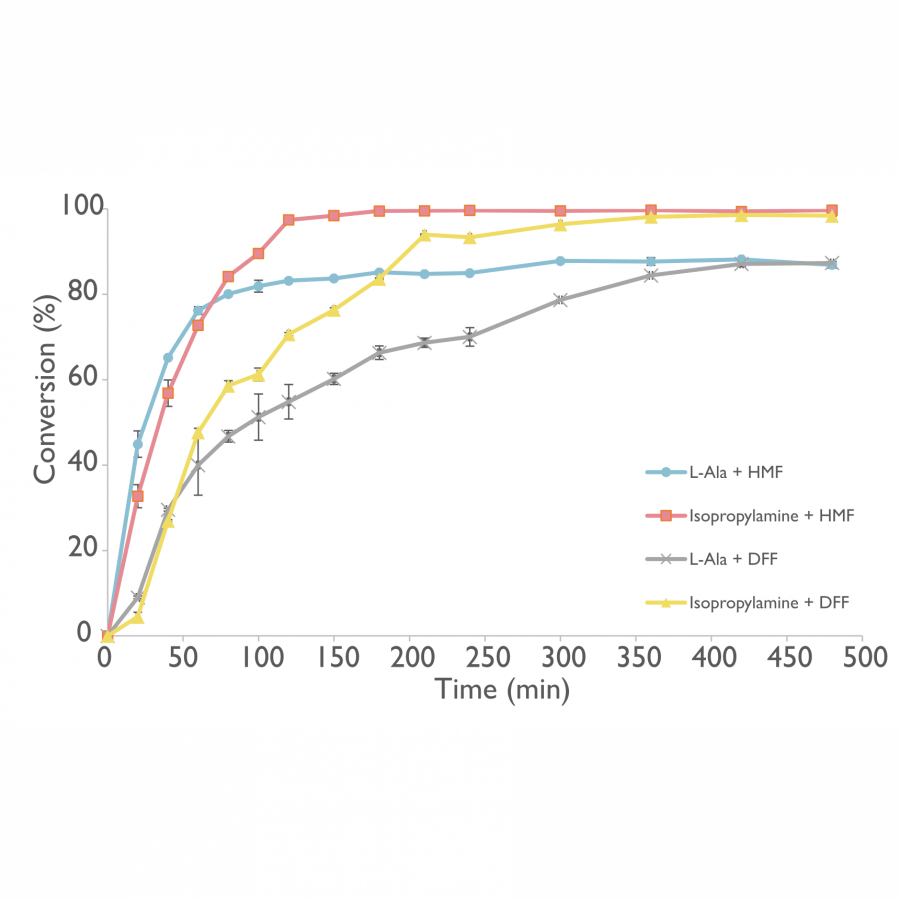 Amination of HMF and DFF by immobilised ATA-Spo in batch-reaction ATA-Spo was immobilized on HAGA-beads and used for the amination of HMF and DFF in 1 mL reaction solution. The reaction was followed by the detection of HMF and DFF. All reactions were performed as duplicates. (●: HMF and L-alanine, ■: HMF and isopropylam |
#1301 | Enzymatic synthesis of hydroxysulfides by ketoreductases |
|
| Presenting author: | Chuhan LI of UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Daniele CASTAGNOLO of UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Chemoenzymatic Cascades / ketoreductase / hydroxysulfides / biocatalytic |
| Purpose: | Chiral hydroxysulfides are an important class of organic compounds which find broad application in organic and pharmaceutical chemistry. Herein we describe the development of novel biocatalytic methods for the enantioselective synthesis of hydroxysulfides by exploiting ketoreductase (KRED) enzymes. Two KREDs were discovered from a pool of 384 enzymes identified and isolated through a metagenomic approach. KRED311 and KRED349 catalysed the synthesis of hydroxysulfides bearing a stereocentre at the C-O bond with opposite absolute configurations and excellent ee values by novel biocatalytic reactions starting from commercially available thiophenols/thiols and vinylketones. Thiocarboxylic acids were also synthesised using nitrilase enzymes. |
| References: | [1] J. Agric. Food Chem. 2015, 63, 36, 8017-8024. [2] J. Agric. Food Chem. 1991, 39, 2249-2252 |
| Figures: | 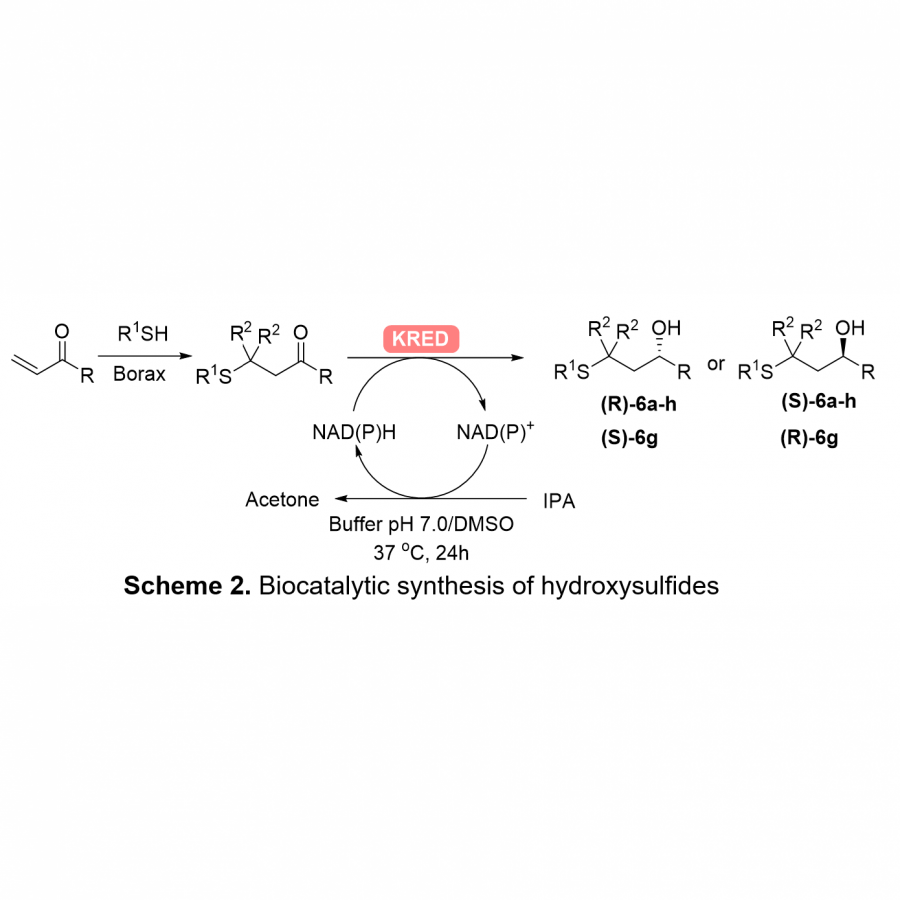 Biocatalytic synthesis of hydroxysulfides Biocatalytic synthesis of hydroxysulfides 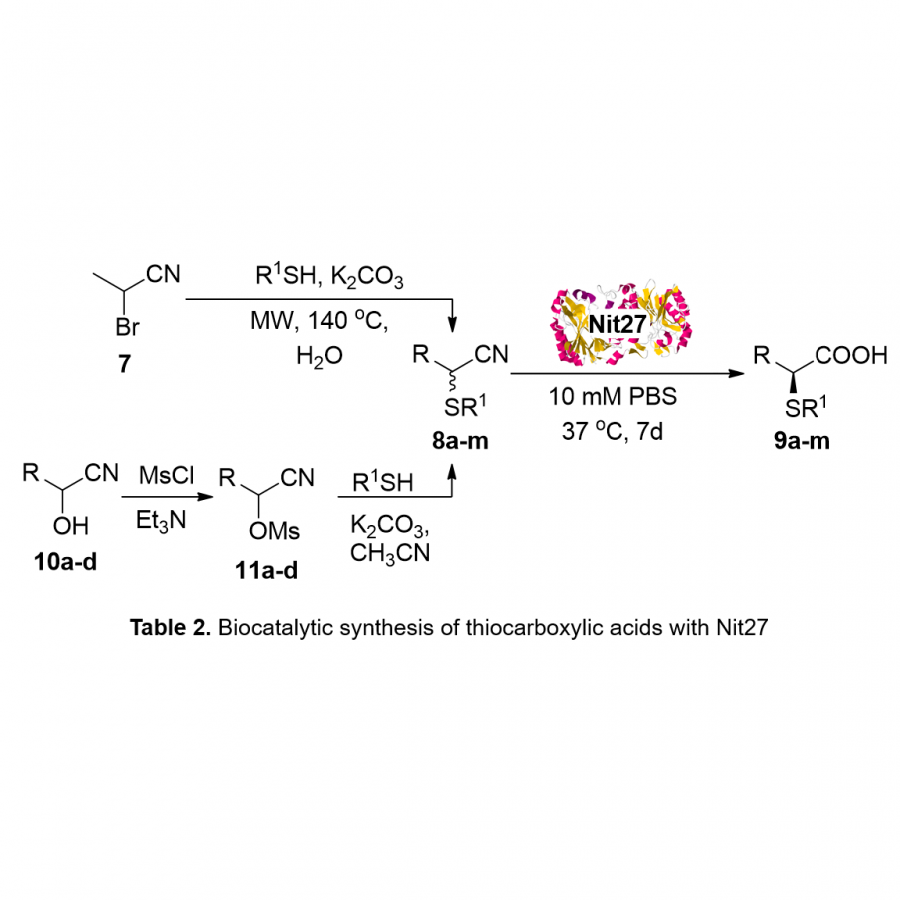 Biocatalytic synthesis of thiocarboxylic acids Biocatalytic synthesis of thiocarboxylic acids with Nit27 |
#1302 | Spheroplasts preparation boosts the catalytic potential of a squalene-hopene cyclase |
|
| Presenting author: | Ana I. BENITEZ-MATEOS of UNIVERSITY OF BERN |
| Corresponding author: | Francesca PARADISI of UNIVERSITY OF BERN |
| Other authors: | Andreas SCHNEIDER of UNIVERSITY OF STUTTGART Eimear HEGARTY of UNIVERSITY OF BERN Bernhard HAUER of UNIVERSITY OF STUTTGART |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Squalene-hopene cyclase / enzyme immobilization / Spheroplasts / terpene cyclazation |
| Purpose: | Squalene-hopene cyclases (SHCs) are a highly attractive class of membrane-bound enzymes to synthesize terpenes which are then used in the fragrance industry and as bioactive molecules [1-3]. Typically, SHCs are used as whole-cell biocatalysts, but they suffer from the outer cell membrane acting as a diffusion barrier for the highly hydrophobic substrate/product. Alternatively, SHCs have been used as purified enzymes but their stability is very poor [1]. In this work, we present the application of SHC spheroplasts [4]. This unexplored strategy for biocatalysis consist of the (partial or total) removal of the outer cell membrane of E. coli cells used as biocatalysts. SHC spheroplasts have improved the enzymatic cyclization of a set of terpenes (squalene, geranyl acetone, farnesol, and farnesyl acetone) up to 100-fold (Figure 1). In addition, we introduce a new concept for the carrier-free immobilization of spheroplasts via crosslinking, which is called CLS (crosslinked spheroplasts). CLS offered the advantage of biocatalyst reusability while maintaining the same activity as the spheroplasts. To sum up, SHC spheroplasts are an innovative approach to overcome the notoriously challenging stereoselective head-to-tail cyclization of terpenes in an straightforward and sustainable manner. |
| References: | [1] A. Schneider, Jegl, P. Hauer, B. Angew. Chem. Int. Ed. 2021, 60: 1325. [2] Hammer, S. C., Marjanovic, A., Dominicus, J. M., Nestl, B. M., Hauer, B. Nat. Chem. Biol. 2015, 11: 121-126. [3] Eichenberger, M. et al. Angew. Chem. Int. Ed. 2021, 60: 26080. [4] A. I. Benítez-Mateos, Schneider, A., Hegarty, E., Hauer, B., Paradisi, F. Nat. Commun. 2022, 13: 6269. |
| Figures: | 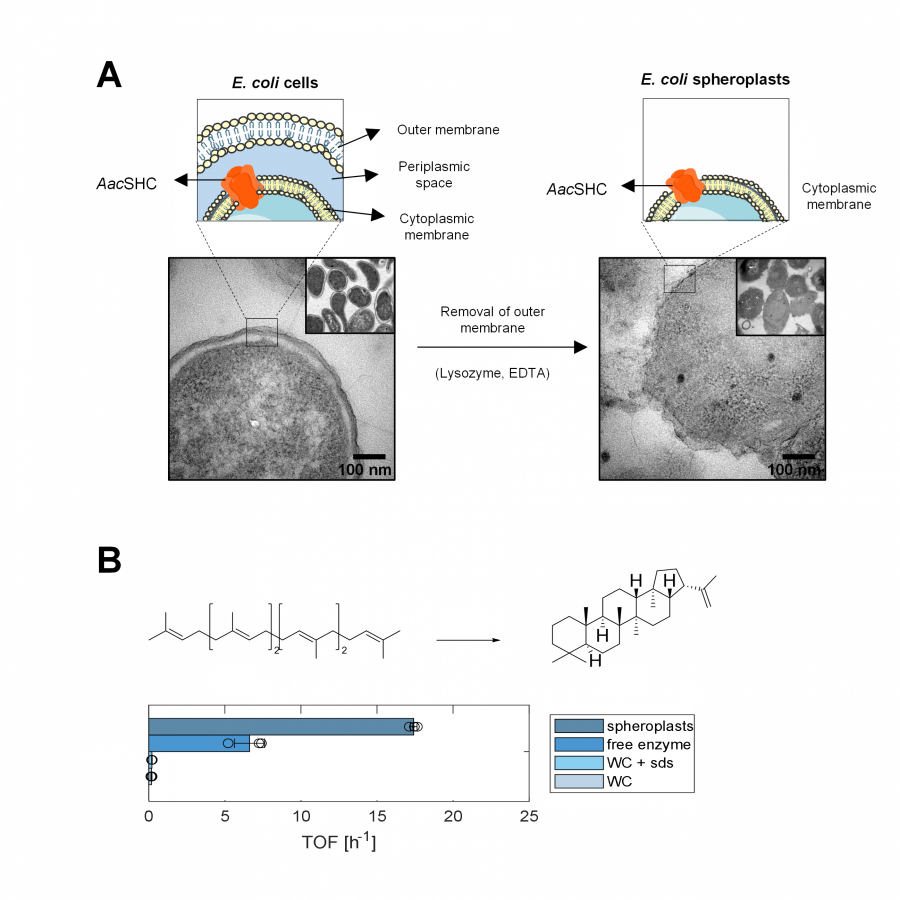 SHC spheroplasts compared to WC (whole cells) biocatalysts. A) Scheme (on the top) and TEM images (at the bottom) of SHC spheroplasts and whole cells. B) SHC-mediated cyclization of squalene by spheroplasts, purified enzyme, WC with SDS and WC. TOF: turnover frequency per hour. |
#1314 | Discovery, functional characterization and synthetic applications of a promiscuous ketoreductase from an Icelandic hotspring metagenome |
|
| Presenting author: | Erica Elisa FERRANDI of SCITEC CNR |
| Other authors: | Ivan BASSANINI of SCITEC CNR Chiara TOGNOLI of SCITEC CNR Susanna BERTULETTI of SCITEC CNR Sergio RIVA of SCITEC CNR Daniela MONTI of SCITEC CNR |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme discovery / biocatalysis / metagenomics / ketoreductases |
| Purpose: | In the search of novel thermostable hydroxysteroid dehydrogenases (HSDHs), enzymes able to regio- and stereoselectively oxidize/reduce steroidal compounds,[1] by (meta)genome mining, we recently discovered in an Icelandic hot spring metagenome a novel Short-chain Dehydrogenase/Reductase (SDR), named Is2-SDR. Surprisingly, despite the high sequence similarity shared with HSDHs, this enzyme showed no activity in the oxidation of steroid substrates, e.g., cholic acid. On the other hand, remarkably, Is2-SDR was able to reduce with high regio- and stereoselectively a diversified panel of carbonylic substrates, including bulky ketones, α- and β-ketoesters, and α-diketones of pharmaceutical relevance, proving to be a very active and versatile ketoreductase.[2-4] Moreover, Is2-SDR showed both a high thermophilicity (Topt = 70 °C) and thermostability (TM = 75 °C), these data being consistent with the environmental conditions of collection of the starting metagenomic DNA (hot spring, 85–90 °C). A broad tolerance to both water-miscible and water-immiscible organic solvents was demonstrated as well, thus, confirming the potential of this new biocatalyst for its synthetic application.[5] Acknowledgments: Financial support from “National Biodiversity Future Center” (grant No. CN00000033, PNRR MUR-M4C2-D. D. n.1034, 17 June 2022) is kindly acknowledged. |
| References: | [1] E. E. Ferrandi, S. Bertuletti, D. Monti, S. Riva, Eur. J. Org. Chem 2020, 2020, 4463-4473. [2] S. Bertuletti, E. E. Ferrandi, S. Marzorati, M. Vanoni, S. Riva, D. Monti, Adv. Synth. Catal. 2020, 362, 2474-2485. [3] S. Bertuletti, I. Bayout, I. Bassanini, E. E. Ferrandi, N. Bouzemi, D. Monti, S. Riva, Eur. J. Org. Chem. 2021, 29, 3992-3998. [4] R. Nasti, I. Bassanini, E. E. Ferrandi, F. Linguardo, S. Bertuletti, M. Vanoni, S. Riva, L. Verotta, D. Monti, ChemBioChem, 2022, 23, e202200105. [5] E. E. Ferrandi, I. Bassanini, S. Bertuletti, S. Riva, C. Tognoli, M. Vanoni, D. Monti, Int. J. Mol. Sci. 2022, 23, 12153. |
#1315 | Engineering (S)-selective ω-transaminases for better thermostability |
|
| Presenting author: | Trishnamoni GAUTOM of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Per BERGLUND of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Other authors: | Bhu-Bhud THONGRAKON of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chiral amine / transaminase / enzyme engineering / biocatalysis |
| Purpose: | Chiral amines are one among the most versatile and valuable building blocks employed for the synthesis of diverse amine containing pharmaceuticals as well as agrochemical compounds. In recent years, sustainable development has driven the evolution of biocatalysis as a green technology for the efficient industrial synthesis of different chiral amine-containing drugs. Amine transaminases (ATAs), the pyridoxal 5’-phosphate (PLP) dependent enzymes, which under mild conditions mediate the highly stereoselective transfer of an amino group from an amino donor to a keto group, have attracted tremendous attention as a powerful biocatalyst for production of enantioenriched chiral amines due to their broad substrate acceptance1. However, industrial applicability of ATAs is still limited owing to their low operational stability as biocatalysis sometimes require enzymes to be employed in unnatural conditions. The key to improving the industrial applicability of ATAs lies in enhancing their operational stability at high temperatures and in the presence of organic solvents, which can transform them into ideal candidates for industrial applications while retaining their enzymatic activity in vitro. To this end, we have characterized several (S)-selective ATAs with respect to their thermodynamic and operational stability. This study presents new data on stability improvements achieved through enzyme engineering, based on the previously discovered ATA inactivation mechanism of ATAs2. By improving the operational stability of ATAs, we aim to enable their wider applications in the industrial synthesis of various chiral amine-containing drugs, contributing to the sustainable development of the pharmaceutical and agrochemical industries |
| References: | 1. Guo, F., & Berglund, P. (2017). Transaminase biocatalysis: optimization and application. Green Chemistry, 19(2), 333-360. 2. Ruggieri, F., Campillo-Brocal, J. C., Chen, S., Humble, M. S., Walse, B., Logan, D. T., & Berglund, P. (2019). Insight into the dimer dissociation process of the Chromobacterium violaceum (S)-selective amine transaminase. Scientific Reports, 9(1), 16946. |
#1317 | Substrate promiscuity of cytidine deaminases |
|
| Presenting author: | Nina URBELIENĖ of INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, V |
| Other authors: | Justas VAITEKŪNAS of INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY Jonita STANKEVIČIŪTĖ of INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY Rolandas MEŠKYS of INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cytidine deaminase / Substrate promiscuity of CDAs / N(S,O)4-substituted pyrimidine nucleosides / pro-drug metabolism |
| Purpose: | Cytidine deaminase (CDA) (EC 3.5.4.5) is an enzyme that catalyses the deamination of cytidine to uridine, which is an important step in nucleotide metabolism. In addition, CDA is known to exhibit substrate promiscuity, meaning that it can catalyse the deamination of other cytidine analogues for example 5-azacytidine, zebularine, gemcitabine, which are used in cancer treatment. Moreover, CDAs could be used for synthesis of N4-hydroxycytidine derivatives with antiviral activity. Our studies have shown that some prokaryotic homo-tetrameric CDAs catalyse the nucleophilic substitution at the 4th position of N4-acyl-, N4-alkyl, N4-alkyloxycarbonyl-cytidines and S4-alkylthio-, O4-alkyl-uridines, converting them to uridine and corresponding amide, amine, carbamate, thiol or alcohol as leaving group. CDA enzymes, which are active with a broad spectrum of substrates, may play a role in salvaging and/or breaking down modified pyrimidine nucleosides. This could include the removal of alkyl groups from mutagen-damaged uridines or the demethylation of S4-methylthiouridine, a molecule that is formed under stress in living organisms. The discovery of this substrate promiscuity of CDAs has expanded our understanding of the cellular turnover of cytidine derivatives, including how pyrimidine-based pro-drugs are activated and/or metabolized in the body. |
| References: | 1. Nina Urbelienė et al., .Sci. Adv.9,eade4361(2023).DOI:10.1126/sciadv.ade4361 |
#1321 | Cooperation between a Laccase and an Amine Transaminase in the Biocatalytic Valorization of 5-(Hydroxymethyl)furfural |
|
| Presenting author: | Nicoletta CASCELLI of UNIVERSIDAD DE OVIEDO |
| Other authors: | Vincenzo LETTERA of BIOPOX SRL Giovanni SANNIA of BIOPOX SRL Vicente GOTOR-FERNANDEZ of UNIVERSITY OF OVIEDO Ivan LAVANDERA of UNIVERSITY OF OVIEDO |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | laccase / transaminase / HMF / biocatalysis |
| Purpose: | 5-(Hydroxymethyl)furfural (HMF) is an essential renewable building block that may be used in various transformations to produce high-value compounds.[1] Its oxidation enable the formation of a variety of derivatives, many of which have uses in the pharmaceutical and polymer industries.[2] Nevertheless, the constant and affordable supply of HMF from renewable resources is still insufficient to move its transformation processes to an industrial scale, being energy demands, operating costs and processes efficiency the main hinders.[3] Scalable and economically advantageous approaches are therefore needed to face these issues. The severe reaction conditions and low yields make inorganic and acid catalysis for furan oxidation less than convincing. However, bioprocesses have demonstrated their ability to successfully offer sustainable solutions for bio-based derivatives from an environmental and economical point of view.[4] Also, the efficient synthesis of amino-furan compounds is highly appealing due to their relevance as precursors of, e.g. pharmaceuticals and polymers. Amongst the most appealing biocatalysts for furan transformations, our focus has been put on amine transaminases,[5] from the transferase enzymatic family, and laccases[6]. Transaminases are considered one of the most versatile biocatalysts for amino furan synthesis, while laccases can mediate the selective oxidation of alcohol and aldehyde functionalities. After proving the combination of laccases and amine transaminases in a first study for the scalable production of furfuryl amine from furfuryl alcohol through furfural as intermediate,[7] herein we propose a similar strategy to effectively transform HMF into 5-aminomethyl-2-furancarboxylic acid (AMFC, Figure 1). This compound was synthesized via 5-formyl-2-furancarboxylic acid (FFCA) accumulation. Reaction conditions were individually investigated, focusing on e.g., pH adjustment and buffering powder influences, for an efficient set-up. |
| References: | [1] K. Saikia, A. K. Rathankumar, P. S. Kumar, S. Varjani, M. Nizar, R. Lenin, J. George, V. K. Vaidyanathan, J. Chem. Technol. Biotechnol. 2022, 97, 409-419. [2] L. Hu, A. He, X. Liu, J. Xia, J. Xu, S. Zhou, J. Xu, ACS Sustain. Chem. Eng. 2018, 6, 15915-15935. [3] M. G. Davidson, S. Elgie, S. Parsons, T. J. Young, Green Chem. 2021, 23, 3154-3171. [4] H. Puetz, E. Puchľová, K. Vranková, F. Hollmann, Catalysts 2020, 10, 952. [5] M. Fuchs, J. E. Farnberger, W. Kroutil, Eur. J. Org. Chem. 2015, 6965-6982. [6] A. D. Moreno, D. Ibarra, M. E. Eugenio, E. Tomás-Pejó, J. Chem. Technol. Biotechnol. 2020, 95, 481-494. [7] N. Cascelli, V. Lettera, G. Sannia, V. Gotor-Fernández, I. Lavandera, ChemSusChem 2023, doi: 10.1002/cssc.202300226. |
| Figures: | 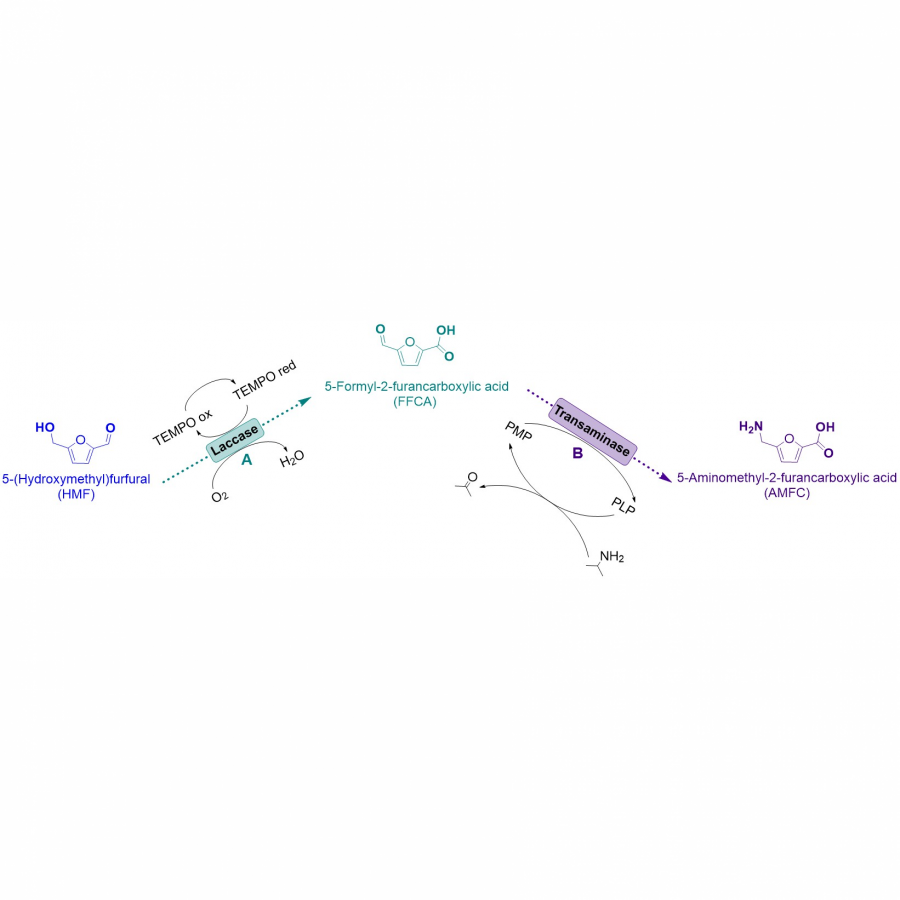 Figure 1. One-pot/two-step catalytic process for the direct synthesis of furan-based amino derivative AMFC from HMF combining a laccase-mediated oxidation (A) and a transamination (B) step. |
#1323 | Mechanistic investigation of the kinetic resolution of α-methyl-substituted phenylacetaldehyde by norcoclaurine synthase |
|
| Presenting author: | Shiqing ZHANG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY, CHI |
| Corresponding author: | Xiang SHENG of TIANJIN INSTITUTE OF INDUSTRIAL BIOTECHNOLOGY CHINESE ACADEMY OF SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | norcoclaurine synthase / reaction mechanism / enantioselectivity / quantum chemical calculation |
| Purpose: | The norcoclaurine synthase from Thalictrum flavum (TfNCS) can stereoselectively catalyze the Pictet-Spengler reaction between dopamine and 4-hydroxyphenylacetaldehyde to give (S)-norcoclaurine which acts as an important intermediate in benzylisoquinoline alkaloid biosynthesis [1,2]. Recently, the kinetic resolution of α-methyl-substituted phenylacetaldehyde was achieved by TfNCS with high stereoselectivity and yields the (1S, 2R) chiral product [3]. However, the reaction mechanisms and the origins of enantiopreference of TfNCS towards α-methyl-substituted phenylacetaldehyde are still unclear. Herein, a cluster model of the enzyme was designed on the basis of the crystal structure and quantum chemical calculations were then performed to obtain the detailed reaction mechanism and energy profiles. Our calculations reveal that the reaction of dopamine with α-methyl-substituted phenylacetaldehyde by TfNCS follows a similar mechanism as the natural substrate, in which the deprotonation of C-H of the cyclized intermediate is the rate-liming step for both R- and S-pathways, and the corresponding energy barriers are 20.1 and 21.6 kcal/mol, respectively. The results are thus able to reproduce the experimental results. Importantly, the calculations could also rationalize the observed enantioselectivity of TfNCS towards the non-natural substrate. By analyzing the geometries of the intermediates and transition states, the M97 and L72 residues are indicated as the important residues in controlling the selectivity of TfNCS on α-methyl-substitute phenylacetaldehyde. The detailed information about the mechanism and the enantioselectivity is helpful to rationally design variants of TfNCS with improved reactivity and selectivity properties for a wider scope of substrates. |
| References: | [1] N. Samanani, D. K. Liscombe, P. J. Facchini. The Plant Journal. 2004, 40, 302-313. [2] H. Minami, E. Dubouzet, K. Iwasa, et al. J. Biol. Chem. 2007, 282, 6274-6282. [3] R. Roddan, G. Gygli, A. Sula, et al. ACS Catal. 2019, 9, 9640-9649. |
| Figures: | 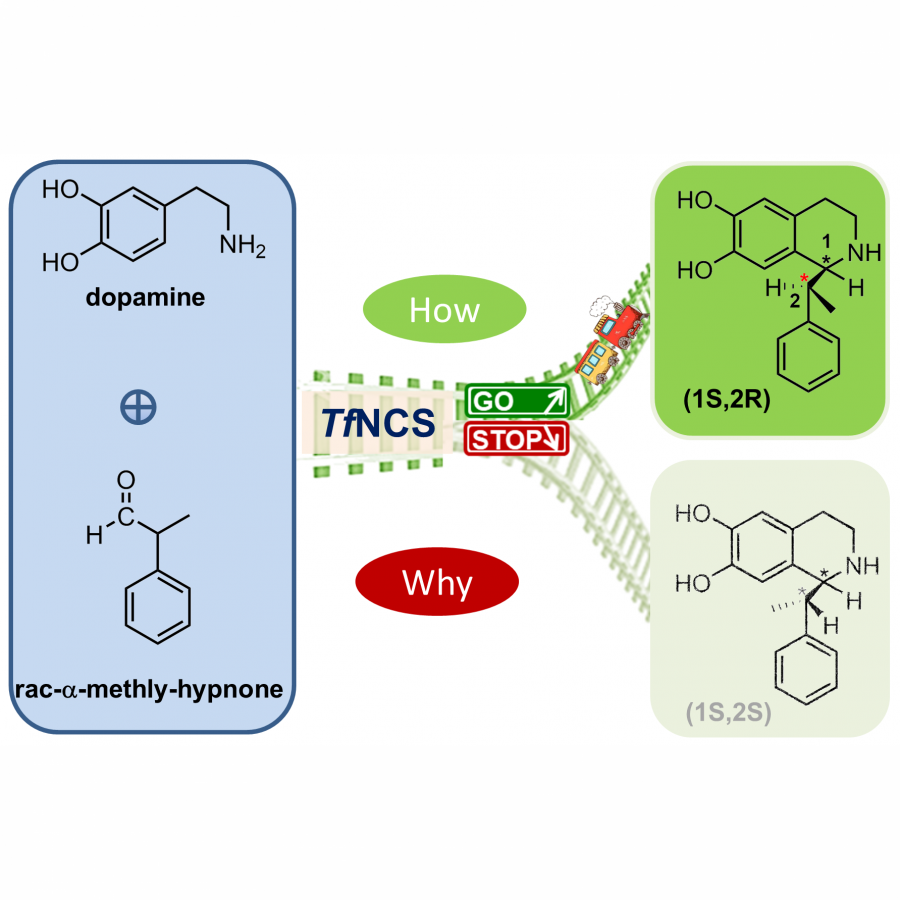 Figure 1 The kinetic resolution reaction catalyzed by TfNCS. |
#1324 | Synthesis and valorization of alpha-hydroxyketones through bio and organocatalysis |
|
| Presenting author: | Camille GADONA of UNIVERSITÉ CLERMONT AUVERGNE, CNRS, INSTITUT DE CHIMIE DE CLERMONT FERRAND |
| Other authors: | Nicolas DUGUET of UNIV LYON, UNIVERSITÉ CLAUDE BERNARD LYON 1, CNRS, INSA-LYON, CPE-LYON, INSTITUT DE CHIMIE ET BIOCHIMIE MOLÉCULAIRES ET SUPRAMOLÉCULAIRES (ICBMS) Franck CHARMANTRAY of UNIVERSITÉ CLERMONT AUVERGNE, CNRS, INSTITUT DE CHIMIE DE CLERMONT FERRAND Laurence HECQUET of UNIVERSITÉ CLERMONT AUVERGNE, CNRS, INSTITUT DE CHIMIE DE CLERMONT FERRAND |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chemo-enzymatic strategy / unsymmetrical alpha-hydroxyketones / transketolase / prodrugs |
| Purpose: | A chemoenzymatic strategy is investigated to obtain highly valuable unsymmetrical alpha-hydroxyketones (acyloins), precursors of vinylene carbonate, useful building blocks for prodrugs synthesis such as medoxomil producing after hydrolysis non-toxic carbon dioxide and alpha-dicarbonyl derivatives.1 In the first enzymatic steps, thermostable Transketolase from Geobacillus stearothermophillus (TKgst)2 catalysing C-C bond formation is used to obtain unsymmetrical alpha-hydroxyketones from alpha-ketoacid donor which brings different functionality (R1 = aliphatic, hydroxyl), while the aldehyde acceptor (biosourced or obtained from cheap and achiral precursor) allows the modulation of the main carbon chain on its R2 group (halogenation, hydroxylation, length variation). The TK reaction enables the irreversible transfer of a ketol group from an alpha-ketoacid donor to an aldehyde acceptor with concomitant decarboxylation of the donor and release of carbon dioxide.3 The alpha-ketoacid can be generated in situ from the corresponding D-aminoacid with a novel D-aminoacid oxidase (DAAO), as already initiated.2 To improve TKgst activity toward the targeted substrates, TKgst variants were designed by rational mutagenesis based on the analysis of TKgst active site.4 Finally, organocatalysts (ORG) such as N-heterocyclic carbenes (NHC) together with a carbonyl source (DPC, DMC or carbon dioxide) could catalyze efficiently the synthesis of vinylene carbonates,5 from unsymmetrical alpha-hydroxyketones never investigated before due to the difficulty to control the regioselectivity by chemical ways. This chemo-enzymatic strategy combining enzymes and organocatalysts in a one-pot could be performed sequentially or simultaneously avoiding the purification of intermediates. |
| References: | [1] R. P. Dash, T. Tichý, V. Veeravalli, J. Lam, J. Alt, Y. Wu, L. Tenora, P. Majer, B. S. Slusher, R. Rais, Enhanced Oral Bioavailability of 2‑(Phosphonomethyl)-pentanedioic Acid (2-PMPA) from its (5-Methyl-2-oxo-1,3-dioxol-4-yl)methyl (ODOL)-Based Prodrugs. Mol. Pharmaceutics 2019, 16, 4292-4301. [2] J. Abdoul Zabar, I. Sorel, V. Hélaine, F. Charmantray, T. Devamani, D. Yi, V. de Berardinis, D. Louis, P. Marlière, W.D. Fessner, L. Hecquet Thermostable Transketolase from G. stearothermophilus: Characterization and Catalytic Properties. Adv. Synth. Catal. 2013, 355, 116-128. [3] H. Casajus, A. Lagarde, M. Leremboure, T. De Dios Miguel, L. Nauton, V. Thery, W-D. Fessner, N. Duguet, F. Charmantray, L. Hecquet. Enzymatic Synthesis of Aliphatic Acyloins Catalyzed by Thermostable Transketolase, ChemCatChem, 2020, 12, 5772-5779. [4] N. Ocal, G. Arbia, A. Lagarde, M. Joly, S. Gittings, K. M. Graham, F.Charmantray, L.Hecquet. Synthesis of Deoxysugars from Aliphatic α-Ketoacids and Aldoses Catalyzed by Thermostable Transketolase Variants from Geobacillus stearothermophilus. Adv. Synth. Catal. 2023, 365, 78-87. [5] K. Onida, A. Haddleton, S. Norsic, C. Boisson, F. D’Agosto, N. Duguet, Organocatalytic synthesis of substituted vinylene carbonates. Adv. Synth. Catal. 2021, 363, 5129-5137. |
| Figures: | 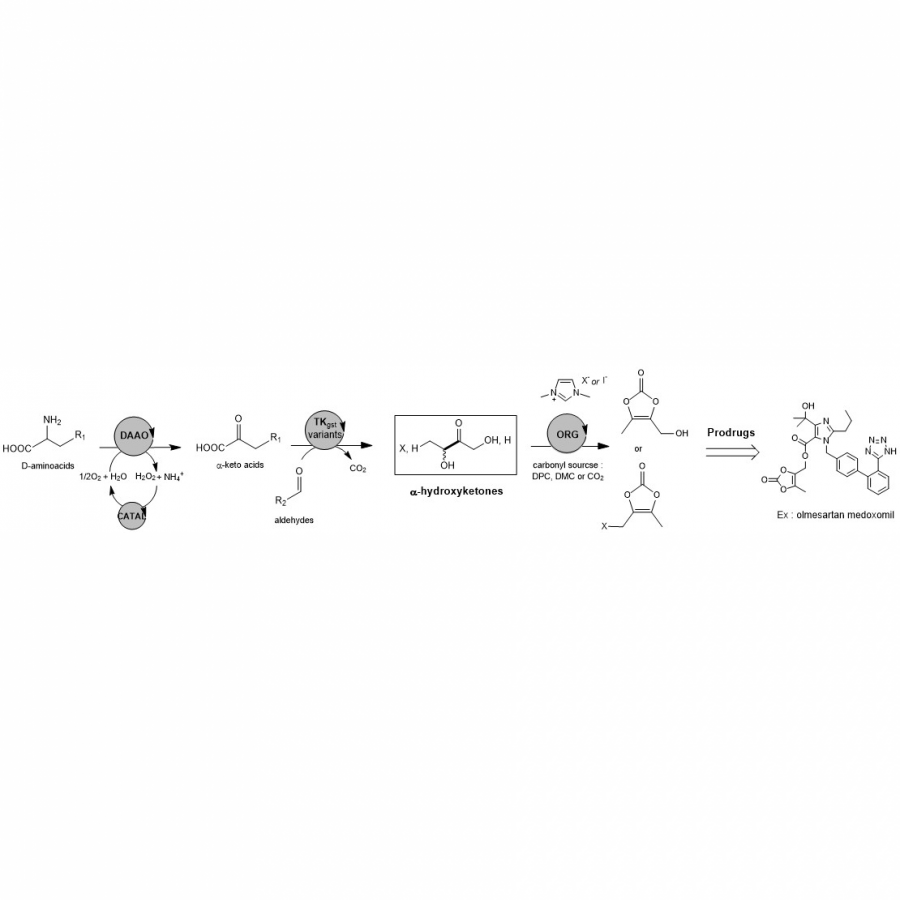 Overview of the chemoenzymatic strategy DAAO : D-aminoacid oxydase
CATAL : Catalase
TKgst : Transketolase de Geobacillus stearothermophilus
ORG : Organocatalyst
NHC : N-heterocyclic carbene
DPC : Diphenyl carbonate
DMC : Dimethyl carbonate |
#1326 | To gel or not to gel: Enzymatic desulfation of carrageenans to modify their rheological properties |
|
| Presenting author: | Alexander FUCHS of TECHNICAL UNIVERSITY OF MUNICH |
| Other authors: | Volker SIEBER of TECHNICAL UNIVERSITY OF MUNICH |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biotransformation / Rheology / Marine Enzymes / Polymer Modification |
| Purpose: | Sulfated biomolecules from the marine environment represent an extraordinary natural source of chemical diversity, ranging from small metabolites to complex polymers with an enormous variety of biological and physicochemical properties. Among the commercially most relevant products from red algae, the sulfated biopolymer carrageenan is of particular interest to the food, cosmetics, and pharmaceutical industries as it can exhibit favorable viscoelastic characteristics [1]. The degree and position of sulfation can have a decisive influence on its rheology and thus determines the polymer’s commercial use [2]. To expand the application scope of carrageenans, the targeted cleavage of sulfate groups catalyzed by sulfatases can therefore be a promising tool to create novel carrageenan variants with promising new physicochemical properties. However, the range of functionally expressed and characterized carrageenan sulfatases so far remains as insufficient as the understanding of their effect on the rheological behavior of the polymers modified by them. To address this issue, we screened several putative carrageenolytic polysaccharide-utilization loci of heterotrophic marine microorganisms for sulfatases active on different types of commercially relevant carrageenans. Using this approach, we were able to heterologously express a series of novel sulfatases in their active form that perform the specific desulfation of ι- and κ-carrageenan molecules to α- and β-carrageenan-moieties or several fine-tuned hybrid structures derived from them. We used one of these variants, a universal G4S-sulfatase, for the preparative biotransformation of ι- and κ-carrageenan in the low gram scale, and rheologically analyzed the products to reveal the unique structure-function relationship of the sulfation architecture on the viscoelastic properties of carrageenans. To evaluate the potential of the enzyme for commercial purposes in a proof-of-concept approach, we applied it to the biocatalytic modification of different carrageenans during their extraction from the most industrially important red algal species for carrageenan production, successfully transferring the new rheological properties and laying the groundwork for further intensification. |
| References: | [1] Necas, J., Vet med, 2013, 58(4):187-205. [2] Cianca, M., Front. Plant Sci., 2020, 11 |
#1327 | Lipase-catalyzed synthesis and physico-chemical characterization of alkyl glycoside fatty acid esters from cheese whey permeate |
|
| Presenting author: | MARCO RABUFFETTI of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY |
| Other authors: | ELEONORA PARGOLETTI of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY GIORGIA BALLABIO of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY LEONARDO GELATI of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY SARA SANGIORGIO of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY MARINA SIMONA ROBESCU of UNIVERSITY OF PAVIA - DEPARTMENT OF DRUG SCIENCES RICCARDO SEMPROLI of UNIVERSITY OF PAVIA - DEPARTMENT OF DRUG SCIENCES DANIELA UBIALI of UNIVERSITY OF PAVIA - DEPARTMENT OF DRUG SCIENCES GIUSEPPE CAPPELLETTI of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY GIOVANNA SPERANZA of UNIVERSITY OF MILAN - DEPARTMENT OF CHEMISTRY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | SUGAR FATTY ACID ESTERS (SFAEs) / BIOSURFACTANTS / CHEESE WHEY PERMEATE / SUSTAINABLE PROCESS |
| Purpose: | Sugar fatty acid esters (SFAEs), usually called sugar esters, are a class of non-ionic surfactants, characterized by excellent emulsifying, stabilizing and detergency properties. These compounds have many advantages over petrochemical-derived surfactants: SFAEs are odorless, tasteless, fully biodegradable, skin-compatible, non-toxic and non-harmful to the environment. Moreover, both the constituent moieties, sugars and fatty acids, can be produced from renewable agricultural resources, i.e. from biomass such as cheese whey permeate and oil wastes, respectively. Depending on carbon chain length, degree of esterification and nature of the sugar head group, SFAEs could cover a wide range of hydrophilic-lipophilic balance (HLB) values, thus resulting in tunable surfactant properties and becoming candidates for many possible applications, such as O/W or W/O emulsifiers, solubilizing agents, lubricants, penetrating enhancers and pore forming agents. Indeed, these biosurfactants are of great interest in many market sectors (i.e. food, detergent, cosmetic and pharmaceutical industry). [1] However, the chemical synthesis of SFAEs is not environmentally friendly as it requires harsh reaction conditions (hazardous solvents, high temperatures, acid or base catalysts) resulting in high energy consumption, formation of undesirable by-products (e.g. due to caramelization of sugars) and low selectivity. [2] Enzymatic strategies appear to be a promising alternative to simplify both product synthesis and downstream. [3] We present here the chemoenzymatic synthesis of a library of alkyl glycoside fatty acid esters (Figure 1) prepared by Fisher glycosylation of glucose and galactose (easily obtained from enzymatic hydrolysis of cheese whey permeate) with naturally occurring alcohols under acidic catalysis (Amberlyst® 15), followed by lipase-catalyzed esterification of the resulting isomeric mixtures of alkyl glycosides (namely α-/β-D-glucosides and α-/β-D-galactosides). CalB (Novozym® 435) was used as a biocatalyst in a highly sustainable and easily scalable solvent-free system. Conversion of glucose and galactose into alkyl glycosides before the esterification reaction played a key role in circumventing the striking different solubility of the two reagents. The influence of the sugar polar head (glucose vs galactose), of the tail chain length (C12 vs C16 vs C18) and of the ring size (pyranosides vs furanosides) on the physico-chemical properties of the synthesized tensides (interfacial tension features, W/O emulsification capability and W/O stability over time) was evaluated. [4] This work was financially supported by Cariplo Foundation (Italy) (call: “Circular Economy for a sustainable future 2020”, project BioSurf, ID 2020-1094, https://www.biosurfproject.it/). |
| References: | [1] a) S. Soultani, S. Ognier, J.-M. Engasser, M. Ghoul, Colloids Surf. A: Physicochem. Eng. Asp. 2003, 227, 35-44; b) J. Kennedy, H. Kumar, P. S. Panesar, S. S. Marwaha, R. Goyal, A. Parmar, S. Kaur, J. Chem. Technol. Biotechnol. 2006, 81, 866-876; c) H. M. El-Laithy, O. Shoukry, L. G. Mahran, Eur. J. Pharm. Biopharm. 2011, 77, 43-55. [2] a) A. M. Gumel, M. S. M. Annuar, T. Heidelberg, Y. Chisti, Process Biochem. 2011, 46, 2079-2090; b) N. R. Khan, V. K. Rathod, Process Biochem. 2015, 50, 1793-1806; c) N.S. Neta, J.A. Teixeira, L.R. Rodrigues, Crit. Rev. Food Sci. Nutr. 2015, 55, 595-610. [3] a) R. Hausmann, M. Henkel, Biosurfactants for the Biobased Economy, Springer Nature Switzerland AG, Cham, 2022; b) J. W. Agger, B. Zeuner, Curr. Opin. Biotechnol. 2022, 78, 102842. [4] a) S. Sangiorgio, E. Pargoletti, M. Rabuffetti, M. Robescu, R. Semproli, D. Ubiali, G. Cappelletti, G. Speranza, Colloids Interface Sci. Commun. 2022, 48, 100630; b) R. Semproli, M. S. Robescu, S. Sangiorgio, E. Pargoletti, T. Bavaro, M. Rabuffetti, G. Cappelletti, G. Speranza, D. Ubiali, ChemPlusChem. 2023, 88, e202200331. |
| Figures: | 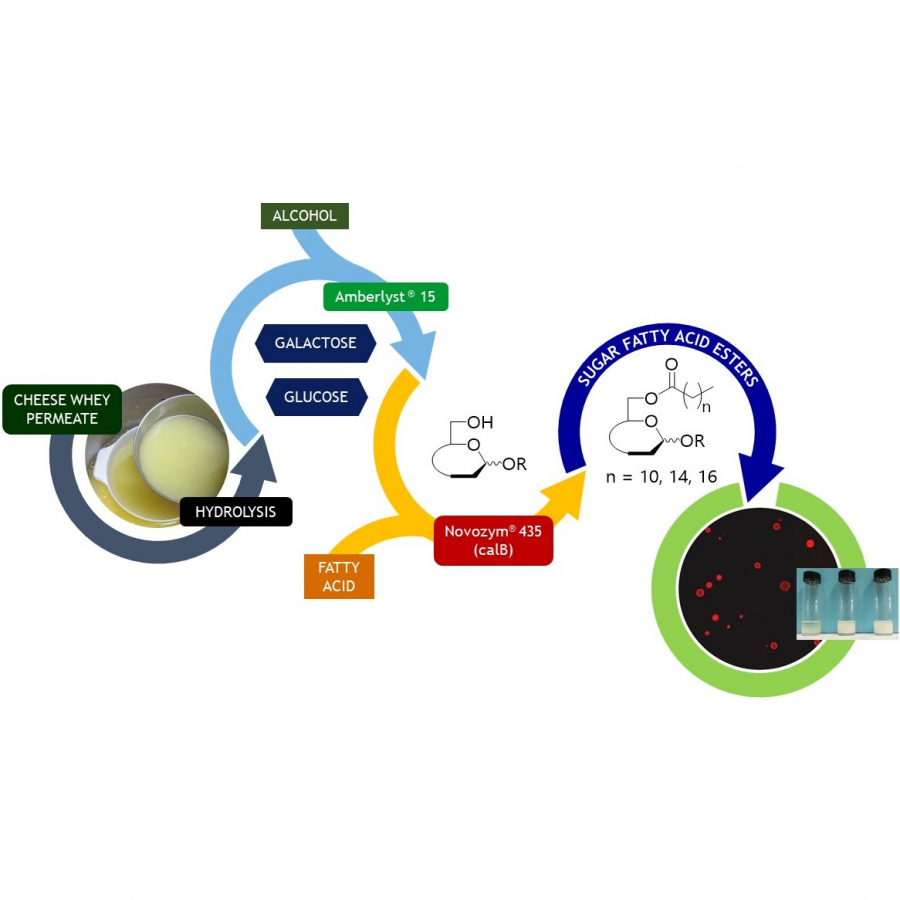 Figure 1 Chemoenzymatic strategy for the synthesis of SFAEs from cheese whey permeate |
#1328 | Conversion of free cyanide to formic acid by a cascade reaction catalyzed by cyanide hydratase and formamidase |
|
| Presenting author: | Barbora KRISTKOVA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Other authors: | Ludmila MARTINKOVA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Natalia KULIK of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Lenka RUCKA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Petr NOVOTNY of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Michal GRULICH of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Miroslav PATEK of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cyanide hydratase / Formamidase / Formate dehydrogenase / Cascade reaction |
| Purpose: | Free cyanide consists of HCN and CN ions in pH-dependent ratios. It is usually found in varying concentrations in wastewaters from gold and silver mining, metal electroplating, jewelry manufacturing, chemical synthesis, coal coking, etc. Today, the treatment of these wastewaters is mainly based on physicochemical principles (precipitation with metals, oxidation to cyanate), followed by a microbial stage. Enzymatic methods are gaining ground, but they are not yet technologically mature. These methods mainly use cyanide dihydratases (CynDs; EC 3.5.5.1), which hydrolyze HCN to formic acid and ammonia, or cyanide hydratases (CynHs; EC 4.2.1.66), which hydrate HCN to formamide. CynHs generally have higher activities and a higher resistance to alkaline pH than CynDs [1]. Nevertheless, the CynH product, formamide, although much less toxic than free cyanide, still poses a significant health risk. Therefore, it is tempting to combine CynH with amidase to convert formamide into formic acid. In this way, the same product is obtained as with CynD, while taking advantage of the CynH´s properties. Previously, a proof-of-concept was demonstrated using immobilized whole cells of wild-type producing strains with low CynH and amidase activities [2]. To increase the efficiency of this type of cascade reaction, we used a different type of amidase, namely formamidase (AmiF; EC 3.5.1.49) with excellent activities for formamide. An AmiF from Bacillus cereus [3] and a CynH from the fungus Exidia glandulosa [4] were produced in Escherichia coli and purified. Both enzymes were functional at pH 9.0 with specific activities on the order of hundreds U/mg protein, although AmiF had a lower pH optimum. The performance of the cascade reaction was optimized by timing the addition of AmiF or changing the pH after the first step. The cascade was performed in batch and continuous modes, the latter with enzymes immobilized on metal affinity resins. The long shelf life of both enzymes at 4 °C is an additional advantage. The same principle could be used for analytical purposes. In this case, NADH was produced in a coupled reaction catalyzed by commercial formate dehydrogenase (EC 1.17.1.9) (Figure 1) and its concentration was used to calculate the concentration of free cyanide. The method is currently being optimized with respect to the CynH/AmiF/FDH ratio, cyanide concentration, and reaction conditions. Acknowledgement: Funded by Czech Science Foundation, grant GA22-06785S. |
| References: | [1] L. Martínková, A. Sedova, P. Bojarová, V. Křen, Crit. Rev. Environ. Sci. Technol. 2023, 53, 416-434. [2] A.G. Campos, P. Pereira, J.C. Roseiro, Enzyme Microb. Technol. 2006, 38, 848-854. [3] P. Soriano-Maldonado, A.I. Martínez-Gómez, M. Andújar-Sánchez, J.L. Neira, et al., Appl. Environ. Microbiol. 2011, 77, 5761-5769. [4] A. Sedova, L. Rucká, P. Bojarová, M. Glozlová, et al., Catalysts 2021, 11, 1410. |
| Figures: | 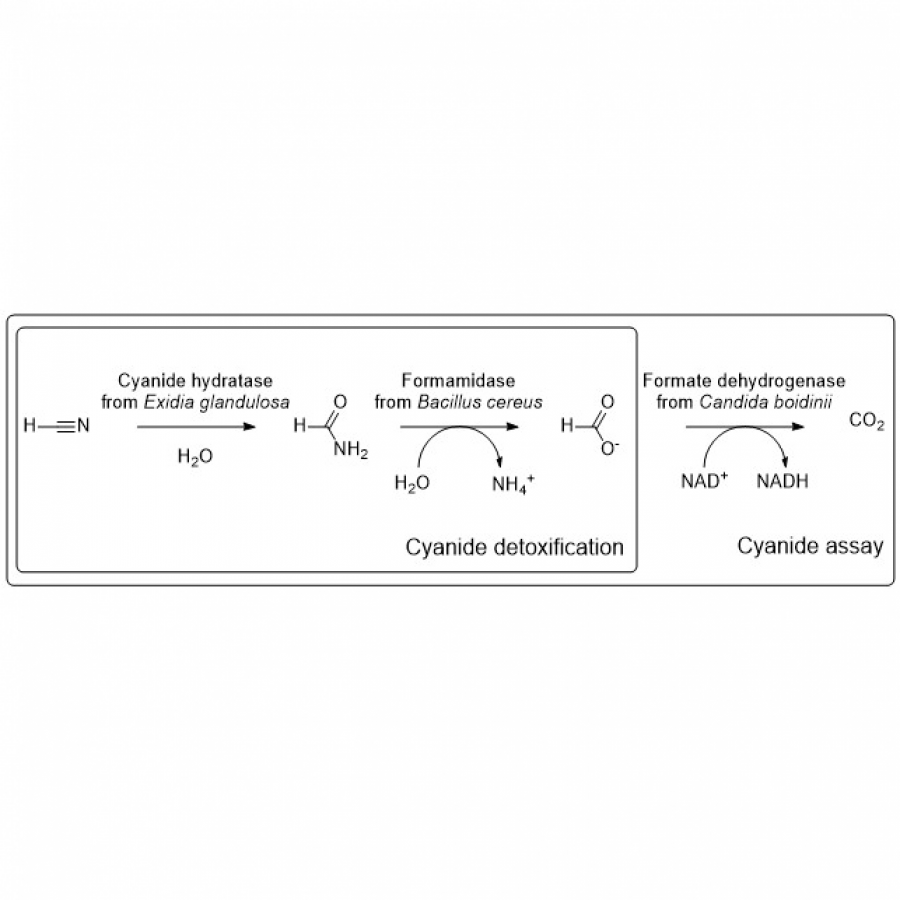 Figure 1 Artificial enzyme cascades for the detoxification and determination of free cyanide. |
#1330 | Unravelling new biological functions of lytic polysaccharide monooxygenases |
|
| Presenting author: | Leonor CARNEIRO of GRAZ UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | lytic polysaccharide monooxygenases / enzyme discovery / pathogenicity / biocatalysis |
| Purpose: | Lytic polysaccharide monooxygenases (LPMOs) are copper-dependent redox enzymes originally identified in saprobiontic fungi and bacteria [1]. The main function of LPMOs is to degrade biopolymers such as cellulose, starch or chitin through an oxidative process that is not yet fully understood [2]. Recent proteomic and biochemical studies have shown that LPMOs fulfil a multiplicity of biological functions beyond biomass degradation. In fact, recent landmark papers have linked LPMO activity to insect resistance in ferns [3] and to the survival of pathogens in human blood [4]. In our ongoing work, a panel of selected LPMOs from pathogenic bacteria is recombinantly produced and biochemically characterised. Importantly, LPMO activity strictly depends on the supply of external reductants and hydrogen peroxide or oxygen. Therefore, we will investigate a range of proteinogenic and other redox-active compounds as possible sources of electrons and hydrogen peroxide for the enzyme in vivo. Using Vibrio cholerae as a model organism, the pathogenetic relevance of its LPMOs will be investigated through serum assays and mouse infection models. Ultimately, the project aims to clarify whether LPMOs found in pathogenic bacteria contribute to infectious processes or if they primarily support the survival of the bacteria outside the host organism. |
| References: | [1] Kracher et al., Science, 2016, vol. 352, 1098-1101. [2] Gaber et al., Biotechnol. Adv., 2020, vol. 43, 107583. [3] Shukla et al., Nature Biotechnology, 2016, vol. 34, 1046-1051. [4] Askarian et al., Nature Communications, 2021, vol. 12, 1-19. |
| Figures: | 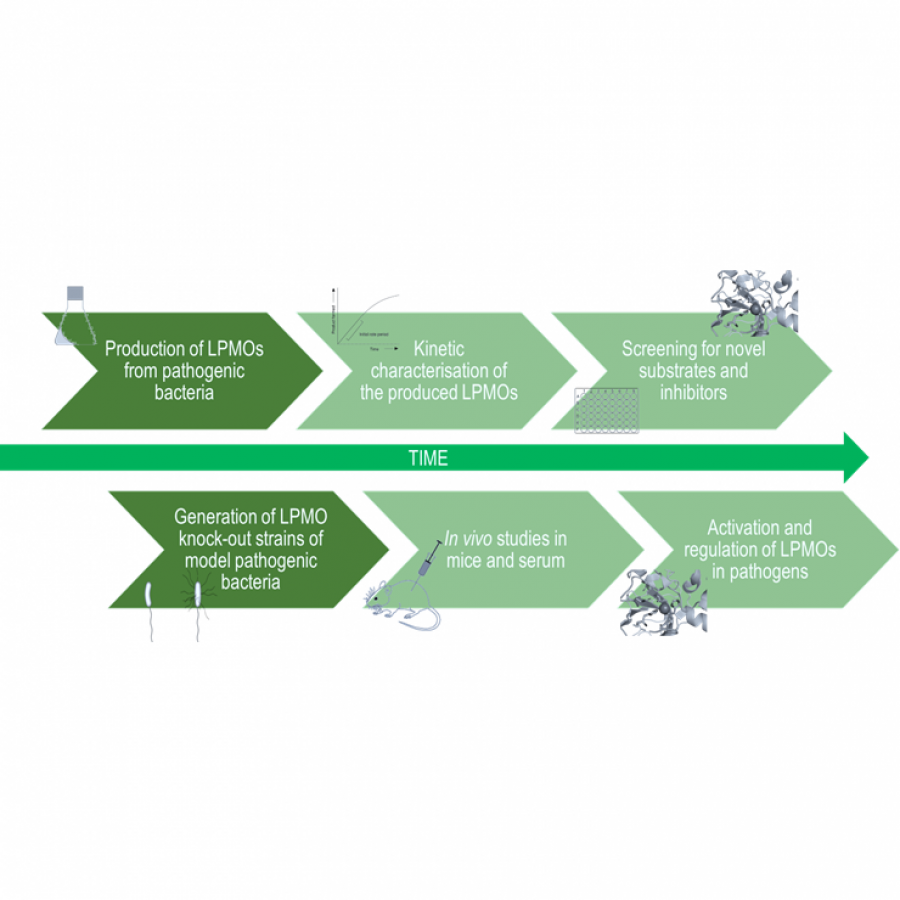 Workflow. Discovery of LPMOs from pathogenic bacteria. The picture depicts the workflow of the ongoing project. |
#1331 | Development and scale-up of an enzymatic process for 2,5-furandicarboxylic acid (FDCA) synthesis. |
|
| Presenting author: | Claudia MATASSA of BIO BASE EUROPE PILOT PLANT |
| Other authors: | Cedric VANDER CRUYSSEN of BIO BASE EUROPE PILOT PLANT Matthias HARTMANN of BIO BASE EUROPE PILOT PLANT Rakesh NAIR of BIO BASE EUROPE PILOT PLANT Tanja MEYER of BIO BASE EUROPE PILOT PLANT Karel DE WINTER of BIO BASE EUROPE PILOT PLANT |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / 2,5-furandicarboxylic acid (FDCA) / Enzyme production / Process intensification |
| Purpose: | The production of bio-based plastics has become an important area of research due to the negative environmental impacts associated with fossil-based polymers. Polyethylene 2,5-furandicarboxylate (PEF) is a bioplastic with superior performance properties, compared to today’s widely used petroleum-based packaging material PET. However, the industrial production of PEF is hampered by the challenges associated with the synthesis of its main building block: 2,5-furandicarboxylic acid (FDCA). A promising alternative to the traditional multi-step chemical routes is the one-step enzymatic oxidation of the biomass-derived 5-hydroxymethyl furfural (HMF) to FDCA. In this study, we report the development, optimization, and scale-up of a process for the production of a FAD-dependent enzyme, which can oxidize HMF to FDCA. A fed-batch fermentation and the associated downstream process (DSP) were developed and optimized at 7 L scale. The fermentation and DSP were scaled up to 1500 L. Finally, the produced enzyme was used for the bioconversion on 150 L scale. After 47 hours, 99.3% of HMF was converted into FDCA, achieving a product concentration of 39.1 g/L. This work demonstrates the feasibility and potential of bio-based process development for the production of PEF and offers an effective and industrially relevant alternative to the chemical synthesis of FDCA. |
| Figures: |  BBEPP process scale-up. Bioreactor (Frings 150 L) used for the enzymatic conversion of HMF to FDCA. 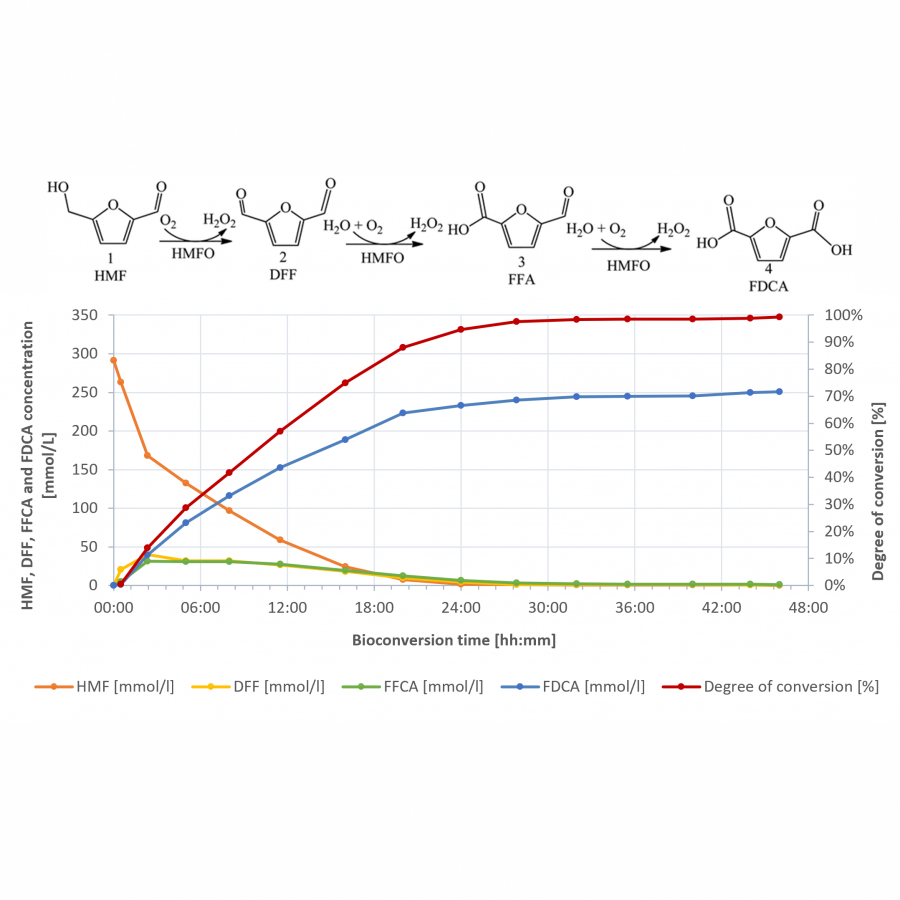 Enzymatic bioconversion of HMF to FDCA (150 L scale). Concentration [mmol/L] of HMF substrate (orange), FDCA product (blue), and the reaction intermediates FFCA/FFA (green), and DFF (yellow) over the time. The degree of HMF conversion [%] over the time is also reported (red). |
#1334 | Citric Acid Production from Wood Sugars: Fermentation and LCA |
|
| Presenting author: | MATIJOŠYTĖ Inga of VILNIUS UNIVERSITY |
| Corresponding author: | Inga MATIJOŠYTĖ of VILNIUS UNIVERSITY LIFE SCIENCES CENTER INSTITUTE OF BIOTECHNOLOGY |
| Other authors: | Lauri VARES of INSTITUTE OF TECHNOLOGY, UNIVERSITY OF TARTU Narie RINKE DIAS DE SOUZA of INDUSTRIAL ECOLOGY PROGRAMME, DEPARTMENT OF ENERGY AND PROCESS ENGINEERING, NORWEGIAN UNIVERSITY OF SCIENCE AND TECHNOLOGY (NTNU) Justinas BABINSKAS of VILNIUS UNIVERSITY LIFE SCIENCES CENTER INSTITUTE OF BIOTECHNOLOGY DOVILĖ DAUNORAITĖ of VILNIUS UNIVERSITY LIFE SCIENCES CENTER INSTITUTE OF BIOTECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | fermentation / LCA / lignocellulose / |
| Purpose: | The need to shift from a fossil-based economy to a more sustainable circular model, forces us to review the materials being used1. Biomass is a potential renewable alternative to replace conventional fossil feedstock in the chemical industry. Currently, a few commercial bio-based chemicals are mainly produced from sugary and starchy crops. Shifting edible feedstock to lignocellulosic residues may alleviate competition for the land for biomass and food production. The citric acid (CA), a natural, non-toxic compound with three carboxylic acid groups, is found in citrus fruits and functions as a natural preservative, flavour enhancer, and natural antioxidant. Industrially produced from sucrose fermentation, it is currently extensively used in the food and pharmaceutical industries. Furthermore, it is also considered one of the potential bio-derived platform chemicals for various polymer and material science applications. Several microorganisms have been shown to produce CA; however, only the white rot fungus Aspergillus niger is currently a microorganism of choice in industry2 In this study, we aimed to compare yeast (Yarrowia lipolytica), bacterial (Bacillus licheniformis) and fungal (Aspergillus niger) strains that can produce CA by employing wood sugars as a carbon source and to develop an optimized fermentation process. It was defined that the composition of the fermentation medium strongly influences the production of CA. The up-scale process up to 1 L was also evaluated for the best-performed process with the yeast and fungi. A proper quantification of sustainability impacts is challenging due to knowledge limitations about industrial-scale performance. Nevertheless, an ex-ante life cycle assessment (LCA) of CA produced from wood sugars from the forest residues was done, implementing a methodology to scale up foreground data of emerging bio-based technologies to higher TRL. After applying the process-based calculations, screening process synergies for material, waste, and energy recovery, and estimating energy requirements, the production chain of CA was evaluated. Acknowledgement: the EEA Grants financial support for the work through the Baltic Research Programme (grant EMP426). The authors thank Karl Peebo (Fibenol, former Graanul Invest) for providing wood sugar samples and valuable suggestions. |
| References: | [1] Sheldon, R.A., ACS Sustain. Chem. Eng. 6, 4464-4480 (2018). [2] Show P.L. et al. Front. Life Sci. 8, 271-283 (2015). |
#1335 | Valorization of pumpkin seeds industrial waste by selective extraction of residual bioactive compounds and their biocatalytic manipulation |
|
| Presenting author: | Daniela MONTI of SCITEC - CNR |
| Other authors: | Lorenzo ZARINI of SCITEC - CNR Alessio MASSIRONI of UNIVERSITÀ DEGLI STUDI DI MILANO Ivan BASSANINI of SCITEC - CNR Erica Elisa FERRANDI of SCITEC - CNR Stefania MARZORATI of UNIVERSITÀ DEGLI STUDI DI MILANO Luisella VEROTTA of UNIVERSITÀ DEGLI STUDI DI MILANO |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | green technologies / oleate hydratases / stereoselectivity / selective extraction |
| Purpose: | Cucurbita pepo L. (pumpkin) seeds are industrially submitted to selective extraction processes to obtain an oil that is commercialized for the treatment of genito-urinary tract pathologies. The residual biomass still contains various bioactive compounds, which were targeted in the framework of the STAIRWAy research project to develop sustainable recovery procedures and, by applying biocatalyzed transformations, to further convert them into high-value derivatives for the nutraceutical, cosmetic and pharmaceutical industry. In the first part of this work, lipophilic extracts, selectively enriched of different classes of compounds, were obtained by supercritical CO2 extraction strategies optimizing process parameters (e.g., pressure and temperature) in an environmentally friendly way, completely avoiding any presence of organic solvents. Free fatty acids, obtained under mild operative conditions, were thus transformed by enzyme-catalyzed reactions into valuable derivatives, such as ethanol amides and, for unsaturated fatty acids, in the corresponding 10-hydroxy fatty acids [1]. Among the hydrophilic components of the C. pepo still present in the residual biomass, the amino acid cucurbitin, whose biological activity is still poorly investigated, was herein extracted in selected aqueous/alcoholic media and enriched by means of ion exchange resins. Furthermore, aimed at a better understanding of the potential bioactivity of cucurbitin and related compounds, the preparation of racemic cucurbitin samples, suitable for the subsequent enzymatic or chemical resolution to enantiomerically enriched species, was investigated by developing a de novo synthetic procedure. Acknowledgments: Financial support from Cariplo Foundation (STAIRWAy project, grant no. 2019–2122) is kindly acknowledged. |
| References: | [1] A. Massironi et al., Molecules, 27, 4783 (2022), DOI: 10.3390/molecules27154783 |
#1336 | Asymmetric Thio-Michael Additions Catalyzed by the Enzyme 4-Oxalcrotonate Tautomerase |
|
| Presenting author: | MICHELE CROTTI of UNIVERSITY OF GRONINGEN |
| Other authors: | Pieter TEPPER of UNIVERSITY OF GRONINGEN Gerrit J. POELARENDS of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / enzyme promiscuity / / |
| Purpose: | *MC Current address: University College London, Chemistry, London, United Kingdom The enzyme 4-oxalcrotonate tautomerase (4-OT) catalyzes multiple promiscuous C-C bond-forming reactions, which find applications in the sustainable synthesis of pharmaceutical precursors and enantioenriched synthons, and proceed via enzyme-bound enamine intermediates. [1–4] Recent protein engineering campaigns unlocked new asymmetric reactions catalyzed by 4-OT proceeding through an enzyme-bound iminium ion intermediate. The C-C bond-forming reaction of Michael-type addition of nitromethane to cinnamaldehydes [5,6] and the C-O bond-forming epoxidation of cinnamaldehyde derivatives using peroxides are examples.[8] In this work, we characterize and optimize a new reactivity of 4-OT, namely the C-S bond-forming Michael-type addition reaction of aliphatic thiols to cinnamaldehyde. Two engineered 4-OT variants were found able to catalyze this addition reaction in a stereospecific manner using a set of aliphatic thiols as nucleophiles and yielding (R)-β-thioenals with conversions up to 97 % and products e.r. up 95:5. Reaction conditions were optimized to avoid the enzyme-catalyzed racemization of the products. |
| References: | [1] M. Rahimi, J. Y. van der Meer, E. M. Geertsema, G. J. Poelarends, ChemBioChem 2017, 18, 1435-1441. [2] M. Saifuddin, C. Guo, L. Biewenga, T. Saravanan, S. J. Charnock, G. J. Poelarends, ACS Catal. 2020, 10, 2522-2527. [3] L. Biewenga, T. Saravanan, A. Kunzendorf, J. Y. Van Der Meer, T. Pijning, P. G. Tepper, R. Van Merkerk, S. J. Charnock, A. M. W. H. Thunnissen, G. J. Poelarends, ACS Catal. 2019, 9, 1503-1513. [4] J. Y. Van Der Meer, H. Poddar, B. J. Baas, Y. Miao, M. Rahimi, A. Kunzendorf, R. Van Merkerk, P. G. Tepper, E. M. Geertsema, A. M. W. H. Thunnissen, W. J. Quax, G. J. Poelarends, Nat. Commun. 2016, 7, 1-16. [5] C. Guo, M. Saifuddin, T. Saravanan, M. Sharifi, G. J. Poelarends, ACS Catal. 2019, 9, 4369-4373. [6] L. Biewenga, M. Crotti, M. Saifuddin, G. J. Poelarends, ACS Omega 2020, 5, DOI 10.1021/acsomega.9b03849. [7] Kunzendorf, A., Xu, G., Saifuddin, M., Saravanan, T., & Poelarends, G. J., Angew. Chemie Int. Ed. 2021, 60(45), 24059-24063. [8] G. Xu, M. Crotti, T. Saravanan, K. Kataja, G. J. Poelarends, Angew. Chemie Int. Ed. 2020, 132(26), 10460-10464. |
#1340 | A novel chemo-enzymatic synthesis of the stable and separable C-C dimeric atropoisomers of the bioactive natural sesquiterpene 8-trans-hydroxycalamenene |
|
| Presenting author: | Chiara TOGNOLI of SCITEC-CNR |
| Other authors: | Stefano SERRA of SCITEC-CNR Ivan BASSANINI of SCITEC-CNR Sergio RIVA of SCITEC-CNR |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chemo-enzymatic synthesis / T. versicolor laccase / terpenoids and natural products / biocatalysis |
| Purpose: | Terpenes are a class of compounds characterized by carbon backbones of a variable number of isoprene units which can be rearranged into cyclic structures. Among these, sesquiterpenes are formed by three isoprene units and display a wide range of biological activities such as antimicrobial, antimycobacterial, analgesic, anti-inflammatory and anti-protozoal properties, as evidenced by in vitro assays of constituents in essential oils extracted from different plant species [1-3]. 8-hydroxycalamenene (mainly in trans configuration) is an important natural product representative of this group, whose (+)-form represents the major toxic constituent of Dysoxylum acutangulum seeds [4]. Moreover, a minor component of the same plant was characterized as an unsymmetrical C-C- dimer of (+)-8-hydroxycalamenene, existing as a discrete atropoisomer [5]. Interestingly, to the best of our knowledge, no further investigations on its chemical synthesis and pharmacological profile have been carried out in more recent years. Thus, a new chemo-enzymatic entry was considered to prepare these peculiar structures characterized by a conformationally-constrained stereogenic axis. Accordingly, the two enantiomers of 8-trans-hydroxycalamenene (1 and 2, Figure 1) were chemically prepared through stereoselective syntheses starting from naturally occurring (-) and (+)-menthol, both easily available from the chiral pool. The obtained sesquiterpene isomers were thus used as substrates in a laccase-mediated oxidative C-C coupling. A synthetic protocol, previously described by us for the preparation of 1 [6], has been successfully adapted for the synthesis of 2. Subsequently, aiming at enzymatically generating the C-C dimers of the two substrates, laccase from Trametes versicolor was selected, based on our expertise with this enzyme in analogues transformations [7]. Laccases are multi-copper oxidases known for being able to activate normally inert Csp2-H bonds of phenol – or aniline – substrates by generating organic radicals at the expense of molecular oxygen, generating water as the only reduction byproduct [8]. The oxidation of 1 and 2 was studied using different reaction media and conditions and, finally, the optimized biotransformations allowed to isolate in good yields the target C-C dimers 3 and 4 (Figure 1), as a stereoenriched mixture of stable and separable atropoisomers. Thanks to this chemo-enzymatic approach, the C-C dimeric metabolites of both the enantiomers 8-trans-hydroxycalamenene were obtained and fully characterized. Their bioactive profiles, starting from potential antiprotozoal activity [2,3,9] and/or cytotoxicity, is now under investigation. |
| References: | [1] A. Masyita, R. M. Sari, A. D. Astuti, B. Yasir et al., Food chem.: X 2022, 100217. [2] R. Mondêgo-Oliveira, J. C. de Sá Sousa, C. J. Moragas-Tellis, P. V. R. de Souza et al., Biomed. Pharmacother. 2021, 133, 111025. [3] C. Kauffmann, A. C. Giacomin, K. Arossi, L. A. Pacheco, et al., Braz. J. Pharm. Sci. 2019, 55. [4] M. Nishizawa, A. Inoue, S. Sastrapradja, Y. Hayashi, Phytochem. 1983, 22(9), 2083-2085. [5] M. Nishizawa, H. Yamada, S. Sastrapradja, Y. Hayashi, Tetrahedron Lett. 1985, 26(12), 1535-1536. [6] S. Serra, C. Fuganti, Tetrahedron Lett. 2005, 46, 4769-4772. [7] S. Ncanana, L. Baratto, L. Roncaglia, S. Riva, S. G. Burton, Adv. Synth. Catal. 2007, 349(8-9), 1507-1513. [8] I. Bassanini, E. E. Ferrandi, S. Riva, D. Monti, Catalysts 2020, 11(1), 26. [9] S. Afoulous, H. Ferhout, E. G. Raoelison, A. Valentin, et al., Molecules 2011, 16(10), 8273-8291. |
| Figures: | 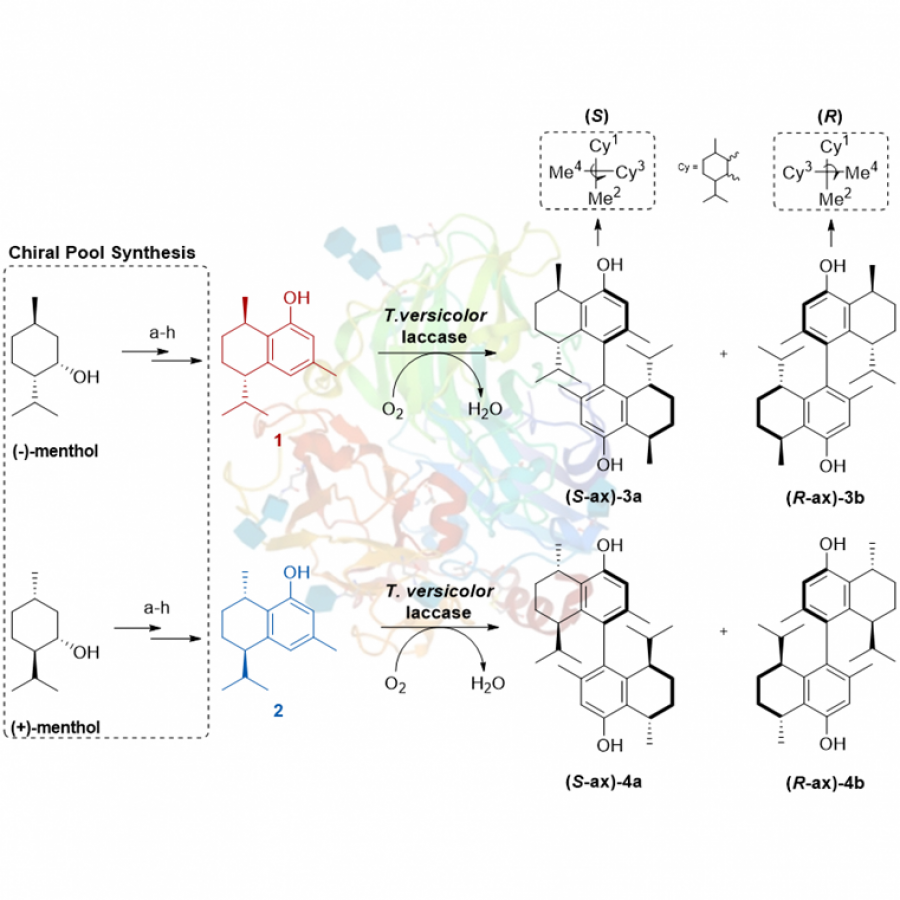 Chemo-enzymatic synthesis of bicalamenene atropoisomers. Reagents and conditions: a) CrO3/H2SO4, Et2O-H2O, 25 deg; b) TsNHNH2, DCM, 0 deg; c) BuLi, hexane/TMEDA, -78 deg - Rt; d) DMF, -78 deg; e) DMS, LDA, THF, -60 deg; f) (CF3CO)2O, TEA, THF, 0 deg; g) LiAlH4, Et2O, 0 deg; h) H2, Pd/C, MeOH |
#1341 | Bringing dead proteins to life; development of an artificial Stetterase. |
|
| Presenting author: | Alice MACAULAY of UNIVERSITY OF EDINBURGH |
| Other authors: | Eva KLEMENCIC of UNIVERSITY OF EDINBURGH Amanda JARVIS of UNIVERSITY OF EDINBURGH Dominic CAMPOPIANO of UNIVERSITY OF EDINBURGH |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Stetter / Genetic code expansion / Artificial enzyme / Biocatalysis |
| Purpose: | Thiamine diphosphate (ThDP, vitamin B1) is one of nature’s versatile organo-catalysts. ThDP-dependent enzymes catalyse a broad range of reactions, the most useful of which are the C-C bond forming biocatalysts that can be used to create chiral products from simple achiral building blocks. Several ThDP-dependent biocatalysts have been used to form α-hydroxyketones in high enantomeric excesses, including benzaldehyde lyases (BAL) that catalyses an enantioselective benzoin condensation. In contrast, only a limited number of ThDP-dependent biocatalysts can catalyse the irreversible formation of 1,4-dicarbonyl compounds from aldehydes and , unsaturated carbonyl containing substrates. These so-called Stetterases have great synthetic potential but are limited in their substrate scope. Here, we aim to expand this pool of Stetterases by creating an artificial biocatalyst using a catalytically-inactive protein scaffold that contains a covalently attached thiamine analogue. Our work utilises a small inactive protein scaffold, the recombinant human steroid carrier protein 2L (SCP-2L). The SCP-2L protein contains a hydrophobic tunnel and has a single cysteine residue engineered via mutagenesis. A thiazolium salt was incorporated into the SCP-2L scaffold by a site-specific, thiol-maleimide linkage to this cysteine residue. This artificial ‘active site’ within the protein successfully catalyses an intramolecular Stetter reaction, in effect bringing a dead protein to life. Moving from the initial human construct to a thermophilic, bacterial version of the SCP scaffold (TT_SCP) we have been able to achieve 12 turnovers with modest e.e. (5%) without further optimisation. We also describe plans for engineering the artificial Stetterase active site, firstly to create an environment which aids deprotonation of the TT_SCP-bound thiazolium moiety. Secondly, we aim to increase the enantioselectivity of our ‘artificial biocatalyst’ as well as broaden its substrate scope. Work on Genetic Code Expansion (GCE) to increase the yield of the functionalised protein is in progress. The chemical synthesis of a thiazolium based unnatural amino acid (UAA) has been achieved and development of a method to incorporate this into a protein scaffold is underway. This proof of concept study paves the way for the in vivo synthesis and incorporation of a thiazolium-based UAAs into any protein scaffold. By combining organo-catalysis with directed evolution, our work provides a platform to design, select and engineer novel biocatalysts. |
| References: | S.Prajapati Current Opinion in Structural Biology, 2022, 76, 102441 |
| Figures: | 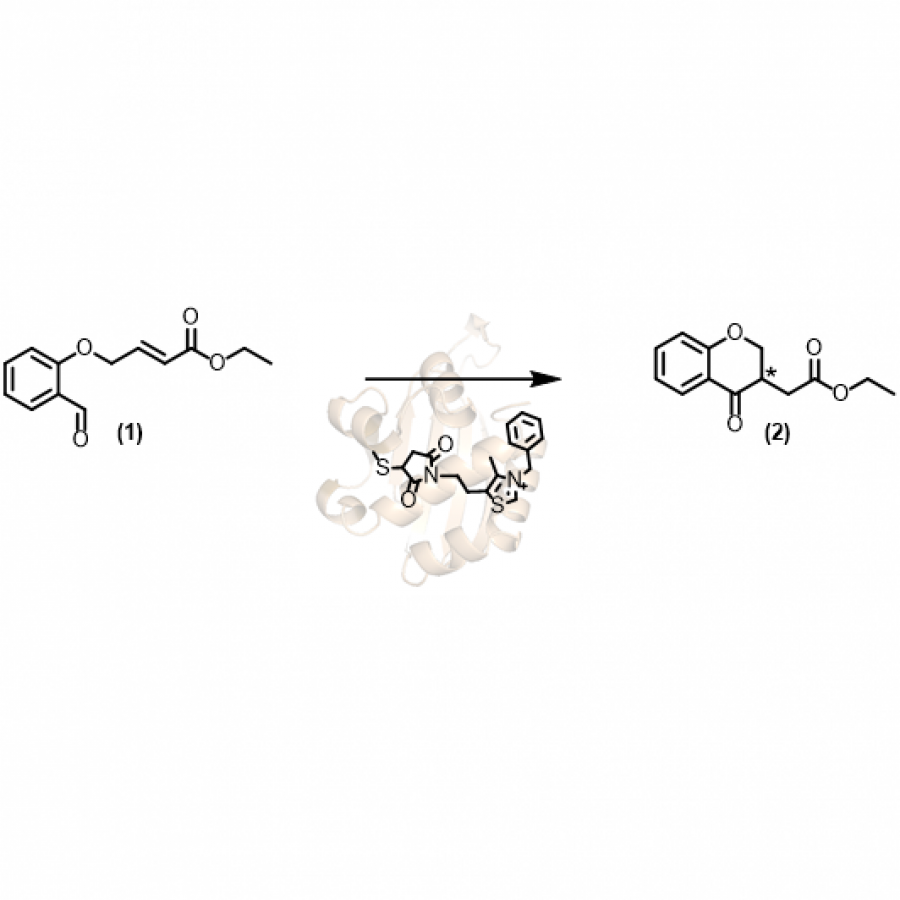 Figure 1 An intramolecular Stetter reaction catalysed by a thiazolium functionalised protein scaffold |
#1342 | Newly discovered metagenomic DERA aldolases catalyse the condensation of furfural and a number of ketones |
|
| Presenting author: | Andrea RIZZO of JOHNSON MATTHEY |
| Other authors: | Carmen ARANDA of JOHNSON MATTHEY Beatriz DOMINGUEZ of JOHNSON MATTHEY Jack JEFFRIES of UNIVERSITY COLLEGE LONDON |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Aldolase / Metagenome / DERA / Furfural |
| Purpose: | The formation of carbon-carbon bonds is a very common reaction in industry, enabling the formation of large molecules starting from small building blocks. In nature, this type of bond is catalysed by several classes of enzymes, among which there are aldolases, which catalyse the aldol addition of aldehydes and ketones into aldols. A sub-class of aldolases named DERA (DEoxyRibose-phosphate Aldolase) proved in the past to be a good candidate for industrial applications due to its broad range of nucleophiles donors and electrophiles acceptors it can use as a substrate1. In this work, we analysed several metagenomes and extracted 56 genes using a sequence-based approach. These sequences were recombinantly expressed in Escherichia coli and screened for activity towards a range of carbon-carbon bond formation reactions. Among these, several enzymes proved to catalyse the aldol addition of furfural with a number of ketones, expanding the known substrate scope of this class. Moreover, furfural gathered interest in recent years for being a platform chemical which can be sustainably produced from lignocellulosic feedstock2, and its condensation with a range of ketones has been reported for the synthesis of jet fuel intermediates3. DERAs could potentially become a sustainable alternative route to chemical processes for the production of these intermediates. |
| References: | [1] Chambre, D., Chem. Commun. 2019, 55(52), 7498-7501. [2] Lee, C.B.T.L., Renew. Sustain. Energy Rev. 2021, 137, 110172. [3] Malkar, R.S., ACS Sustain. Chem. Eng. 2019, 7(19), 16215-16224. |
| Figures: |  Project summary Summary of the project. DERA sequences were extracted from the analysis of metagenomes from salt mines, bioreactors, compost, and hydrothermal vent, and were used to catalyse the aldol addition of furfural with several ketones |
#1345 | Outstanding substrate specificity of rutinosidase from Aspergillus niger |
|
| Presenting author: | Katerina BRODSKY of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Other authors: | Pavla BOJAROVÁ of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Vladimír KŘEN of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Katerina BRODSKY of DEPARTMENT OF BIOCHEMISTRY AND MICROBIOLOGY, UNIVERSITY OF CHEMISTRY AND TECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Rutinosidase / Quercitrin / Substrate specificity / Diglycosidase |
| Purpose: | Rutinosidases (6-α-L-rhamnosyl-β-D-glucosidases) are retaining diglycosidases that cleave the glycosidic bond between the disaccharide rutinose and the respective aglycone (Figure 1). One of the best studied rutinosidases is a rutinosidase from Aspergillus niger (AnRUT) [1]. Its crystal structure has been previously published [2], and yet the substrate specificity of AnRUT is intriguing due to its diversity. Contrary to expectations, rutinosidase from A. niger showed higher hydrolytic activity with isoquercitrin (quercetin-3-β-O-glucopyranoside) as substrate, indicating that it has a broad substrate specificity and can be considered as a β-D-glucosidase. The structure of this enzyme shows strong and specific binding of the aromatic aglycone (flavonoid) and high tolerance at the glycone binding site (Figure 2) [3], especially at the C-6' position of the glucose moiety. Not all aromatic aglycones bind properly to the active site, and then the glycone plays an important role. The enzyme activity may be higher for a disaccharide substrate than for a glucoside if the aglycone only partially fits into the active site. In this study, we investigated the effects of glycone modifications on enzyme activity. We tested a group of potential substrates, including aromatic diglycosides with different types of glycosidic bonds between the respective carbohydrate moieties (e.g. p-nitrophenyl β-D-lactoside, p-nitrophenyl β-D-maltoside, p-nitrophenyl β-D-isomaltoside, amygdalin, and p-nitrophenyl β-D-rutinoside) as well as various 6'-O-acyl derivatives of isoquercitrin synthesized under lipase catalysis [3] and evaluated as (potential) substrates for AnRUT. In addition to its hydrolytic activity, AnRUT also possesses distinct synthetic (transglycosylating) capabilities [1]. By transferring rutinosyl and β-D-glucopyranosyl residues to various alcohols and even to phenolic acceptors [3], which are generally difficult to glycosylate by glycosidases, new compounds can be created. Supported by Czech Science Foundation projects 21-01948L and 22-00197K. |
| References: | [1] Šimčíková, D. Adv. Synth. Catal. 2015, 357, 107-117 [2] Pachl, P. FEBS J., 2020, 287, 3315-3327 [3] Brodsky, K. Int. J. Mol. Sci. 21, 5671 [4] Vavříková, E. Int. J. Mol. Sci. 2016, 17, 899 |
| Figures: |  Rutin hydrolysis catalyzed by AnRUT Hydrolysis products are quercetin and disaccharide rutinose.  AnRUT active site model Interactions between enzyme amino acids and the substrate (rutin) in the active site of AnRUT |
#1346 | Fusion Proteins for Biocatalyst-Based Polymer Degradation |
|
| Presenting author: | Lydia SUCHY of INSTITUTE OF APPLIED SYNTHETIC CHEMISTRY TU WIEN |
| Other authors: | Florian RUDROFF of INSTITUTE OF APPLIED SYNTHETIC CHEMISTRY TU WIEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | polymer degradation / hydrophobin / fusion protein / surface analysis |
| Purpose: | Currently, the accumulation of plastic waste due to insufficient/inefficient methods for the degradation of commodity polymers represents a growing environmental problem. In this regard, the use of polymer-degrading biocatalysts has been subject to many studies [1, 2, 3]. Polymers such as polyesters, polyamides, polyurethanes, and polyolefins are highly hydrophobic and water insoluble molecules. However, a biocatalyst needs an aqueous system in order to be active. This difference in hydrophilicity prevents the adsorption of high amounts of biocatalyst to the polymer surface. To maximize biocatalytic efficiency, methods to improve protein-substrate interaction have been explored over the last few years. One possibility represents the fusion of a substrate-binding domain that acts as a plastic-recognition element (a hydrophobic anchor) and increases the protein’s binding affinity. Hydrophobins, which are small fungal proteins that self-assemble on lipophilic surfaces, represent one option to enhance substrate binding [4]. In previous research, PET hydrolysis efficiency has been improved by the fusion of hydrophobins to a PET hydrolase [5]. In this research, the fusion of several hydrophobic elements into a polymer-degrading biocatalyst is being studied. Different constructs consisting of a hydrophobin (class II hydrophobins from T. reesei and a mutant class I hydrophobin from G. frondosa) or a small hydrophobic peptide connected to the biocatalyst were constructed via molecular cloning and produced in E. coli. For the analysis of the fusion protein – polymer interaction, either commercially available polymer samples were used or thin films were prepared via spin-coating. The assembly on the hydrophobic polymer surface and the efficiency of the polymer-degrading biocatalyst is being analyzed via different surface analysis methods such as water contact angle measurement, surface plasmon resonance (SPR), attenuated total reflection Fourier transform infrared spectroscopy (ATR-FTIR), x-ray photoelectron spectroscopy (XPS) and atomic force microscopy (AFM). |
| References: | [1] P. Demarche, C. Junghanns, R. R. Nair, S. N. Agathos, Biotechnol. Adv., 2012, 30, 933-953. [2] N. Mohanan, Z. Montazer, P. K. Sharma, D. B. Levin, Front. Microbiol., 2020, 11, 580709. [3] B. Zhu, D. Wang, N. Wei, Trends Biotechnol., 2022, 40, 22-37. [4] B. W. Berger, N. D. Sallada, J. Biol. Eng., 2019, 13, 1-8. [5] a) L. Espino-Rammer, D. Ribitsch, A. Przylucka, A. Marold, K. J. Greimel, E. Herrero Acero, G. M. Guebitz, C. P. Kubicek, I. S. Druzhinina, Appl. Environ. Microbiol., 2013, 79, 4230-4238. b) D. Ribitsch, E. Herrero Acero, A. Przylucka, S. Zitzenbacher, A. Marold, C. Gamerith, R. Tscheließnig, A. Jungbauer, H. Rennhofer, H. Lichtenegger, H. Amenitsch, K. Bonazza, C. P. Kubicek, I. S. Druzhinina, G. M. Guebitz, Appl. Environ. Microbiol., 2015, 81, 3586-3592. |
| Figures: | 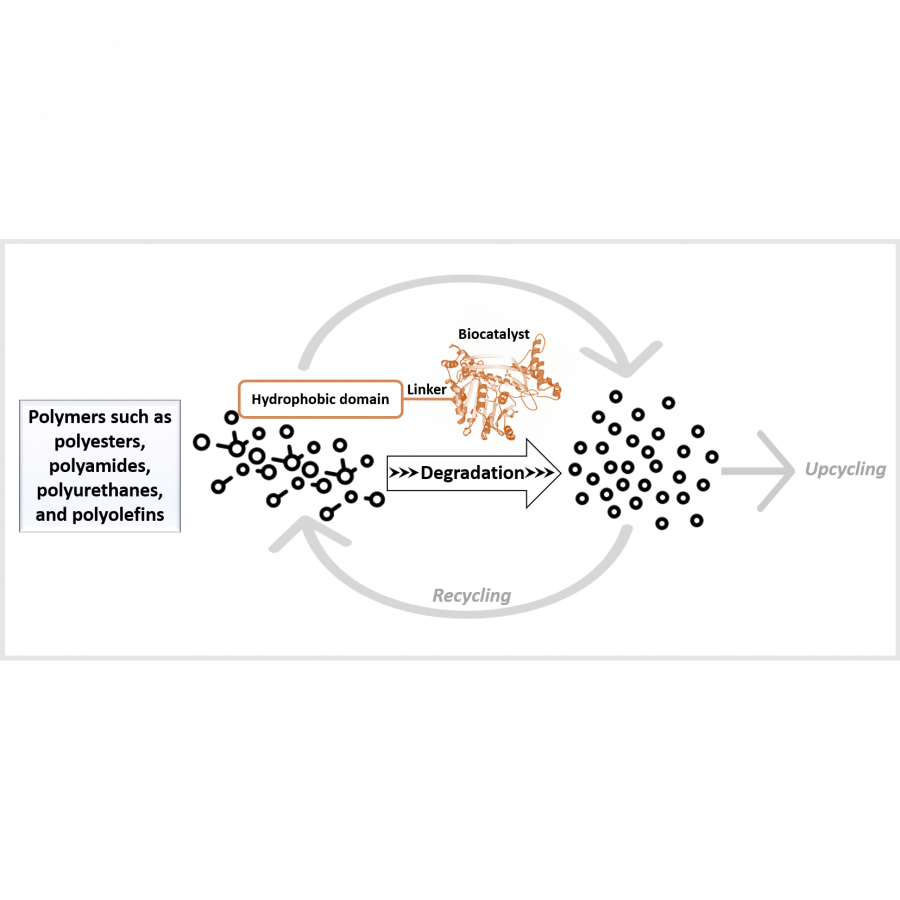 Schematic representation of the research topic A hydrophobic domain (hydrophobin or a small peptide consisting of hydrophobic amino acids) is linked to a polymer-degrading biocatalyst. |
#1350 | Biocatalytical diversification of 4'-thiouridine gives access to halogenated pyrimidine and purine derivates |
|
| Presenting author: | Sarah WESTARP of TECHNISCHE UNIVERSISTÄT BERLIN |
| Other authors: | Jonas MOTTER of TECHNISCHE UNIVERSITÄT Caecilie BENCKENDORFF of KEELE UNIVERSITY Mieke GUINAN of KEELE UNIVERSITY Peter NEUBAUER of TECHNISCHE UNIVERSITÄT Gavin MILLER of KEELE UNIVERSITY Anke KURRECK of TECHNISCHE UNIVERSITÄT |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | 4'-thianucleoside / Nucleoside Phosphorylase / biocatalysis / drug diversification |
| Purpose: | The key role of nucleosides and their analogues in cell proliferation and viral replication renders this class of small molecules prominent drug candidates. While a wide range of nucleoside analogues are approved drugs, only a small number features 4’-modified sugar residues, namely the 4’-C derivates Entecavir and Ticagrelor and the 4´-N derivative Forodesine (De Jonghe and Herdewijn, 2022). Recently, the interest in 4’-thio nucleosides analogues was revived, and a new up-scalable chemical synthesis route for pyrimidine derivates was developed (Guinan et al., 2022). While this methodology gives access to a wide range of 2’-modified 4’-thiopyrimidines, obtaining 4’-thiopurine nucleosides remains a challenge. To fill this gab, 4’-thiouridine was chosen as starting compound in a proof-of-concept study focussing on biocatalytical diversification of the uracil base. To this end, we applied thermostable nucleoside phosphorylases to cleave this nucleoside analogue into uracil and 4’-thioribo-1’-phosphate. The latter served as substrate for a second reaction, where diverse modified bases were attached. The synthesis of 4’-thio-5-Iodouridine was up-scaled to 100 mg product to enable additional chemical diversification reactions. While enzymatic synthesis on itself is not new, as Van Draanen and colleagues (Van Draanen et al. 1996) used it before to produce 2’-deoxy-4’-thio-6-Cl-2-aminopurine nucleoside, we were focussing on a directed reaction optimization by characterizing the two reaction steps thermodynamically. This enabled the development of an ecological up-scaling process by avoiding usage of disproportional amounts of substrate. |
| References: | De Jonghe and Herdewijn, Current Protocols, 2, e376 Guinan et al., Org. Biomol. Chem., 2022, 20, 1401-1406 Van Draanen et al., J. Med. Chem. 1996, 39, 2, 538-542 |
| Figures: | 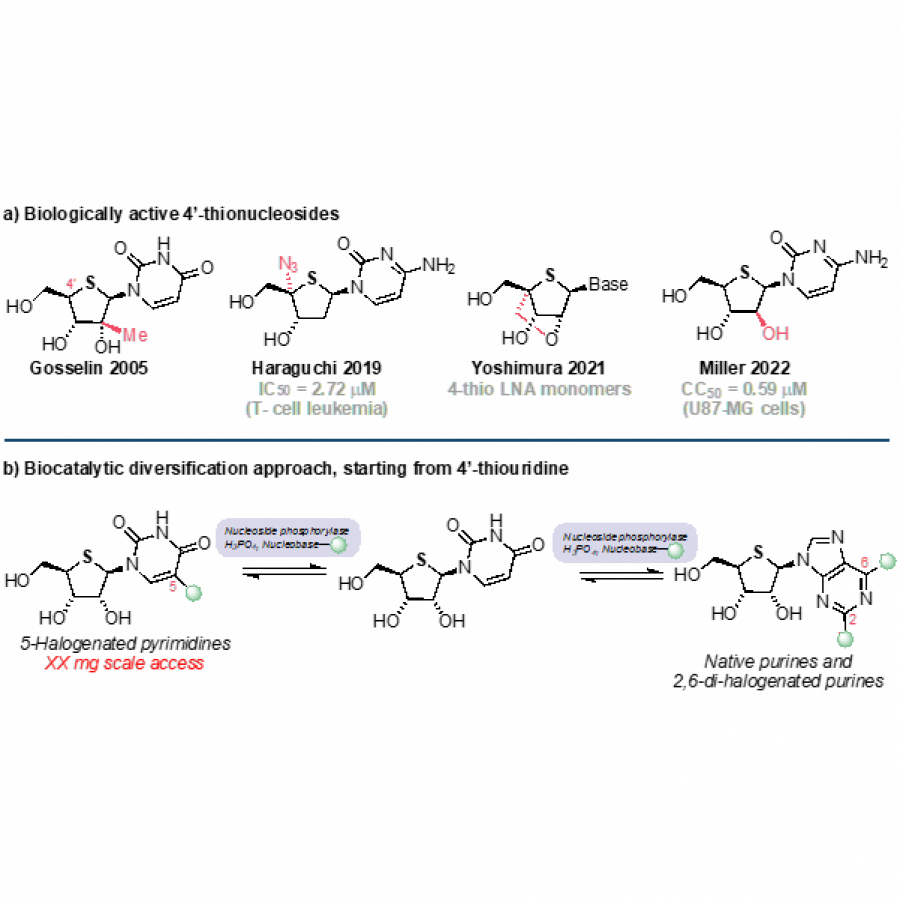 Concept a) Examples of biologically relevant 4’-thionucleoside analogues, with furanose ring modification replacing oxygen with sulfur, and additional structural modifications (shown in red); b) Strategy undertaken here to deliver heterobase-modified 4’-thionucle 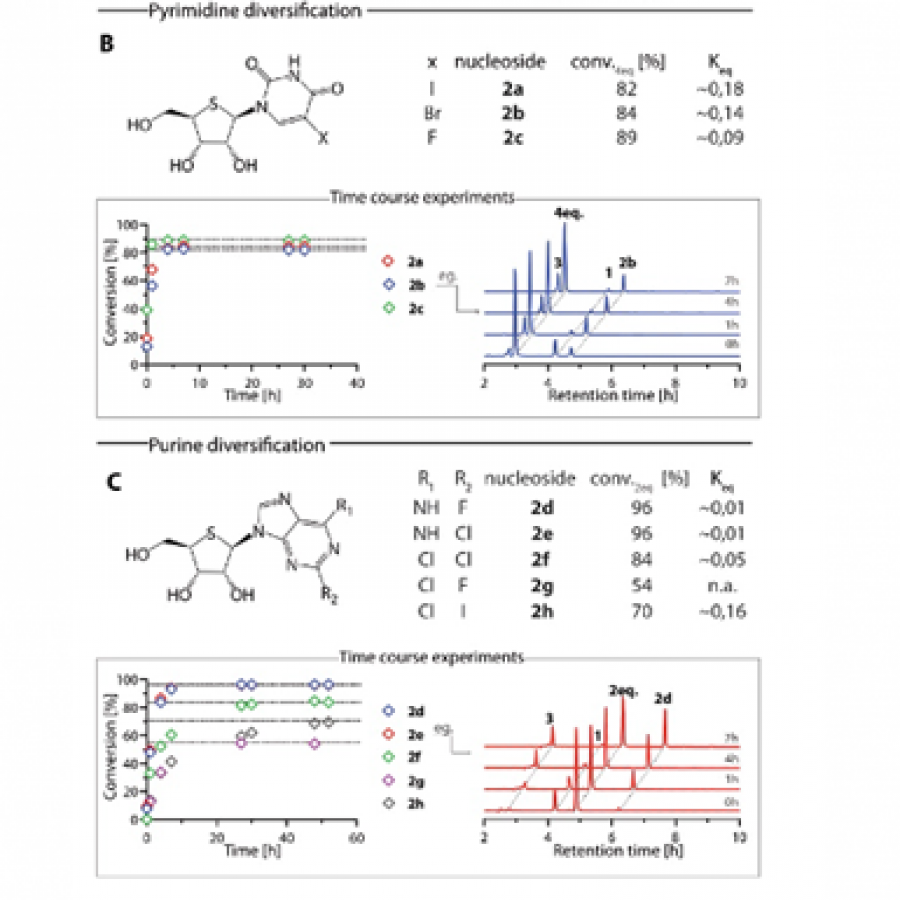 Diversification Results Reaction equilibrium and apparent K_eq of the target molecules. HPLC chromatograms highlight absence of unwanted side products, and the time course shows reaching of reaction equilibrium under chosen conditions for halogenated B) Pyrimidines and C) Purin |
#1351 | Dative coordination of a di-rhodium cofactor in a de novo protein |
|
| Presenting author: | Florian LEISS-MAIER of TECHNICAL UNIVERSITY MUNICH, TUM SCHOOL OF NATURAL SCIENCES, DEPARTMENT OF BIOSCIENCE & CENTER FOR FUNCTIONAL PROTEIN ASSEMBLIES (CPA) |
| Corresponding author: | Cathleen ZEYMER of TECHNICAL UNIVERSITY MUNICH, TUM SCHOOL OF NATURAL SCIENCES, DEPARTMENT OF BIOSCIENCE & CENTER FOR FUNCTIONAL PROTEIN ASSEMBLIES (CPA) |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzymes / / / |
| Purpose: | Metal catalysts facilitate challenging chemical reactions such as C-C bond formations [1]. However, their regio- and enantioselectivity must often be dictated by large ligands that usually require multi-step syntheses. Combining a powerful metal catalyst such as di-rhodium(II,II) with the chiral environment of a protein to yield an artificial metalloenzyme allows for easier fine-tuning of the metal’s microenvironment and is the basis for a large field of research [2]. Previous work has reported novel catalysts that consist of (i) Rh2 coordinated to peptides [3] or (ii) proteins linked to a Rh2 catalyst [4], which both exhibited improved regio- or enantioselectivity compared to conventional ligands. Here we show a de novo designed protein scaffold with four central glutamic acid residues [5] that form an ideal coordination geometry to harbor a di-rhodium cofactor. By datively coordinating the metal to the amino acids of the protein, we envision a direct control over the reactivity of the metal by altering its second and third coordination spheres. Analytical methods, such as dynamic light scattering and analytical ultracentrifugation, show distinct changes in the shape of the protein upon metal addition. Inductively coupled plasma mass spectrometry indicates binding of the metal to the protein that cannot be observed in knockout variants. We aim to demonstrate the correct placement of the metal by elucidating the structure of the holo-protein and through mass spectrometric analysis. Additionally, computational redesign of the protein surface will be used to minimize off-target binding of the metal. The envisioned reactions catalyzed by this artificial metalloenzyme include reactions not observed in natural enzymes, such as cyclopropanations or heteroatom-H insertions. |
| References: | [1] Chen, Z. et al., Organic Chemistry Frontiers 2015, 2, 1107-1295 [2] Klein, A. S. & Zeymer, C., Protein Engineering, Design and Selection 2015, 34, gzab003 [3] Sambasivan, R. & Ball, Z. T., Angew. Chem. Int. Ed. 2012, 51, 8568-8572 [4] Srivastava, P., Yang, H., Ellis-Guardiola, K. & Lewis, J. C., Nature Communications 2015, 6, 7789 [5] Caldwell, S. J. et al. Proceedings of the National Academy of Sciences 2020, 117, 30362-30369 |
| Figures: | 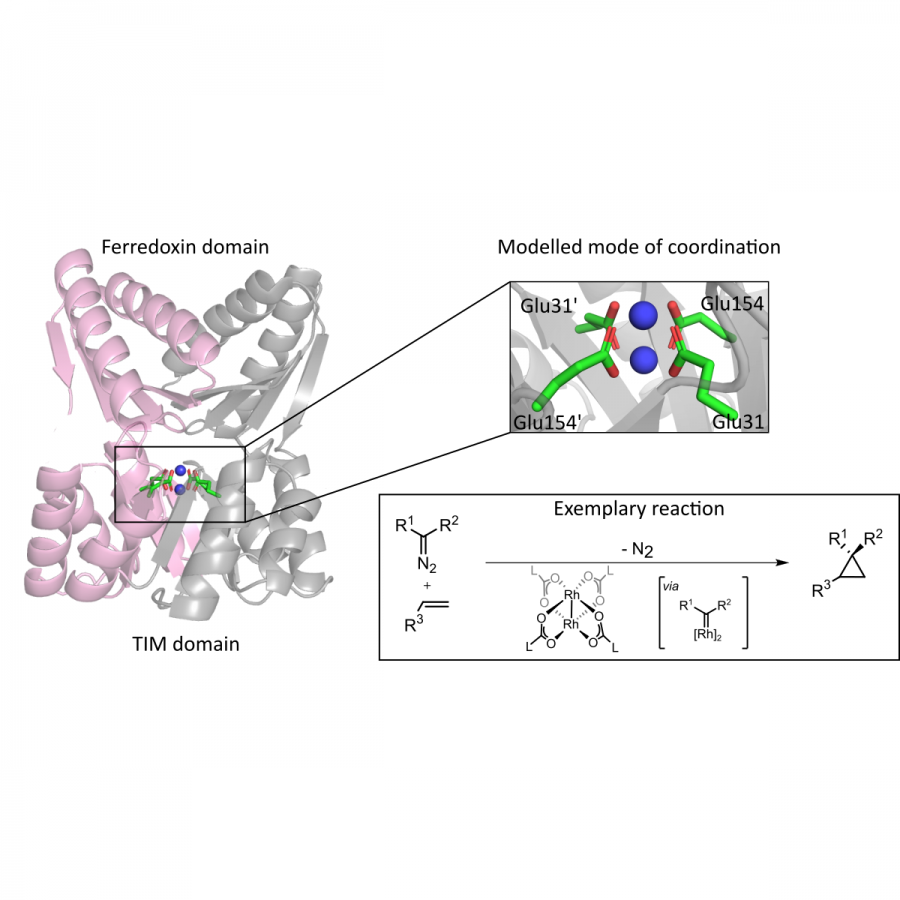 Artificial di-rhodium enzyme Left: Model of the protein with a central di-rhodium cofactor. Bottom right: Exemplary reaction. |
#1352 | Enzyme Optimization and Process Development for a Scalable Synthesis of Methoxymandelic Acid |
|
| Presenting author: | Laura DORNAN of ALMAC SCIENCES |
| Corresponding author: | Stefan MIX of ALMAC SCIENCES |
| Other authors: | Mark SCOTT of GILEAD ALBERTA ULC Xiaotian WANG of GILEAD ALBERTA ULC Luke HUMPREYS of GILEAD ALBERTA ULC Michael GEIER of GILEAD ALBERTA ULC Balamurali KANNAN of GILEAD ALBERTA ULC Johann CHAN of GILEAD ALBERTA ULC Gareth BROWN of ALMAC SCIENCES Daniel DOURADO of ALMAC SCIENCES Darren GRAY of ALMAC SCIENCES Aliaksei PUKIN of ALMAC SCIENCES |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Nitrilase / Enzyme Engineering / Process Development / Biocatalysis |
| Purpose: | Enzyme engineering and process development are two key tools towards industrial biocatalytic solutions. Almac has experience of developing commercially successful biocatalytic solutions.[1-3] Herein we detail a parallel approach to process development and engineering of a nitrilase enzyme (Burkholderia cenocepacia J2315 (BCJ2315)) to achieve an economically viable process for the one-pot, enantioselective, dynamic kinetic resolution towards (R)-2-methoxymandelic acid.[4] Through a combination of molecular docking and B-factor analyses, a focused library of mutants was identified and screened to improve nitrilase selectivity, activity, and stability. Initial optimization revealed that the addition of sodium bisulfite prevented enzyme deactivation by the aldehyde starting material, removing the need to isolate the cyanohydrin and allowing for development of a one-pot process for mutant screening. Further optimization of the process with the preferred mutants revealed subtle interactions between temperature, pH, substrate loading, and enzyme source which ultimately led to development of a suitable whole cell process for scale-up, affording (R)-2-methoxymandelic acid in 97% ee and 70% isolated yield on multigram scale. |
| References: | [1] A. T. P. Carvalho, D. F. A. R. Dourado, T. Skvortsov, M. de Abreu, L. Ferguson, D. J. Quinn, T. S. Moody, M. Huang, Phys. Chem. Chem. Phys., 2017, 19, 26851-26861. [2] D. F. A. R. Dourado, S. Pohle, A. T. P. Carvalho, D. S. Dheeman, J. M. Caswell, T. Skvortsov, I. Miskelly, R. T. Brown, D. J. Quinn, C. C. R. Allen, M. Huang, T. S. Moody, ACS Catal., 2016, 6, 7749-7759. [3] G. Brown, D. Mangan, I. Miskelly, T. S. Moody, Org. Process Res. Dev., 2011, 15, 1036-1039. [4] M. E. Scott, X. Wang, L. D. Humphreys, M. J. Geier, B. Kannan, J. Chan, G. Brown, D. F. A. R. Dourado, D. Gray, S. Mix, A. Pukin, Org. Process Res. Dev., 2022, 26, 849-858 |
| Figures: | 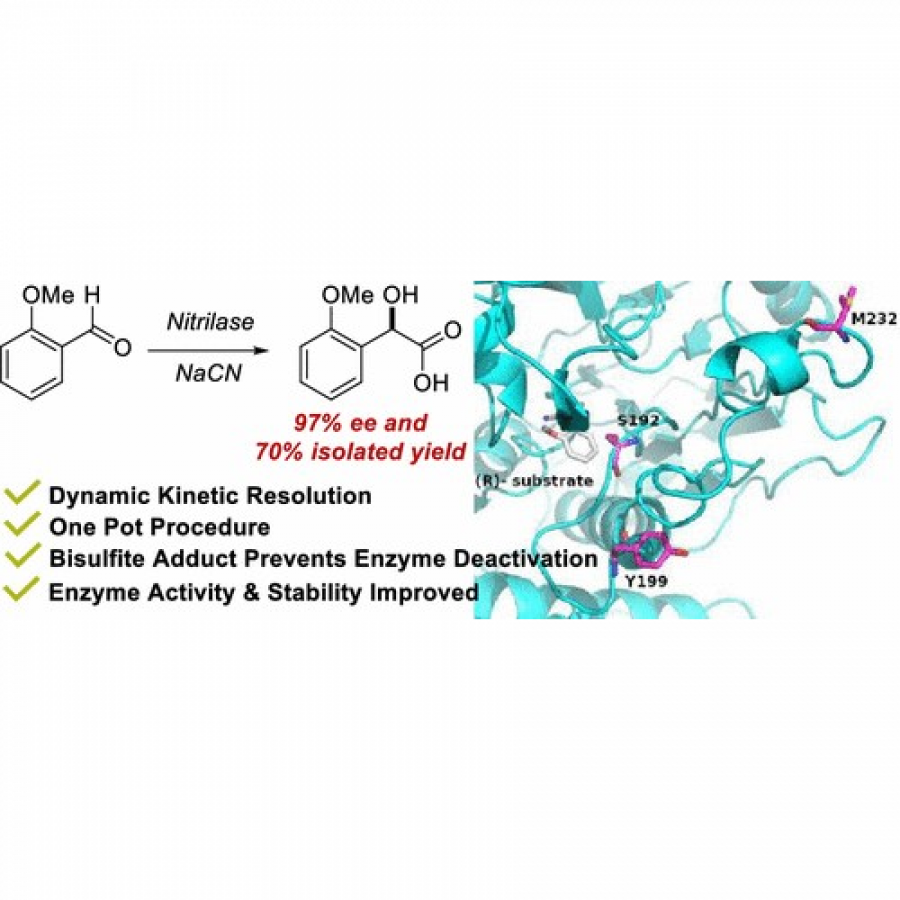 Nitrilase-mediated DKR for (R)-2-methoxymandelic acid |
#1359 | Unveiling the structural determinants for substrate recognition and catalysis of bacterial C-glycoside oxidases |
|
| Presenting author: | André TABORDA of ITQB NOVA |
| Corresponding author: | Lígia O. MARTINS of ITQB NOVA |
| Other authors: | Tomás FRAZÃO of ITQB NOVA Miguel RODRIGUES of ITQB NOVA Xavier FERNÁNDEZ-LUENGO of DEPARTMENT OF CHEMISTRY Ferran SANCHO of ZYMVOL BIOMODELING Carlos FRAZÃO of ITQB NOVA Rita VENTURA of ITQB NOVA Laura MASGRAU of DEPARTMENT OF CHEMISTRY Patricia BORGES of ITQB NOVA LIgia MARTINS of ITQB NOVA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Flavoenzymes / Carbohydrate oxidases / Auxiliary active family AA3 / Glycosides |
| Purpose: | C-glycosides are a class of natural products with biological activities but are recalcitrant to degradation, making their catabolic pathway a topic of interest. C-glycoside 3-oxidases (G3Oxs) are a newly identified group of bacterial flavo-oxidases from the glucose-methanol-choline (GMC) superfamily, which catalyze the oxidation of C-glycosides with the concomitant reduction of O2 to H2O2. Physiologically, this oxidative step is followed by the C-C cleavage through the action of a deglycosylation complex, which releases the sugar and the free aglycone. Despite the high sequence similarity between G3Oxs and the well-studied pyranose-2-oxidases (P2Oxs), which oxidize monosaccharides at the C2 position, these two groups belong to distinct phylogenetic clades. This work shows that a bacterial G3Ox, previously known as AsP2Ox [1], has a 4-order magnitude higher catalytic efficiency (kcat/Km) to the C-glycoside mangiferin compared to D-glucose, which is the preferred substrate of fungal P2Ox. NMR analysis shows that mangiferin is oxidized to 3-keto-mangiferin, and we have renamed AsP2Ox to PsG3Ox [2]. The crystal structure was solved, showing a higher similarity to previously characterized bacterial monomeric G3Oxs [3] than to fungal tetrameric P2Oxs. The combination of mutagenesis, X-ray studies, molecular simulations, and substrate dockings revealed (i) a substrate loop located inside the active site that modulates substrate regioselective recognition, and (ii) a new structural motif, an insertion segment, with a key role for the catalysis. The joint articulation of these two regions is essential for the access and proper accommodation of the substrates in the active site. This work advances our understanding of the structure-function relationships among this family of enzymes and the mechanisms underlying the different substrate specificity between P2Ox and G3Ox. Aknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia, Portugal, by project grants (PTDC/BII-BBF/29564/2017, EXPL/BIA-BQM/0473/2021 and 2022.02027.PTDC), and PhD fellowships for AT (2020.07928.BD), TF (2022.13872.BDANA) and MVR (2022.09426.BD). B-Ligzymes (GA 824017) from the European Union’s Horizon 2020 Research and Innovation Program is acknowledged for funding T.F. secondment at Zymvol. |
| References: | 1. Mendes, S., et al., Characterization of a bacterial pyranose 2-oxidase from Arthrobacter siccitolerans. Journal of Molecular Catalysis B: Enzymatic, 2016. 133: p. S34-S43. 2. Taborda, A., et al., Mechanistic Insights into Glycoside 3-Oxidases Involved in C-Glycoside Metabolism in Soil Microorganisms. PREPRINT (Version 1) available at Research Square 2023. 3. Kumano, T., et al., FAD-dependent C-glycoside-metabolizing enzymes in microorganisms: Screening, characterization, and crystal structure analysis. Proceedings of the National Academy of Sciences, 2021. 118(40): p. e2106580118. |
| Figures: | 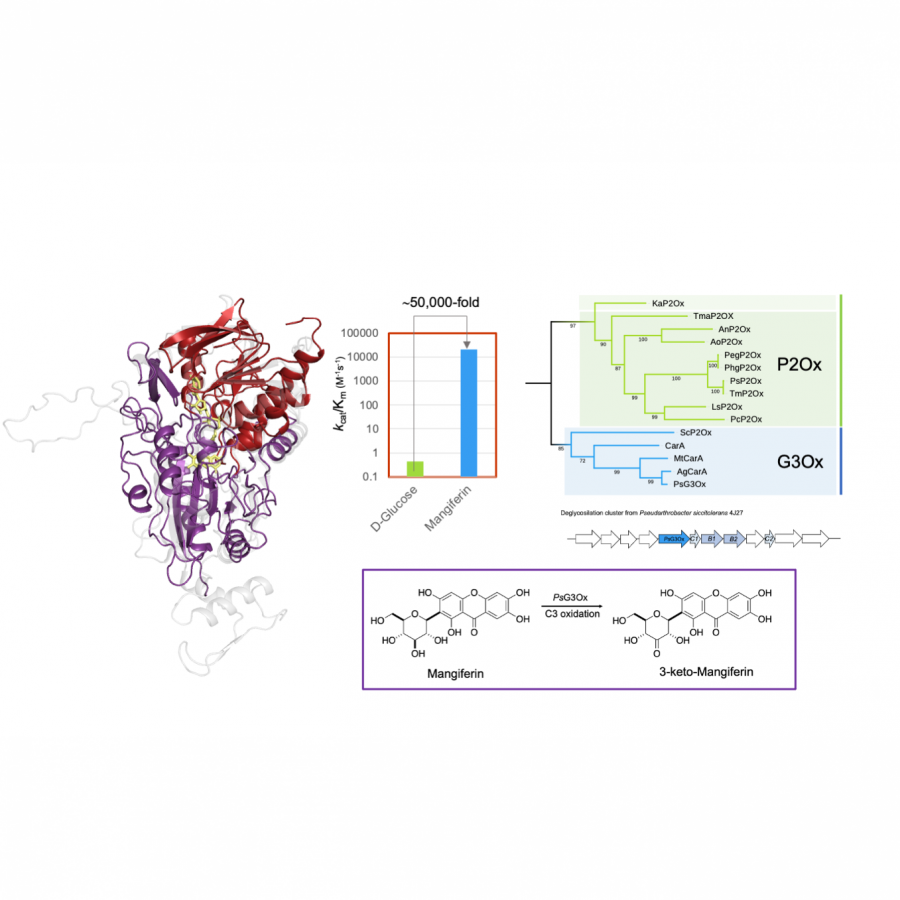 Glycoside 3-oxidases: New Bacterial Enzymes With Distinctive Phylogenetic, Functional And Structural Features .
|
#1361 | A biocatalytic cascade for the synthesis of ephedrine ana-logues |
|
| Presenting author: | Fabio SANGALLI of POLITECNICO DI MILANO |
| Other authors: | Noemi FRACHIOLLA of POLITECNICO DI MILANO Stefania PATTI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE “G. NATTA” (SCITEC) Erica FERRANDI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE “G. NATTA” (SCITEC) Daniela MONTI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE “G. NATTA” (SCITEC) Pierpaolo GIOVANNINI of UNIVERSITA` DI FERRARA Fabio PARMEGGIANI of POLITECNICO DI MILANO Elisabetta BRENNA of POLITECNICO DI MILANO Davide TESSARO of POLITECNICO DI MILANO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | norephedrine / dichloro-phenolindophenol oxidoreductase / / transaminase |
| Purpose: | Nor(pseudo)ephedrines are vicinal amino alcohols possessing a sympathomimetic function in the human body; due to the presence of two 1,2 chiral centers, they constitute valuable intermediates and chiral building blocks for the organic synthesis of active pharmaceutical ingredients[1]. Their conventional chemical asymmetric syntheses often involve long multi-step procedures, frequently with the use of expensive and harmful metal catalysts or organic precursors/intermediates, in which high yields and optical purities are very difficult to achieve overall. For this reason, biocatalysis can be a promising solution in the development of a stereoselective synthesis of analogues of (1S,2S)-norpseudoephedrine and (1S,2R)- norephedrine. A two-steps biocatalytic cascade was therefore designed and carried out, consisting of a benzoin-type condensation catalysed by the (S)-selective acetoin:dichloro- phenolindophenol oxidoreductase (Ao:DCPIP OR, EC 2.3.1.190)[2] and a reductive amination mediated by an amine transaminase ((S)- or (R)-ATA, EC 2.6.1.x)[3][4]. A multistep, non-selective chemical synthesis for nor(pseudo)ephedrines was also performed in order to obtain reference material for evaluating the performance of the biocatalysed reactions. The novel bienzymatic synthesis afforded the desired products with acceptable yields and good diastereo- and enantiomeric excesses, thus opening the way for a greener production of such important building blocks. |
| References: | [1] Sehl, T.; Maugeri, Z.; Rother, D. J. Mol. Catal. B Enzym. 2015, 114, 65-71, doi:10.1016/J.MOLCATB.2014.12.005 [2] Giovannini, P.P.; Lerin, L.A.; Müller, M.; Bernacchia, G.; Bastiani, M. De; Catani, M.; Di Carmine, G.; Massi, A. Adv. Synth. Catal. 2016, 358, 2767-2776, doi:10.1002/ADSC.201600359 [3] Ferrandi, E.E.; Spasic, J.; Djokic, L.; Vainshtein, Y.; Senthamaraikannan, R.; Vojnovic, S.; Grumaz, C.; Monti, D.; Nikodinovic-Runic, J. Catal. 2021, Vol. 11, Page 919 2021, 11, 919, doi:10.3390/CATAL11080919 [4] Ferrandi, E.E.; Previdi, A.; Bassanini, I.; Riva, S.; Peng, X.; Monti, D. Appl. Microbiol. Biotechnol. 2017, 101, 4963-4979, doi:10.1007/s00253-017-8228-2 |
| Figures: | 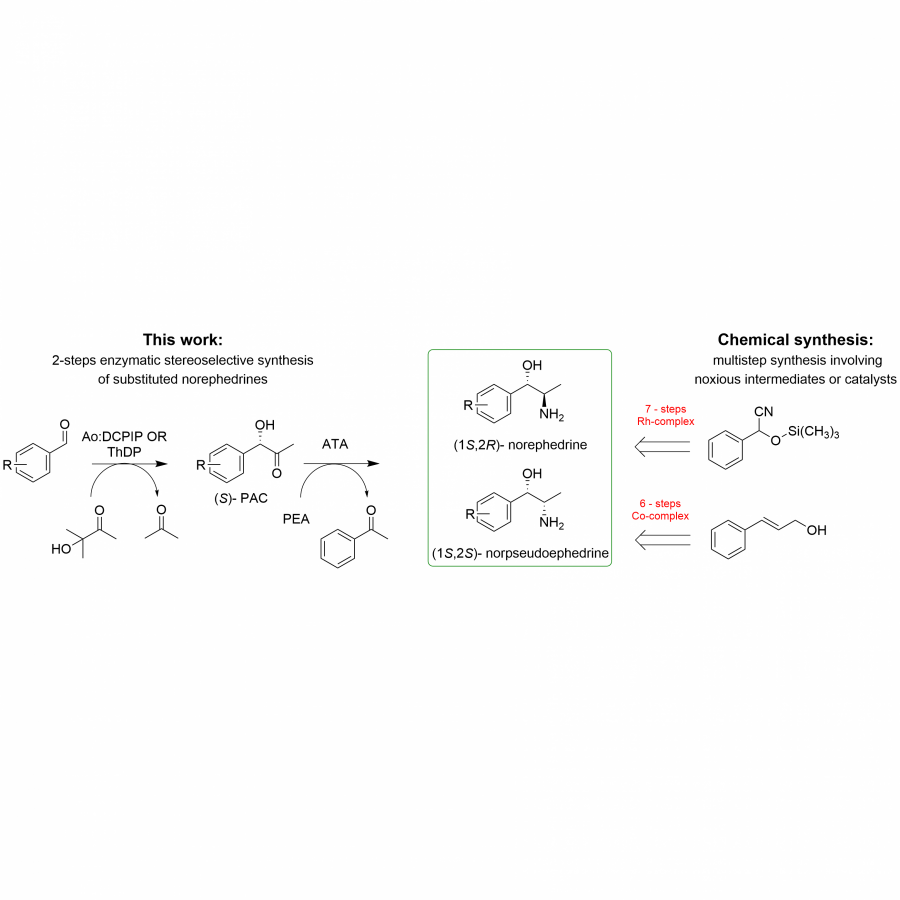 Abstarct Enzymatic two-step synthesis of norephedrine in comparison with literature chemical synthesis. |
#1364 | Upcycling of CO2 to ethyl formate in a hybrid process |
|
| Presenting author: | Nina KLOS of FORSCHUNGSZENTRUM JÜLICH, IBG-1 |
| Other authors: | Lars M. BLANK of INSTITUTE OF APPLIED MICROBIOLOGY Walter LEITNER of MAX PLANCK INSTITUTE FOR CHEMICAL ENERGY CONVERSION Dörte ROTHER of FORSCHUNGSZENTRUM IBG-1 BIOKATALYSE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | #biocatalysis / #hybrid process / #CARs / #CO2 usage |
| Purpose: | In recent years, greenhouse gas emissions have increased and therefore the integration of CO2 in the production of chemicals or fuels is beneficial. When using CO2 as a substrate, small molecules are synthesized in the first step, such as formic acid from an electrochemical conversion. The further valorization of small molecules with well established, traditional chemistry methods have often high energy demands and do not completely fit into the concept of green chemistry. A biocatalyic conversion using enzymes in various formulations provides an alternative. This bears the advantages that enzymes work e.g. under mild conditions and have high selectivity, which could increase the process sustainability and the value of gained products simultaneously [1]. Within the Fuel Science Center, an cluster of excellence at RWTH Aachen University, our colleagues have already developed a combined microbial and chemo-catalytical one-pot synthese process for the production of formic acid and bioethanol from renewable resources [2]. In this biphasic process an aqueous yeast fermentation is used for the production of bioethanol and CO2. The CO2 is captured together with H2 using a ruthenium catalyst in an organic phase above the fermentation broth in the same vessel to synthesize formic acid. This process should now be extended with a biocatalytic step to increase the product spectrum again in the same vessel. In detail we want to upgrade the product to ethyl formate, which is industrially applicable in the food industry [3]. The biocatalytic esterification reaction from ethanol and formic acid to ethyl formate should be performed by using carboxylic acid reductases (CARs) (E.C.1.2.1.30) (see Figure 1). Carboxylic acid reductases (CARs), work under aqueous conditions and are a highly potent class of enzymes identified in recent years. Recently, we established methods for the expression of high titers of actively und solubly produced CARs for the reduction of aromatic carboxylic acids to aldehydes [4,5]. Apart from the physiologically known reaction, they catalyzed the reduction from carboxylic acid to aldehydes, amidation, thioesterification and esterification reactions [6-8]. Pongpamorn et al. have already described esterification reactions using CARs (only A-domain) with aromatic carboxylic acids as substrates, however short-chain aliphatic acids, such as formic acid, have not yet been used [8]. We expect that the overall process concept will combine the advantages of chemocatalysis, microbial catalysis, and biocatalysis with the goal of developing a simple, sustainable process that can compete with existing fossil resource-based processes. |
| References: | [1] Alcántara, Biocatalysis as Key to Sustainable Industrial Chemistry, ChemSusChem, 15, 9, e202102709 (2022). [2] Guntermann, Bio-energy conversion with carbon capture and utilization (BECCU): integrated biomass fermentation and chemo-catalytic CO2 hydrogenation for bioethanol and formic acid co-production, Green chemistry, 23, 9680 - 9864 (2021). [3] Simpson, Effects of ethyl formate on fruit quality and target pest mortality for harvested strawberries, Postharvest Biology and Technology, 34, 3, 313-319, (2004). [4] Weber, Production of the Carboxylate Reductase from Nocardia otitidiscaviarum in a Soluble, Active Form for in vitro Applications, ChemBioChem, 22, 10, 1823 - 1832, (2021). [5] Weber, Multi-enzyme catalysed processes using purified and whole-cell biocatalysts towards a 1,3,4-substituted tetrahydroisoquinoline, RSC Adv., 13, 10097-10109 (2023). [6] Wood, Adenylation Activity of Carboxylic Acid Reductases Enables the Synthesis of Amides, Angew. Chem. Int. Ed., 56, 46, 14498 - 14501 (2017). [7] Schnepel, Thioester-mediated biocatalytic amide bond synthesis with in situ thiol recycling, Nature Catalysis, 6, 89-99 (2023). [8] Pongpamorn, Carboxylic Acid Reductase Can Catalyze Ester Synthesis in Aqueous Environments, Agric. Angew. Chem. Int. Ed., 60, 11, 5749 - 5753 (2021). |
| Figures: | 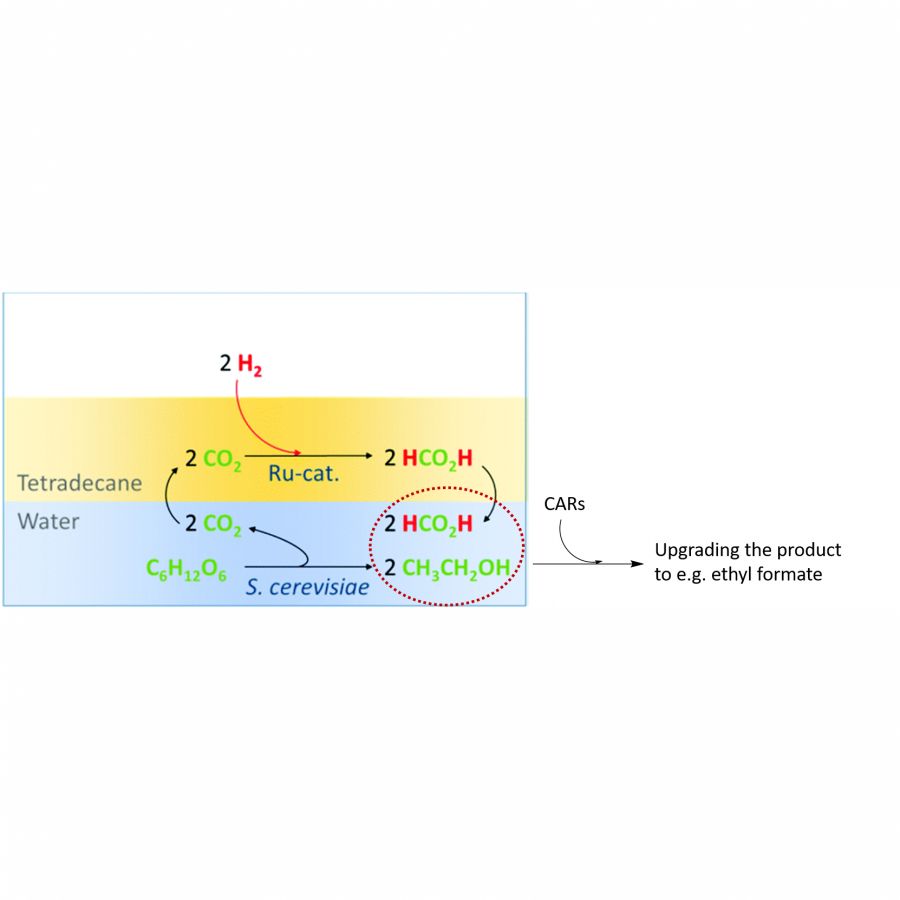 Figure 1. One-pot, one-step process from glucose and CO2 to formic acid and bioethanol [2]. By adding CARs the product spectrum can be increased. |
#1366 | Production and characterization of bacterial aminoacylases by an experimental/numerical approach for their implementation in a green N-acylation process |
|
| Presenting author: | CHEVALOT Isabelle of UNIVERSITE DE LORRAINE LRGP |
| Other authors: | Stéphane DELAUNAY of UNIVERSITE DE LORRAINE, CNRS, LRGP Laurence HOTEL of DYNAMIC, FACULTÉ DES SCIENCES ET TECHNOLOGIES Bertrand AIGLE of DYNAMIC, FACULTÉ DES SCIENCES ET TECHNOLOGIES Isabelle CHEVALOT of UNIVERSITE DE LORRAINE, CNRS, LRGP Xavier FRAMBOISIER of UNIVERSITE DE LORRAINE, CNRS, LRGP Yann GUIAVARC'H of UNIVERSITE DE LORRAINE, CNRS, LRGP Catherine HUMEAU of UNIVERSITE DE LORRAINE, CNRS, LRGP Laureline GENNESSEAUX of LRGP UL |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | N-acylation enzymatic process / aminoacylase / production of recombinant enzymes / molecular modelling |
| Purpose: | The N-acylation reaction leads to the addition of an acyl group to the amine function of a molecule which can be an amino acid or a peptide. These acylated derivatives have a high application potential because of their technofunctional properties, among others emulsifying and foaming, or their biological activities. On an industrial scale, they are produced by the Schotten-Baumann reaction which is highly efficient but non-selective and polluting. These acylated derivatives can also be produced by an enzymatic way, in particular using aminoacylases. Aminoacylase activities were discovered in the culture supernatant of Streptomyces ambofaciens [1]. Four genes of S. ambofaciens were identified, presenting a very high percentage of homology with the gene sequences of aminoacylases identified and characterized in Streptomyces mobaraensis by Koreishi's team [2]. The construction of deletion mutants of S. ambofaciens confirmed the activity of the enzymes encoded by the four genes [3]. The characterization of these enzymes began using culture extracts [4] and revealed the particular interest of SamAA that catalyzes the acylation of the α amine function of lysine selectively, which is rare in the world of enzymes. First attempts to produce this enzyme by heterologous expression showed a significant production under the form of inclusion bodies. To workaround this, a strategy based on the modification/combination of various experimental conditions was adopted for the production of SamAA. Recombinant SamAA was produced by the Escherichia coli BL21 Gold (DE3) strain with an optimized gene, for 24 hours on an LB medium supplemented or not with cobalt. Induction was done with the addition of IPTG (0.1 mM) at 0.6 or 0.4 of Do(600 nm), respectively in the absence or in the presence of cobalt in the culture medium. SDS-PAGE analyzes did not reveal any visible band of over-expression at the expected size of SamAA in the soluble fractions obtained after lysis with a French press. The presence of the recombinant protein is going to be confirmed by Western blot analysis. Unpurified concentrated soluble fractions were used for a synthesis of N-α-lauryol-L-lysine at 45°C and pH8 in 48h, which showed an apparent specific activity of 710 mg/L of product/ 100mg of protein/ L of crude extract with cobalt in the culture medium while the activity is zero without cobalt in the culture medium. Purification tests on IMAC column are in progress. Along with this experimental study, a numerical approach was implemented to gain knowledge of the 3D structure and the catalytic mechanism of SamAA that remain unknow until now. Templates that are defined as proteins having high percentages of identity and similarity with the target sequence and whose structures are known were identified using Blastp and Swiss Model’s « Search for templates » tools. The structures of PDB entries 1Q7L, 5VO3, 4PPZ, 4RUH and 7LGP were identified as SamAA templates with identity percentages between 17 and 33%. Basing on these templates, 3D homology models were built using Modeller from Discovery Studio suite, Swiss Model [5] and AlphaFold2 [6] and compared. The best 3D structure of SamAA according to energy calculations and presence of conserved residues was shown to be that obtained by ColabFold (Figure 1). Based on the literature survey relative to the templates [7], metal ions were added and structural characteristics were determined : SamAA would be a homodimeric protein with two zinc atoms per subunit. The His88, Asp120, Glu155, Glu182 et His416 residues would be responsible for the binding of divalent ions. The potential catalytic residues are Asp90 and Glu154: the latter would play the acid/base residue essential to the catalytic mechanism. Energy minimization is applied to the retained models. Docking simulations are in progress aiming to determine the key residues and interactions that are responsible for the selectivity of SamAA towards the lysine α amino group. |
| References: | [1] Ferrari F. Thesis Université de Lorraine 2014 [2] Koreishi M., Nakatani Y., Ooi M., Imanaka H., Imamura K. et Nakanishi K. Biosci. Boitechnol. Biochem. 2009, 73 (9), 1940-1947 [3] Dettori L., Ferrari F., Framboisier X., Paris C., Guiavarc’h Y., Hôtel L., Aymes A., Leblond P., Humeau C., Kapel R., Chevalot I., Aigle B. et Delaunay S. Eng. Life Sci. 2018, 18, 589-599 [4] Bourkaib M.C., Delaunay S., Framboisier X., Hôtel L., Aigle B., Humeau C., Guiavarc’h Y. et Chevalot I. Enzyme Microb. Tech. 2020, 137, 109536 [5] Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F.T., de Beer T.A.P., Rempfer C., Bordoli L., Lepore R. et Schwede T. Nucleic Ancids Res. 2018, 46 (W1), W216-W303 [6] Mirdita M., Schütze K., Moriwaki Y., Heo L., Ovchinnikov S. et Steinegger M. Nat. Methods, 2022 [7] Nocek B.P., Gillner D.M., Fan Y., Holz R.C. et Joachimiak A, J. Mol. Biol. 2010, 397, 617-626 |
| Figures: |  3D structure of SamAA Structure of SamAA obtained by ColabFold after adding metal ions |
#1368 | EXPLORING THE EVOLUTIONARY LANDSCAPE OF PSGALOX, A BACTERIAL GALACTOSE OXIDASE |
|
| Presenting author: | Tiago LOPES of ITQB NOVA |
| Corresponding author: | Lígia MARTINS of ITQB NOVA |
| Other authors: | André TABORDA of ITQB NOVA Carolina DIAS of ITQB NOVA João COSTA of ITQB NOVA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Copper Radical Oxidases / Enzyme engineering / Directed evolution / Galactose oxidase |
| Purpose: | Galactose oxidases (GalOxs) are highly attractive enzymes because they efficiently combine oxygen with high specificity and regioselectivity toward carbohydrate substrates.[1] These monomeric metalloenzymes have a copper ion as a cofactor and are secreted by various filamentous fungi, with GalOx from Fusarium graminearum being the best-studied representative. These enzymes’ potential include biotechnological applications in small molecule synthesis, oxygen removal, biosensors, and cell surface glycoprotein modification.[2] While galactose oxidases from fungal origins have been vastly studied, there is a growing interest in GalOxs from bacterial origin due to its ease of production and engineering. PsGalOx, an enzyme from Pseudoarthrobacter siccitolerans, is the only known bacterial galactose oxidase. This work unravels the properties of PsGalOx while tailoring its properties through enzyme engineering approaches. The research seeks to improve the understanding of PsGalOx's catalytic and stability mechanisms by examining the structure-to-function and characterizing the wild-type and variants. To accomplish this, directed evolution was applied recurring to random mutagenesis through error-prone PCR (epPCR). The workflow was developed recurring to two high-throughput screening methodologies, 'activity-on-plate' and 96-well plate liquid screening, which were successfully optimized and validated. Additionally, it was also explored the improvement of PsGalOx‘s solubility and production yields. Multiple rounds of engineering led to the identification of the variant 11C7 which shows close to 20-fold higher protein production yields as compared with the wild-type and 10-fold increased catalytic efficiency (kcat/Km) for D-galactose. We are currently continuing with the evolution of this enzyme to further enhance its catalytic activity and exploring further the effects of directed evolution on its solubility. These findings will contribute to a better understanding of GalOx from different organisms, revealing novel or improved properties to be explored in various fields |
| References: | 1. Savino, S. and Fraaije, M. W. (2021) The vast repertoire of carbohydrate oxidases: An overview. Biotechnol Adv. 51 2. Pedersen, A. T., Birmingham, W. R., Rehn, G., Charnock, S. J., Turner, N. J. and Woodley, J. M. (2015) Process Requirements of Galactose Oxidase Catalyzed Oxidation of Alcohols. Org Process Res Dev. 19, 1580-1589 |
#1372 | ALKALI- AND HYPERTHERMO-STABLE ENGINEERED LACCASE FOR LIGNIN-RELATED PHENOLICS. |
|
| Presenting author: | Vania BRISSOS of ITQB NOVA |
| Corresponding author: | Ligia O. MARTINS of ITQB NOVA |
| Other authors: | Patricia BORGES of ITQB NOVA Ricardo ESTEVINHO of ITQB NOVA Magdalena LEJMEL of ITQB NOVA Maria Paula ROBALO of CENTRO DE QUÍMICA ESTRUTURAL, IST, UL Ligia O. MARTINS of ITQB NOVA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Hyperthermophiles / Directed evolution / Enzyme catalysis / Lignin Valorization |
| Purpose: | Laccases catalyze the oxidation of various substrates, aromatic amines, phenols, and metal ions, coupled with the reduction of molecular oxygen to water. Laccases have a remarkable redox ability and are in the front line for establishing enzymatic bioconversions of aromatics [1]. We have previously reported a laboratory evolution approach that led to the improved efficiency of the Aquifex aeolicus McoA hyperthermophile bacterial metallo-oxidase for the typical laccase substrate ABTS (2,2’-azinobis-(3-ethyl-benzothiazoline-6-sulfonic acid)) while showing an enhanced kinetic and thermodynamic thermostability [2]. In this study, we have used a combination of directed evolution (DNA-shuffling and error-prone PCR followed by high-throughput screening) and rational design to evolve the McoA 2B3 variant for 2,6-dimethoxyphenol (syringol), a lignin-related phenolic substrate. These experiments led to the identification of the hit variant, McoA 15E6-ΔMetloop, carrying seven mutations, and showing a melting temperature 15ºC higher than that of wild-type (Tm ~105ºC), and 400-fold higher kcat/Km for syringol when compared to 2B3 variant. A comprehensive biochemical analysis of hit variants from the in vitro evolution enabled to differentiate among beneficial, neutral and deleterious mutations, showing the path to construct a variant, PheLac, carrying only four mutations (three of them near the Cu centers) and with a higher enzymatic activity. We have investigated the catalytic potential of this variant using a set of representative lignin-related phenolic compounds, including syringyl, guaiacyl, and hydroxybenzene derivatives at pH 8. The results show that this evolved variant has similar or higher turnover numbers than the model CotA-laccase, with the advantage of being significantly more thermostable. The scale-up reaction using the best substrate for McoA hit, sinapyl alcohol, was performed, and syringaresinol, a product with medicinal value and as a building block of biopolymers, was identified using NMR. So far, no studies report this product's synthesis using bacterial laccases. Enzymatic valorization of lignin monomers from phenolic platform chemicals is envisaged as one of the potential environmentally friendly breakthrough applications to valorize lignin bio-wastes. Acknowledgments: This work was supported by the Fundação para a Ciência e Tecnologia, Portugal, by project grants (PTDC/BII-BBF/29564/2017, EXPL/BIA-BQM/0473/2021 and 2022.02027.PTDC). B-Ligzymes (GA 824017) from the European Union’s Horizon 2020 Research and Innovation Program is also acknowledged for funding a secondment of P. Borges to Zymvol, Barcelona, SP. |
| References: | 1-Sousa, A. C., Martins, L. O. and Robalo, M. P. (2021) Laccases: Versatile Biocatalysts for the Synthesis of Heterocyclic Cores. Molecules. 26, 1-26 2-Brissos, V., Ferreira, M., Grass, G. and Martins, L. O. (2015) Turning a hyperthermostable metallo-oxidase into a laccase by directed evolution. ACS Catal. 5, 4932-4941 |
#1375 | Mirror, mirror on the wall: Application of an N-methyltransferase for preparative scale enzymatic kinetic resolution |
|
| Presenting author: | Benjamin CHAPPLE of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF |
| Other authors: | Pascal SCHNEIDER of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF Mona HAASE of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF Jörg PIETRUSZKA of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / methylation / kinetic resolution / pyrroloindolines |
| Purpose: | The attachment of a single methyl group can increase the potency of a pharmaceutical by up to three orders of magnitude – primarily due to conformational “gridlocking” – making the “magic methyl” effect intensely sought after. However, chemical methylation often lacks the selectivity needed to generate compounds of interest, underlining the need for new methylation methods. [1,2] Over the past years, methyltransferases have been making their way into the organic chemist’s toolbox, offering high selectivity under benign reaction conditions. [3–7] Their practical use has been, e.g., exemplified in the enantiopure generation of various physostigmine analogues, [8,9] which bear the pyrroloindoline scaffold, a motif found in numerous naturally occurring alkaloids with similarly numerous biological activities. [10,11] Being able to access both enantiomers of pyrroloindolines would be of additional use, as some enantiomeric drug pairs do not only differ in efficacy but may also have different targets and mechanisms of action. [12] In this work, we have characterised and applied an N-methyltransferase for the first time in a kinetic resolution of pyrroloindolines, in accord with the implementation of a supply system for the cofactor S-adenosyl methionine. Using two easily obtainable catalyst formulations, the enzymatic reaction can be performed on a preparative scale. The system can potentially be used for the selective chemoenzymatic access to the enantiomeric pair (+)-Posiphen and (-)-Phenserine, two synthetic pyrroloindolines of medical importance [13,14] and with separate biological targets and distinct mechanisms of action. [12] |
| References: | [1] S. Sun, J. Fu, Bioorg. Med. Chem. Lett. 2018, 28, 3283-3289. [2] H. Schönherr, T. Cernak, Angew. Chem. Int. Ed. 2013, 52, 12256-12267. [3] C. Liao, F. P. Seebeck, Nat. Catal. 2019, 2, 696-701. [4] S. Mordhorst, J. Siegrist, M. Müller, M. Richter, J. N. Andexer, Angew. Chem. Int. Ed. 2017, 56, 4037-4041. [5] B. J. C. Law, A.-W. Struck, M. R. Bennett, B. Wilkinson, J. Micklefield, Chem. Sci. 2015, 6, 2885-2892. [6] L. L. Bengel, B. Aberle, A. Egler‐Kemmerer, S. Kienzle, B. Hauer, S. C. Hammer, Angew. Chem. Int. Ed. 2021, 60, 5554-5560. [7] B. Aberle, D. Kowalczyk, S. Massini, A.-N. Egler-Kemmerer, S. Gergel, S. Hammer, B. Hauer, Angew. Chem. Int. Ed. 2023, e202301601. [8] P. Schneider, B. Henßen, B. Paschold, B. P. Chapple, M. Schatton, F. P. Seebeck, T. Classen, J. Pietruszka, Angew. Chem. Int. Ed. 2021, 60, 23412-23418. [9] D. A. Amariei, N. Pozhydaieva, B. David, P. Schneider, T. Classen, H. Gohlke, O. H. Weiergräber, J. Pietruszka, ACS Catal. 2022, 12, 14130-14139. [10] F. R. de Sá Alves, E. J. Barreiro, C. A. M. Fraga, Mini Rev. Med. Chem. 2009, 9, 782-793. [11] C. Sun, W. Tian, Z. Lin, X. Qu, Nat. Prod. Rep. 2022, 39, 1721-1765. [12] S. Mikkilineni, I. Cantuti-Castelvetri, C. M. Cahill, A. Balliedier, N. H. Greig, J. T. Rogers, Park. Dis. 2012, 2012, 142372. [13] R. E. Becker, N. H. Greig, L. S. Schneider, C. Ballard, D. Aarsland, D. K. Lahiri, D. Flanagan, R. Govindarajan, M. Sano, D. Kapogiannis, L. Ferrucci, Curr. Alzheimer Res. 2018, 15, 883-891. [14] A. F. Teich, E. Sharma, E. Barnwell, H. Zhang, A. Staniszewski, T. Utsuki, V. Padmaraju, C. Mazell, A. Tzekou, K. Sambamurti, O. Arancio, M. L. Maccecchini, Alzheimer's Dement.: Transl. 2018, 4, 37-45. |
| Figures: | 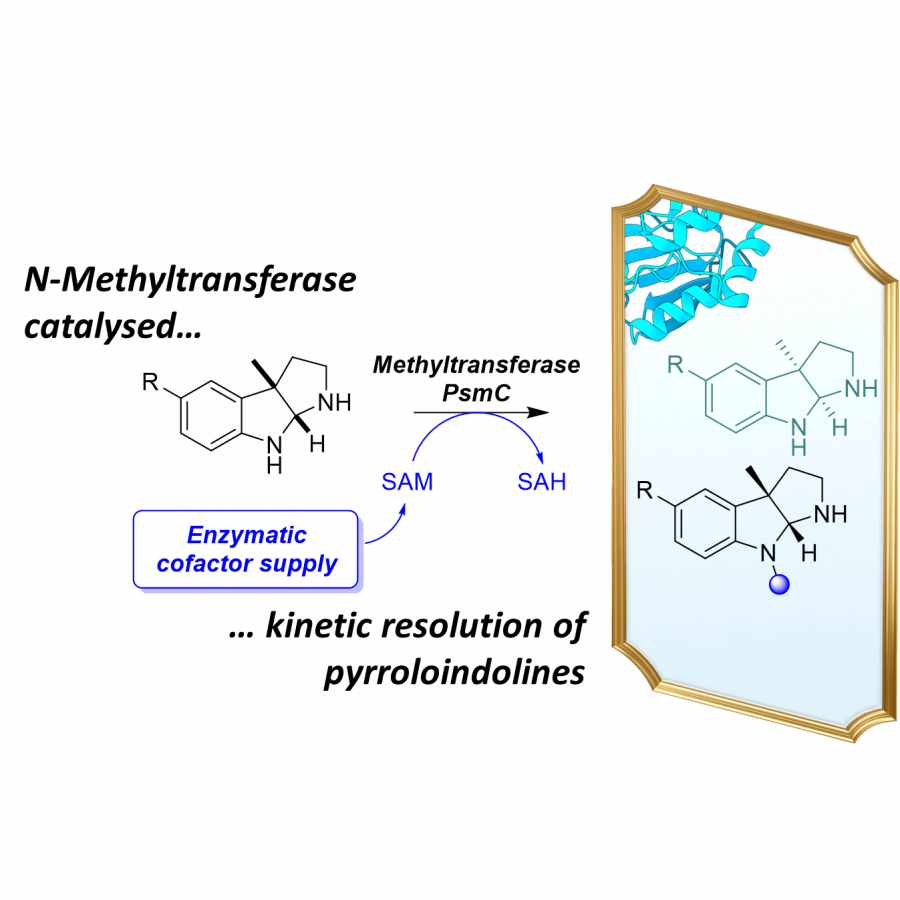 Enzymatic kinetic resolution by PsmC The N-methyltransferase PsmC can be used for preparative scale enzymatic kinetic resolution of pyrroloindoles. SAM: S-adenosyl methionine; SAH: S-adenosyl homocysteine. |
#1378 | Application of linoleate 13-hydratase in stereoselective hydration of 18-carbon unsaturated fatty acids |
|
| Presenting author: | Joanna GACH of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Stefano SERRA of C.N.R. SCITEC ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE Stefano MARZORATI of C.N.R. SCITEC ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE Teresa OLEJNICZAK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | fatty acid hydratase / substrate specifity / stereoselectivity / heterologous expression |
| Purpose: | Fatty acid hydratases are one of the most common enzymes able to catalyze the water addition to the non-activated double bonds. Their physiological function in the microorganisms presumably may be linked with the process of detoxification of fatty acids or alteration in host-microbe interactions [1]. From the industrial point of view, the resulting products, namely the hydroxy fatty acids, find use as starting materials for production of signaling molecules, biopolymers, emulsifiers, stabilizers and aroma compounds [2-5]. In this work, we investigated the biocatalytic potential of the linoleate 13-hydratase from Lactobacillus acidophilus, which is able to catalyze the addition of water to the C-12 double bond of linoleic acid. For this purpose, we overexpressed the latter enzyme using Escherichia coli BL21(DE3) as a heterologous host. Then, the obtained biocatalyst was purified in order to study its catalytic properties. More specifically, we evaluated both the activity and the regio- and stereoselectivity of the hydration reaction using different C18 unsaturated fatty acids as substrates. Beside linoleic acid, which is the natural substrate of 13-hydratase from L. acidophilus, we have tested the substrate specificity using other C18 Δ-12 unsaturated fatty acids. In particular, we employed the synthetically derived C18:1 Δ-12 fatty acid and two isomeric C18:3 polyunsaturated fatty acids, namely Δ-9,12,15 (linolenic acid) and C18:3 Δ-6,9,12 (γ-linolenic acid). The hydration of C18:1 Δ-9 (oleic acid) was also checked as negative control. The obtained hydroxyl derivatives were extracted from the biotransformation mixture, purified using column chromatography and characterized by nuclear magnetic resonance (1H NMR and 13C NMR). The gas chromatography analysis coupled with mass spectrometry (GC-MS) allow determining the regioselectivity of the hydration reaction. The enantiomeric excess of the products was measured by NMR analysis after derivatization of the hydroxy-fatty acids to their corresponding (S)-O-acetylmandelate esters. Overall, we established that the aforementioned enzyme catalyzes the hydration of the unsaturated fatty acids with complete regioselectivity with exclusive formation of the 13-hydroxy derivatives in very high enantioselectivity (ee >95%), regardless of the kind of substrate used. This substrate flexibility points to the potential use of 13-hydratase from Lactobacillus acidophilus for the preparative synthesis of different 13-hydroxy-acid derivatives. |
| References: | [1] Löwe, J.; Gröger, H., Catalysts 2020, 10, 287. [2] Kang, W.-R.; Park, C.-S.; Shin, K.-C. et al., JAOCS 2016, 93, 649-655. [3] Oh, H.-J.; Kim, S.-U.; Song, J.-W. et al., Adv. Synth. Catal. 2015, 357, 408-416. [4] Park, J.-Y.; Lee, S.-H.; Kim, K.-R. et al., J. Biotechnol. 2015, 208, 1-10. [5] Serra, S.; De Simeis, D.; Marzorati, S.; Valentino, M., Catalysts 2021, 11, 1051. |
#1379 | Development of Sensitive, Specific, Isothermal One-pot Fluorescence RNA Detection Assay Based on Cell-free Synthetic Biology |
|
| Presenting author: | Chang Ha WOO of SCHOOL OF INTERDISCIPLINARY BIOSCIENCE & BIOENGINEERING(I-BIO)/ POSTECH |
| Corresponding author: | Jeong Wook LEE of POSTECH |
| Other authors: | Da-ae GWON of CHEMICAL ENGINEERING / POSTECH Donghyeon KIM of CHEMICAL ENGINEERING / POSTECH |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cell-free synthetic biology / Molecular Diagnostics / Isothermal, One-pot RNA detection / Point-Of-Care test |
| Purpose: | The COVID-19 pandemic has highlighted the crucial necessity for efficient and accurate diagnostic tools to identify infectious diseases caused by RNA viruses. The epidemic has reinforced the significance of rapid and sensitive detection of viral RNA in order to detect and control outbreaks. Consequently, there is an urgent need for the advancement and implementation of new RNA detection methods. Current gold-standard RNA detection technique as known as reverse transcription-polymerase chain reaction (RT-PCR) has played a critical role in COVID-19 diagnosis. However, it necessitates specialized equipment and highly skilled technicians, posing challenges to its usage in resource-constrained settings. Furthermore, RT-PCR technique often entails lengthy sample preparation, amplification, and analysis, leading to delays in patient diagnosis and treatment. To address these issues, we developed a cell-free, one-step assay for detecting pathogen-derived RNAs based on fluorescence. The assay uses a ligation-dependent one-pot isothermal reaction cascade. It consists of two simple enzymatic reactions: single-stranded DNA ligation reaction by SplintR ligase and subsequent transcription reaction by T7 RNA polymerase. The ligation of the adjacent single-stranded DNA probes, namely the promoter probe and the reporter probe, occurs upon hybridization with a complementary RNA, which is used as a splint. Following the ligation process, the resulting product contains a T7 promoter sequence. It is recognized by T7 RNA polymerase, leading to the transcription reaction. The transcript forms an RNA aptamer that binds a cognate fluorogenic dye and emits fluorescence only when target RNA exists in a sample. Our assay was able to detect methicillin-resistant Staphylococcus aureus (MRSA) RNA in 30 min with a limit of detection of 0.1 aM (6 copies RNA per reaction). Merely by altering the hybridization regions of the probes, we were able to employ this assay to detect a diverse array of pathogens, comprising Vibrio vulnificus, Escherichia coli O157:H7, Middle East respiratory syndrome-related coronavirus (MERS-CoV), Influenza A viruses and severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2). Our research indicates that our assay has the potential for detecting various pathogen RNA markers by simply redesigning the probe sequences. This can be easily achieved using readily available web-based software such as primer-blast and NUPACK, requiring minimal computational effort. Through a clever use of the straightforward probe design, we pushed the boundaries of our assay’s capacity to facilitate the detection of two separate target RNAs simultaneously within a one-pot, single reaction. This is crucial in accurately mitigating the risks of false positives and false negatives, improving diagnostic precision. By leveraging the high specificity of our probes and unique spectral properties of light-up RNA aptamers, we substantiated the effectiveness of dual detection of multiple regions within the RdRp gene of SARS-CoV-2. Finally, we culminated in the successful validation of our assay as a reliable tool for detecting SARS-CoV-2 virus from clinical samples, underscoring its potential as a valuable asset in the arsenal of diagnostics assay. We were able to display a remarkable diagnostic performance with 95% of sensitivity and 100% specificity achieved within a mere hour without the requirement of RNA extraction. In conclusion, the presented diagnostic assay has the potential to revolutionize pathogen detection. The future automation of probe design will be essential for the rapid development of assays for emerging pathogens. Additionally, expanding its utility into portable, field-deployable devices that offer isothermal incubation and fluorescence measurement could be a significant step toward creating simple and transportable paper-based or lateral-flow-type tests. |
| References: | 1. Jin. J et al, Nucleic Acids Res. 2016, 44(13), e116 2. Ying, Z.-M et al, Chem. Commu. 2018, 54, 3010-3013 3. Woo et al, Nat. Biomed. Eng. 202, 4, 1168-1179 |
| Figures: | 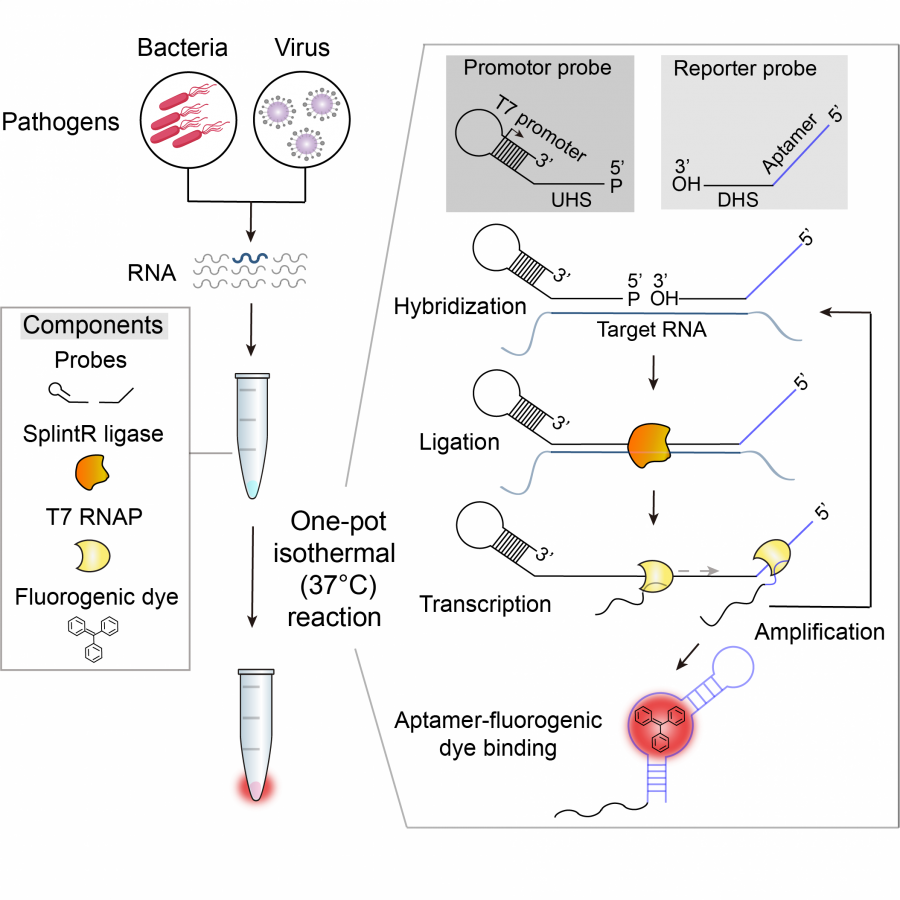 Graphical scheme of SENSR SENSR, a one-pot isothermal reaction cascade for the RNA detection. 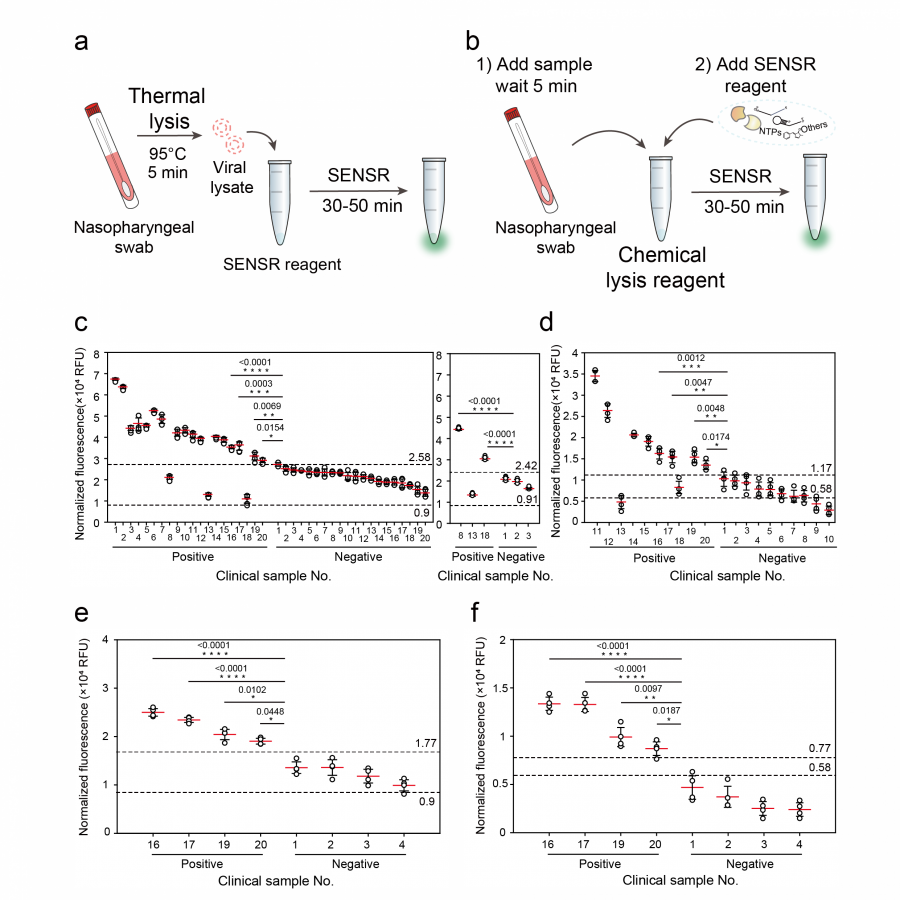 Schematics of SARS-CoV-2 detection from clinical samples via SENSR Clinical samples in UTM were treated by either thermal (a, c and e) or chemical lysis (b, d and f) and mixed directly with the SENSR mixture. c,d, SARS-CoV-2 detection from clinical samples. e,f, Detection of SARS-CoV-2 by 30-min SENSR reaction. |
#1380 | Whole-cell catalysis in green and blue: Synechococcus PCC11901 as a new workhorse? |
|
| Presenting author: | Thomas ROHR of TU WIEN |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cyanobacteria / Whole-cell catalysis / Oxygenating Enzymes / |
| Purpose: | BIOCATALYSIS IN GREEN AND BLUE: SYNECHOCOCCUS PCC11901 AS A NEW WORKHORSE? Thomas Rohr1, Fanny Kormout1, Ilka M. Axmann2, Florian Rudroff1 1Getreidemarkt 9 OC-163, Vienna Austria. Institute of Applied Synthetic Chemistry, TU Wien – thomas.rohr@tuwien.ac.at; 2Universitätsstrasse 1 Gebäude 22.06, Düsseldorf, Germany Institute of Synthetic Microbiology, Heinrich-Heine-Universität -Ilka.axmann@hhu.de;1 Can we exploit Photosynthesis for Whole-cell-catalysis? In oxygenic, photoautotrophic organisms, light energy provides redox-energy and oxygen accumulates. An ideal “reaction-vessel” for oxygenating enzyme-catalyzed reactions. Fast co-factor recycling without the need for sacrificial co-substrates and thus for whole-cell catalysis1. Cyanobacteria, ancient photoautotrophic prokaryotes, have gained interest by the biotechnology community for good reasons. They grow faster than other photosynthetic active organisms and their comparatively simple genetic built render them accessible to genetic engineering. Despite their advantages, Cyanobacteria lag behind more commonly commonly used, heterotrophic microbes, as they grow more slowly and less densely and reliable high expression of enzymes is often difficult to achieve2. In this project, we wanted establish whole-cell catalysis in a cyanobacterial strain, that could overcome some of the current drawbacks of cyanobacteria by implementing an apt two-step enzymatic reaction. We planned to test a two-step enzymatic reaction from ferulic acid to vanillin in the newly discovered Synechococcus PCC 11091. This strain is one of the fastest cyanobacteria in terms of growth and reach the highest cell density ever measured in cyanobacteria grown in a shake flask3. The enzymes of interest turn ferulic acid into 4-vinylguaicol and release one molecule of CO2 and in the second step react with O2 to Vanillin, releasing formaldehyde. Our hypothesis was, that by removing CO2 and providing O2, the photosynthetic metabolism of Synechococcus PCC11901 boosts the reaction efficiency4. While the first enzyme of this mini-cascade (Phenolic Acid Decarboxylase (PAD)) was stably inserted into the hosts genome and showed high reaction rates, the second enzyme (Aromatic dioxygenase (ADO)) could not be stably inserted into the hosts genome. We also tested a newly discovered enzyme, called mapADO (not published). This enzyme showed a higher activity and better expression in E. coli. but continued to evade our attempts to be inserted into the cyanobacterium’s genome. As this idea approached a dead-end, we tried a “work-around”. While E. coli expresses our enzyme of interest, we added Cyanobacteria to the reaction, in hope to improve yield by shifting the reaction equilibrium. In addition to substrate and product concentration, we also monitor oxygen content and chlorophyll content. With this, we hope to find the best conditions for our reactions to occur and prove the additional benefits, Synechococcus PCC11901 has on whole cell catalysis. |
| Figures: | 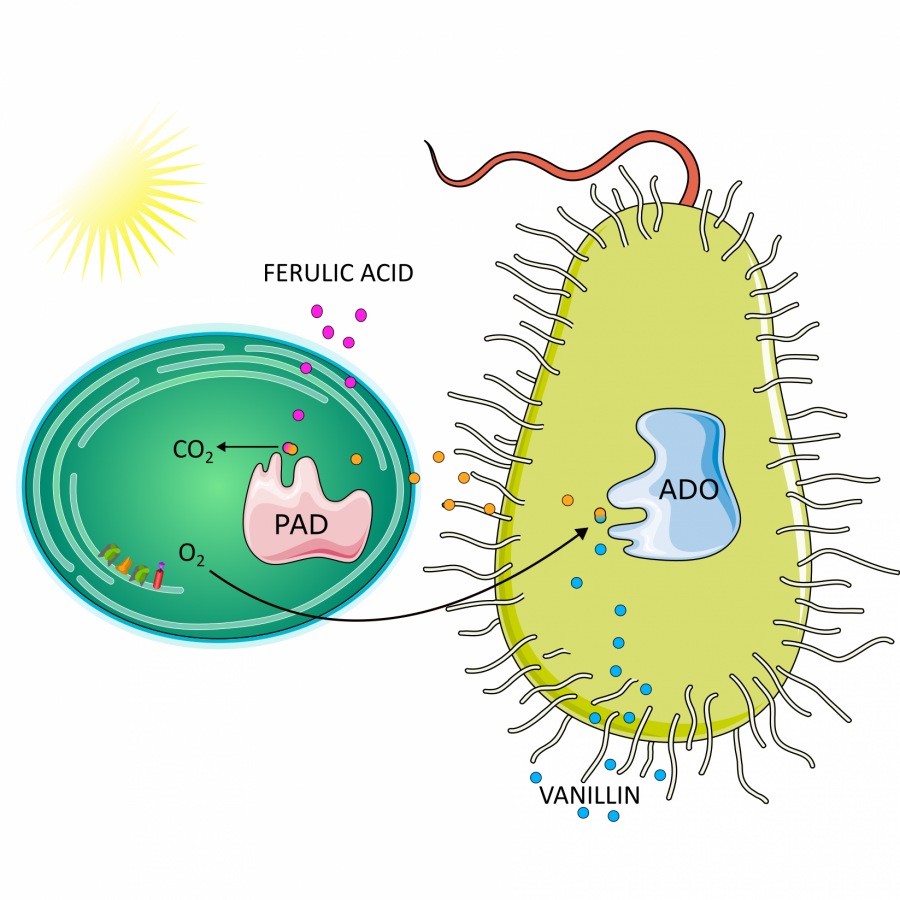 Schematic of two-whole-cell catalysis E. coli (right) expresses the two necessary enzymes (PAD and ADO) to turn Ferulic acid into 4-Vinylguaicol and subsequently into Vanillin. Synechococcus elongatus PCC11901 (left) consumes the produced CO2 and produced CO2 and produces O2 to shift the reac |
#1381 | Convergent biocatalytic mediated synthesis of siRNA |
|
| Presenting author: | Stephanie PAUL of ALMAC SCIENCES |
| Corresponding author: | Jill CASWELL of ALMAC SCIENCES |
| Other authors: | Darren GRAY of ALMAC SCIENCES Jill CASWELL of ALMAC SCIENCES Joshua BROOKS of ALNYLAM PHARMACEUTICALS INC Wenjie YE of ALNYLAM PHARMACEUTICALS INC Thomas S. MOODY of ALNYLAM PHARMACEUTICALS INC Roumen RADINOV of ALNYLAM PHARMACEUTICALS INC Lubomir NECHEV of ALNYLAM PHARMACEUTICALS INC |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | RNA ligase / oligonucleotide synthesis / biocatalysis / process development |
| Purpose: | The application of biocatalysis in pharmaceutical development continues to grow with the strong technology base being applied to develop enzymatic methods for oligonucleotide synthesis resulting in improved yields and purity profiles compared to traditional methods. Biocatalytic approaches are alleviating the pressures on existing solid phase capacity and resulting in more convergent syntheses. Herein we report a convergent biocatalytic synthesis strategy for an Alnylam model siRNA. The siRNA chemical structure includes several of the unnatural modifications and conjugations typical of siRNA drug substances. Using Almac’s 3-2-3-2 hybrid RNA ligase enzyme strategy that sequentially ligates short oligonucleotide fragments (blockmers), the target siRNA was produced to high purity at 1 mM concentration. Additional strategies were investigated including the use of polynucleotide kinase phosphorylation and the use of crude blockmer starting materials without chromatographic purification1. These findings highlight a path towards a convergent synthesis of siRNAs for large scale manufacture marrying both enzymatic liquid and classical solid phase synthesis. |
| References: | Stephanie Paul, Darren Gray, Jill Caswell, Joshua Brooks, Wenjie Ye, Thomas S. Moody, Roumen Radinov, and Lubomir Nechev ACS Chemical Biology Article ASAP DOI:10.1021/acschembio.3c00071 |
| Figures: | 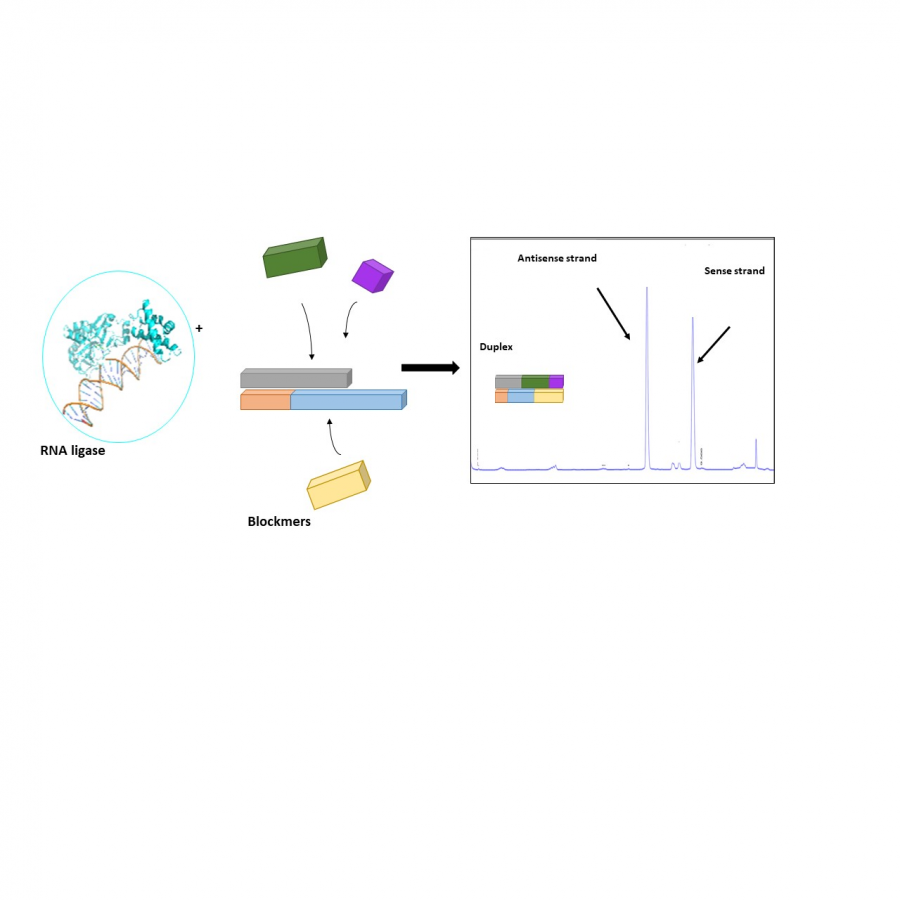 Almacs 3-2-3-2 hybrid RNA ligase enzyme strategy Almac’s 3-2-3-2 hybrid RNA ligase enzyme strategy used to produce 1 mM of high purity siRNA. |
#1382 | Enzymes boost a circular polyurethane economy |
|
| Presenting author: | Georg Andreas ROEHLING of RWTH AACHEN CHAIR OF BIOTECHNOLOGY |
| Corresponding author: | Lukas REISKY of COVESTRO DEUTSCHLAND AG |
| Other authors: | Daniela KNOPP of COVESTRO DEUTSCHLAND AG Simone GOEBBELS of COVESTRO DEUTSCHLAND AG Linda PASTOR of RWTH AACHEN CHAIR OF BIOTECHNOLOGY Gernot JAEGER of COVESTRO DEUTSCHLAND AG Ulrich SCHWANEBERG of RWTH AACHEN CHAIR OF BIOTECHNOLOGY |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymatic recycling / Polyurethane recycling / Circular economy / |
| Purpose: | The synthetic polymer industry is a versatile sector, whose products can be found in nearly every other industry ranging from special coatings for fibers to insulation materials for fridges and houses to packages for daily life products. Industrial polymer production started in the mid-20th century. Since then, it has been growing with an expected annual global production of 1.1 billion tons metric tons by 2050 [1]. Despite the great advantages of synthetic polymers, the major drawback can be seen all over the world. Since yet, no efficient waste management exist for the enormous number of heterogenous plastics. Consequently, the majority of them end up in landfills or is incinerated causing greenhouse gas emission or fine dust pollution. Only 15 % of the annually produced plastics are recycled [2]. There are still no efficient recycling technologies for many plastic types to generate valuable monomers which can be re-introduced into the production loop [1, 3]. This is one central aim of the circular economy concept – keeping waste streams at a minimum by recycling them as often as possible. A circular polymer economy could enable the sustainable and responsible usage of synthetic polymers. As one of the leading manufacturers of high-tech polymer materials, Covestro wants to drive the transformation to a circular economy. For this, a cooperation with the Institute of Biotechnology of the RWTH Aachen was introduced aiming at the development of enzymatic systems for recycling of polyurethanes (PUs), whose precursors are produced by Covestro. The enzymatic recycling could have many potential advantages over conventional recycling methods: lower energy consumptions due to mild reaction conditions, avoidance of potentially toxic organic solvents during the process, high selectivity and specificity. So, enzymes could enable the depolymerization of PU into its monomers which can then re-enter the production loop [4, 5]. We have obtained the first promising results of enzymatic depolymerization of a urethane by a proprietary urethanase, releasing valuable monomers which can be used in the production of new polymers. |
| References: | 1. Geyer, R., Production, use, and fate of synthetic polymers, in Plastic waste and recycling. 2020, Elsevier. p. 13-32. 2. OECD, Improving Markets for Recycled Plastics. 2018. 3. Leslie, H., et al., Propelling plastics into the circular economy—weeding out the toxics first. Environment international, 2016. 94: p. 230-234. 4. Branson, Y., et al., Urethanases for the enzymatic hydrolysis of low molecular weight carbamates and the recycling of polyurethanes. Angewandte Chemie International Edition, 2023. 62(9): p. e202216220. 5. Islam, S., et al., Targeting microplastic particles in the void of diluted suspensions. Environment International, 2019. 123: p. 428-435. |
#1383 | Phosphates from renewable raw materials for the functionalization of plastic consumer goods |
|
| Presenting author: | Patrick LENZ of INSTITUTE OF BIOTECHNOLOGY, RWTH AACHEN UNIVERSITY |
| Corresponding author: | Ulrich SCHWANEBERG of INSTITUTE OF BIOTECHNOLOGY, RWTH AACHEN UNIVERSITY AND DWI LEIBNIZ-INSTITUTE FOR INTERACTIVE MATERIALS |
| Other authors: | Johanna STOTZ of INSTITUTE OF BIOTECHNOLOGY, RWTH AACHEN UNIVERSITY Anna Joëlle RUFF of INSTITUTE OF BIOTECHNOLOGY, RWTH AACHEN UNIVERSITY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | phytase / phosphorus recovery / / |
| Purpose: | The element phosphorus (P) is a finite resource which is gained exclusively by rock mining outside Europe[1]. Phosphorus is used in various industrial sectors, applications and in numerous everyday products - often in the form of polyphosphate[2]. Due to the continuously growing demand, our consumption far exceeds the natural regeneration rates. Despite the importance of this element, few to no adequate recycling technologies have been used to date, resulting in a strong dependency associated with this scarce resource[3]. The Department of Biotechnology and the Institute of Applied Microbiology at RWTH Aachen University have developed and patented a two-step process for the biotechnological production of (bio)polyphosphate from deoiled seeds or bran[4]. The principle storage form of phosphate in plants is phytate (inositol hexakisphosphate). In the first process step, this stored phosphate is enzymatically mobilized from these renewable raw materials, which represent side streams of food industry. For the complete mobilization of all phosphates, a phytase blend is used. The enzymatic process allows to mobilize about 30 g P/kg deoiled rape seeds or rye bran. This is a P-content reduction by up to 90 %. In the second process step, the recovered phosphate is then accumulated and isolated using yeasts in the form of industrially relevant (bio)polyphosphate. Promising approaches for upscaling of the process as well as phytase production were investigated to coat bioplastic surfaces. As part of the competence center Bio4MatPro, the two institutes, together with the DWI-Leibniz Institute for Interactive Materials, have set themselves the goal within the ProPhos project (BoostLab1-5) of using the biotechnologically produced (bio)polyphosphate in a novel application for the functionalization of everyday objects made of bioplastics, e.g. for bacteriostatic effectiveness. The developed processes thus contribute to the circular phosphorus (bio)economy. Funding: We acknowledge the financial support by the German Federal Ministry of Education and Research (BMBF) for the project ProPhos within the “Bio4MatPro - Competence Center for Biological Transformation of Materials Science and Production Engineering” program (grant no. 031B1143A). |
| References: | [1]: Herrmann, K. R., Ruff, A. J., Infanzón, B., & Schwaneberg, U. (2019). Engineered phytases for emerging biotechnological applications beyond animal feeding. Applied microbiology and biotechnology, 103, 6435-6448. [2]: Christ, J. J., Smith, S. A., Willbold, S., Morrissey, J. H., & Blank, L. M. (2020). Biotechnological synthesis of water‐soluble food‐grade polyphosphate with Saccharomyces cerevisiae. Biotechnology and bioengineering, 117(7), 2089-2099. [3]: Widderich, N., Mayer, N., Ruff, A. J., Reckels, B., Lohkamp, F., Visscher, C., Schwaneberg, U., Kaltschmitt, M., Liese, A., Bubenheim, P. (2022). Conditioning of feed material prior to feeding: Approaches for a sustainable phosphorus utilization. Sustainability, 14(7), 3998. [4]: Herrmann, K. R., Fees, J., Christ, J. J., Hofmann, I., Block, C., Herzberg, D., Bröring, S., Reckels, B., Visscher, C., Blank, L. M., Schwaneberg, U., Ruff, A. J. (2023). Biotechnological production of food-grade polyphosphate from deoiled seeds and bran. EFB Bioeconomy Journal, 3, 100048. |
#1385 | EnzymeML-based workflow for FAIR enzyme kinetics |
|
| Presenting author: | Max HÄUSSLER of INSTITUTE OF BIOCHEMISTRY AND TECHNICAL BIOLOGY |
| Corresponding author: | Juergen PLEISS of INSTITUTE OF BIOCHEMISTRY AND TECHNICAL BIOLOGY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | FAIR / Kinetic parameter estimation / modeling / |
| Purpose: | Data management according to FAIR data principles is an emerging best practice in science.[1] In biocatalysis, the EnzymeML format implements these principles, enabling storing and working with biocatalytical (meta)data.[2] Fur- thermore, modeling results can be stored together with data on which the results are based on. Together with PyEnzyme, a Python interface for EnzymeML, powerful analysis workflows can be implemented, which exceed the capabilities of analysis in spreadsheet applications. Thus, the EnzymeML infrastructure supports the field by providing structured documentation to experimental data. Hence, enabling the implementation of FAIR analysis pipelines. Here we present an EnzymeML-based workflow for kinetic parameter esti- mation, enabling a continuous data flow from raw data to kinetic parameters. Thereby, analytical raw data together with calibration data is used for accurate concentration calculation, enabling description of linear and non-linear relation- ships between the analytical signal and the analyte concentration. After cali- bration, the respective information is directly applied to the EnzymeML data model containing analytical raw data and yielding an EnzymeML data model with concentration data. Thereafter, concentration data from the EnzymeML data model is mapped to EnzymePynetics, a tool for kinetic parameter estima- tion based on time-course measurement data. EnzymePynetics enables time- course analysis of enzymatic reactions, assessing the inhibition constant Ki for potential substrate and product inhibition or an enzyme inhibitor apart from substrate and product. Furthermore, different enzyme inactivation models are applied. After fitting of different model combinations, the best fitting model is selected based on different statistical criteria and thus provides an insight into the kinetic mechanism of the enzymatic catalysis. Finally, the modeling results are written back to the EnzymeML data model and serialized to the standardized EnzymeML format. |
| References: | [1] Wilkinson, M. D., Dumontier, M., Aalbersberg, I. J., Appleton, G., Axton, M., Baak, A., ... & Mons, B. . The FAIR Guiding Principles for scientific data management and stewardship. Scientific data, 3(1), 1-9 (2016). [2] Lauterbach, S., Dienhart, H., Range, J. et al. EnzymeML: seamless data flow and modeling of enzymatic data. Nat Methods 20, 400-402 (2023). |
| Figures: | 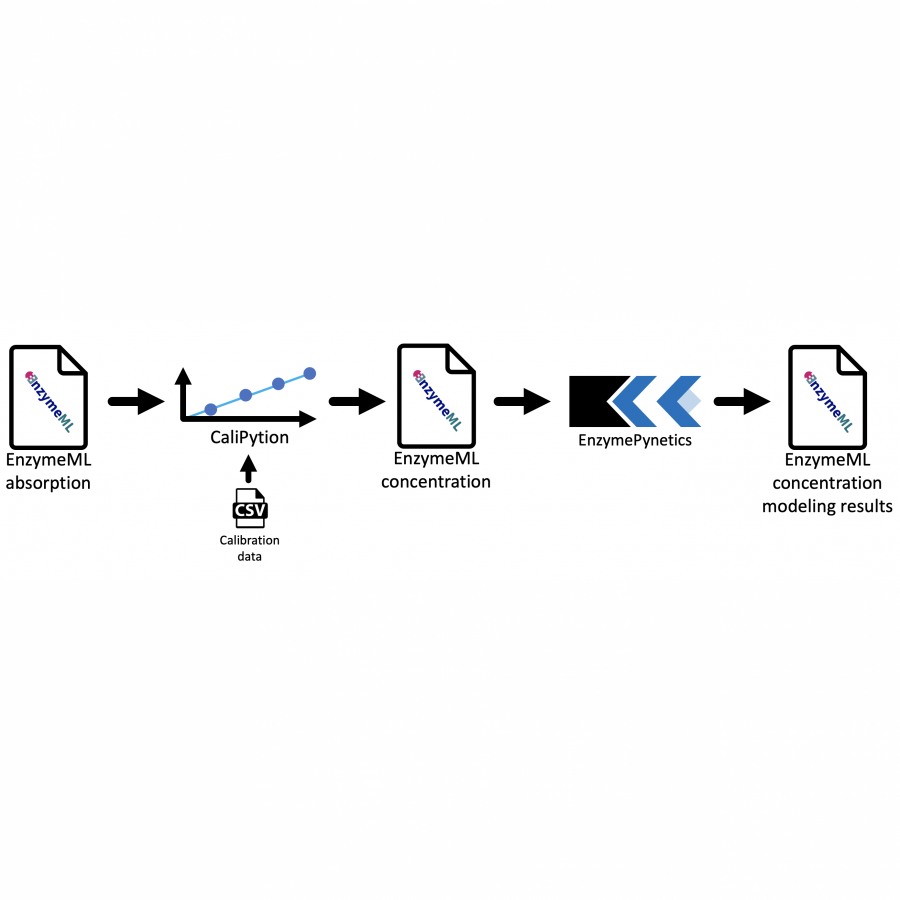 Workflow from raw data to kinetic parameters Components of the kinetic parameter estimation workflow for enzyme reaction data. |
#1386 | Comparison of recombinant protein production in Escherichia coli and Pichia pastoris |
|
| Presenting author: | Tatiana PETROVIČOVÁ of INSTITUTE OF BIOTECHNOLOGY, SLOVAK UNIVERSITY OF TECHNOLOGY |
| Other authors: | Kristína KÁNTOROVÁ of INSTITUTE OF BIOTECHNOLOGY, SLOVAK UNIVERSITY OF TECHNOLOGY Zuzana HEGYI of INSTITUTE OF BIOTECHNOLOGY, SLOVAK UNIVERSITY OF TECHNOLOGY Martin REBROŠ of INSTITUTE OF BIOTECHNOLOGY, SLOVAK UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | recombinant protein / P. pastoris / non-conventional yeasts / fermentation |
| Purpose: | Since the 1980s, E. coli has been preferred as a microbial expression host due to its ability to fast-growing in inexpensive complex media, well-characterized genetics, physiology, and metabolism, quick and straightforward transformation with foreign DNA, and easy-to-scale fermentation process. However, despite this acquired knowledge, difficulties (i.e., the formation of inclusion bodies due to poor solubility) have been faced by many researchers in expressing a biocatalyst from a eukaryotic origin. On the other hand, the efficiency of yeast as a host for recombinant protein expression has proven and has become one of the most abundant alternatives for large-scale protein production [1]. One of them is Pichia pastoris which expresses recombinant proteins both intracellularly and extracellularly, which simplifies protein isolation from the production media. In addition, the concentration of produced proteins is quite high, and the posttranslational modifications are superior to those in prokaryotic expression systems. The gene encoding the protein is mostly located under the control of the alcohol oxidase (AOX) promoter, which is induced by methanol and repressed by glycerol [2]. Engineered monoamine oxidase gene originated from Aspergillus niger was successfully expressed intracellularly in both E. coli BL21(DE3) and P. pastoris CBS7435 MutS, while the productivity of cell extract from P. pastoris was 83 times higher than E. coli extract [3]. Optimised extracellular upscale recombinant protein production protocol for P. pastoris KM71H MutS strain [2] was used for expression of the polymerase enzyme, which is used in RT-qPCR detection of virus RNA molecules, such as SARS-CoV-2 virus. The aim of this work was the comparison of the yield and activity of enzymes produced in E. coli as well as in P. pastoris. Acknowledgments: This work was funded by the Slovak Research and Development Agency under contract No. PP-COVID-20-0056 and by the OP Integrated Infrastructure, the project: Research on COVID-19 progressive diagnostic methods and biomarkers useful in early detection of individuals at increased risk of severe disease, ITMS: 313011ATA2, co-financed by the European Regional Development Fund. This work was also supported by COST Action “Non-Conventional Yeasts for the Production of Bioproducts”- YEAST4BIO (CA18229). |
| References: | [1] Duman-Özdamar, Z. E.; Binay, B. Protein J 2021, 40, 367-376 [2] Markošová, K.; Wegnerová, L.; Rosenberg, M.; Křen, V.; Rebroš, M. Front Microbiol 2015, 6, 1140 [3] Markošová, K.; Camattari, A.; Rosenberg, M.; Glieder, A.; Turner, N.J.; Rebroš, M. Biotechnol Lett 2018, 40, 127-133 |
#1387 | Cheese, wine and durian flavour components produced by carboligases |
|
| Presenting author: | Valentina JURKAŠ of TU GRAZ, INSTITUTE OF MOLECULAR BIOTECHNOLOGY |
| Other authors: | Hana DOBIAŠOVÁ of STU BRATISLAVA, INSTITUTE OF CHEMICAL AND ENVIRONMENTAL ENGINEERING Kvetoslava VRANKOVÁ of AXXENCE SLOVAKIA S.R.O. Florian RUDROFF of TU WIEN, INSTITUTE OF APPLIED SYNTHETIC CHEMISTRY Dörte ROTHER of FORSCHUNGSZENTRUM JÜLICH GMBH Margit WINKLER of TU GRAZ, INSTITUTE OF MOLECULAR BIOTECHNOLOGY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | carboligase / flavour / acyloins / α-hydroxypentanone |
| Purpose: | 2-Hydroxy-3-pentanone (flavour: truffle, peanut; odour: buttery, hay-like) and its isomer, 3-hydroxy-2-pentanone (flavour: herbaceous, truffle; odour: caramel-sweet, buttery), are volatile acyloins that have been identified as flavour components of cheese, wine, durian, honey, butter, soy sauce, sherry and other foods [1]. Acyloins are typical products of the enzymatic acyloin-type condensation reaction catalysed by thiamine diphosphate (ThDP)-dependent carboligases. The reaction usually includes a decarboxylation of an α-keto acid and its subsequent ligation to an aldehyde, leading to acyloins [2]. By choosing complementary substrates the reaction can be directed towards one or the other isomer: pyruvate and propanal condense to 3-hydroxypentane-2-one (Figure 1), whereas 2-oxobutyric acid and acetaldehyde give the 2-hydroxypentan-3-one (Figure 2). Screening a panel of carboligases identified the pyruvate dehydrogenase from E. coli (EcPDH E1) as the most suitable, reaching product formation of up to 90%. Therefore, we decided to explore the sequence space by looking for uncharacterized analogues of EcPDH E1, in a search for higher activity and tolerance to high concentrations of substrates. To pre-screen the analogues, a colorimetric assay detecting the decarboxylation activity using DCPIP (2,6-dichloroindophenol) was used [3]. |
| References: | [1] F. Neuser, H. Zorn, R. G. Berger, J. Agric. Food Chem. 2000, 48, 6191-6195. [2] M. Müller, D. Gocke, M. Pohl, The FEBS Journal 2009, 276, 2894-2904. [3] S. R. Marsden, D. G. G. McMillan, U. Hanefeld, Int. J. Mol. Sci. 2020, 21(22), 8641. |
| Figures: | 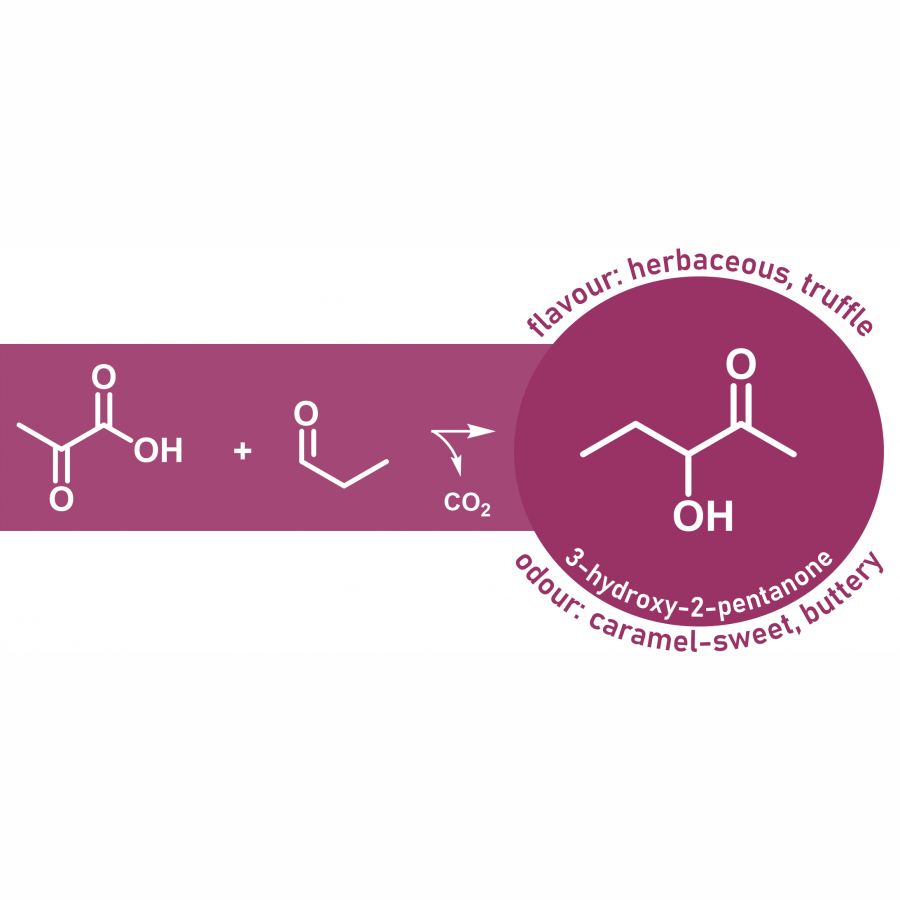 Carboligation reaction for preparation of 3-hydroxy-2-pentanone. 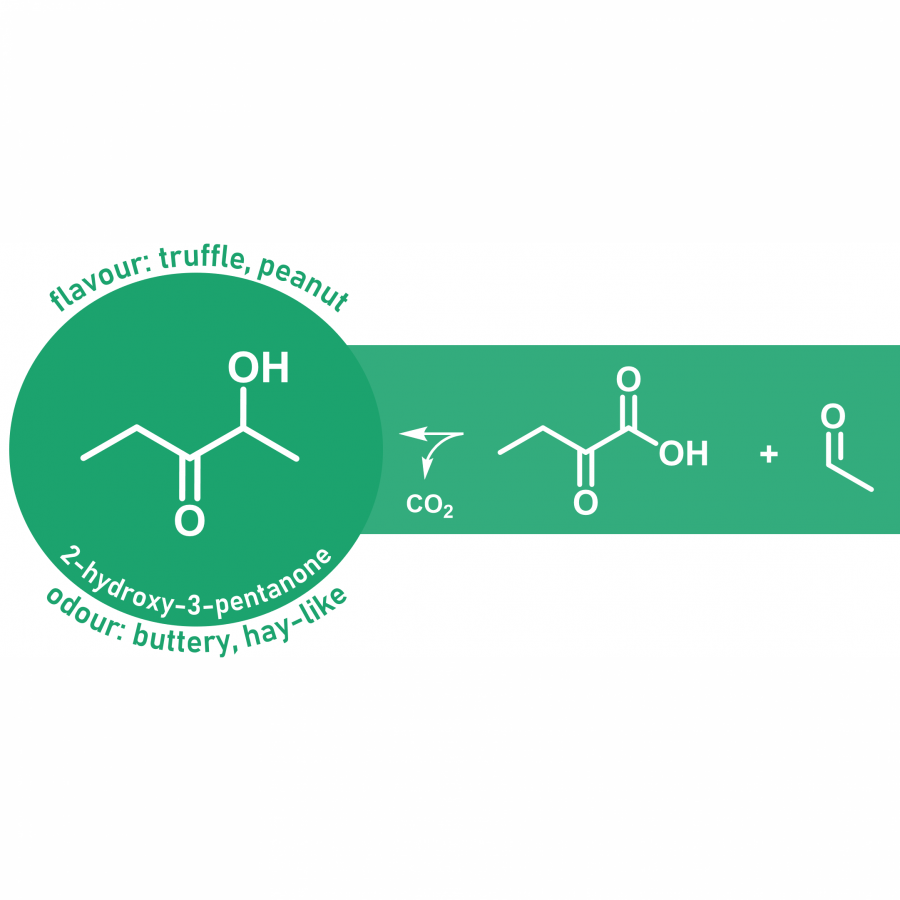 Carboligation reaction for preparation of 2-hydroxy-3-pentanone. |
#1389 | Deep Eutectic Solvents for the Enzymatic Synthesis of Sugar Esters: A Generalizable Strategy? |
|
| Presenting author: | Daniela UBIALI of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES |
| Other authors: | Riccardo SEMPROLI of UNIVERSITY OF PAVIA, DEPARTMENT OF DRUG SCIENCES Selin KARA of AARHUS UNIVERSITY, DEPARTMENT OF BIOLOGICAL AND CHEMICAL ENGINEERING Santiago Nahuel CHANQUIA of AARHUS UNIVERSITY, DEPARTMENT OF BIOLOGICAL AND CHEMICAL ENGINEERING Jan Philipp BITTNER of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF THERMAL SEPARATION PROCESSES Simon MÜLLER of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF THERMAL SEPARATION PROCESSES Pablo DOMÍNGUEZ DE MARÍA of SUSTAINABLE MOMENTUM S. L |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | deep eutectic solvents / lactose esters / molecular dynamics simulation / COSMO-RS |
| Purpose: | Sugar Fatty Acid Esters (SFAEs) (or just “sugar esters”) are non-ionic surfactants with excellent emulsifying, stabilizing, and detergent properties. Moreover, they are tasteless, odourless, non-toxic, benign to the environment, fully biodegradable, and can be derived from renewable sources. The preparation of these industrially relevant compounds is cumbersome due to the opposite solubility profile of the substrates, i.e. the polar head (sugar) and the hydrophobic tail (fatty acid), which forces the use of environmentally unfriendly reaction media and harsh operational conditions. As a result, high energy consumption, formation of byproducts, and low regioselectivity are encountered. [1,2] In this context, using nonconventional (benign) media and an enzyme-based synthesis can overcome the above-mentioned drawbacks. Whereas glucose and saccharose esters have been widely studied in this frame, lactose has been underinvestigated despite its large availability, being the most abundant component of cheese whey, the main waste stream of the dairy industry. Herein, [3] deep eutectic solvents (DESs) were considered as promising reaction media, starting from the evidence that glucose-based esters can be obtained by lipase-based (trans)esterification of this monosaccharide with different acyl donors in classic DESs like choline chloride/urea (ChCl/U) as well as in hydrophobic DESs. [4,5] However, when lactose is used, enzymes cannot efficiently perform (trans)esterifications in a number of DESs (ChCl/U, ChCl/Lac.H2O, lidocaine/palmitic acid), while the same reactions can proceed in “conventional” mixtures like pyridine/tetrahydrofuran (Py/THF). Computational solubility studies and molecular dynamics simulations (Figure 1) [3] of both ChCl/U and Py/THF systems played a pivotal role to rationalize this output, highlighting two effects: (i) on the one hand, large acyl donors (more than C10) display poor solubility in DESs and (ii) on the other hand, lactose interacts with DES components. Thus, the DES affects the conformation of lactose (compared to that observed in the Py/THF mixture), in such a way that the enzymatic reaction results impaired. Despite that classic DESs (e.g., ChCl/U) may not be useful for generalizing their use in saccharide ester synthesis, we hope that the theoretical understanding of the reaction here achieved will pave the way for designing novel DESs that can broaden the use of these solvents in sugar chemistry. Acknowledgements Cariplo Foundation (Italy) is acknowledged for the grant “Integrated platform for the sustainable production of bio-based surfactants from renewable resources (BioSurf)” to D.U. (ID 2020-1094). S.K. thanks to Novo Nordisk Foundation, Light-BioFuels project, grant no. NNF19OC0057522, and Deutsche Forschungsgemeinschaft (DFG), grant no. KA 4399/3-2 and SM 82/25-2. Full affiliation for R.S., J.P.B., and S.K.: Aarhus University, Department of Biological and Chemical Engineering (R.S.), Leibniz University Hannover, Institute of Technical Chemistry (J.P.B. and S.K.) |
| References: | [1] V.S. Nagtode, C. Cardoza, H.K.A. Yasin, S.N. Mali, S.M. Tambe, P. Roy, K. Singh, A. Goel, P.D. Amin, B.R. Thorat, J.N. Cruz, A.P. Pratap, ACS Omega 2023, 8, 11674-11699. [2] B. Pérez, S. Anankanbil, Z. Guo, in Fatty Acids 2017, 329-354. [3] R. Semproli, S.N. Chanquia, J.P. Bittner, S. Müller, P. Domínguez de María, S. Kara, D. Ubiali, ACS Sustainable Chem. Eng, in press, https://doi.org/10.1021/acssuschemeng.2c07607 [4] R. Hollenbach, B. Bindereif, U.S. Van der Schaaf, K. Ochsenreither, C. Syldatk, Front. Bioeng. Biotechnol., 2020, 8, 382. [5] R. Hollenbach, K. Ochsenreither, C. Syldatk, Int. J. Mol. Sci., 2020, 21, 4342. |
| Figures: |  Figure 1 Snapshots of the configuration of α-lactose in the DES ChCl/U (1:2) (5% w/w water) (a,b) and in Py/THF (1:1 v/v) (c,d) from the MD simulations and two replica simulations each. α-Lactose is displayed as licorice, while the oxygen atom that is part of the |
#1392 | Toward Reproducible Enzyme Modeling with Isothermal Titration Calorimetry |
|
| Presenting author: | Felix OTT of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Gudrun GYGLI of WEGA INFORMATIK AG |
| Other authors: | Kersten S. RABE of KARLSRUHE INSTITUTE OF TECHNOLOGY Christof M. NIEMEYER of KARLSRUHE INSTITUTE OF TECHNOLOGY Gudrun GYGLI of WEGA INFORMATIK AG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | (S)-stereoselective ketoreductase / kinetic model / FAIR/F.A.I.R / reproducibility |
| Purpose: | To apply enzymes in technical processes, a detailed understanding of the molecular mechanisms is required. Kinetic and thermodynamic parameters of enzyme catalysis are crucial to plan, model, and implement biocatalytic processes more efficiently. While the kinetic parameters, Km and kcat, are often accessible by optical methods, the determination of thermodynamic parameters requires more sophisticated methods. Isothermal titration calorimetry (ITC) allows the label-free and highly sensitive analysis of kinetic and thermodynamic parameters of individual steps in the catalytic cycle of an enzyme reaction. However, since ITC is susceptible to interferences due to denaturation or agglomeration of the enzymes, the homogeneity of the enzyme sample must always be considered, and this can be accomplished by means of dynamic light scattering (DLS) analysis. The here presented ITC-dependent workflow[1] was used to determine both the kinetic and the thermodynamic data for a cofactor-dependent enzyme. Using a standardized approach with the implementation of sample quality control by DLS, we obtained high-quality data suitable for the advanced modeling of the enzyme reaction mechanism. Specifically, we investigated stereoselective reactions catalyzed by the NADPH-dependent ketoreductase Gre2p under different reaction conditions. The results revealed that this enzyme operates with an ordered sequential mechanism in which the cofactor NADPH binds first, resulting in Gre2pholo, and that only Gre2p(holo) can then bind the substrate NDK. In addition, the enzyme was found to be affected by substrate or product inhibition depending on the reaction buffer. Data reproducibility, a mandatory prerequisite to achieve robust modeling, is ensured by specifying standard operating procedures, using programmed workflows for data analysis and storing all data in a F.A.I.R. (findable, accessible, interoperable, and reusable) repository. Our approach highlights the utility for combined binding and kinetic studies for such complex multisubstrate reactions. Because it is amenable to automation and scale-up for high-throughput, the combination of such diverse approaches will provide the high-quality data needed for the engineering of enzymes and biocatalytic processes through machine learning to accelerate future development of industrial biocatalysis. |
| References: | [1] ott, f., rabe, k. s., niemeyer, c. m., gygli, g., ACS Catalysis 2021, 11, 10695-10704. |
| Figures: |  Workflow to create high-quality data for robust enzyme reaction modeling Traditional approaches to enzyme kinetic data (blue) primarily use spectrophotometric methods. The implementation of DLS and ITC for quality control and mechanistic insights, respectively, leads to an increase in data quality to enable robust modeling.  Proposed mechanistic model for Gre2p Gre2p(apo) (black), Gre2p(holo) (blue), fully reactive Gre2p with both NADPH and NDK (purple) and Gre2p after the reaction (yellow).
The crossed arrow (red) indicates that NDK cannot bind to Gre2p unless NADPH is already bound (Gre2p(holo)). |
#1393 | Formulation and characterisation of enzyme-based biomaterials for µFluidic experiments |
|
| Presenting author: | Julian S. HERTEL of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Corresponding author: | Christof M. NIEMEYER of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Other authors: | Kersten S. RABE of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | hydrogels / immobilization / flowbiocatalysis / microreactors |
| Purpose: | The fundamental principle of biological compartmentalisation of cellular life provides the basis for space-time resolved reaction processes. Based on this, intensive work is currently done on the use of interconnected, continuously flowing reaction chambers in order to improve the reaction control and efficiency of chemical syntheses, especially with integrated biocatalysts (so called flow biocatalysis). To this end we have developed formulations of enzyme-based biomaterials based on a genetically encoded coupling system.[1] The resulting enzyme fusions are being used as modular building blocks for the assembly of catalytically active materials, which can be formulated as self-assembling all-enzyme hydrogels (AEHs) or thin films, and characterized in terms of their immobilization and biocatalytic activity in miniaturized flow reactors. The concept of self-assembling AEHs is compatible with a wide range of enzymes.[2-7] For example, our initial work established a material made of the two homotetrameric enzymes, a (R)-selective alcohol dehydrogenase (ADH) and the cofactor regenerating glucose 1-dehydrogenase (GDH), which are genetically fused with either the SpyCatcher (SC) or the SpyTag (ST).[1] This site-specific mediated enzyme conjugation allows the formulation of a very stable and highly active biomaterial which consists almost exclusively of enzymes, therefore making optimal use of the reaction space compared to other carrier-based immobilization techniques. The novel material was initially characterised via dynamic light scattering (DLS) and scanning electron microscopy (SEM) in terms of their physicochemical behaviour. Moreover, the gels showed excellent stereoselectivity, stable conversion rates, high space-time yields (STY) and cofactor retention for more than seven days in continuous flow experiments. Therefore, AEH is a highly promising material whose high application potential for sustainable and environmentally friendly industrial biocatalysis will be further enhanced by innovative formulations. |
| References: | [1] t. peschke, p. bitterwolf, s. gallus, y. hu, c. oelschlaeger, n. willenbacher, k. s. rabe, c. m. niemeyer, angew. chem. 2001, 113, 3443-3453 2018, 57, 17028-17032. [2] p. bitterwolf, s. gallus, t. peschke, e. mittmann, c. oelschlaeger, n. willenbacher, k. s. rabe, c. m. niemeyer, chem sci 2019, 10, 9752-9757. [3] p. bitterwolf, f. ott, k. s. rabe, c. m. niemeyer, micromachines 2019, 10, 783. [4] e. mittmann, s. gallus, p. bitterwolf, c. oelschlaeger, n. willenbacher, c. m. niemeyer, k. s. rabe, micromachines 2019, 10, 795. [5] t. peschke, p. bitterwolf, S. Hansen, J. Gasmi, K. S. Rabe, C. M. Niemeyer, Catalysts 2019, 9, 164. [6] T. Peschke, P. Bitterwolf, K. S. Rabe, C. M. Niemeyer, Chemical Engineering & Technology 2019, 42, 2009-2017. [7] P. Bitterwolf, A. E. Zoheir, J. Hertel, S. Kröll, K. S. Rabe, C. M. Niemeyer, Chemistry - A European Journal 2022, 28, e202202157. |
| Figures: |  Design, formation and morphological characterization schematic illustration of the enzymatic builing blocks, that can self-assemble to a hydrogel upon mixing via SC/ST-mediated conjugation and representative SEM image. |
#1394 | Solving NAD(P) Recycling Challenges in Organic Solvent Biocatalysis with Enzyme Immobilization |
|
| Presenting author: | Antía PINTOR LABANDEIRA of ENGINZYME |
| Other authors: | Iván LAVANDERA of UNIVERSITY OF OVIEDO Vicente GOTOR-FERNÁNDEZ of UNIVERSITY OF OVIEDO Alexey VOLKOV of ENGINZYME |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme immobilization / organic solvent / cofactor recycling / ERED, ADH |
| Purpose: | Ene-reductases (EREDs) are enzymes that have gained significant attention as valuable catalysts for the (stereoselective) reduction of activated alkenes.1,2 The poor solubility of target compounds in aqueous systems usually hamper the implementation of biocatalytic process on industrial scale,3 and in fact, there is currently limited knowledge available on ERED enzymes performance in organic solvents.4,5 One of the major challenges of using redox enzymes in organic solvents is the supply of the required NAD(P)(H) cofactor for transformations. The recycling of NAD(P) in these media is particularly challenging due to the insolubility of the nicotinamide cofactor, severely hindering the hydride equivalent transfer between the reductase and the recycling enzyme in a coupled-enzyme system. To address these challenges, the use of an ERED from Zymomonas mobilis has been fully investigated using cyclohex-2-en-1-one as model substrate. Special attention has been given to searching for efficient alcohol dehydrogenases (ADHs) for NAD(P) cofactor recycling purposes, as well as optimizing the (co)immobilized catalytic system in organic solvents. With this aim, ADH from Thermoanaerobacter ethanolicus was found to be a suitable enzyme, and both ERED and ADH were immobilized on different types of EziG polymer-coated controlled porosity glass carrier materials. The results of this study demonstrate the potential of using redox enzymes in organic solvents and provide insights into their optimization for future industrial applications. |
| References: | 1. T. K. Roy, R. Sreedharan, P. Ghosh, T. Gandhi, D. Maiti, Chem. Eur. J., 2022, 28, e202103949. 2. F. Parmeggiani, E. Brenna, D. Colombo, F. G. Gatti, F. Tentori, D. Tessaro, ChemBioChem, 2022, 23, e202100445. 3. M. M. C. H. van Schie, J.-D. Spöring, M. Bocola, P. Domínguez de María, D. Rother, Green Chem., 2021, 23, 3191-3206. 4. Y. Yanto, C. K. Winkler, S. Lohr, M. Hall, K. Faber, A. S. Bommarius, Org. Lett., 2011, 13, 2540-2543. 5. D. Clay, C. K. Winkler, G. Tasnádi, K. Faber, Biotechnol. Lett., 2014, 36, 1329-1333. |
#1397 | Chemoenzymatic synthesis of enantioenriched (R)- and (S)- aryloxyalkanoic herbicides |
|
| Presenting author: | Celeste NOBBIO of POLITECNICO DI MILANO |
| Corresponding author: | Fabio PARMEGGIANI of POLITECNICO DI MILANO |
| Other authors: | Danilo COLOMBO of POLITECNICO DI MILANO Davide TESSARO of POLITECNICO DI MILANO Francesco Gilberto GATTI of POLITECNICO DI MILANO Maria Elisabetta BRENNA of POLITECNICO DI MILANO Alessia ALBERGATI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE GIULIO NATTA CONSIGLIO NAZIONALE DELLE RICERCHE Erica FERRANDI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE GIULIO NATTA CONSIGLIO NAZIONALE DELLE RICERCHE Daniela MONTI of ISTITUTO DI SCIENZE E TECNOLOGIE CHIMICHE GIULIO NATTA CONSIGLIO NAZIONALE DELLE RICERCHE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Chemo-enzymatic catalysis / Enzymes / Green chemistry / Industrial Biocatalysis |
| Purpose: | Aryloxyalkanoic herbicides are a class of agrochemicals of widespread use for the protection of cereals and monocotyledonous crops from broad-leaf weeds. The production method of this class of herbicides changed a lot during the past decades. Indeed, chemical industry and in particular the agrochemical sector, has been subjected to a consistent and urgent pressure to develop and adopt more sustainable manufacturing processes, with particular regard to the reduction of greenhouse gas emission, waste output and replacement of toxic and dangerous reagents [1]. The extensive chemical development led to aryloxy alkanoic acids and derivatives with decreased toxicity and environmental hazard, and these molecules are currently sold as mixture of both (R) and (S) enantiomers although usually one of the two is more active than the other. Dichloroprop and mecoprop are sold as racemate but only the (R)-enantiomer is responsible for the biological activity, conversely herbicide (S)-beflubutamid is 1000 time more active than its corresponding (R)-enantiomer. The stereoselectivity of an enzymatic transformation in this context appear as an elite strategy for the synthesis of the more active enantiomer [2,3]. In this work, starting from simple and commercially available materials, the combination of a biocatalytic asymmetric C=C reduction with a simple sequence of chemical transformations was implemented in a new chemo enzymatic synthesis of various substituted Aryloxyalkanoic acids (Figure 1) [4]. Stereoselective bioreduction is mediated by ene-reductases (ERs) belonging to the family of Old Yellow Enzymes (OYEs), versatile biocatalysts for the enantiospecific reduction of C=C double bonds activated by electron withdrawing groups [5]. By careful selection of the biocatalyst, either enantiomer of the product could be obtained in good yield and moderate to good ee without chromatographic purifications and by using crude enzyme preparations [4]. |
| References: | [1] C. M. Wieben, U.S. Geological Survey data release 2020, 2013-17, ver. 2.0 [2] H. S. Toogood, et al. ACS Catal. 2018,8, 3532-3549 [3] G. Tasnadi, et al, Catal. Sci. Technol. 2012, 2, 1548-1552 [4] D. Colombo, et al, Eur. J. Org. Chem. 2022, 25, e202200609 [5] F. Parmeggiani, et al, ChemBioChem 2022, 23, e202100445 |
| Figures: | 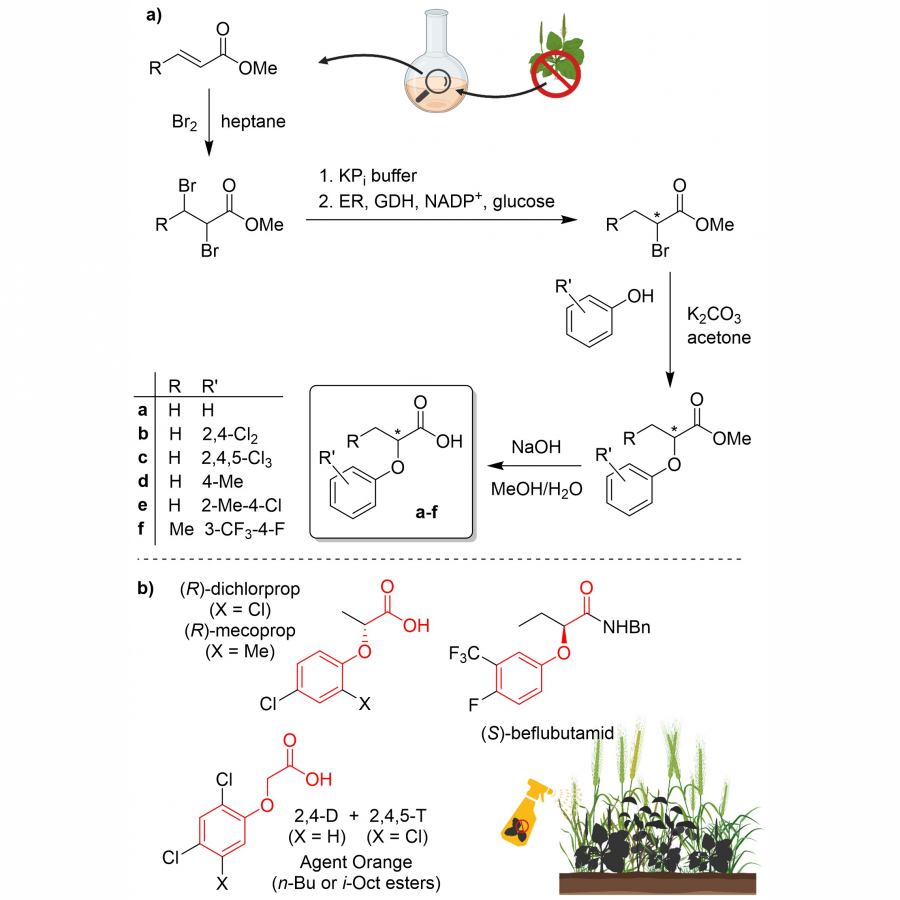 Figure 1 (a) Chemoenzymatic synthesis of enantioenriched aryloxyalkanoic herbicides.
(b) Representative examples of commercial chiral aryloxyalkanoic herbicides in their most active enantiomeric form (the common phenoxy-acetic acid skeleton is shown in red). |
#1398 | Two anti-Prelog and NAD-dependent alcohol dehydrogenases with broad substrate scope and excellent enantioselectivity |
|
| Presenting author: | Matteo DAMIAN of UNIVERSITY OF AMSTERDAM |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Alcohol dehydrogenase / Enantioselectivity / anti-Prelog / |
| Purpose: | BIOTRANS 2023 Type of communication requested: poster Communication title: Two anti-Prelog and NAD-dependent alcohol dehydrogenases with broad substrate scope and excellent enantioselectivity Topic (select up to two): (chemo)enzymatic strategy Authors: Matteo Damian1, Tanja Knaus1, Francesco G. Mutti1 Affiliations: 1Van’ t Hoff Institute for Molecular Sciences, HIMS-Biocat, University of Amsterdam, Science Park 904, 1098 XH, The Netherlands The synthesis of enantiopure molecules such as alcohols is pivotal to the chemical fields. Especially, the possibility to access both enantiomers of a molecule with high enantioselectivity and chemoselectivity is of great interest for the synthesis of intermediates for pharmaceuticals, agrochemicals, flavour and fragrances, and fine chemicals.1 In this context, biocatalysis offers great opportunities for the highly efficient and enantioselective synthesis of molecules by exploiting the exquisite selectivity of the enzymes. Through alcohol dehydrogenases (ADH), alcohols can be obtained via the enzymatic reduction of carbonyls.2 The majority of the available ADHs lead to the formation of one of the two enantiomers following the Prelog rule.3 A limited number of wild-type anti-Prelog ADH have been reported in the literature so far and most of them are NADPH-dependent.4 This cofactor dependence increases the costs for the application of these anti-Prelog ADHs, thereby making them less attractive for the chemical industry. Herein, we investigated two NADH-dependent anti-Prelog ADHs from Candida maris and Pichia finlandica,5 which are capable of performing the reduction of a broad range of ketones with good yields and excellent enantioselectivity. References (1) Goldberg, K.; Schroer, K.; Lütz, S.; Liese, A. Appl. Microbiol. Biotechnol. 2007, 76, 237-248. (2) Hollmann, F.; Arends, I. W.; Holtmann, D. Green Che. 2011, 13 (9), 2285-2314. (3) Musa, M. M. ChemistryOpen 2022, 11 (4), e202100251. (4) Bradshaw, C. W.; Hummel, W.; Wong, C. H. J. Org. Chem. 1992, 57 (5), 1532-1536. DOI: 10.1021/jo00031a037. Leuchs, S.; Greiner, L. Chem. and Biochem. Eng. Q. 2011, 25 (2), 267-281. Nie, Y.; Xu, Y.; Mu, X. Q. Org. Process Res. Dev. 2004, 8 (2), 246-251. DOI: 10.1021/op0341519. Meng, F.; Xu, Y. P. Biotechnol. Lett. 2010, 32, 533-537. Wang, Y.-J.; Ying, B.-B.; Chen, M.; Shen, W.; Liu, Z.-Q.; Zheng, Y.-G. World J. Microbiol. and Biotechnol. 2017, 33, 1-11. Orrego, A. H.; Andrés-Sanz, D.; Velasco-Lozano, S.; Sanchez-Costa, M.; Berenguer, J.; Guisan, J. M.; Rocha-Martin, J.; López-Gallego, F. Catal. Sci.Technol. 2021, 11 (9), 3217-3230. (5) Yamamoto, H.; Kudoh, M. Appl. Microbiol. Biotechnol. 2013, 97, 8087-8096. Kawano, S.; Yano, M.; Hasegawa, J.; Yasohara, Y. Biosci. Biotechnol. Biochem 2011, 75 (6), 1055-1060. |
| References: | (1) Goldberg, K.; Schroer, K.; Lütz, S.; Liese, A. Appl. Microbiol. Biotechnol. 2007, 76, 237-248. (2) Hollmann, F.; Arends, I. W.; Holtmann, D. Green Che. 2011, 13 (9), 2285-2314. (3) Musa, M. M. ChemistryOpen 2022, 11 (4), e202100251. (4) Bradshaw, C. W.; Hummel, W.; Wong, C. H. J. Org. Chem. 1992, 57 (5), 1532-1536. DOI: 10.1021/jo00031a037. Leuchs, S.; Greiner, L. Chem. and Biochem. Eng. Q. 2011, 25 (2), 267-281. Nie, Y.; Xu, Y.; Mu, X. Q. Org. Process Res. Dev. 2004, 8 (2), 246-251. DOI: 10.1021/op0341519. Meng, F.; Xu, Y. P. Biotechnol. Lett. 2010, 32, 533-537. Wang, Y.-J.; Ying, B.-B.; Chen, M.; Shen, W.; Liu, Z.-Q.; Zheng, Y.-G. World J. Microbiol. and Biotechnol. 2017, 33, 1-11. Orrego, A. H.; Andrés-Sanz, D.; Velasco-Lozano, S.; Sanchez-Costa, M.; Berenguer, J.; Guisan, J. M.; Rocha-Martin, J.; López-Gallego, F. Catal. Sci.Technol. 2021, 11 (9), 3217-3230. (5) Yamamoto, H.; Kudoh, M. Appl. Microbiol. Biotechnol. 2013, 97, 8087-8096. Kawano, S.; Yano, M.; Hasegawa, J.; Yasohara, Y. Biosci. Biotechnol. Biochem 2011, 75 (6), 1055-1060. |
| Figures: | 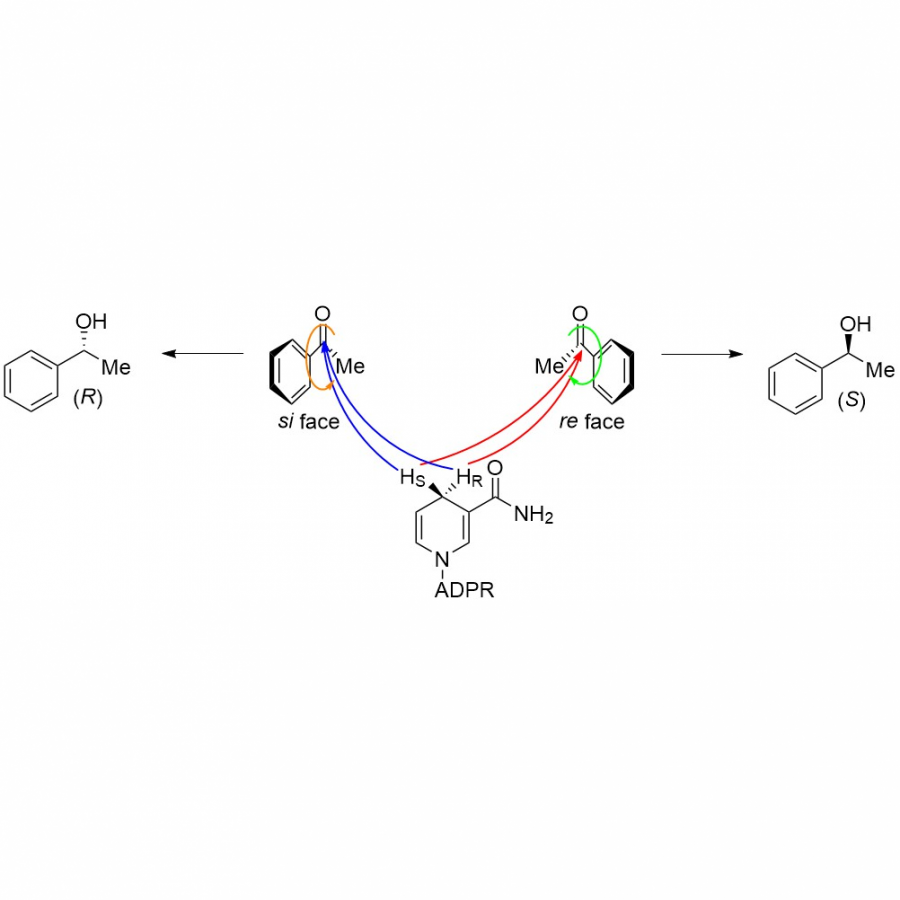 Prelog and anti-Prelog selectivity Typical Prelog selectivity with most of the alcohol dehydrogenases (ADHs) and anti-Prelog selectivity with two ADHs from Pichia finlandica and Candida Maris |
#1399 | Standardizing the methodology for transformation of anaerobic bacteria as hosts for recombinant enzyme production and development of fast activity screening for tungsten-dependent aldehyde oxidoreductases |
|
| Presenting author: | Ivana ALEKSIC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY |
| Other authors: | Gabriela OLEKSY of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY Kai KRÄMER of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Dominik HEGE of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Yvonne GEMMECKER of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Johann HEIDER of FACULTY OF BIOLOGY, UNIVERSITY OF MARBURG, GERMANY Maciej SZALENIEC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transformation / enzyme expression / Aromatoleum evansii / activity assay |
| Purpose: | The development of new techniques for the expression of various enzymes is a critical task in biotechnology. Not all valuable enzymes are easy to produce in standard bacterial hosts such as E. coli, due to their complex structures and strict requirements for folding and post-translational modification or special requirements for activation or cofactor insertion. The aim of this research was to overcome the challenges of expressing and producing enzymes from anaerobic metabolic pathways that are typically hard to obtain using traditional expression hosts. Our approach involved standardizing the methodology for the transformation of the bacterial cells, optimizing growth conditions, and selecting appropriate vectors for gene expression. For this study, we have chosen the facultatively anaerobic bacterium Aromatoleum evansii KB 740 as a host strain for the expression of three different enzymes from related species, i.e., benzylsuccinate synthase (BSS) from Thauera aromatica K172, tungsten-dependent aldehyde oxidoreductases (AOR) from Aromatoleum aromaticum and molybdenum steroid C25 dehydrogenase (S25DH) from Sterolibacterium denitrificans. BSS requires activation to a glycine radical state by a special activating enzyme, while both AOR and S25DH require the synthesis and correct incorporation of tungsten or molybdopterin cofactors, respectively. Compared to T. aromatica and A. aromaticum, A. evansii has a shorter generation time, can switch its metabolism from anaerobic to aerobic swiftly, and grows better on solid agar media. However, no molecular biology protocols for the genetic transformation of A. evansii have been developed, leaving conjugation as the only functional way of recombinant enzyme engineering. We have compared classical conjugation and direct transformation of A. evansii for developing recombinant enzyme expression systems with a broad-host plasmid pASG3_mob+ [1]: i) conjugation was achieved with a 2,6-diaminopimelic acid (DAP)-auxotrophic donor strain, E. coli WM3064 mixed with A.evnasii, and ii) direct transformation conditions of A. evansii with plasmid DNA were developed by electroporation. The former strategy, albeit time-consuming, yielded recombinant systems for AOR and BSS and enabled mutational studies of both enzymes. On the other hand, electroporation turned out to be much faster than conjugation. It was carried out under aerobic conditions, with 1 mM MOPS buffer, but using lower voltage and significantly increased incubation times compared to other established protocols. This improved methodology for electroporation significantly increased our success rate of obtaining recombinant A. evansii, while simultaneously cutting down the amount of time necessary for obtaining and purifying the desired clones. Comparing electroporation to conjugation outcomes, we can state that electroporation had a clear advantage on conjugation, providing recombinant clones faster and limiting the possibility of getting a contaminated culture. Additionally, we have developed a fast-screen colorimetric assay on Petri dishes for the early detection of AOR production. Overproduced enzyme activity was visualised through a reaction of benzaldehyde oxidation with benzyl viologen. Our findings demonstrate the feasibility of using non-standard bacteria as expression systems for difficult enzymes, providing a potential avenue for the development of novel biocatalysts. Also, the assay developed for the fast detection of enzyme activity proved to be cost-effective and useful for high-throughput screening while making a plasmid library. Acknowledgment: This work was supported by DFG/National Science Center Poland Beethoven Life grant (2018/31/F/NZ1/01856 and He2190/13-1). |
| References: | [1] I. Salii, M. Szaleniec, A. Alhaj Zein, D. Seyhan, A. Sekuła, K. Schühle, I. Kaplieva-Dudek, U. Linne, R. Meckenstock, J. Heider., ACS Catal. 2021, 11, 6, 3361-3370 |
#1400 | DIRECTED EVOLUTION OF NOV1 DIOXYGENASE TO ENHANCE THE PRODUCTION OF LIGNIN-DERIVED VANILLIN |
|
| Presenting author: | Mario DE SIMONE of ITQB-UNIVERSIDADE NOVA DE LISBOA |
| Other authors: | Lígia O. MARTINS of ITQB-UNIVERSIDADE NOVA DE LISBOA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Protein engineering / Biocatalysis / / |
| Purpose: | NOV1 from Novosphingobium aromaticivorans is a non-heme iron-dependent dioxygenase that has activity on lignin-derived phenolics, such as isoeugenol. NOV1 oxidatively cleaves the C=C bond of the propenyl functional group of isoeugenol and produces vanillin without adding external coenzymes. The enzymatic conversion of lignin-derived isoeugenol to vanillin, catalyzed by NOV1, can potentially replace the current petroleum-based synthesis and simultaneously valorize lignin biowaste. In this work, we applied directed evolution (DE) to improve NOV1 activity towards isoeugenol and its thermal stability, tailoring a more suitable enzyme for industrial applications. Libraries of variants were constructed by error-prone PCR and high throughput screening assays in 96-well plates were developed and validate to test their activity and stability. The best hits from each generation were purified to characterize their kinetic parameters and assess kinetic stability. After three rounds of DE, the best hit 16G3, harbouring 4 non-synonymous mutations, shows a 4-fold increased turnover rate (kcat) and 2.5-fold higher catalytic efficiency (kcat/Km), as compared to wild-type. During the evolution, the low kinetic stability of NOV1 wild-type, which previous studies determined to be linked to the iron cofactor loss [1], was also improved. 16G3 variant displays about 40-fold longer half-life time at 25°C, compared to the wild-type. Overall, these preliminary results show the potential of applying DE to engineer the NOV1 enzyme for tailored properties and to elucidate the structure-function relationship underlying the effect of each mutation. Acknowledgments: This project has received funding from the Bio-based Industries Joint Undertaking (JU) under the European Union’s Horizon 2020 research and innovation program under grant agreement No 837890. MDS acknowledges a Ph.D. grant (2020.08246.BD) from Fundação para a Ciência e Tecnologia. |
| References: | [1] De Simone M, Alvigini L, Alonso-Cotchico L, Brissos V, Caroli J., Lucas MF, Monza E, Pinho Melo E, Mattevi A, and Martins LO. Rationally Guided Improvement of NOV1 Dioxygenase for the Conversion of Lignin-Derived Isoeugenol to Vanillin. Biochemistry 2023 62 (2), 419-428 DOI: 10.1021/acs.biochem.2c00168. |
#1406 | The first crystal structure of a tannase-like feruloyl esterase in complex with a hydroxycinnamic acid |
|
| Presenting author: | Maria DIMAROGONA of UNIVERSITY OF PATRAS |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | feruloyl esterase / tannase superfamily / p-coumaric acid / X-ray crystallography |
| Purpose: | Feruloyl esterases (FAEs) are enzymes implicated in plant biomass decomposition, by hydrolyzing the ester bond between hydroxycinnamic acids and arabinose residues [1]. They present biotechnological interest, not only for biomass saccharification but also for the generation of potent antioxidant compounds such as ferulic acid. Here, we report the crystal structure of an FAE from Fusarium oxysporum (FoFaeC) at 1.7 Å resolution in complex with p-coumaric acid (PCA), which is the first ligand-bound structure of a tannase-like FAE [2]. PCA is sandwiched between the lid and catalytic domain, with its carboxyl group being oriented towards the catalytic triad that is comprised of Ser201, His452 and Asp412 (Figure 1). A close inspection of the substrate binding site reveals two conformations of Ser201, possibly indicating a “resting” and an “active” state of the enzyme. Significant alterations are also observed in the hydrophobic core of the lid domain, the most pronounced ones being Met124 and Tyr351 side chains that shift towards the bound phenolic ring. The FoFaeC structure is discussed in relation to other known FAEs, but also to a bacterial mono-(2-hydroxyethyl) terephthalate esterase (MHETase) from Ideonella sakaiensis, which is its third closest structural homologue, and is implicated in polyethylene terephthalate (PET) degradation [3]. The enrichment of structural data of the tannase superfamily assists towards our fundamental understanding of the underlying mechanisms of these biotechnologically relevant enzymes. |
| References: | [1] Puchart, V. & Biely, P. Essays Biochem. 2022, 67(3), 479-491. [2] Ferousi, C., Kosinas, C., Nikolaivits, E., Topakas, E., & Dimarogona, M. FEBS Lett. 2023, in press [3] Knott, B. C., Erickson, E., Allen, M. D., Gado, J. E., Graham, R., Kearns, F. L., Pardo, I., Topuzlu, E., Anderson, J. J., Austin, H. P., Dominick, G., Johnson, C. W., Rorrer, N. A., Szostkiewicz, C. J., Copié, V., Payne, C. M., Woodcock, H. L., Donohoe, B. S., Beckham, G. T. & McGeehan, J. E. PNAS, 2020, 117, 25476-25485. |
| Figures: | 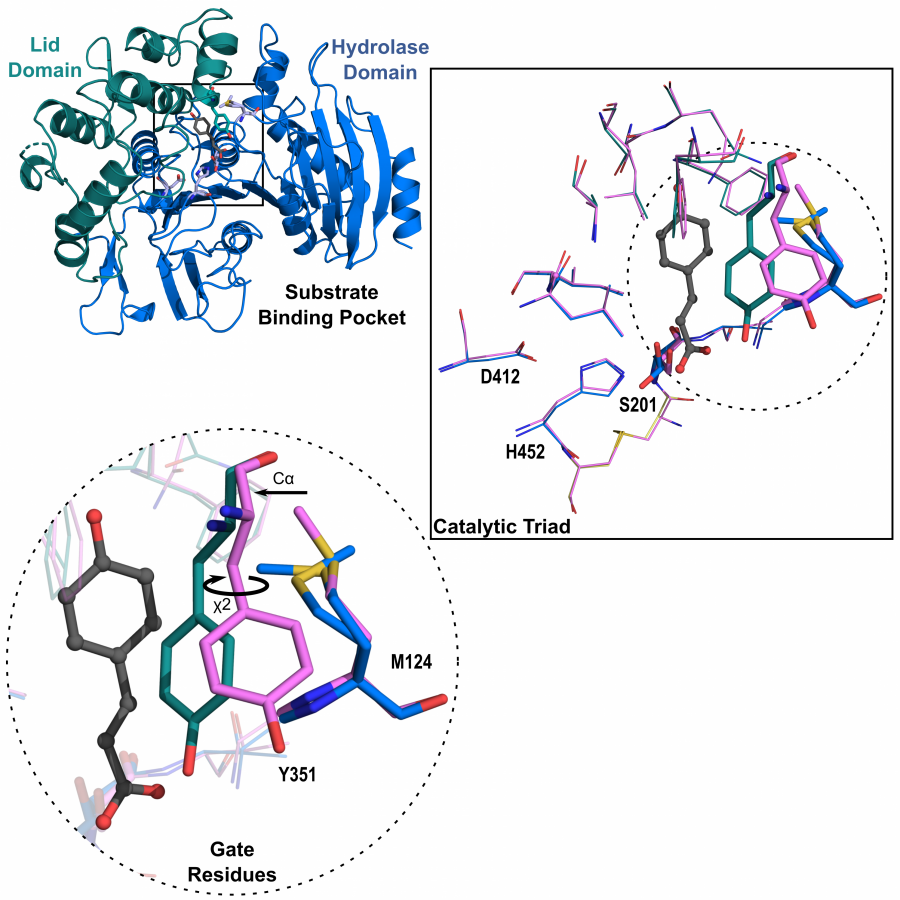 Figure 1. Cartoon representation of FoFaeC monomer in complex with p-coumaric acid and close-up view of the substrate binding pocket. |
#1408 | Transaminase-catalyzed crystallization assisted synthesis of enantiopure beta-methylphenethylamine |
|
| Presenting author: | Feodor BELOV of UNIVERSITY OF ROSTOCK |
| Corresponding author: | Jan VON LANGERMANN of OTTO-VON-GUERICKE-UNIVERSITY MAGDEBURG |
| Other authors: | Hannah BORK of UNIVERSITY OF BIELEFELD Harald GRÖGER of UNIVERSITY OF BIELEFELD Jan VON LANGERMANN of OTTO-VON-GUERICKE-UNIVERSITY MAGDEBURG |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | beta-chiral amines / crystallization / transaminase / |
| Purpose: | The synthesis of β-chiral amines with biocatalytic methods has been an object of study for some time. A typical example of such a β-chiral amine, possessing applicability as a pharmaceutical building block, is β-methylphenethylamine (β-MPEA). The attempts for synthesis of this amine range from multi-step chemocatalytic approaches [1],[2] to enzymatic cascades [3]. However, only two enzymatic one-step approaches in recent years achieved β-MPEA yields higher than 10 mM, namely one utilizing IREDs [4] and one utilizing transaminases (TAs) [5] in the synthesis process. In this study, we synthesize β-MPEA in a one-step reaction utilizing another well-studied transaminase from Ruegeria pomeroyi (Rpo-ATA, pdb:3HMU), thus widening the substrate scope of this enzyme with β-chiral amines. The reaction is characterized high yields utilizing high substrate concentrations, which are further improved by the integration of simultaneous in-situ product crystallization (ISPC) of the amine product to overcome product inhibition (Figure 1). By trying to produce further derivates of β-MPEA, we aim to show the potential for further engineering of this well-studied enzyme towards new substrate scopes. |
| References: | [1] Y. Ding, S. Luo, C. Weng, J. An, J. Org. Chem. 2019, 84(23), 15098-15105. [2] M. Liu, D. Kong, M. Li, G. Zi, G. Hou, Adv.Synth.Catal.2015,357,3875-3879. [3] R. Xin, W.W. L. See, H. Yun, X. Li, Z. Li, Angew. Chem.Int. Ed.2022,61(28),e202204889. [4] P. Matzel, S. Wenske, S. Merdivan, S. Gunther, M. Hohne, ChemCatChem 2019, 11, 4281-4285. [5] C. S. Fuchs, M. Hollauf, M. Meissner, R. C. Simon, T. Besset, J. N. H. Reek, W. Riethorst, F. Zepeck, W. Kroutil, Adv. Synth. Catal. 2014, 356, 2257 - 2265. |
| Figures: | 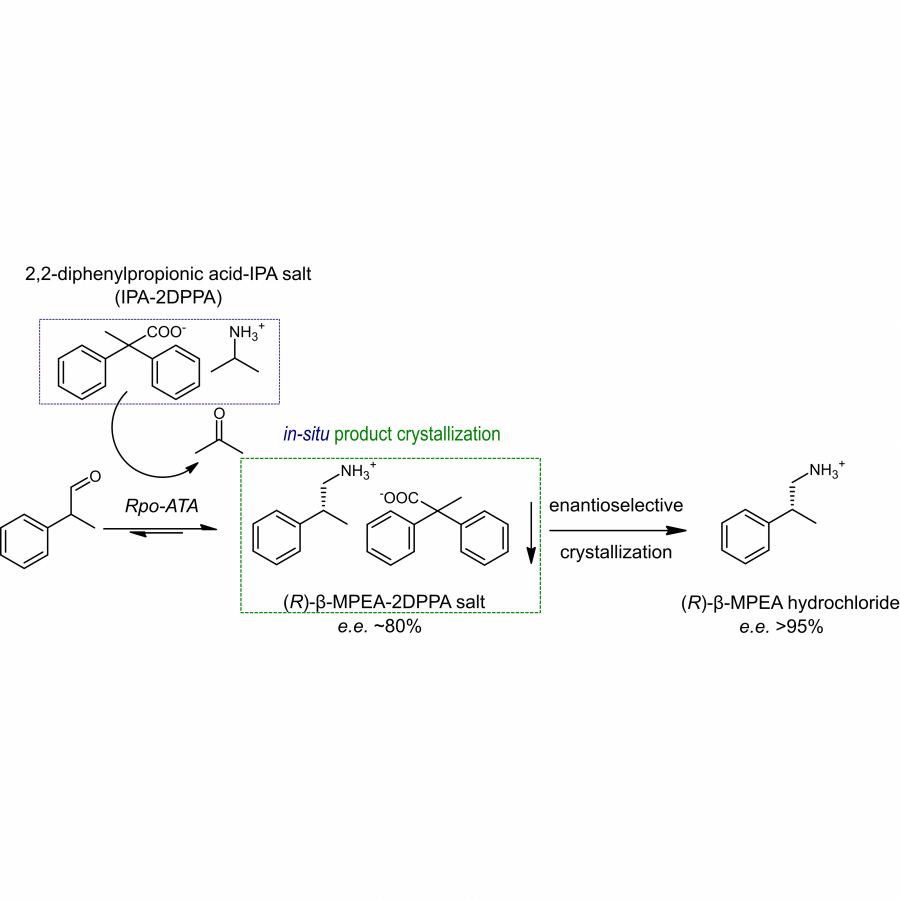 Reaction scheme of Rpo-ATA yielding β-MPEA with integrated ISPC Reaction scheme of Rpo-ATA yielding β-MPEA with integrated ISPC |
#1409 | Activity, stability and applications of aldoxime dehydratase biocatalysts immobilized on metal affinity resins |
|
| Presenting author: | Barbora KRISTKOVA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Other authors: | Ludmila MARTINKOVA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Robert RADISCH of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Lenka RUCKA of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Natalia KULIK of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES Margit WINKLER of ACIB GMBH AND GRAZ UNIVERSITY OF TECHNOLOGY Miroslav PATEK of INSTITUTE OF MICROBIOLOGY OF THE CZECH ACADEMY OF SCIENCES |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Aldoxime dehydratase / (E,Z)-Phenylacetaldoxime / Phenylacetonitrile / Metal affinity resin |
| Purpose: | Aldoxime dehydratase (Oxd) is a valuable tool in organic synthesis, for producing nitriles from aldoximes in aqueous media at near-neutral pH and moderate temperatures, and, furthermore, with enantioselectivity to certain chiral aldoximes. However, the robustness of the Oxds needs to be increased to fully exploit their potential. One of the ways to achieve this goal is enzyme immobilization, but this has rarely been used for Oxds (e.g. in the form of whole cells immobilized in polyacrylic acid and used in organic solvents [1]). In this study, we immobilized an Oxd on metal affinity resins (Talon, Ni-NTA) for the first time. Previously, this type of method was used, e.g., for the immobilization of a carboxylate reductase [2]. Here, we used OxdFv (an Oxd from Fusarium vanettenii), which we previously overproduced in Escherichia coli. This enzyme is of great interest, because it does not require anaerobic conditions. We also optimized the reaction to maximize the nitrile yield [3]. OxdFv was applied as a crude extract on Talon and Ni-NTA resins, and the unbound protein was washed out. The activities and stabilities of the immobilized Oxd catalysts were studied using a model reaction of (E,Z)-phenylacetaldoxime to phenylacetonitrile. The catalysts could be used for at least five cycles with over 40% conversion in each (Figure 1). The same approach can be used for preparative reactions, which increases the cost efficiency of the process in terms of catalyst productivity. Other arylacetonitriles, and alkylnitriles can be prepared in an analogous way. Acknowledgements: This research was funded by Czech Science Foundation (grant number GF20-23532L), and the Austrian Science Fund FWF (grant number I 4607-N). |
| References: | [1] A. Hinzmann, N. Adebar, T. Betke, M. Leppin, H. Gröger, Eur. J. Org. Chem. 2019, 6911-6916. [2] M.M. Maphatsoe, C. Hashem, J.G. Ling, M. Horvat, K. Rumbold, F.D. Abu Bakar, M. Winkler, J. Biotechnol. 2022, 345, 47-54. [3] B. Křístková, R. Rädisch, N. Kulik, M. Horvat, L. Rucká, M. Grulich, F. Rudroff, A. Kádek, M. Pátek, M. Winkler, L. Martínková, Enzyme Microb. Technol. 2023, 164, 110187. |
| Figures: | 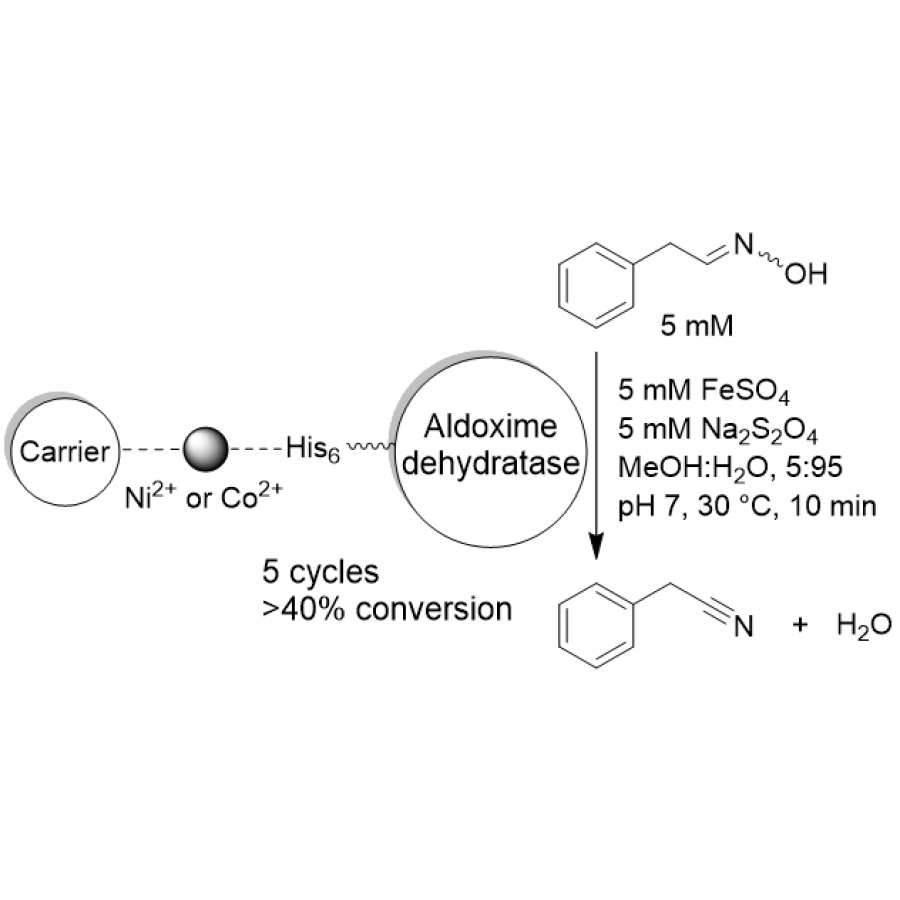 Figure 1 Conversion of (E,Z)-phenylacetaldoxime to phenylacetonitrile by aldoxime dehydratase immobilized on nickel or cobalt affinity resin. |
#1410 | Crystallization-based downstream processing of ω-transaminase- and amine dehydrogenase-catalyzed reactions |
|
| Presenting author: | Feodor BELOV of UNIVERSITY OF ROSTOCK |
| Corresponding author: | Jan VON LANGERMANN of OTTO-VON-GUERICKE-UNIVERSITY MAGDEBURG |
| Other authors: | Andrea MILDNER of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG Tanja KNAUS of UNIVERSITY OF AMSTERDAM Francesco MUTTI of UNIVERSITY OF AMSTERDAM Jan VON LANGERMANN of OTTO-VON-GUERICKE-UNIVERSITY MAGDEBURG |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transaminase / amine dehydrogenase / downstream processing / crystallization |
| Purpose: | Biocatalytic synthesis is a powerful and frequently chosen method to produce chiral amines.[1] Unfortunately, these biocatalytic reactions often result into complex mixtures, bearing many components aside the main product amine such as residual co-substrates, co-products, cofactors and buffer salts. This issue typically requires an additional effort during downstream-processing towards the isolation of the desired chiral amine.[2],[3] For instance, transaminase- and amine dehydrogenase-catalyzed reactions, which often use high surpluses of amine or ammonia co-substrates, face complications in removing the residual amine donor or unreacted substrate and salts from the isolated amine products, thus complicating and increasing the costs of the process of product isolation and purification.[4], [5] This study explores the selective removal of chiral amines from model amine transaminase and amine dehydrogenase-catalyzed reactions via a salt-based specific crystallization step (see Figure 1). The product amine is precipitated directly in one step from the reaction mixture as a product ammonium salt, which can easily be filtered off the reaction mixture, while the other reactants remain unchanged in solution for a potential re-use. |
| References: | [1] S. Wu, R. Snajdrova, J.C. Moore, K. Baldenius and U.T. Bornscheuer, Angew Chem Int Ed Engl, 2021, 60(1), 88. [2] C. Matassa, D. Ormerod, U.T. Bornscheuer, M. Höhne and Y. Satyawali, Process Biochemistry, 2019, 80, 17. [3] I. Slabu, J.L. Galman, C. Iglesias, N.J. Weise, R.C. Lloyd and N.J. Turner, Catalysis Today, 2018, 306, 96. [4] J.L. Galman, I. Slabu, N.J. Weise, C. Iglesias, F. Parmeggiani, R.C. Lloyd and N.J. Turner, Green Chem., 2017, 19(2), 361. [5] V. Tseliou, T. Knaus, M.F. Masman, M.L. Corrado and F.G. Mutti, Nat Commun, 2019, 10(1), 3717. |
| Figures: | 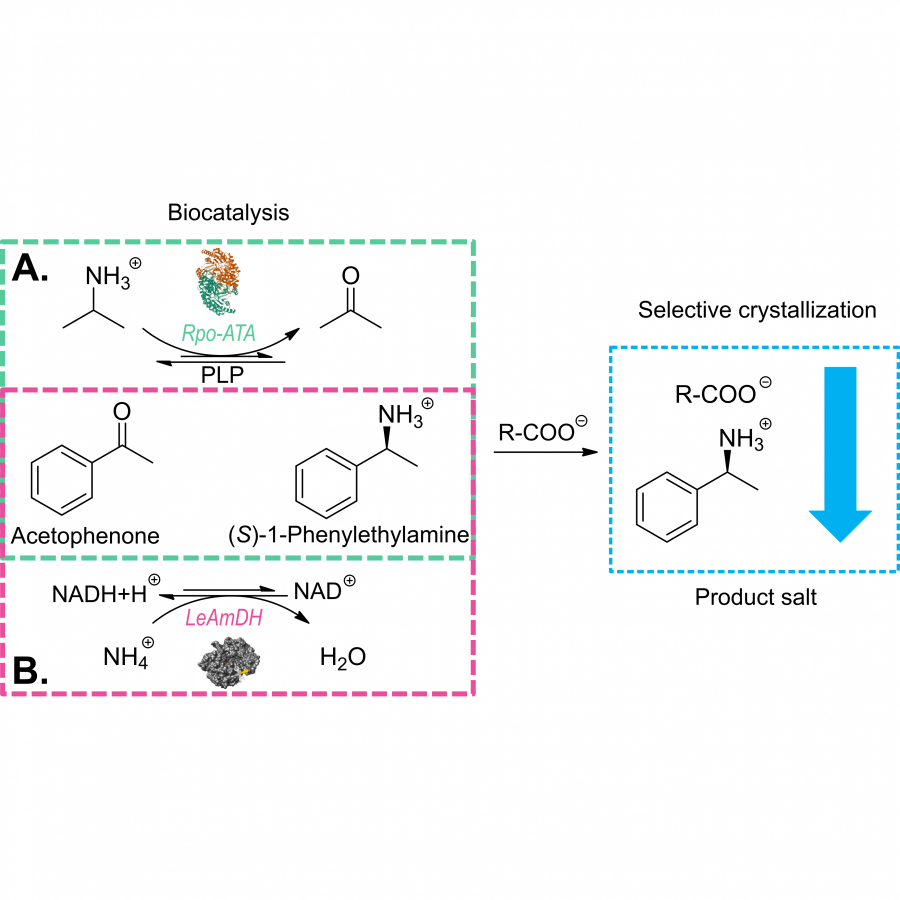 Reaction scheme of selective (S)-1-Phenylethylamine crystallization obtained from Rpo-ATA and LeAmDH catalyzed reactions. Reaction scheme of selective (S)-1-Phenylethylamine crystallization obtained from Rpo-ATA and LeAmDH catalyzed reactions. |
#1411 | A fluorogenic kinetic assay for the discovery of N-acetylneuraminic acid synthase and aldolase |
|
| Presenting author: | John MARTINEZ of UAM |
| Corresponding author: | Aurelio HIDALGO of UNIVERSIDAD AUTONOMA DE MADRID |
| Other authors: | Simon CHARNOCK of PROZOMIX LIMITED Wolf-Dieter FESSNER of TECHNISCHE UNIVESITÄT DARMSTAD Aurelio HIDALGO of UNIVERSIDAD AUTONOMA DE MADRID |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymes / Biocatalysis / Microfluidics / Sialic acid |
| Purpose: | N-acetylneuraminic acid (Neu5Ac) and its variants constitute a diverse group of nine-carbon monosaccharides involved in cell communication and recognition processes. Due to their structural and functional diversity, there is a growing interest on the discovery of enzymes that catalyse their synthesis for the industrial production of pharmaceutical and nutritional compounds. In this matter, prokaryotic Neu5Ac synthase and aldolase have been considered potential candidates. However, conventional activity-based screening of metagenomic libraries requires a considerable use of economic resources and time. Therefore, (ultra)high-throughput (HTP) methods are needed for a more efficient biocatalyst discovery [1, 2]. Thus, the aim of the current project is to develop a droplet based ultra-HTP screening assay for the functional discovery of Neu5Ac synthases and aldolases. The proposed droplet-based screening method for discovery of Neu5Ac synthases and aldolases consisted in the coupling of Neu5Ac synthase, Neu5Ac aldolase, lactate dehydrogenase (LDH) and a ketoreductase (KRED) to link the main reaction to the development of a fluorescent signal [3]. In order to find a selective ketoreductase able to catalyse the oxidation of a fluorogenic substrate, in silico screening was performed. The coupling of Neu5Ac synthase, Neu5Ac aldolase, and LDH was achieved first in microtiter plate format. Then, a specific KRED was selected from a library of metagenomic sequences and the full coupled assay was evaluated for its use in microfluidic droplet scale. Acknowledgements The current work has received funding from the European Union’s Horizon 2020 and Horizon Europe Research and Innovation Programmes under Grant Agreement number 956631 and 101081957 respectively. |
| References: | sheludko, y., v and fessner, w.-d. curr. opin. struct. biol. 2020, 63, 123-133. jacque, p. et al. bioprocess biosyst. eng. 2017. 40. 161-180. thai, y., c. et al. bioorganic med. chem. 2018. 26. 1320-1326. |
#1413 | Sulphotransferases: Enabling green and effective sulphation of bioactive complex molecules |
|
| Presenting author: | Elena TOMARELLI of UNIVERSITY OF PERUGIA |
| Other authors: | Tanja KNAUS of UNIVERSITY OF AMSTERDAM Antimo GIOIELLO of UNIVERSITY OF PERUGIA Francesco MUTTI of UNIVERSITY OF AMSTERDAM |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sulphation / enzymatic cascade / steroid / cofactor regeneration |
| Purpose: | The sulphation of biomolecules is a widespread process in nature, which occurs in various organisms, ranging from prokaryotes to multicellular species, and numerous biological functions are associated with this crucial transformation.1 The enzymes responsible for sulphation are sulphotransferases, which transfer the sulphate group from the 3′-phosphoadenosine-5′-phosphosulfate (PAPs) cofactor to the substrate of interest.2 Among the different molecules which could be sulphated, steroids represent an intriguing class since these compounds play a significant role in several pathophysiological processes.3 Given the increasing interest in these molecules and the limitation of conventional chemical sulphation—that involves the use of toxic reactants, harsh conditions, and results in mediocre selectivity—4 alternative biocatalytic methods are highly desirable. That being said, enzymatic sulphation has been poorly investigated to date: the high cost and instability of the PAPs required cofactor make the biocatalytic approach indeed extremely challenging. To address this issue and expand the previously limited substrate scope, inspired by nature we developed a biocatalytic sulphation approach that employs an enzymatic cascade for in-situ synthesis and regeneration of the cofactor from cheap and readily available ATP and p-nitrophenol. Furthermore, novel sulphotransferases that are active on steroids have been identified through a survey of the biochemistry literature as well as an analysis of protein and genome databases. These enzymes have been recombinantly expressed in E. coli in good yields and obtained in highly purified form. In conclusion, this biocatalytic strategy along with a new toolbox of sulphotransferases would allow the sulphation not only of steroids but also of many structurally diverse molecules. 1 Chapman, E.; Best, M. D.; Hanson, S. R.; Wong, C.-H. Angew.Chem.Int.Ed. 2004, 43, 3526 –3548. 2 a) Duffel, M. W. Chapter 4.18, Sulfotransferases. In Comprehensive Toxicology (Second Edition), 2010; 4, 367-384 b) Bojarová, P.; Williams, S. J. Curr. Opin. Chem. Biol. 2008, 12, 573-581. 3 a) Mueller, J. W.; Gilligan, L. C.; Idkowiak, J.; Arlt, W.; Foster, P. A. Endocr. Rev., 2015, 36, 526–563; (b) Foster, P. A.; Mueller, J. W. J. Mol. Endocrinol., 2018, 61, T271–T283. c) Pounina, T.A.; Gloriozova, T.A.; Savidov, N.; Dembitsky, V.M. Mar. Drugs 2021, 19, 240. d) Carvalhal, F.; Correia-da-Silva, M.; Sousa, E.; Pinto, M.; Kijjoa, A. J. Mol. Endo., 2018, 61, 2, T211–T231. 4 Badri, A.; Williams, A.; Xia, K.; Linhardt, R. J.; Koffas, M. A. G. Biotechnol. J. 2019, 14, 1800436. |
| References: | 1 Chapman, E.; Best, M. D.; Hanson, S. R.; Wong, C.-H. Angew.Chem.Int.Ed. 2004, 43, 3526 -3548. 2 a) Duffel, M. W. Chapter 4.18, Sulfotransferases. In Comprehensive Toxicology (Second Edition), 2010; 4, 367-384 b) Bojarová, P.; Williams, S. J. Curr. Opin. Chem. Biol. 2008, 12, 573-581. 3 a) Mueller, J. W.; Gilligan, L. C.; Idkowiak, J.; Arlt, W.; Foster, P. A. Endocr. Rev., 2015, 36, 526-563; (b) Foster, P. A.; Mueller, J. W. J. Mol. Endocrinol., 2018, 61, T271-T283. c) Pounina, T.A.; Gloriozova, T.A.; Savidov, N.; Dembitsky, V.M. Mar. Drugs 2021, 19, 240. d) Carvalhal, F.; Correia-da-Silva, M.; Sousa, E.; Pinto, M.; Kijjoa, A. J. Mol. Endo., 2018, 61, 2, T211-T231. 4 Badri, A.; Williams, A.; Xia, K.; Linhardt, R. J.; Koffas, M. A. G. Biotechnol. J. 2019, 14, 1800436. |
#1414 | Application of Self-Immobilizing Decarboxylases: From Biogenic Nanoparticles to Monolithic All-Enzyme Hydrogels in Flow |
|
| Presenting author: | Astrid WINTERHALTER of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY |
| Corresponding author: | Kersten S. RABE of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY |
| Other authors: | Esther MITTMANN of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY Martin PENG of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY Christof M. NIEMEYER of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY Kersten S. RABE of INSTITUTE FOR BIOLOGICAL INTERFACES (IBG-1), KARLSRUHE INSTITUTE OF TECHNOLOGY, KARLSRUHE, GERMANY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / enzyme immobilization / phenolic acid decarboxylase / genetic engineering |
| Purpose: | Introduction and Research Concept: The beneficiation of lignin as the largest regenerative source for aromatic compounds is in the focus of scientific research. For this matter, the application of biocatalysts such as phenolic acid decarboxylases (PADs) are required to valorize lignin product streams. Biocatalytic flow reactor systems enable the synthesis of industrially relevant styrenes. However, high and stable conversion is limited by inefficient immobilization of enzymes. Hence, new strategies providing precise temporal and spatial reaction control in flow reactors are needed.[1] Given the tremendous impact of continuous reactor formats, we developed decarboxylative flow reactors based on self-immobilizing PADs and evaluated their performance to boost productivity. Methods and Results: Carrier-free enzyme immobilization technologies are a major development in the field of streamlined continuous processes. These were expanded to PADs in the form of monolithic, self-assembling all-enzyme hydrogels, generating highly active materials with tunable rheological properties. Arranged in microfluidic reactors, the biocatalytic gels reached near quantitative conversion (> 90%) along with high robustness under process and storage conditions.[2] Complementary, isolated biogenic magnetosomes displaying SpyCatcher moieties enable a flexible magnetic reactor design.[3] Following in vivo expression of such constructs, Spy-Tag-equipped PAD could be immobilized on the nanoparticles in vitro, demonstrating the simplicity and versatility of the application in biocatalytic processes. Commercially available particles with similar functionalizations were surpassed by the stable substrate conversion for prolonged times by our nanobiocatalysts. In a parallel approach we demonstrate the utility of thermostable enzymes in the generation of biocatalytic agarose-based inks for a simple temperature-controlled 3D printing process for a two-step sequential biotransformation in a fluidic setup.[4] Conclusion: Genetically encodable immobilization tags enable self-assembling biocatalytic reactor concepts with unprecedented elegant simplicity and efficiency. Especially in combination with magnetosomes, they enable a completely biologically produced alternative to commercial solutions. |
| References: | [1] T. Peschke, P. Bitterwolf, S. Hansen, J. Gasmi, K. S. Rabe, C. M. Niemeyer (2019) "Self-Immobilizing Biocatalysts Maximize Space-Time Yields in Flow Reactors", Catalysts, 9, 164, doi: 10.3390/catal9020164. [2] E. Mittmann, S. Gallus, P. Bitterwolf, C. Oelschlaeger, N. Willenbacher, C. M. Niemeyer, K. S. Rabe (2019) "A Phenolic Acid Decarboxylase-Based All-Enzyme Hydrogel for Flow Reactor Technology", Micromachines, 10, doi: 10.3390/mi10120795. [3] E. Mittmann, F. Mickoleit, D. S. Maier, S. Y. Stäbler, M. A. Klein, C. M. Niemeyer, K. S. Rabe, D. Schüler (2022) "A Magnetosome-Based Platform for Flow Biocatalysis", ACS Appl. Mater. Interfaces, doi: 10.1021/acsami.2c03337. [4] M. Peng, E. Mittmann, L. Wenger, J. Hubbuch, M. K. M. Engqvist, C. M. Niemeyer, K. S. Rabe (2019) "3D-Printed Phenacrylate Decarboxylase Flow Reactors for the Chemoenzymatic Synthesis of 4-Hydroxystilbene", Chem-Eur. J., 25, 15998-16001, doi: 10.1002/chem.201904206. |
| Figures: | 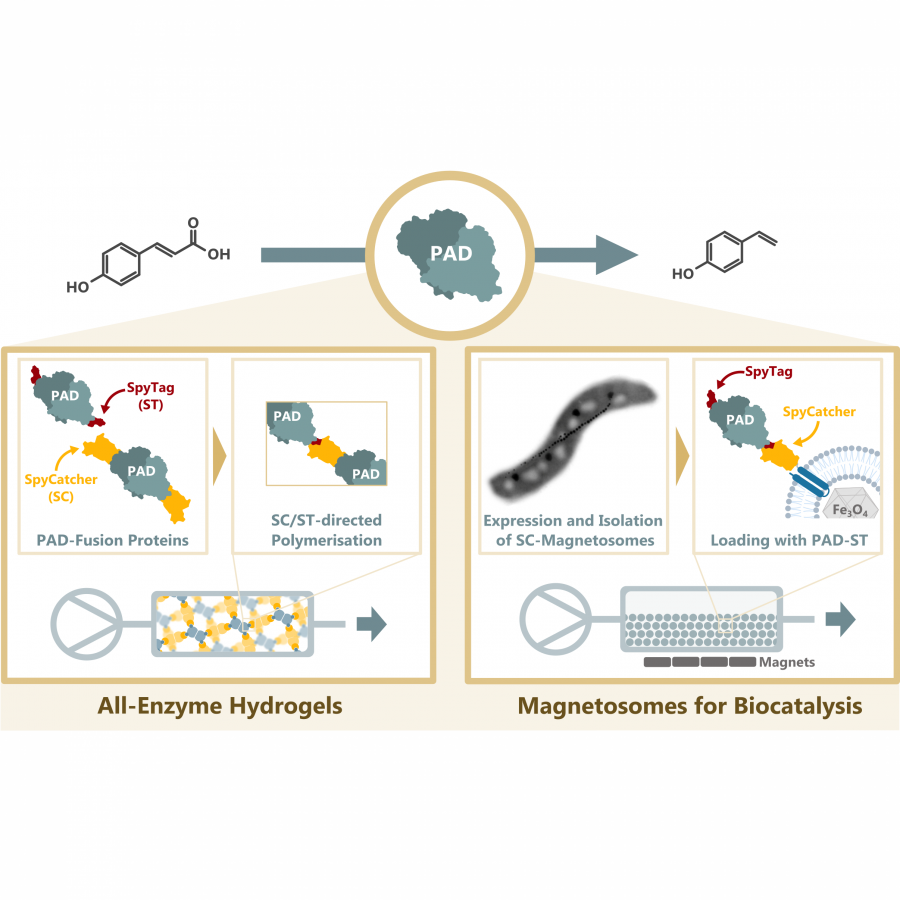 Immobilizing Strategies for PAD in Flow Reactors Continuous flow production of p-hydroxystyrene from p-coumaric acid by a phenolic acid decarboxylase (PAD) immobilized as an all-enzyme hydrogel (left) or on functionalized magnetosomes using the SpyCatcher-SpyTag system. |
#1415 | Model-based Optimization of Neu5Ac Synthesis |
|
| Presenting author: | Mehmet Mervan ÇAKAR of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB |
| Other authors: | Theofania Pagona ANDREADAKI of PROZOMIX LIMITED Simon CHARNOCK of PROZOMIX LIMITED Wolf-Dieter FESSNER of INSTITUTE OF ORGANIC CHEMISTRY AND BIOCHEMISTRY, TECHNISCHE UNIVERSITAET DARMSTADT Zvjezdana FINDRIK BLAŽEVIĆ of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Neu5Ac synthase / Enzyme kinetics / Optimization / Biocatalysis |
| Purpose: | Sialic acids (SAs) are a group of more than 50 structurally distinct α-keto acids found in viruses, mammalian cells, and microorganisms1. They control various biological functions such as development, recognition, cell signaling, cell-cell interactions, and adhesion2. Sialylation of neural cell adhesion molecules during embryonic development is crucial for proper neural tissue development, while in certain cancers, sialylation is correlated with tumorigenesis and metastasis3. Pathogenic bacteria use sialylated glycoproteins and glycolipids to mask their presence from the host’s immune system. There are over fifty molecules of sialic acid that carry diverse substituents at hydroxyl or amino groups, and their distribution is strongly regulated on a gene level and varies depending on the species of animal and cell's function4. N-Acetylneuraminic acid (Neu5Ac) is the most commonly occurring and studied SA, and its biosynthesis is controlled by Neu5Ac synthase, which catalyzes the aldol-like condensation of phosphoenolpyruvate (PEP) to N-Acetylmannosamine (ManNAc) to yield Neu5Ac. In this work, a thorough kinetic analysis of Neu5Ac synthase (NeuB) catalysed synthesis of Neu5Ac was done in order to build the kinetic model and use it to find the bottlenecks of the system and optimize the reaction. The interdependent relationships between process variables of NeuB from Neisseria meningitidis and a promising NeuB homologue from metagenomic library were successfully investigated and evaluated for their potential use in SA synthesis5. Acknowledgements: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 956631 |
| References: | [1] Varki, A. Glycobiology. 1993, 3, 97-130. [2] Cunningham, B. A., Hoffman, S., Rutishauser, U., Hemperly, J. J. & Edelman, G. M. PNAS USA. 1983, 80, 3116-3120. [3] Gorelik, E., Galili, U. & Raz, A. Cancer Metastasis Rev. 2001, 20, 245-277. [4] Schauer, R. Carbohydrates in Chemistry and Biology. 2008, 3-4, 227-243. [5] Sudar, M. & Blažević, Z. F. Enzyme Cascade Design and Modelling, Springer International Publishing, 2021, 91-108. |
| Figures: | 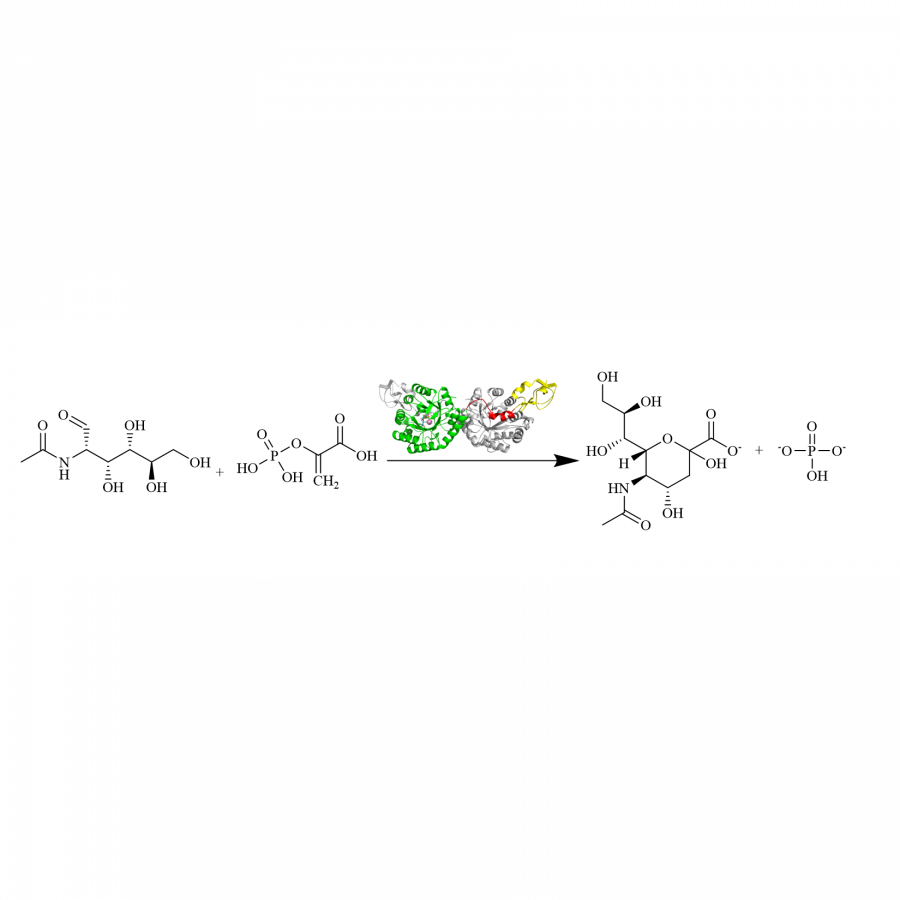 Scheme 1. NeuS catalyzed Neu5Ac synthesis from ManNAc and PEP via aldol-like addition |
#1416 | The synergistic relationships between main chain-acting and debranching enzymes towards the efficient biodegradation of xylan |
|
| Presenting author: | ZERVA Anastasia of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Corresponding author: | Evangelos TOPAKAS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | Anastasia ZERVA of NATIONAL TECHNICAL UNIVERSITY OF ATHENS Christina PENTARI of NATIONAL TECHNICAL UNIVERSITY OF ATHENS Evangelia MYLONA of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biorefinery / lignocellulose decomposition / enzyme cocktails / xylan degradation |
| Purpose: | Enzymatic utilization of lignocellulosic biomass for industrial applications has been proposed to require a wide spectrum of biocatalytic activities. Hemicellulose is a complex heteropolymer that confers rigidity on biomass structures and recalcitrance against biodegradation. Xylan in particular, mostly encountered in hardwood biomass, carries a variety of substitutions that promote molecular interactions with cellulose and lignin. Acetic acid, ferulic acid, arabinosyl and glucuronoyl residues are some of the most common substitutions that occupy positions O-2, O-3 or even O-4 of the xylopyranosyl residue (Xylp), and has been shown to inhibit enzymatic hydrolysis of the xylan main chain by xylanases and xylosidases. Therefore, the addition of accessory enzymes, such as acetyl-esterases, ferulic acid esterases, glucuronoyl esterases, α-L-arabinofuranosidases, α-glucuronidases and lytic polysaccharide monooxygenases, can help overcome the recalcitrance of hemicellulose by either removing the decorations of xylan or disassociating xylan from other biomass components and thus enabling xylanases to proceed with hydrolysis. In this work, the synergistic effect between a number of accessory enzymes and a set of hemicellulose main-chain degrading enzymes has been examined, in order to gain some insight regarding the modifications of the biomass required in order to promote their activity. During hydrolysis of arabinoxylan-containing substrates, the removal of arabinofuranosyl residues was investigated using two arabinofuranosidases of distinct specificities, TtAbf43 and AnAbf51, in combination with xylanases of different glycoside hydrolase (GH) families. TtAbf43 removes the side-group from position O-3 of doubly arabinofuranosyl-substituted Xylps, whereas AnAbf51 hydrolyzes arabinose substitutions from positions O-2 or O-3 of singly substituted xyloses. The disassociation of glucuronoxylan form lignin moieties was investigated with the use of two glucuronoyl esterases of the carbohydrate esterase (CE) family 15, TlGE15 and AeGE15, by testing activity on the liquid and the solid fraction of the xylanase-hydrolyzed pretreated beechwood samples. The activity of accessory enzymes on hemicellulose resulted to increased hydrolysis efficiency by the xylanases. During hydrolysis of polymeric arabinoxylan, TtAbf43 was able to unblock the xylobiohydrolase activity of TtXyn30A, while in the presence of both arabinofuranosidases the xylobiose release was further increased. During hydrolysis of pretreated wheat bran, TtAbf43 and AnAbf51 exhibited synergistic relationships with xylanases of the GH10, GH11 and GH30 families, where both xylanase and arabinofuranosidase activities were enhanced. Regarding the degradation of glucuronoxylan-lignin complexes, TlGE15 and AeGE15 exhibited activity both on the xylanase-derived hydrolysate and the residual biomass fraction. Overall data suggests that a cautious design of enzymatic cocktails, bearing in mind the composition of each substrate and the exact specific activities of the enzymes to be involved, could significantly promote biomass biodegradation. In this way, exploitation of residual biomass gains potential for the production of value-added carbohydrate-based chemicals, while minimizing the enzyme load of the treatment, contributing thus to the concept of circular bioeconomy. This research was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “2nd Call for H.F.R.I. Research Projects to support Post-Doctoral Researchers” – Project ‘ARSIS’ (Project Number: 00328) |
| References: | [1] Katsimpouras, C. et al., Biotech Biofuels, 2019, 12, 120 [2] Malgas, S. et al., Molecules, 2021, 26(22), 6770 [3] Pentari, C. et al., Carbohydr Polym, 2023, 305, 120527 [4] Pouvreau, L. et al., Enzyme Microb Technol., 2011, 48, 4-5, 397-403 [5] Zerva, A. et al., Bioresour Technol, 2021, 342, 126058 [6] Zong, Z., et al., Nat Commun, 2022, 13. 1449 |
| Figures: | 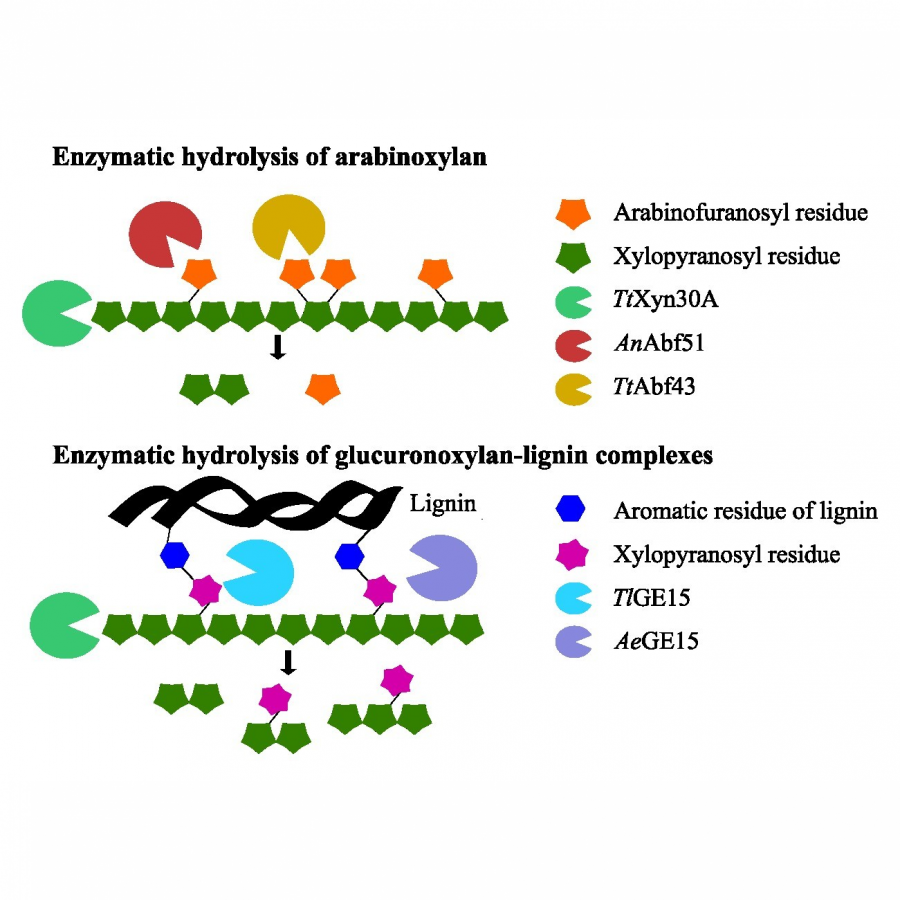 Enzymatic hydrolysis of arabino-and glucuronoxylan Fig. 1. Enzymatic hydrolysis of decorated arabino- and glucuronoxylan  Acknowledgements Hellenic Foundation for Research and Innovation |
#1417 | Controlling the selectivity of peroxygenases via pH |
|
| Presenting author: | PABLO VELAZQUEZ of TU DELFT |
| Topic: | Reaction design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | PaDa-I / Biocatalytic halogenation / Halogenation selectivity / |
| Purpose: | Unspecific peroxygenases from fungal species (UPOs) are well known for their capability to catalyze oxyfunctionalization reactions by using H2O2 as oxidant and have the unique ability to perform a broad range of reactions, including epoxidation, hydroxylation, demethylation, and even oxygenation of non-activated C-H bonds. The first described UPO (AaeUPO) was also reported to present haloperoxidase activity with the halogenation of MCD with a specific activity of 354,3 U mg -1 (1). For this reason, we decided to use the rAaeUPO (PaDa-I variant) for the generation of OBr - and catalyze halohydroxylation reactions. In this work, we chose styrene as a substrate and our goal is to show that the rAaeUPO can catalyze its halohydroxylation into 2-bromo-1 phenylethanol. |
| References: | 1. Hofler, G. T.; But, A.; Hollmann, F. Haloperoxidases as catalysts in organic synthesis, Organic & Biomolecular Chemistry 2019, 17, 9267-9274. 2 Fernandez-Fueyo, E.; Wingerden, M.; Renirie, R.;Wever, R; Ni, Y.; Holtmann, D.; Hollmann, F. Chemoenzymatic Halogenation of Phenols by using the Haloperoxidase from Curvularia inaequalis, ChemCatChem 2015, 7,4035-4038 |
| Figures: | 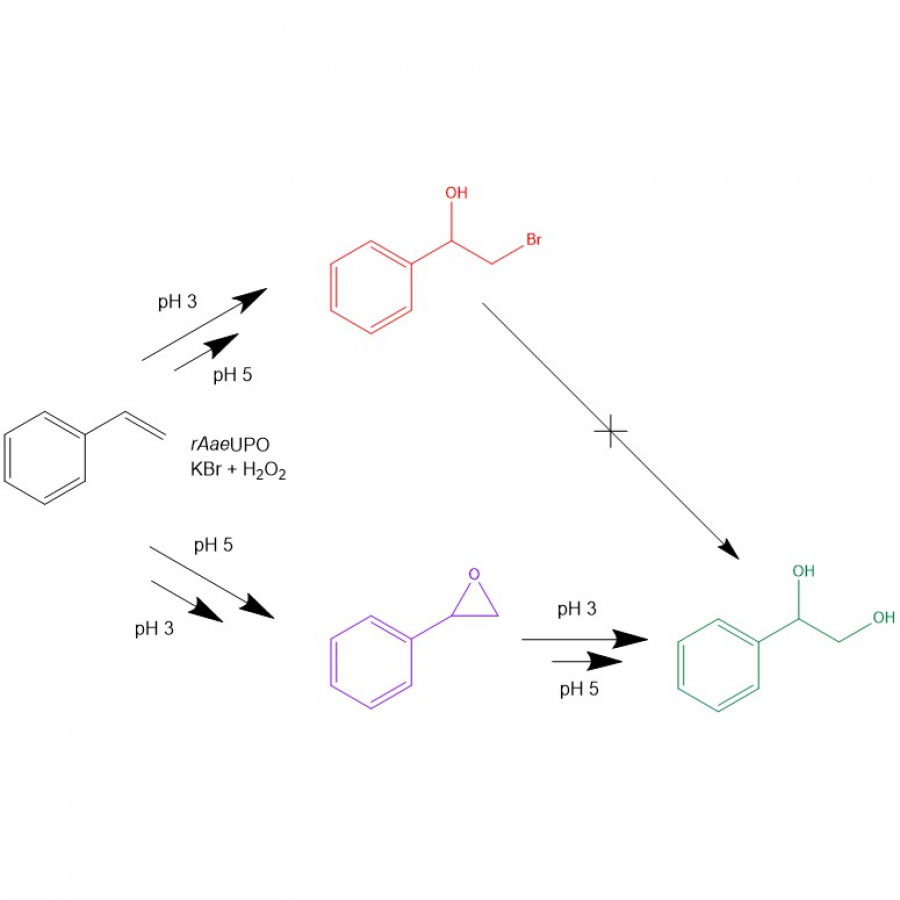 Reaction scheme Fig. 2. Modulating the reaction selectivity via pH |
#1418 | Electrophile promiscuity of KdoS and AroS phosphoenolpyruvate (PEP) dependent aldolases in aldol addition reactions |
|
| Presenting author: | Mehmet Mervan ÇAKAR of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB |
| Other authors: | Karel HERNÁNDEZ of CATALONIA INSTITUTE FOR ADVANCED CHEMISTRY-IQAC-CSIC, DEPT. CHEMICAL BIOLOGY AND MOLECULAR MODELLING Theofania Pagona ANDREADAKI of PROZOMIX LIMITED Simon CHARNOCK of PROZOMIX LIMITED Zvjezdana FINDRIK BLAŽEVIĆ of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY, UNIVERSITY OF ZAGREB Pere CLAPÉS of CATALONIA INSTITUTE FOR ADVANCED CHEMISTRY-IQAC-CSIC, DEPT. CHEMICAL BIOLOGY AND MOLECULAR MODELLING |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | PEP-Dependent Synthase / Enzyme Promiscuity / Aldol addition / Biocatalysis |
| Purpose: | The formation of carbon–carbon bonds is strategic reaction in organic synthesis as it allows for the construction of the carbon scaffolds of organic molecules to generate more complex compounds from simpler ones1. 2-keto-3-deoxyoetosonic acid 8-phosphate synthase (KdoS, EC 2.5.1.55) and 3-deoxy-D-arabino-heptulosonic acid 7-phosphate synthase (AroS, EC 2.5.1.54) catalyze the irreversible addition of phosphoenolpyruvate (PEP) to D-arabinose 5-phosphate (Ara5P) and D-erythrose-4-phosphate (Ery4P) to yield 3-deoxy-D-manno-octulosonate-8-phosphate (KDO-8-P) and 3-deoxy-D-arabino-heptulosonic acid 7-phosphate (DAHP), respectively. In this communication, metagenomic libraries of KdoS and AroS were screened to identify promising candidates capable of catalyzing the promiscuous aldol addition of phosphoenolpyruvate to aliphatic aldehydes (e.g. 2-(benzyloxy)acetaldehyde, 2-phenylacetaldehyde and 3-phenylpropanal)3 (Scheme 1). Acknowledgements: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 956631 |
| References: | [1] Schmidt, N. G., Eger, E. & Kroutil, W. B. ACS Catal. 2016, 6, 4286-4311. [2] Fessner, W.-D. & Walter, C. 1996, 184, 97-194. [3] Moreno, C. J., Hernández, K., Charnok, S. J., Gittings, S., Bolte, M., Joglar, J., Bujons, J., Parella, T., Clapés, P. ACS Catal. 2021, 11, 4660-4669. |
| Figures: | 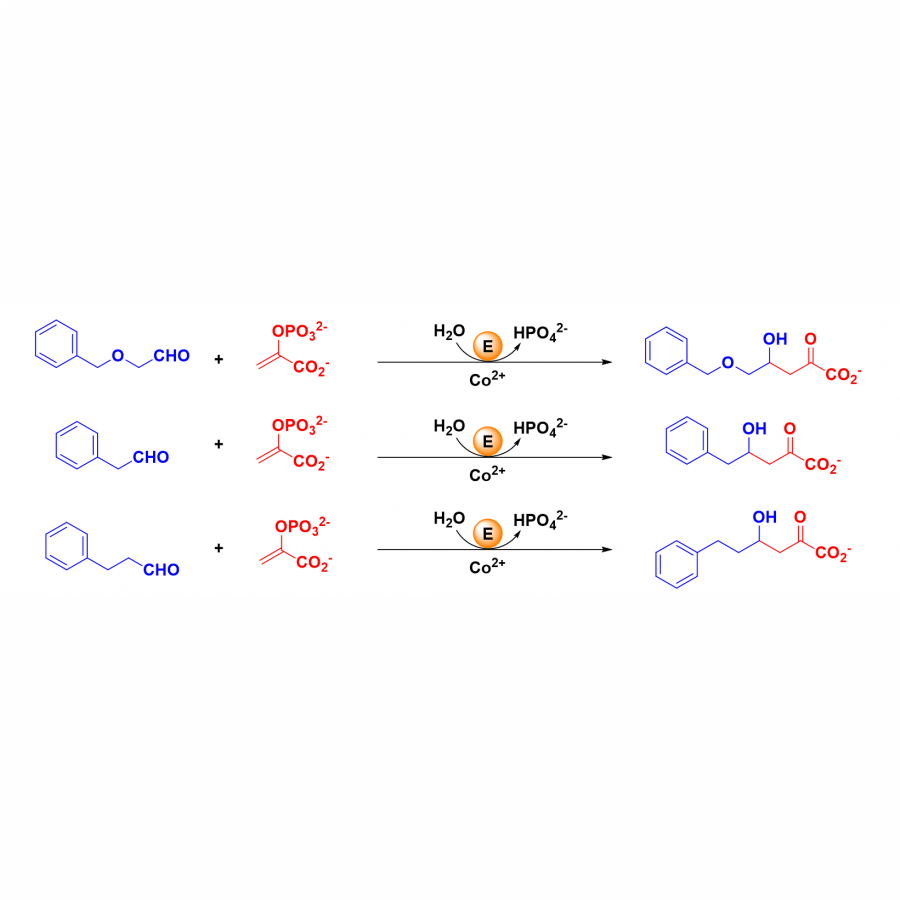 Scheme 1. KdoS and AroS homologues from Prozomix panels catalyzed promiscuous aldol addition of phosphoenolpyruvate to aliphatic aldehydes. |
#1420 | Biocatalysis - A Bio-logical Approach to Catalysis in the Pharmaceutical Industry |
|
| Presenting author: | Theo PESCHKE of NOVARTIS AG |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Proteinengineering / Enzyme Immobilization / Pharmaceutical industry / |
| Purpose: | Biocatalysis has become a routine tool for the manufacturing of pharmaceuticals allowing to increase capital efficiency while lowering the carbon dioxide footprint. Having started this journey with well-established commercial enzymes in non-GMP steps, Novartis nowadays applies engineered enzymes across all scales (see Figure 1). [1,2] Showcases like the transaminase applied in the synthesis of sacubitril [3] or a phenylalanine ammonia lyase (PAL) for EMA401 [4], clearly shows the power of biocatalysis in shortening synthesis routes. |
| References: | References [1] G. Mann and F. V. Stanger, A Bio-logical Approach to Catalysis in the Pharmaceutical Industry, Chimia (2020) 407-417. [2] E. Siirola et al., Evolution of Biocatalysis at Novartis over Last 40 Years, submitted [3] WO2017/098430; WO2018/231462; J. Org. Chem. 2020, 85, 6844-6853 [4] L. A. Hardegger et al., Toward a Scalable Synthesis and Process for EMA401, Part III: Using an Engineered Phenylalanine Ammonia Lyase Enzyme to Synthesize a Non-natural Phenylalanine Derivative, Org. Process Res. Dev. (2020), 24, 9, 1763-1771 |
| Figures: | 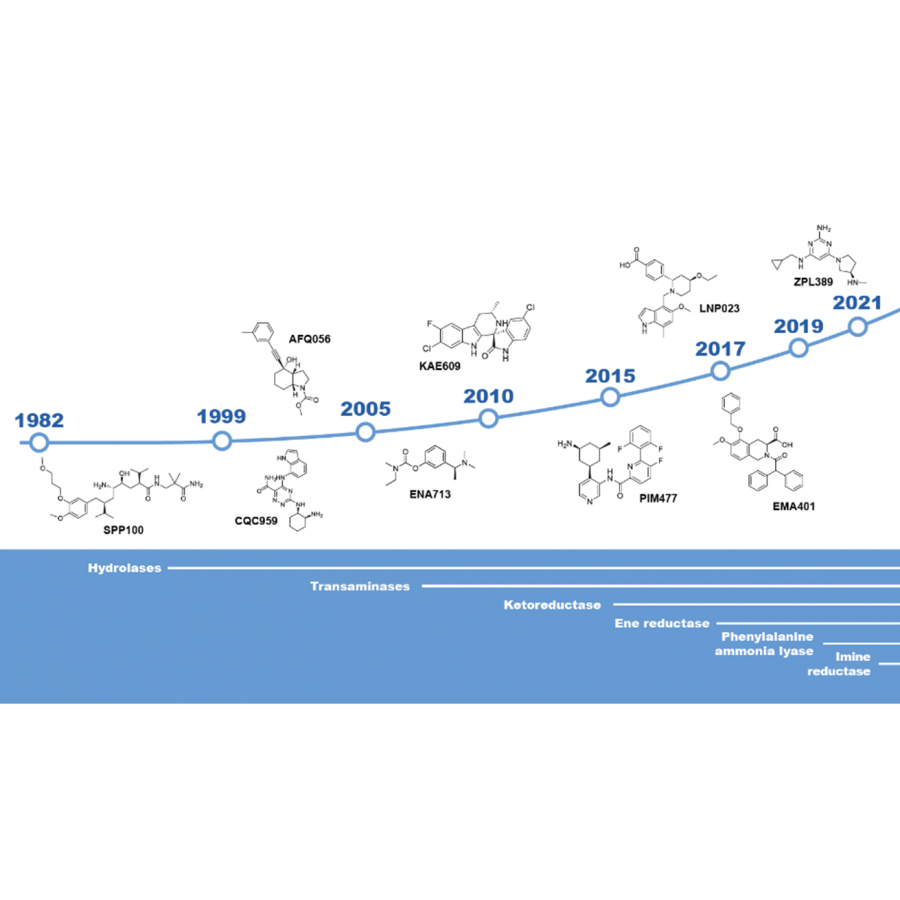 Fig. 1: Evolution of the enzymatic toolbox over the past 4 decades at Novartis. [2] |
#1422 | Optimizing the reaction conditions of enzyme – catalysed raspberry ketone production |
|
| Presenting author: | Emerik LEAKOVIC of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY |
| Other authors: | Isabel THIER of ANALYTICON DISCOVERY GMBH Karsten SIEMS of ANALYTICON DISCOVERY GMBH Zvjezdana FINDRIK BLAŽEVIĆ of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY Ana VRSALOVIĆ PRESEČKI of FACULTY OF CHEMICAL ENGINEERING AND TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Raspberry ketone is a natural phenolic compound found in the red raspberry (Rubus Idaeus) and is widely used in the food and cosmetic industries. [1] The amount of raspberry ketone in the fruit is low, which makes direct isolation of the compound economically unviable. [2] A promising production method is a biocatalytic process that uses as substrate rhododendrol glycosides, precursors found in the inner birch bark. Hydrolysis of rhododendrol glycosides results in the (R)- and (S)- rhododendrol, which can be enzymatically converted to raspberry ketone (4-(4-hxdroxyphenyl)-butan-2-one). [3] In this study, the reaction of (R)–rhododendrol (4-[(3R)-3- hydroxybutyl]phenol) oxidation by R-selective alcohol dehydrogenase (ADH) in a batch reactor was studied (Figure 1.). NAD(P)H oxidase (NOX) was employed for the coenzyme regeneration. The optimal pH for the reaction was examined. The kinetic parameters were estimated under the optimal conditions. The kinetics for both enzymes were described by double substrate Michaelis Menten kinetics. Based on the kinetic studies, a mathematical model was developed and validated in the batch reactor. |
| References: | [1] C. Morimoto et al., Life Sciences 77 (2005) 194-204 [2] M. Larsen et al., Acta Agric. Scand 41 (1991) 447-454 [3] Becker et al., Appl. Microb. and Biotechnol.105 (2021) 4189-4197. |
| Figures: | 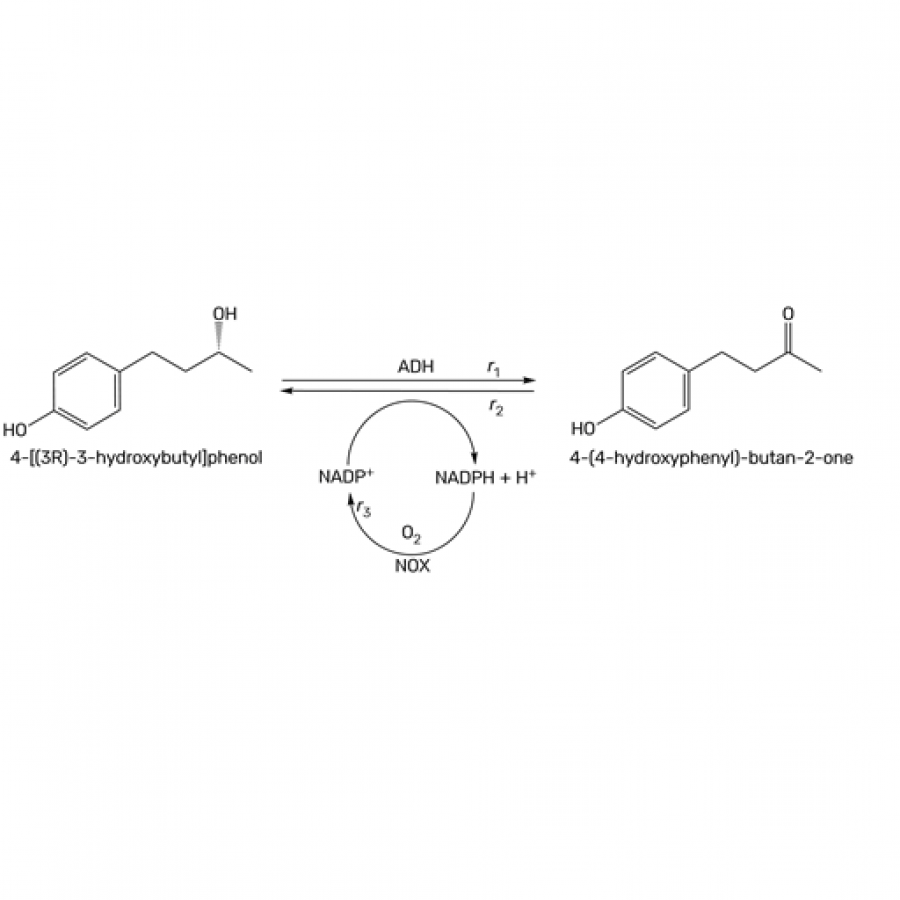 Figure 1. The reaction scheme |
#1423 | Exploring alternative materials for the preparation of Enzybeads in HTE Screening |
|
| Presenting author: | Alexander FEJZAGIC of BOEHRINGER INGELHEIM PHARMA |
| Other authors: | Marco SANTAGOSTINO of BOEHRINGER INGELHEIM PHARMA Matthias SCHMID of BOEHRINGER INGELHEIM PHARMA |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | HTE screening / biocatalysis / immobilization / |
| Purpose: | Finding a suitable catalyst for a chemical transformation is key in developing more efficient and greener processes on larger scale. To increase data density while reducing required amounts of catalyst and starting materials, screening in a HTE environment gives significant advantages compared to traditional screening approaches. One way to enable automated dosing in Multititerplates (MTPs) of low amounts of catalyst is the adsorption of the catalyst on inert glass beads.[1] Since then, this technique has been successfully used for HTE screening in pharmaceutical industry. To expand this technique to biocatalysts, an adapted procedure has been published under the name of Enzybeads.[2] However, all methods require the use of a Resodyn Acoustic Mixer (RAM) or manual milling of the crude lysate powders. Besides the cost of this device (ca. 60.000 $), the acoustic mixing process leads to heat generation in the samples, requiring a strict temperature control to avoid enzyme degradation. We aimed to investigate alternative materials for Enzybeads to enhance high-throughput experimentation (HTE) screening of biocatalysts. The beads are prepared starting from enzyme liquid formulations, thus circumventing the use of the RAM and facilitating the process. Different commercial resins as materials for Enzybeads were investigated and evaluated based on their ability to adsorb enzymes, retaining activity and stability under different conditions. The most promising materials were used to prepare Enzybeads of different enzyme classes, which were tested for their performance using model reactions. The beads can not only be used with automated dispensing machines but also using a custom-made manual parallel powder dosing apparatus, enabling a broader use of this technique for enzymatic screenings. By developing this preparation method a significant decrease in material required for screenings will be possible and allow more frequent implementation of biocatalysis in chemical processes in pharmaceutical industry as well as in high-troughput screenings in enzyme discovery. As biocatalysts usually convey high selectivities by using mild conditions and aqueous media, the integration of more biocatalysts in chemical synthesis of APIs may improve the green chemistry metrics of future processes. |
| References: | [1] Tu, N. P. et al., Angew Chem-ger Edit 131, 8071-8075 (2019) [2] Brown, J. T. C., Tu, N. P. & Phelan, R. M. Solid, Org Process Res Dev 25, 337-341 (2021). |
| Figures: | 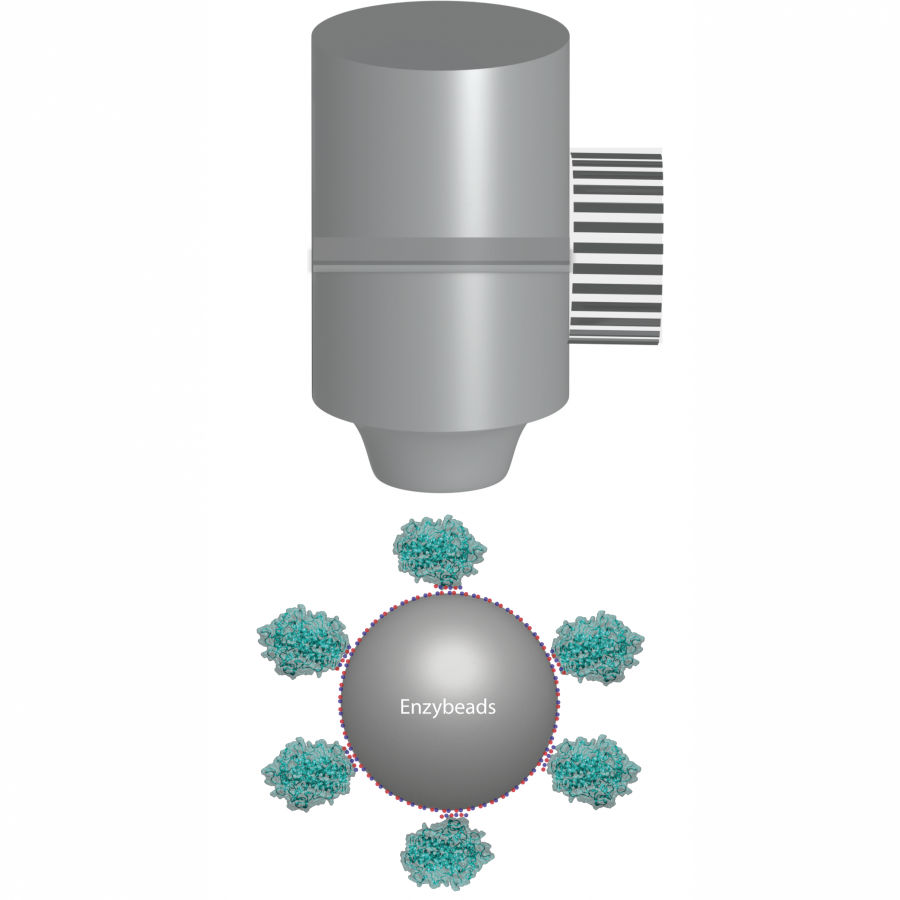 Enzybeads Schematic representation of Enzybeads |
#1424 | The Biocatalytic Synthesis of Indoles from Indolines |
|
| Presenting author: | Krisztina PISAK of UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Daniele CASTAGNOLO of UNIVERSITY COLLEGE LONDON |
| Other authors: | FEI ZHAO of UNIVERSITY COLLEGE LONDON DOMIZIANA MASCI of CATHOLIC UNIVERSITY OF THE SACRED HEART Nicholas TURNER of MANCHESTER INSTITUTE OF BIOTECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | indole / monoamine oxidase / chemoenzymatic cascade / biocatalysis |
| Purpose: | The indole scaffold is a prominent structure in organic chemistry due to its prevalent presence in both natural products and in the pharmaceutical industry [1]. Examples of its use as a building block for pharmaceutical compounds include frovatriptan, indomethacin, sumatriptan and rizatriptan [2]. Indole- containing natural products include examples such as vincamine, psilocin, melatonin and tryptophan [3]. The evolution of methods to synthesise this important heterocycle has been a key concern for organic chemists for years. However, many methods that have been developed, such as the well-known Fischer synthesis of indoles [4] utilise harsh reaction conditions and often require the use of rare metals and stoichiometric amounts of oxidising agents. This is unappealing in a world striving towards green chemistry. An alternative approach which has scarcely been explored in literature is the use of chemo-enzymatic and biocatalytic methods inspired by nature to synthesise indoles and their derivatives [5]. Enzymes are now widely exploited to construct stereospecific chemical bonds, however their use as biocatalysts has seldom been explored. This work has been inspired by natural metabolic transformations and demonstrates the aromatization of indoline derivates into indoles biocatalytically, exploiting monoamine oxidase (MAO-N) enzymes. MAO-N D11 whole cell biocatalyst was used to chemoselectively aromatize indoline substrates. The substrates for biocatalysis were prepared either by arylative dearomatization of unsubstituted indoles or by photocatalytic cyclisation of arylaniline precursors. The biocatalytic mechanism and experimental results were explained by computational docking studies of the indoline substrates in the MAO-N active site. The findings of this study show an efficient way to biocatalytically synthesise nonchiral aromatic molecules under mild reaction conditions and the huge potential of MAO-N enzymes to be used as aromatizing biocatalysts. |
| References: | 1. de Sá Alves, F.R., E.J. Barreiro, and C.A. Fraga, From nature to drug discovery: the indole scaffold as a 'privileged structure'. Mini Rev Med Chem, 2009. 9(7): p. 782-93. 2. Kochanowska-Karamyan, A.J. and M.T. Hamann, Marine Indole Alkaloids: Potential New Drug Leads for the Control of Depression and Anxiety. Chemical Reviews, 2010. 110(8): p. 4489-4497. 3. Heravi, Majid M., et al., Fischer indole synthesis applied to the total synthesis of natural products. RSC Advances, 2017. 7(83): p. 52852-52887. 4. Fischer, E. and F. Jourdan, Ueber die Hydrazine der Brenztraubensäure. Berichte der deutschen chemischen Gesellschaft, 1883. 16(2): p. 2241-2245. 5. Parmeggiani, F., et al., One-Pot Biocatalytic Synthesis of Substituted d-Tryptophans from Indoles Enabled by an Engineered Aminotransferase. ACS Catalysis, 2019. 9(4): p. 3482-3486. |
#1428 | EziG®: A Universal Immobilisation Technology for Enhanced Biocatalytic Processes |
|
| Presenting author: | Sebastian GERGEL of ENGINZYME AB |
| Other authors: | Ashley MATTEY of ENGINZYME AB Diego ACOSTA GARCÍA of ENGINZYME AB |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme immobilisation / Flow biocatalysis / Industrial biocatalysis / |
| Purpose: | Immobilised enzymes have emerged as a critical tool in industrial biocatalysis, providing a range of benefits such as biocatalyst reusability, streamlined downstream processing, and foremost improved enzyme stability. The optimal choice of immobilisation support materials, however, is often a time-consuming and expensive process. In this poster presentation, we introduce our advanced EziG® technology – a universal immobilisation matrix that offers a simple and cost-effective solution for a wide range of biocatalytic applications. EziG® is designed to achieve superior enzyme loadings, high activities, and stabilities with a wide range of enzymes, making it an efficient solution for immobilised enzyme-based processes, helping to meet the growing demand for sustainable industrial processes. At EnginZyme, we have successfully used EziG® to develop new process solutions for the production of in-demand chemicals ranging from pharmaceutical intermediates to food and cosmetic ingredients. Here, we will showcase the versatility, efficiency, and reliability of EziG®-based biocatalytic processes, highlighting their optimisation and scale-up with a case study as an example. |
#1432 | Lypolytic potential of licuri fruit (Syagrus coronata (Mart.) Becc.) as self-immobilized biocatalyst source |
|
| Presenting author: | César ALMEIDA RODRIGUES of CNRS |
| Other authors: | Milena CHAGAS LISBOA of UCLOUVAIN, IMCN/BSMA Moulay BACHIR SAHAKA MAHAMAN SALAH of CNRS Frédéric CARRIÈRE of CNRS Álvaro SILVA LIMA of UNIT/ITP Cleide MARA FARIA SOARES of UNIT/ITP |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Licuri / Delipidation / Self-immobilized / Biocatalyst |
| Purpose: | Despite the extensive range of microbial lipolytic enzymes being applied in oils and fats biotransformation, it is important to search alternatives with lower cost, easier market acceptance and direct application as a biocatalyst with partial purification, being naturally immobilized, designated as self-immobilized biocatalyst, such as enzymes obtained from oilseeds delipidation [1]. Although the oil application as product itself, it becomes also interesting to evaluate the lipolytic potential of its proteins, since the presence of oil also suggest the presence of endogenous enzymes, as part of the metabolic path on oil seed germination [2-4]. This kind of evaluation can be interesting to add value to oil production sector, giving potential use for its residues. Licuri fruit (Syagrus coronata (Mart.) Becc.) is good candidate for such application since it has a high amount of oil (49%) on its endosperm, being well used as food supply in Brazil Northeast [5]. In this context, the aim of this work is to evaluate the potential of the delipidated majoritarian parts of licuri fruit as self-immobilized biocatalyst. Licuri fruit parts were delipidated using cold hexane to delipidate the endosperm or mesocarp (ECH and MCH). The catalytic activity of the potential self-immobilized biocatalysts obtained in this work was evaluated in the hydrolysis reaction of olive oil, tributyrin and commercial licuri endosperm oil using pHStat and represented by means of relative activity. As result, summarized in Fig 1, in one hand MCH obtained an initial hydrolytic activity in olive oil of 10.5% (pH 7, 3% gum arabic) being optimized for 78.9 % (pH 8, 1% gum arabic) and 37.2 % (pH 8, 1% gum arabic) in the hydrolysis of licuri endosperm oil. In another hand, ECH showed an initial hydrolytic activity in olive oil of 0.8 % (pH 7, 37°C), with an optimal value of 2.8 U/g (pH 3, 50°C). MCH also could performed well on tributyrin hydrolytic activity, with an initial hydrolytic activity of 60.5 % (pH 7, 0.5 mM NaTDC), optimized for 100 U/g (pH 7, 10 mM of NaTDC) while ECH displayed no activity. After a storage of 1-year MCH maintained 75.43% of its initial hydrolytic activity. When comparing those results with the hydrolytic activity of others vegetable lipolytic enzymes, we can see that castor bean and palm oil lipases (CBL and POL, respectively) also have a higher hydrolytic activity using tributyrin as substrate when compared to olive oil [6], [7]. However, MCH showed a higher potential to hydrolyze olive oil (Fig. 2), since the drop of activity among tributyrin and olive oil was only 21.1% while CBL had a drop of 96.5 and POL of 64.6 % according to EBONGUE et al., (2006)[7] and CAVALCANTI et al., (2007) [6], respectively. Thus, we conclude that MCH presents better potential to catalyze reactions with TAGs with a longer carbon chain, commonly present in human digestion, representing great biotechnological value. The authors are grateful for the financial support by the Coordenaçao de Aperfeiçoamento de Pessoal de Nível Superior - Brasil (CAPES), specially for the PhD scholarship provided for Milena Chagas Lisboa (PROSUP - 88882.365551/2019e01, PDSE 41/2018e88881.3 61582/2019e01). |
| References: | [1] S. Seth, D. Chakravorty, V. K. Dubey, and S. Patra, “An insight into plant lipase research - Challenges encountered,” Protein Expr. Purif., vol. 95, pp. 13-21, 2014. [2] V. Verma, G. Singhal, S. Joshi, M. Choudhary and N. Srivastava. Plant extracts as enzymes. In Plant Extracts: Applications in the Food Industry (pp. 209-223) (2022). Academic Press [3] A. L. Quettier and P. J. Eastmond, “Storage oil hydrolysis during early seedling growth,” Plant Physiol. Biochem., vol. 47, no. 6, pp. 485-490, 2009. [4] M. Barros, L. F. Fleuri, and G. A. Macedo, “SEED LIPASES : SOURCES , APPLICATIONS AND PROPERTIES - A REVIEW,” vol. 27, no. 01, pp. 15-29, 2010. [5] I. C. Crepaldi, L. B. De Almeida-Muradian, M. D. G. Rios, M. D. V. C. Penteado, and A. Salatino, “Composição nutricional do fruto de licuri (Syagrus coronata (Martius) Beccari),” Rev. Bras. Botânica, vol. 24, pp. 155-159, 2001. [6] E. D. C. Cavalcanti, F. M. Maciel, P. Villeneuve, R. C. A. Lago, O. L. T. Machado, and D. M. G. Freire, “Acetone powder from dormant seeds of Ricinus communis L: Lipase activity and presence of toxic and allergenic compounds,” Appl. Biochem. Biotechnol., vol. 137-140, no. 1-12, pp. 57-65, 2007. [7] G. F. N. Ebongue, R. Dhouib, F. Carrière, P.-H. A. Zollo, and V. Arondel, “Assaying lipase activity from oil palm fruit (Elaeis guineenses Jacq.) mesocarp,” 2006. |
| Figures: | 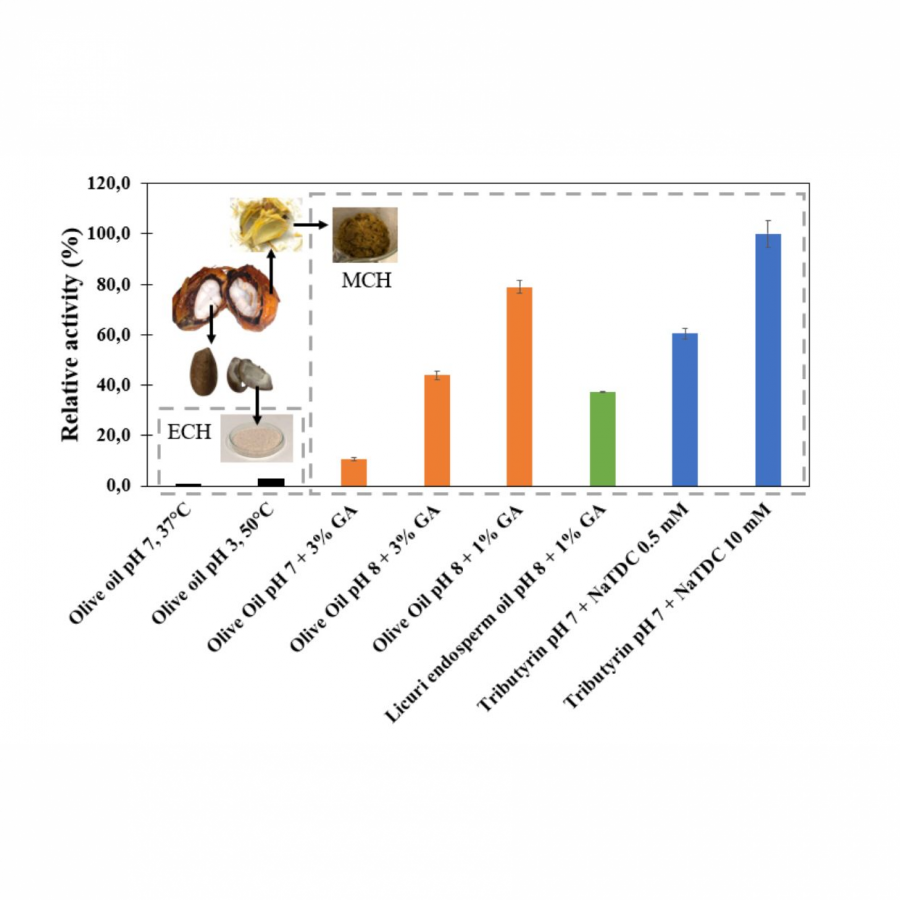 Fig.1 Optimization and comparison among ECH and MCH in hydrolysis activity against different substrates with different carbon chain sizes. 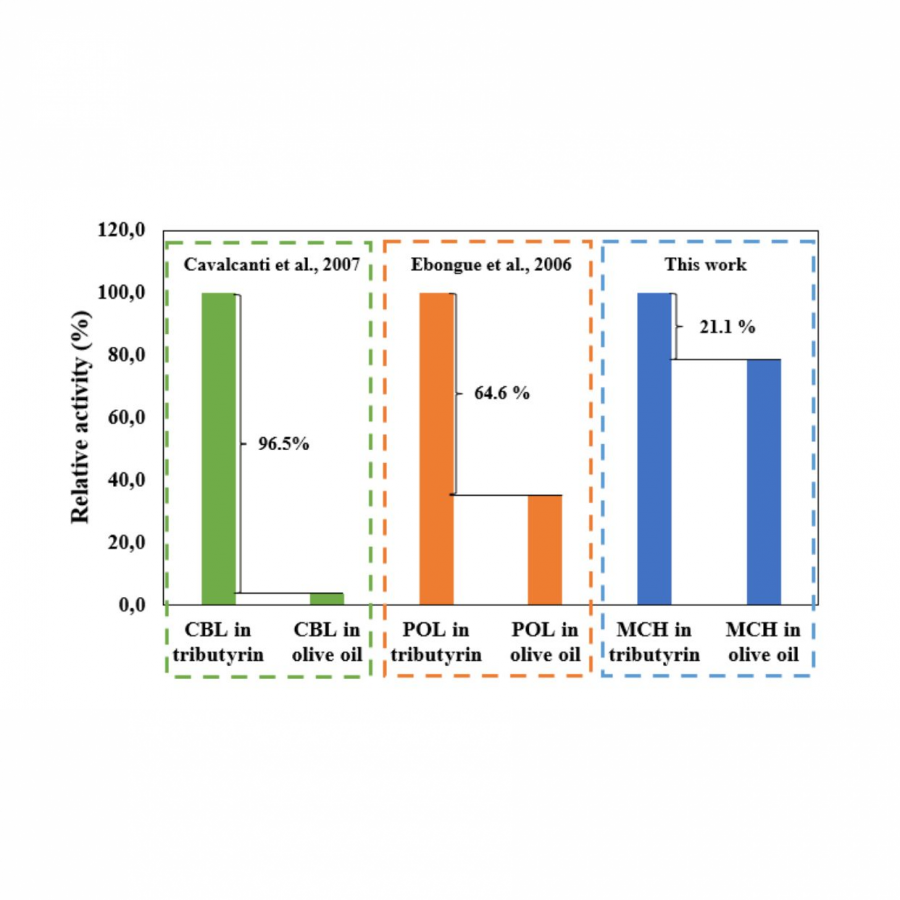 Fig 2. MCH potential to catalyze long-chain oils (olive oil) compared to other self-immobilized vegetable lipases from castor bean (Cavalcanti et al., 2007 [6]) and palm oil (Ebongue et al., 2006 [7]) |
#1436 | Experimental and computational studies of chalcone synthase |
|
| Presenting author: | Mohammad YOUSEFI of LEIBNIZ INSTITUTE OF PLANT BIOCHEMISTRY |
| Other authors: | Mehdi D. DAVARI of LEIBNIZ INSTITUTE OF PLANT BIOCHEMISTRY Martin DIPPE of LEIBNIZ INSTITUTE OF PLANT BIOCHEMISTRY Ludger A. WESSJOHANN of LEIBNIZ INSTITUTE OF PLANT BIOCHEMISTRY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Polyketide synthases / molecular docking / Naringenin / |
| Purpose: | Polyketide synthases (PKS) are key enzymes for biosynthesis of flavonoid/isoflavonoid pathway in bacteria, fungi, and plants. PKS based on differences in nucleotide sequences, primary structure and catalytic mechanisms fall into three subclasses (type I,II, and III) [1, 2]. Chalcone synthase (CHS) is a member of type III, and are homodimers including two identical domains of 41-44 kDa polypeptide. CHS uses p-coumaroyl-CoA as a starter molecule and three malonyl-CoA as extender molecules to form a tetraketide intermediate that is cyclized into 2',4,4’,6’-tetrahydroxychalcone (Chalcone). The chalcone scaffold is an essential for producing a wide range of biologically important compounds, that can act e.g. for antimicrobial, pollen fertility, biotic and abiotic stress management [3, 4]. These secondary metabolites also have a wide range of applications in industry, medicine and pharmacy, e.g. anticancer, antiparasitic, and antioxidant compounds [5]. Furthermore, CHS like other type III PKSs, due to the spacious binding pocket, can accept a wide range of substrates including aromatic and aliphatic CoA, and also thioesters non-physiological substrates. All of these applications make PKS as a target for bioengineering to improve its activity and yield. The binding pocket of CHS consists of substrate binding pocket, catalytic site, and cyclization site [4, 6]. Four residues (Cys167, His306, Asn339, and Phe218), which are highly conserved, form the catalytic center of CHS enzyme [4]. Derailment of intermediates on the path of chalcone production lead to production of side products, which can be a barrier for large-scale in vitro application of CHS. Specific activity of TaCHS was defined as micromole conversion of p-coumaorly-CoA (substrate) per minute under standard conditions per milligram of total protein. Specific activity was determined by conducting a spectrophotometric assay at 410λnm using Ellman reaction. The HPLC was used to determine the main product (chalcone naringenin) as main product and CTAL as by product. In order to understand the role of key residues in the active site of enzyme, we performed covalent molecular mechanism-based docking with natural substrate (4-coumaroyl-CoA). Molecular docking studies revealed that Asn339 play an important role to stabilize the substrate. In addition, after three steps elongation with malonyl-CoA, the formation of the tetraketide intermediate alters the substrate conformation, thereby facilitating cyclization steps. As the substrate conformation changes, Phe218 and Phe268 act as gatekeepers to prevent the entry of additional malonyl-CoA and further elongation, and also navigate the substrate toward the cyclization residues. To identify the rate-determining step, we performed semi-empirical quantum chemical calculations using the PM7 method [7]. Based on calculations for the first intermediate, we can infer that the nucleophilic attack of Cys167 on the carbonyl with an activation energy of 27 kcal/mol is the rate-determining step. |
| References: | 1. Das, A. and C. Khosla, Biosynthesis of aromatic polyketides in bacteria. Accounts of chemical research, 2009. 42(5): p. 631-639. 2. Shen, B., Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Current opinion in chemical biology, 2003. 7(2): p. 285-295. 3. Dao, T., H. Linthorst, and R. Verpoorte, Chalcone synthase and its functions in plant resistance. Phytochemistry Reviews, 2011. 10(3): p. 397-412. 4. Austin, M.B. and J.P. Noel, The chalcone synthase superfamily of type III polyketide synthases. Natural product reports, 2003. 20(1): p. 79-110. 5. Bisht, R., et al., An overview of the medicinally important plant type III PKS derived polyketides. Frontiers in Plant Science, 2021: p. 2155. 6. Ferrer, J.-L., et al., Structure of chalcone synthase and the molecular basis of plant polyketide biosynthesis. Nature structural biology, 1999. 6(8): p. 775-784. 7. Dutra, J.D.L., et al., Sparkle/PM7 lanthanide parameters for the modeling of complexes and materials. Journal of chemical theory and computation, 2013. 9(8): p. 3333-3341. |
| Figures: | 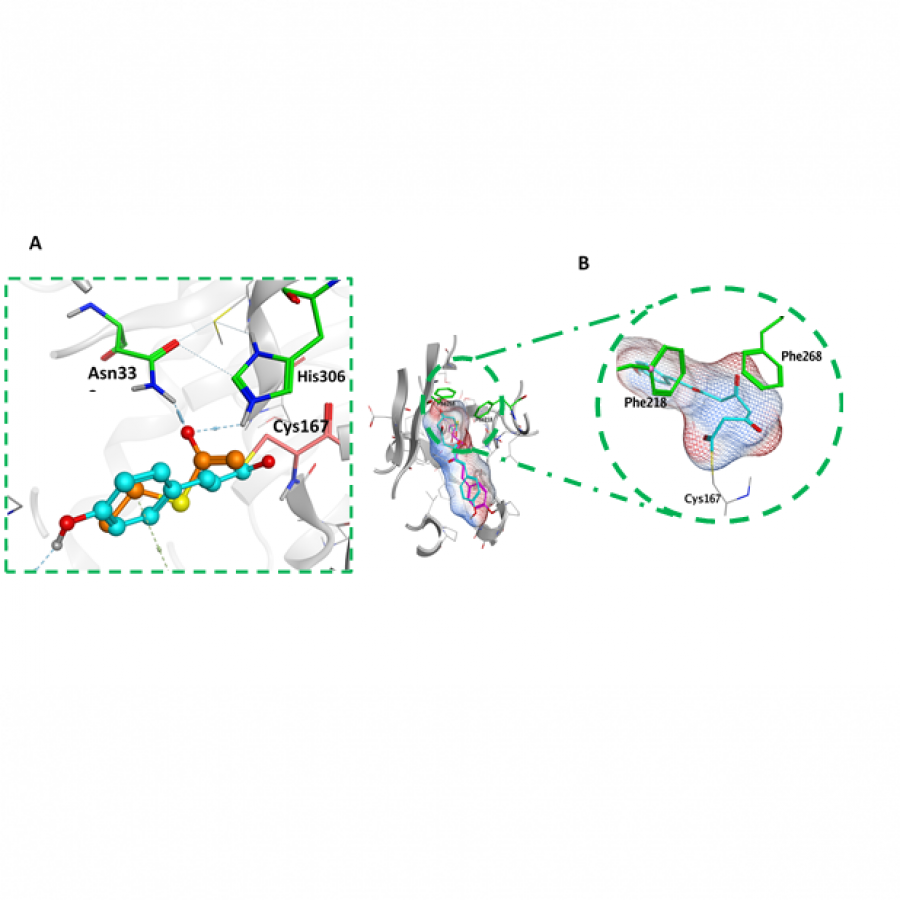 Covalent mechanism-based docking of CoA-ester substrates in the TaCHS enzyme A) Catalytically competent docking pose; 4-coumaroyl-CoA and malonyl-CoA are shown in cyan and golden, respectively, with a ball-stick representation. Catalytic residues are displayed in green, while Cys167, which is covalently bonded to the substrate is |
#1437 | Discovery of novel split transketolases for application in biocatalysis |
|
| Presenting author: | Alessia TONOLI of UNIVERSITY COLLEGE LONDON, DEPARTMENT OF BIOCHEMICAL ENGINEERING |
| Other authors: | Jack JEFFRIES of UNIVERSITY COLLEGE LONDON, DEPARTMENT OF BIOCHEMICAL ENGINEERING Helen HAILES of UNIVERSITY COLLEGE LONDON, DEPARTMENT OF CHEMISTRY John WARD of UNIVERSITY COLLEGE LONDON, DEPARTMENT OF BIOCHEMICAL ENGINEERING |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / transketolase / metagenomics / |
| Purpose: | Biocatalysis represents today a major resource in the industrial sector for several reasons, including the possibility of implementing more sustainable chemical processes, the high stereoselectivity of enzymes, and their potential to be modified through protein engineering for new biocatalytic reactivities. Transketolases (TKs) are thiamine pyrophosphate dependent enzymes with huge potential in the field of industrial biocatalysis, due to their ability to catalyse the transfer of carbon units in a stereoselective manner. The most well-known and characterised transketolases are encoded as proteins of around 650 amino acids. However, in many organisms from the archaeal and bacterial domains transketolases consists, instead, of two proteins of around 300 amino acids, aligning one with the N-terminal half of full-length TKs, and the other with their C-terminal half. For this reason, these enzymes are also named as ‘split-gene’ transketolases (or split TKs). Up to now only one example of split TK has been biochemically and structurally characterised [1]. The reconstituted biosynthetic pathway for the production of a phosphonate with herbicidal properties has been found to involve a split TK homologue [2]. Understanding characteristics and peculiarities of split TKs could be beneficial to explore their applicability in biocatalysis and possibly discover novel reactivities, as well as to gain insights about their structure-function relationship, in comparison to full-length transketolases. In this project genome and metagenome mining techniques were applied to broaden the panel of split transketolases available for investigation and use in biocatalysis. Thus far, 70 putative split TKs were retrieved both from (meta)genomic online repositories and from 4 different in-house metagenomes. These were assembled into a phylogenetic tree to investigate their spread across archaeal and bacterial domains and their relatedness to full-length transketolases. From a first selected group, 11 putative split TKs were cloned and expressed in Escherichia coli in soluble form. The preliminary characterisation of some of these biocatalysts, including the reaction with glycolaldehyde and lithium hydroxypyruvate for the production of erythrulose and their temperature optimum, allowed the identification of interesting hits to be further explored in the future and exploited for application in biocatalysis. |
| References: | [1] P. James, M.N. Isupov, S.A. De Rose, C. Sayer, I.S. Cole, J.A. Littlechild, Front Microbiol 11 (2020) 2752. [2] Y. Zhu, T. Shiraishi, J. Lin, K. Inaba, A. Ito, Y. Ogura, M. Nishiyama, T. Kuzuyama, J Am Chem Soc 144 (2022) 16715-16719. |
| Figures: | 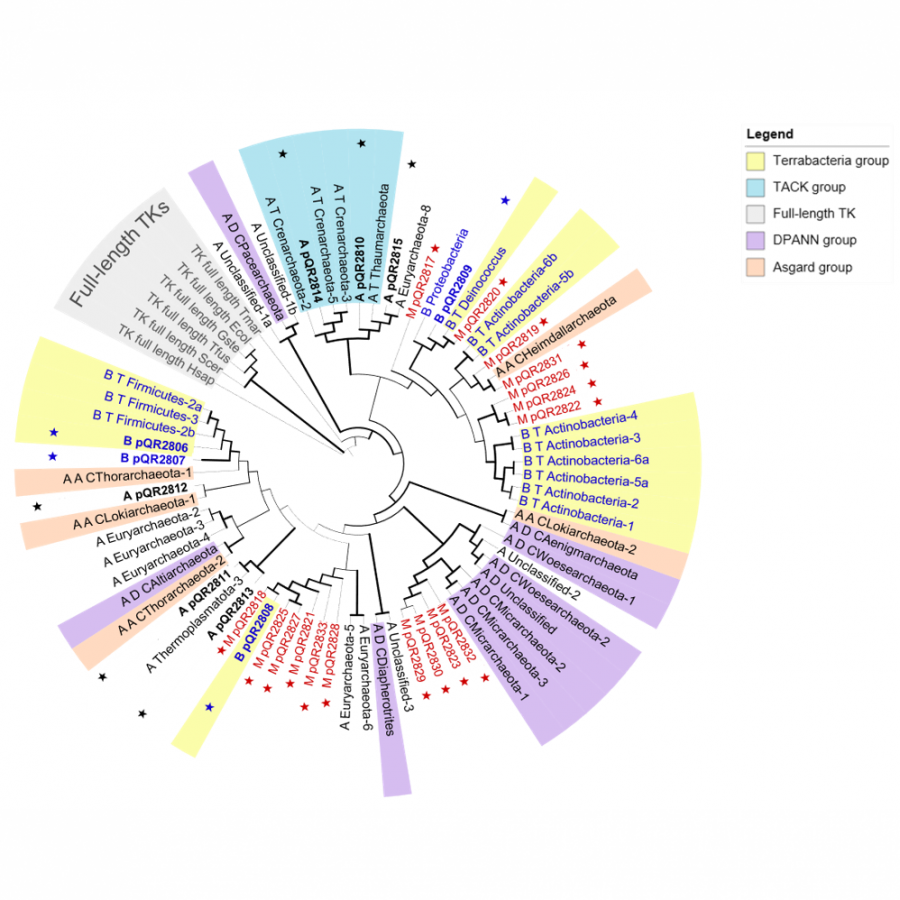 Phylogenetic tree including 76 sequences of different transketolases, mainly split transketolases. Black: split TK from Archaea. Blue: split TK from Bacteria. Red: split TK from in-house metagenome. Gray: full-length TK. ★: split TK selected for expression and ordered as synthetic gene. |
#1439 | Almac’s INSIGHTTM: From Genes to GMP |
|
| Presenting author: | Emma BARNARD of ALMAC |
| Corresponding author: | Derek QUINN of ALMAC |
| Other authors: | Alexandra CARVALHO of ALMAC Jane MUELLER of ALMAC Christine FLEMING of ALMAC Aliyu IBRAHIM of ALMAC Matthew BOYD of QUEEN'S UNIVERSITY BELFAST Xiangwen WANG of QUEEN'S UNIVERSITY BELFAST Tom MOODY of ALMAC |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | machine learning / smart enzyme discovery / in silico design / metagenomes |
| Purpose: | Almac’s INSIGHTTM: From Genes to GMP Biocatalysis offers a sustainable approach to chemical and GMP API manufacture, however the processes involved in traditional enzyme selection and screening can be resource heavy and limited to currently available sequenced genomes. Almac’s INSIGHTTM platform streamlines enzyme discovery, engineering and development. The initial step of INSIGHTTM involves smart enzyme discovery which utilises molecular modelling tools, machine learning, and bioinformatic algorithms to mine available and/or customer proprietary metagenomes. This allows for the identification of genes to subsequently build a panel of highly specific and selective enzymes. Following this, INSIGHTTM protein in-silico design approaches are employed to improve enzyme activity, selectivity and/or stability, with a targeted approach towards the needs of the customer. Selected panels of enzymes are subsequently cloned, expressed and screened in vitro for the reaction of interest. Additional offerings from Almac’s INSIGHTTM program includes a range of proprietary expression systems and hosts with the potential to improve the levels of soluble expression and activity of recombinant proteins. INSIGHTTM is focussed on development of better enzymes to be deployed in a cornucopia of reaction types. INSIGHTTM can be deployed from Gene to GMP. |
| Figures: | 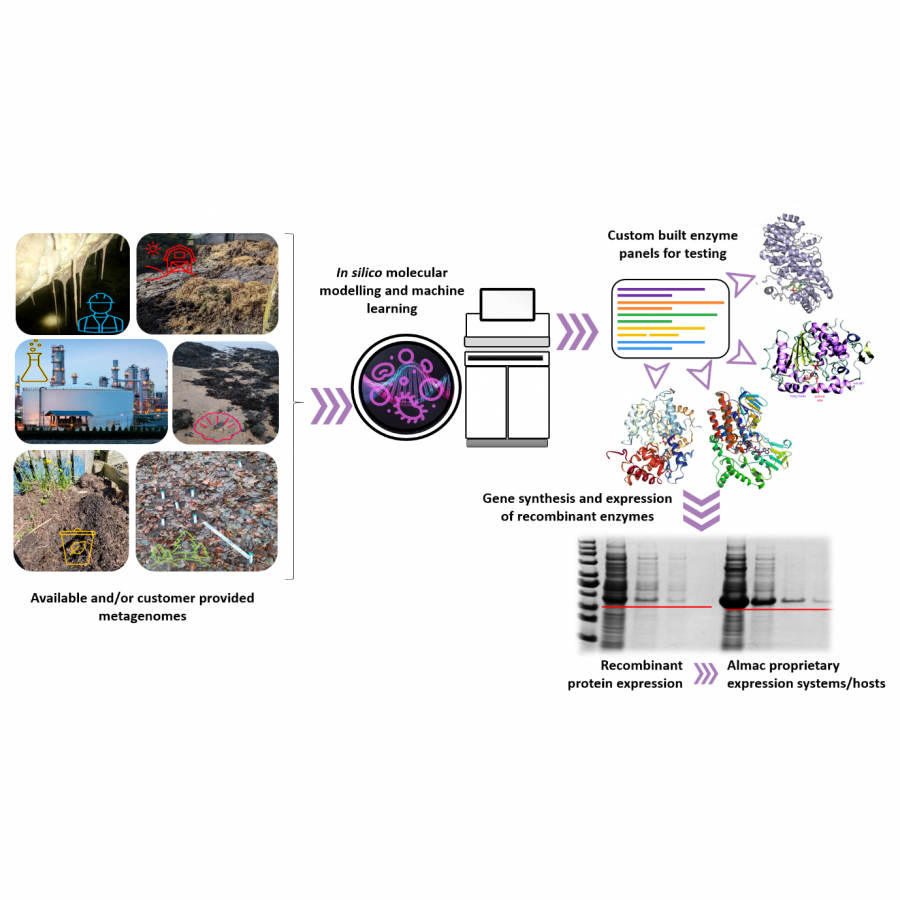 |
#1440 | Photosynthesis-driven whole-cell biocatalysis using Synechocystis sp. PCC 6803 for the conversion of cyclohexane to cyclohexanone |
|
| Presenting author: | Nina SIEBERT of LEIPZIG UNIVERSITY |
| Corresponding author: | Rohan KARANDE of LEIPZIG UNIVERSITY |
| Other authors: | Adrian TÜLLINGHOFF of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Bruno BÜHLER of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Rohan KARANDE of LEIPZIG UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Cyanobacteria / Redox-biocatalysis / Cascade reaction / Metabolic engineering |
| Purpose: | Photosynthesis-driven whole-cell biocatalysis holds tremendous potential for developing sustainable and environmentally friendly processes [1]. Photoautotrophic organisms, such as cyanobacteria, convert inorganic carbon (CO2) to organic carbon by using water, light energy, and various nutrients. The light energy absorbed by the cyanobacterial cell in the photosynthetic apparatus is used to split water molecules and produce activated reduction equivalents and O2, which can serve as co-substrates for oxygenase-catalyzed reactions [2]. This study aims to design a recombinant photoautotrophic strain and gain insight into photosynthetically driven redox biocatalysis using a 2-step heterologous enzyme cascade, consisting of a cytochromeP450 monooxygenase and a cyclohexanol dehydrogenase. Both enzymes originate from the soil bacterium Acidovorax and are part of the initial cyclohexane degradation pathway [3]. In this poster, our cloning strategy to design the recombinant Synechocystis sp. PCC 6803 strain and the preliminary data for converting cyclohexane to cyclohexanone driven by photosynthetic water oxidation will be presented. |
| References: | [1] J. Toepel, R. Karande, S. Klähn, & B. Bühler, Current Opinion in Biotechnology 2023, 80, art. 102892 [2] A. Hoschek, B. Bühler, & A. Schmid, Angew. Chem.-Int. Edit. 2017, 56 (47), 15146 - 15149. [3] R. Karande, D. Salamanca, A. Schmid, & K. Bühler, Biotechnol. Bioeng. 2018, 115 (2), 312 - 320. |
| Figures: | 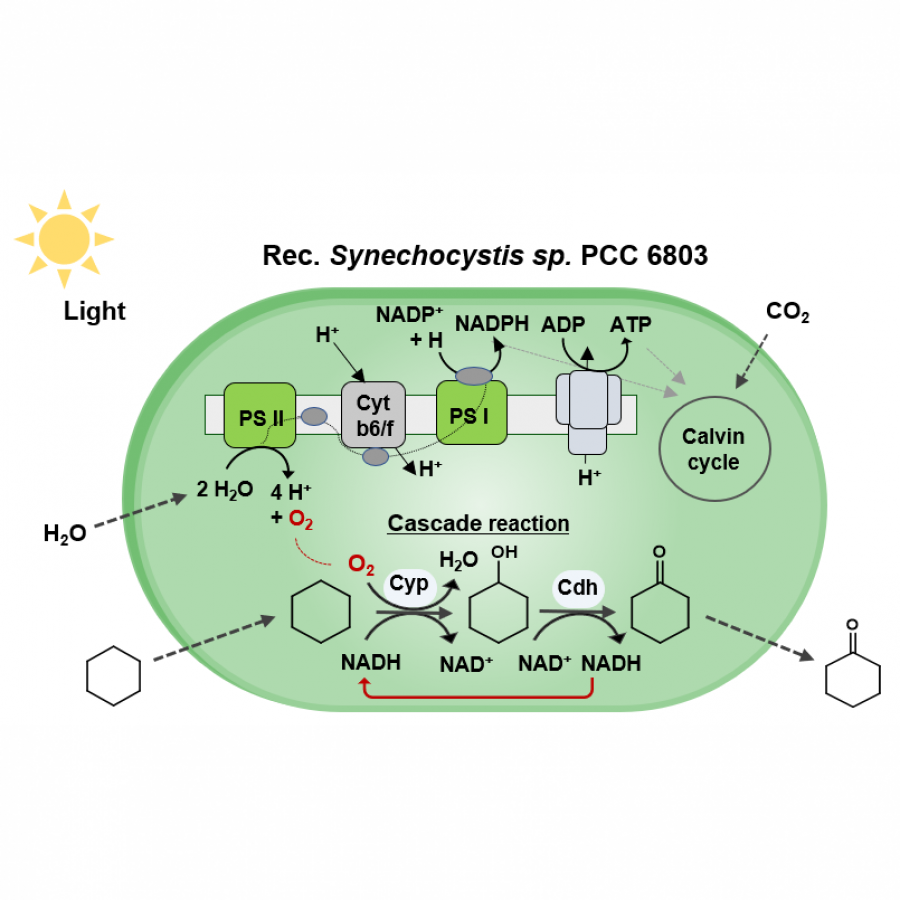 Schematic presentation of 2-step enzyme-cascade in Synechocystis sp. PCC 6803 for conversion of cyclohexane to cyclohexanone Cyp-CytochromeP450 monooxygenase, Cdh-Cyclohexanol dehydrogenase |
#1441 | From academia to industry: Process intensification for the biocatalytic synthesis of metaraminol employing a 2-step enzymatic cascade |
|
| Presenting author: | Lisa SEIBT of FORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE |
| Other authors: | Nina KLOS of ORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE Sarah SCHMITZ of ORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE Torsten SEHL of ORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE Laura GRABOWSKI of ORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE Dörte ROTHER of ORSCHUNGSZENTRUM JÜLICH, IBG-1, BIOKATALYSE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / enzymatic cascades / transaminase / process intensification |
| Purpose: | Biocatalysis is a growing research field with the opportunity to support, compete with or replace traditional chemical synthesis on an industrial scale. In recent years, intense research has enabled the community to draw on a versatile enzymatic toolbox to find ever new pathways leading to industrially relevant pharmaceutical building blocks or fine chemicals [1]. However, these processes are usually performed on a laboratory scale (micrograms – milligrams), rather than generating industrially utilisable quantities such as grams or even kilograms (Figure 1). To boost the potential of biocatalysis in an industrial context, it is of the utmost importance to investigate the potential to scale the biocatalytic process up. However simple it might seem to scale up a reaction, various aspects need to be taken into account in order to intensify the process, thus making a scale up feasible. For example the production of the expensive biocatalyst needs to be considered, as well as the need for special equipment (larger vessels for the reaction and downstream processing). To investigate and overcome these challenges, it is sensible to trial a well-studied system and use it as a. The enzymatic synthesis of metaraminol, which is used for the treatment of hypotension, is such a well characterised reaction [2, 3, 4, 5]. By employing a 2-step 2-enzyme cascade starting from the inexpensive aldehyde 3-hydroxy-benzaldehyde, the product metaraminol can be obtained via combination of two enzymes in consecutive steps (Figure 1) [2]. The cascade features a carboligation reaction (Enzyme 1, Figure 2) and is followed by a transaminase step (Enzyme 2, Figure 2) to form metaraminol. This cascade can even be altered to obtain a variety of analogues and their stereoisomers. Furthermore, an extension to tetrahydroquinolinones with multiple applications is feasible (Figure 2) [3]. With this work we are focusing on the scalability of the transamination step, as it provides a good example for the challenges hindering application in industry. The transamination step is characterised by an unfavourable reaction equilibrium. Another obstacles in the process is an inhibition caused by the substrate 3-hydroxy-phenylacetylcarbinol (3-HO-PAC) and the product metaraminol. Therefore, the major challenge is to design the system in a way that neither the substrate nor the product inactivate the expensive catalyst, while pushing the reaction equilibrium and recovering high yields of valuable product without complex DSP. We are therefore exploring different ideas including for example in-situ product removal/crystallisation (ISPR/ISPC) [6] to simultaneously shift the equilibrium and remove the inhibitor metaraminol. A further exemplary study concerns the choice of amine donor [7] for pushing the reaction equilibrium while keeping DSP easy. |
| References: | [1] Finnigan, W. et al. (2021) Nat Catal. 2021 Feb; 4(2): 98-104 [2] Labib, M., Grabowski, L. et al. (2022). ACS Sustainable Chem. Eng., 10, 16, 5117-5128. [3] V. Erdmann, Angew.Chem. Int. Ed., (2017), 56, 12503-12507. [4] Mack, K., Doeker, M., Grabowski, L., Jupke, A., Rother, D. (2021). Green Chem., 23, 4892-4901. [5] Doeker, M., Grabowski, L., Rother, D., Jupke, A. (2022). Green Chem., 24, 295-304. [6] Hülsewede, D., Dohm, J-M., von Langermann, J. (2019) Adv. Synth. Catal. 2019, 361, 2727- 2733 [7] McKenna, C. A. et al. (2021) Green Chem., 2022, 24, 2010-2016 |
| Figures: | 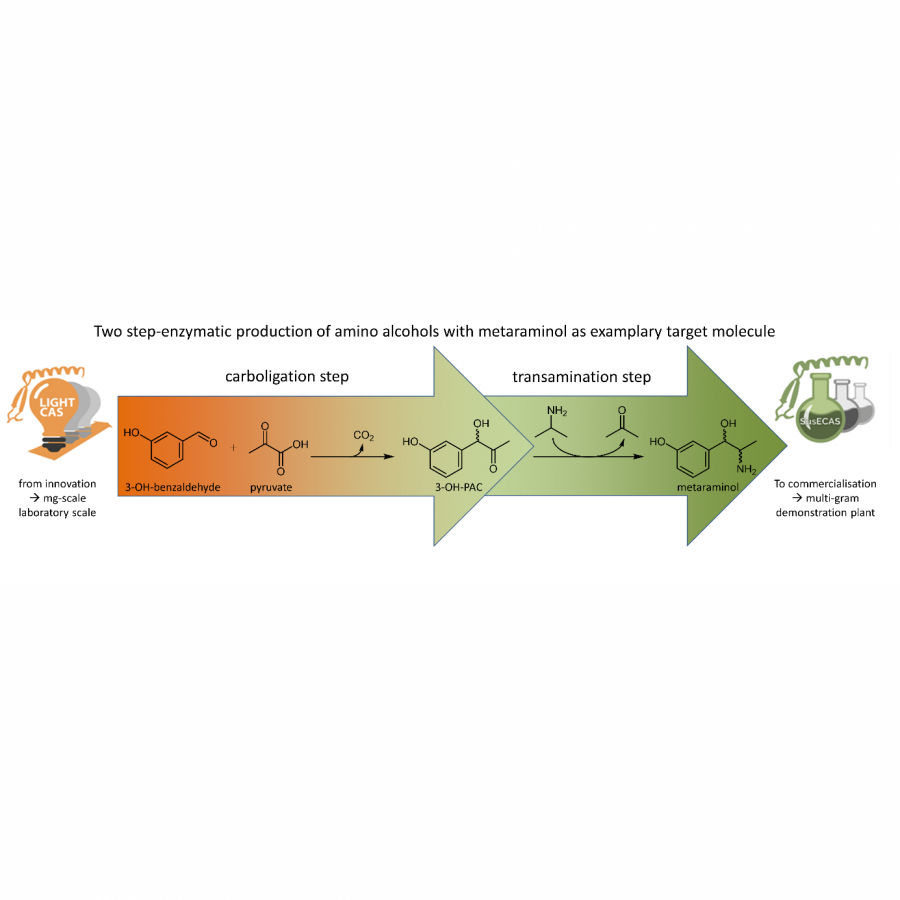 Figure 1 Enzymatic 2-step cascade for synthesis of amino alcohols developed in the ERC Starting Grand `LightCAS´ on mg-scale. In the ERC proof-of concept project `SusECAS DemoPlant´, we aim for a gradual commercialization of products derived from this synthesis. 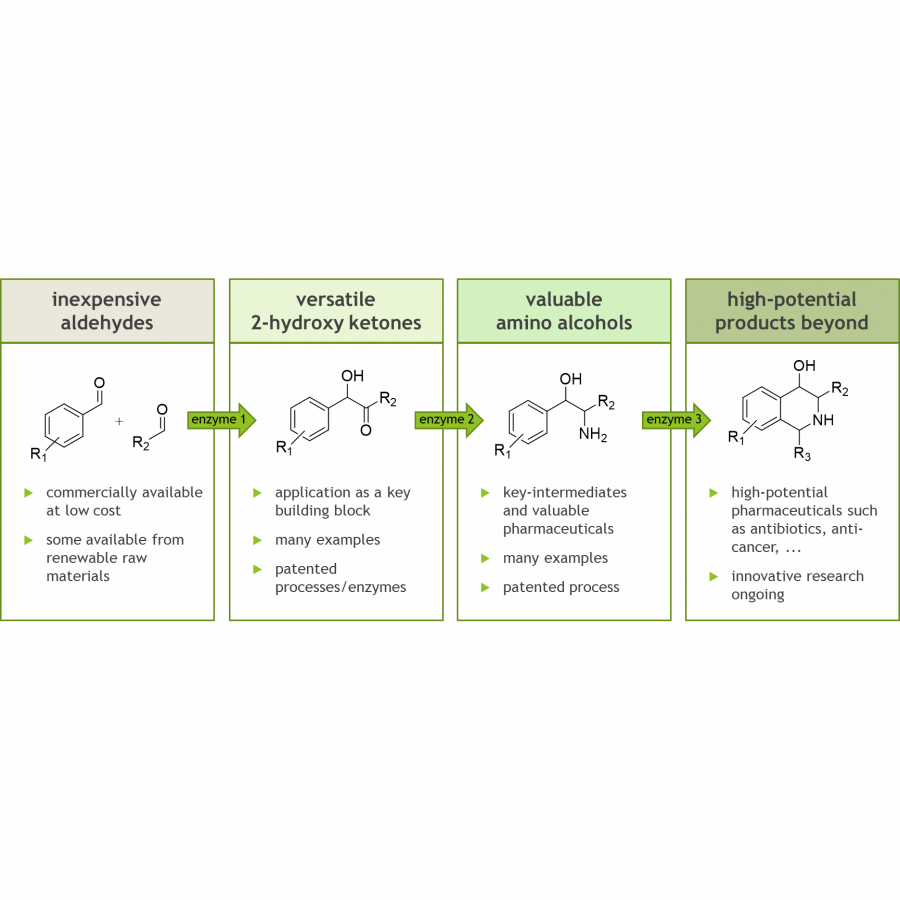 Figure 2 Two-step enzymatic cascade towards metaraminol consisting of a carboligation (catalysed by enzyme 1) and reductive amination (catalysed by enzyme 2) step, or even further towards high value THIQs (catalysed by enzyme 3). |
#1442 | Enzymatic hydrolysis of polyamide and elastane for textile recycling |
|
| Presenting author: | Pilar CHAVEZ LINARES of UNIVERSITY OF LORRAINE, CNRS, LRGP |
| Other authors: | Aigerim KONYSBAY of UNIVERSITY OF LORRAINE, CNRS, LRGP Sandrine HOPPE of UNIVERSITY OF LORRAINE, CNRS, LRGP Isabelle CHEVALLOT of UNIVERSITY OF LORRAINE, CNRS, LRGP |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme hydrolysis / degradation / polyamide / elastane |
| Purpose: | Currently, the textile industry is one of the most polluting in the world. The mixture of synthetic fibers and chemical additives makes recycling management difficult. However, the use of biotechnological tools could implement a greener, closed-loop recycling process. The present study focuses on two synthetic fibers, polyamide and elastane fibers, which are widely used in the daily clothing. These two fibers are very often closely combined and have different physico-chemical properties that make them difficult to separate and recycle. Therefore, a process based on depolymerization catalyzed by enzymes is proposed in order to recover initial monomers . Polyamide fibers contain amino and carboxylic groups that can be cleaved with hydrolytic enzymes such as proteases and specific amidases [1]–[3]. However, the crystalline structure of polyamide makes enzymatic attack difficult. The hydrolytic degradation by using enzymes starts on the surface towards amino groups which are present as end-group chains and led to the releasing of soluble oligomers [4]. Elastane fiber is a complex chemical structure composed of two segments, a soft segment containing polyols and a hard segment containing urethanes groups. These two segments are linked by chain extenders of low molecular weight (diols or diamines). This fiber is difficult to degrade with microorganisms and there are still no studies showing hydrolytic degradation with the use of enzymes. In the present work, two commercial proteases and enzymatic extracts from fungi sp were used and their performances in hydrolytic degradation have been evaluated. Hydrolyses of polyamide and elastane textile fibers (film and yarn) were quantified by measuring amino functional groups released during the enzymatic treatment (Figure 1). For this purpose, the ortho-phthalaldehyde (OPA) assay, based on a reaction with primary amino groups, has been used. Surface degradation of the solid residue has been followed by scanning electron microscopy (SEM) [5] and the modification of chemical structures has been investigated by spectroscopy infrared (FT-IR). Thermal analyses such as differential scanning calorimetry (DSC) were carried out in order to measure the degree of degradation in the amorphous phase of the polymer. It has been demonstrated that the commercial enzymes are able to hydrolyze amide and urethane bonds of polyamide and elastane, respectively, with degradation only on the surface (Figure 2). The comparison of the hydrolytic degradation of polyamide and elastane synthetic fibers (film and yarn) shows the impact of the polymer surface area. The degradation rate depends on the surface area of the polymeric material. Other studies for new enzymes isolation from fungi are in progress in order to produce more specific and more efficient biocatalysts. |
| References: | [1] S. Heumann et al., Biotechnol. Bioeng., 2009, 102, 1003-1011 [2] M. P. Gashti et al, Prep. Biochem. Biotechnol., 2013, 43, 798-814, 2013. [3] H. R. Kim and H. Y. Seo, Text. Res. J., 2013, 83, 1181-1189. [4] C. Silva and A. Cavaco-Paulo, Biocatal. Biotransformation, 2004, 22, 357-360,. [5] A. Magnin et al., Waste Manag., 2019, 85, 141-150. |
| Figures: | 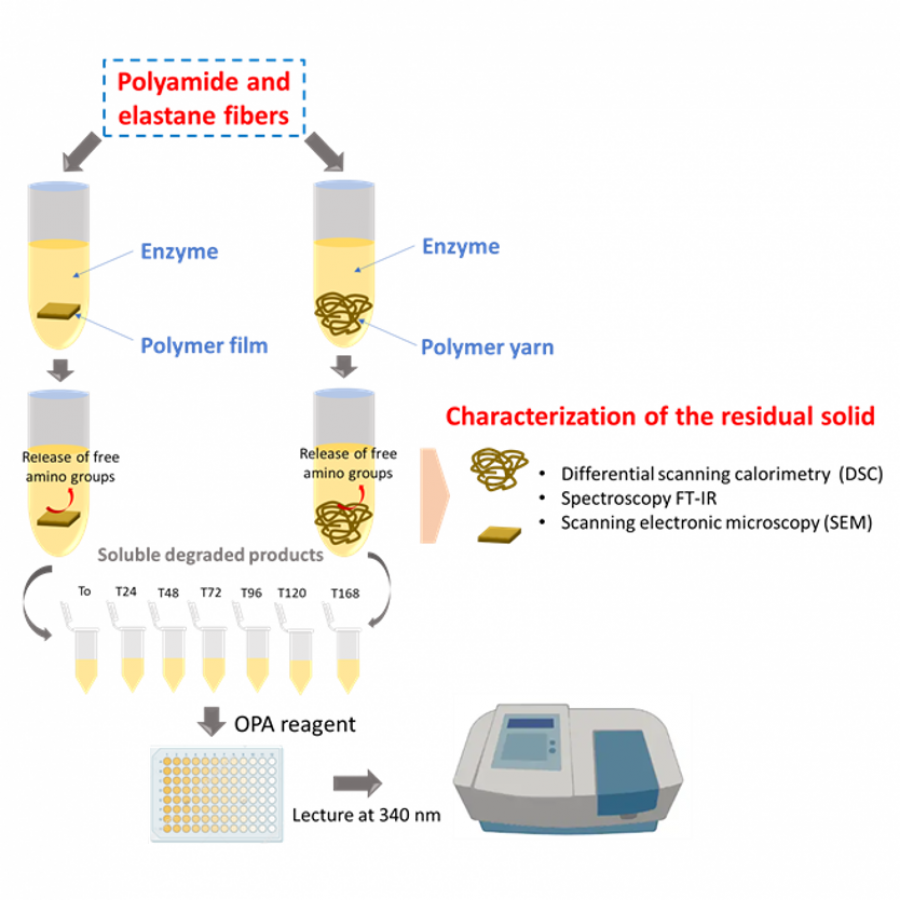 Methodology of surface enzyme hydrolysis of polymer film and yarn Surface hydrolyses of polyamide and elastane textile fibers (film and yarn) is quantified by measuring amino functional groups released during the enzymatic treatment by OPA assay at 340nm. The residual solid is characterized by thermal analyses, FT-IR sp 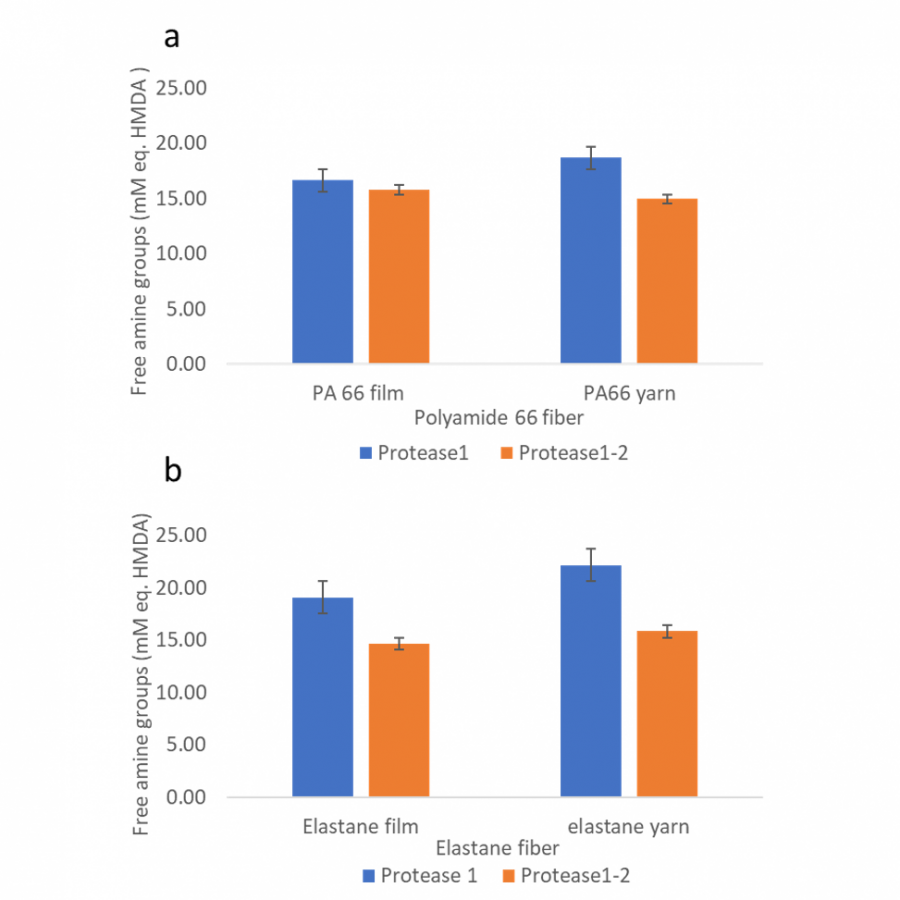 Quantification of total of amines released with OPA A) Surface enzymatic hydrolysis of nylon and B) Elastane fiber |
#1443 | Artificial Metalloenzyme-Catalyzed Enantioselective Amidation via Nitrene Insertion in Unactivated C(sp3)-H Bonds |
|
| Presenting author: | Kun YU of UNIVERSITY OF BASEL |
| Corresponding author: | Thomas WARD of UNIVERSITY OF BASEL |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial Metalloenzyme / Nitrene insertion / C-H activation / Chemogenetic optimization |
| Purpose: | Enantioselective C-H amidation offers attractive means to assemble C-N bonds to synthesize high-added value, nitrogen-containing molecules. In the past few decades, complementary enzymatic and homogeneous-catalytic strategies for C-H amidation have been reported. Herein, we report on an Artificial Metalloenzyme (ArM) resulting from anchoring a biotinylated Ir-complex within streptavidin (Sav). The resulting ArM catalyzes the enantioselective amidation of unactivated C(sp3)-H bonds. Chemogenetic optimization of the Ir cofactor and Sav led to significant improvement in both activity and enantioselectivity. Up to > 600 TON and 92% ee for the amidation of unactivated C(sp3)-H bonds was achieved. X-ray analysis of the artificial nitrene insertase (ANIase) along the evolutionary trajectory sheds light on critical second coordination sphere contacts leading to improved catalytic performance. |
| References: | [1] Hong, S. Y.; Park, Y.; Hwang, Y.; Kim, Y. B.; Baik, M. H.; Chang, S. Science 2018, 359, 1016-1021 [2] Wang, H.; Park, Y.; Bai, Z.; Chang, S.; He, G.; Chen, G. J. Am. Chem. Soc. 2019, 141, 7194-7201. [3] Wilson, M. E.; Whitesides, G. M. J. Am. Chem. Soc. 1978, 100, 306-307. [4] Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y. F.; Pellizzoni, M. M.; Lebrun, V.; Reuter, R.; Kohler, V.; Lewis, J. C.; Ward, T. R. Chem. Rev. 2018, 118, 142-231. |
| Figures: | 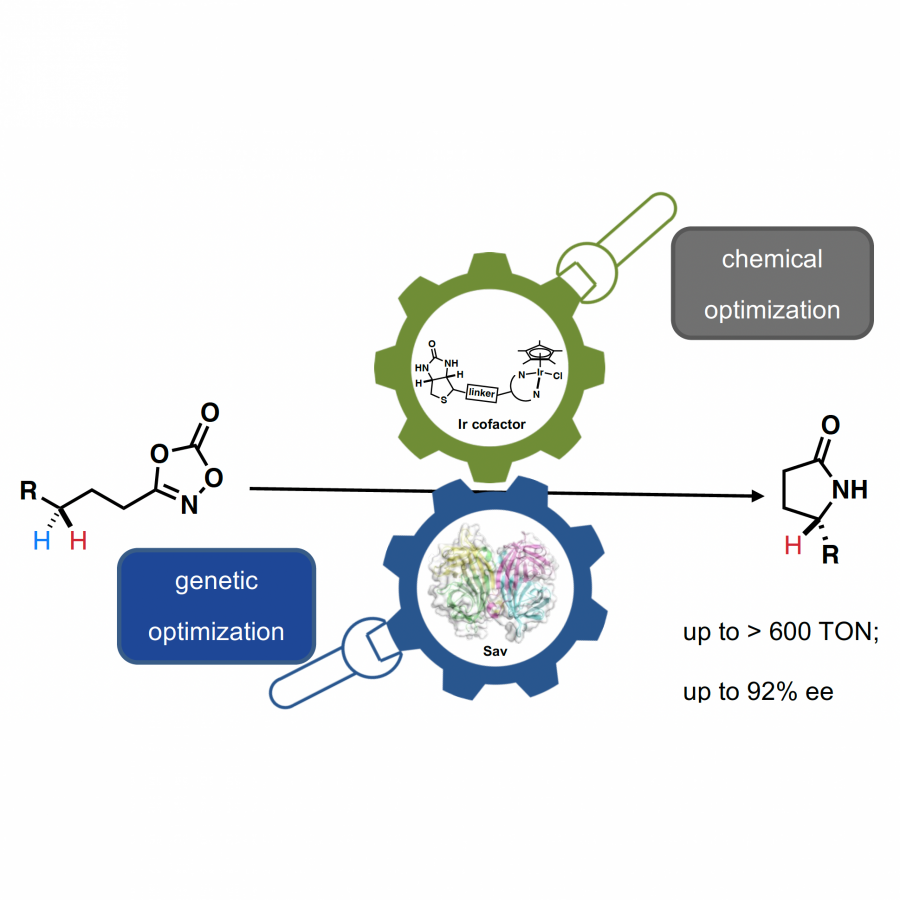 Artificial Metalloenzyme-Catalyzed Enantioselective Amidation via Nitrene Insertion in Unactivated C(sp3)-H Bonds The Artificial Nitrene insertase (ANIase) resulting from anchoring an Ir complex within Streptavidin is highly efficient for nitrene insertion in unactivated C(sp3)-H bonds to afford chiral γ-lactams. Chemogenetic optimization of the ANIase gave up to >60 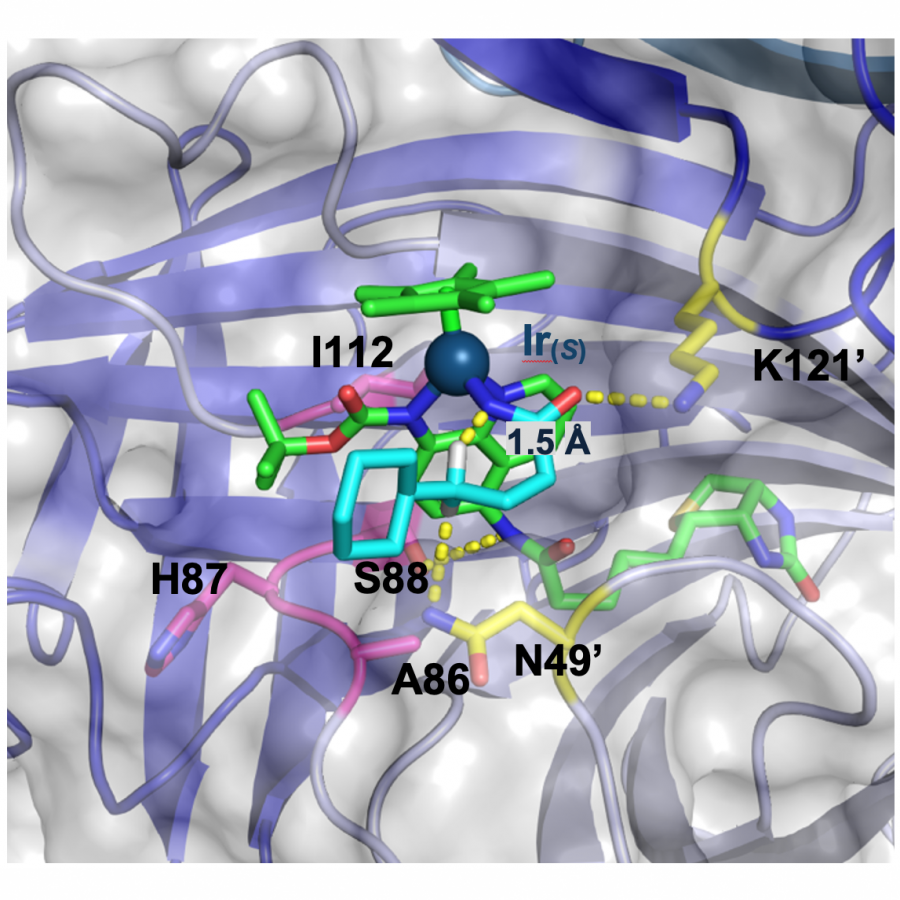 Computational study QM-MM calculations of the reaction path leading to the enantiopure product based on crystal structures of the ANIase. |
#1444 | Enchanted enzyme environments: The biocatalytic stereoconvergent cation-olefin cyclization |
|
| Presenting author: | Andreas SCHNEIDER of UNIVERSITY OF STUTTGART |
| Corresponding author: | Bernhard HAUER of UNIVERSITY OF STUTTGART |
| Other authors: | Christian CURADO of UNIVERSITAT DE GIRONA Sergi RUIZ-BARRANGAN of UNIVERSITAT DE GIRONA Thomas LYSTBAEK of UNIVERSITY OF STUTTGART Silvia OSUNA of UNIVERSITAT DE GIRONA & ICREA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / cation-olefin cyclization / stereoconvergence / squalene-hopene cyclase |
| Purpose: | The stereocontrolled cationic cyclization cascade is one of nature’s most elegant tools for generating molecular complexity in terpene metabolism and is unmatched by man-made catalysts. Moreover, it remains a major challenge for chemistry to harness the unique catalytic power of the enzymes that control such reactions for synthesis. Here we report the tailoring of the squalene-hopene cyclase (SHC) for cationic cyclization catalysis beyond the current state-of-the-art. Based on the promiscuous cyclization of homofarnesol to (–)-ambroxide as a model reaction, we demonstrated a rational enzyme engineering and application strategy that allowed us to exceed 105 catalytic turnovers. Moreover, we created a locally electron-enriched, active site that enables an unprecedented stereoconvergent cationic cyclization of a 50:50, 3E:3Z-homofarnesol mixture to sole (–)-ambroxide. Our results demonstrate how enzyme engineering can unlock and even extend the chemical wizardry of terpene cyclases. |
| References: | [1] A. Schneider, C. Curado, T. B. Lystbaek, S. Osuna, B. Hauer, ChemRxiv 2022, DOI 10.26434/CHEMRXIV-2022-XWCQJ. [2] A. Schneider, C. Curado, T. B. Lystbaek, S. Osuna, B. Hauer, Angew. Chem. Int. Ed. 2023, e202301607. |
| Figures: |  Scheme 1 The stereoconvergent head-to-tail cyclization of 3E/3Z-homofarnesol in an electronically enriched confined active site of the SHC results in the formation of sole (-)-ambroxide. |
#1445 | Enzymatic synthesis of the chiral neurotransmitter noradrenaline |
|
| Presenting author: | MIGKENA ZOUPALI of NORTHUMBRIA UNIVERSITY |
| Other authors: | MARK MULDOWNEY of STERLING PHARMA SOLUTIONS JOSE MUNOZ of NORTHUMBRIA UNIVERSITY AT NEWCASTLE WARISPREET SINGH of NORTHUMBRIA UNIVERSITY GRAEME TURNBULL of NORTHUMBRIA UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzymatic synthesis / biocatalysis / computational analysis and molecular docking / drug development |
| Purpose: | Green chemistry and biocatalysis has become a core field in the industrial biotechnology world, due to the need for sustainable and affordable methods for producing pharmaceuticals, fine chemicals and polymers, but most importantly due to enzymes’ excellent catalytic efficiency. The aim of our project is to identify target enzymes and structurally related homologues to be cloned, expressed, and used in biocatalytic assays against target compounds at Sterling Pharma Solutions. We will perform docking studies of chemical compounds within the enzyme models, and enzyme engineering to develop and enhance substrate specificity and overall yield. Enzyme immobilisation will be applied to enhance stability and performance. Finally, we aim to identify enzymes that are not commercially available from the open databases and produce them in-house, while lead candidate enzymes with great potential in biocatalysis and customer interest will be used in scale-up productions. Currently, our focus is on the development of the ketoreductase (KRED) superfamily. It has been demonstrated that KREDs are incredibly selective in the reduction of a variety of ketones, including bridged ring systems and rings with five, six, and seven members. One very known function common to the KRED superfamily is the transformation of ketones to chiral alcohols. At Sterling, our initial efforts are to synthesise the chiral neurotransmitter noradrenaline (compound 3, fig. 1) which can be accessed from the chloroketone (1) after treatment with hexamine. At the fundamental level, this project will offer the expertise and tools necessary to quickly produce customised enzyme solutions, made possible by the emerging fields of computational biochemistry and molecular biology, and will establish where biocatalysis can be offered as an alternative solution instead of the chemical synthesis. This is in line with Sterling’s future to onboard biocatalysis and produce enzymes in-house, which will result in the development of a new core technology platform entitled Sterling Platform for Enzyme Engineering and Development (SPEED). |
| Figures: |  Fig. 1. Proposed route to noradrenaline, 3, from commercially available chloroketone, 1. Proposed route to noradrenaline, 3, from commercially available chloroketone, 1. |
#1447 | High-throughput pH-shift assay for the screening of new PEP-dependent carboligase activities |
|
| Presenting author: | Melissa CONTE of TU DARMSTADT - DEPT. OF ORGANIC CHEMISTRY & BIOCHE |
| Other authors: | Wolf-Dieter FESSNER of TECHNISCHEN UNIVERSITÄT DARMSTADT |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | High-throughput / Carboligation / Screening / Neuraminic acid synthase |
| Purpose: | Neuraminic acid synthases (NeuS) are a family of PEP-dependent enzymes catalyzing an carbon-carbon bond formation, which is irreversible due to the concomitant release of phosphate. This renders these enzymes superior to related carboligases such as aldolases, which catalyze an equilibrium addition.[1] The identification of enzyme orthologs, or variants engineered by knowledge-guided smart mutagenesis, that are suitable for synthetic applications with non-native substrates requires a reliable high-throughput screening method.[2] With this aim, we have developed an assay that exploits the formal hydrolytic cleavage of the enol phosphate moiety with release of one equivalent of acid. The resultant drop in pH can be measured using a pH-sensitive dye such as Cresol Red, which changes its color from purple to yellow in an acidic environment.[3] This allows a semi-quantitative determination of the enzyme activity by reading the decrease in absorption at 570 nm. The pH assay has been tested on both cell-free extract obtained by sonication or chemical lysis, confirming its applicability for large-scale library screening and avoiding lengthy purification processes. Lastly, the sensitivity of the pH dependent assay has been validated by measuring the activity of the neuraminic acid synthase from Neisseria meningitidis (NeuSnme.)[4] against its native substrate N-acetyl-D-mannosamine and structurally analogous sugars such as D-Mannose. This project is funded by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Grant Agreement no. 956631 – CC-TOP |
| References: | [1] Fessner, W.-D., He, N., Yi, D., Unruh, P., Knorst, M. Enzymatic Generation of Sialoconjugate Diversity. In Cascade Biocatalysis: Integrating Stereoselective and Environmentally Friendly Reactions (Riva, S., Fessner, W.D., eds.) Wiley-VCH, Weinheim 2014, pp 361 - 392. [2] Sheludko, Y.V., Fessner, W.-D. Winning the numbers game in enzyme evolution - fast screening methods for improved biotechnology proteins. Curr. Opin. Struct. Biol., 2020, 63, 123-133. [3] Chapman, E., Wong, C.-H. A pH sensitive colorometric assay for the high-throughput screening of enzyme inhibitors and substrates: a case study using kinases. Bioorg. Med. Chem. 2002, 10, 551-555. [4] Gunawan J., Simard D., Gilbert M., Lovering A. L., Wakarchuk W. W., Tanner M. E., et al. Structural and Mechanistic Analysis of Sialic Acid Synthase NeuB From Neisseria Meningitidis in Complex With Mn2+, Phosphoenolpyruvate, and N-Acetylmannosaminitol. J. Biol. Chem. 2005, 280, 3555-3563. |
#1448 | The biotechnological potential of a novel CE16 exo-deacetylase from Thermothelomyces thermophilus |
|
| Presenting author: | CHRISTINA PENTARI of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Corresponding author: | EVANGELOS TOPAKAS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | ANASTASIA ZERVA of NATIONAL TECHNICAL UNIVERSITY OF ATHENS CHRISTOS KOSINAS of UNIVERSITY OF PATRAS PANAGIOTA KARAMPA of UNIVERSITY OF PATRAS MARIA DIMAROGONA of UNIVERSITY OF PATRAS VLADIMIR PUCHART of SLOVAK ACADEMY OF SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | CE16 acety esterase / Mode of action / Synergies / CE16 crystal structure |
| Purpose: | Biotechnological utilization of hemicellulose is an enticing concept, hindered by the recalcitrance of this biomaterial against biodegradation. Acetic acid substitutions of the main-chain carbohydrates contribute to the recalcitrance of biomass, posing a severe inhibitory effect on a number of hemicellulose-targeting enzymes such as xylanases, β-xylosidases and α-glucuronidases. Glucuronoxylan, in specific, is heavily substituted by acetic acid at positions O-2 and O-3, or even O-4 of the non-reducing-end xylopyranosyl residue (Xylp). The acetyl esters of xylan degraded by carbohydrate esterases (CE) which are grouped in the families CE1-CE7 and CE16. The latter family 16 is a growing group of enzymes, involving acetyl esterases, that exhibit an exceptional diversity regarding substrate specificity, regioselectivity and preference on oligomeric or polymeric substrates. However, further insight into the CE16 family is required for their efficient biotechnological exploitation. In this study, the acetyl esterase TtCE16B from Thermothelomyces thermophilus was heterologously expressed in Pichia pastoris and purified using immobilized metal affinity chromatography. The esterase was biochemically characterized and its mode of action was determined using monoacetates of 4-nitrophenyl β-D-xylopyranosides and multiply acetylated methyl β-D-xylopyranosides as substrate. The first crystal structure of a CE16 representative was determined, in apo- and product (acetate) bound form to 1.9 and 1.42 Å resolution, respectively. TtCE16B structure was solved by molecular replacement, using an AlphaFold prediction as starting model. Finally, the synergistic effect of TtCE16B activity with a number of hemicellulose-targeting enzymes, such as a CE6 acetylxylan esterase, xylanases of the GH10 and GH30 families and a GH43 β-xylosidase, during hydrolysis of delignified beechwood, was investigated. TtCE16B is an exo-acting deacetylase that performs optimally on oligosaccharides. The esterase targets the non-reducing-end Xylp, removing acetyl groups from positions O-3 and O-4, given that the other vicinal hydroxyl group is free, while a second acetylation at position O-2 does not obstruct the esterase activity. Catalyzing the O-4-deacetylation TtCE16B exhibits complementary regiospecificity to CE6 esterases, which are unable to deesterify this position. Regarding deconstruction of pretreated beechwood, TtCE16B acts in synergy with the β-xylosidase, by deacetylating the non-reducing-end Xylp of the xylooligomers and thus making them accessible to the exo-glycosidase. A minor synergy was also observed when TtCE16B was combined with the xylanases, although their activity is much more enhanced by the CE6 esterase efficiently deesterifying internal acetylated Xylps. Overall, the presence of TtCE16B resulted in increased hydrolysis efficiency of the pretreated biomass. The discovery of novel biocatalysts with distinctive specificities could become an asset for the design of efficient hemicellulolytic cocktails and further assist in the upcycling of residual biomass. This research was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “2nd Call for H.F.R.I. Research Projects to support Post-Doctoral Researchers” – Project ‘ARSIS’ (Project Number: 00328), by Grant (81074) from the Research Committee of the University of Patras via “C. CARATHEODORI” program and by the Slovak Research and Development Agency under the Contract no. APVV-20-0591. This work benefited from access to PETRAIII (EMBL Hamburg) και BESSY II (Helmholtz-Zentrum Berlin) and has been supported by iNEXT-Discovery, project number 871037, funded by the Horizon 2020 program of the European Commission. C. Pentari would like to thank the State Scholarship Foundation (IKY) of Greece for providing a PhD fellowship (NSRF 2014–2020) through the program “Development of human resources, education and lifelong learning”. |
| References: | [1] Gupta et al., Cellulose, 2021, 28, 3327-3345 [2] Puchart et al., Journal of Biotechnology, 2016, 233, 228-236 [3] Puchart et al., Carbohydrate Polymers, 2019, 205, 217-224 [4] Zerva et al., Bioresource Technology, 2021, 342-126058 |
| Figures: | 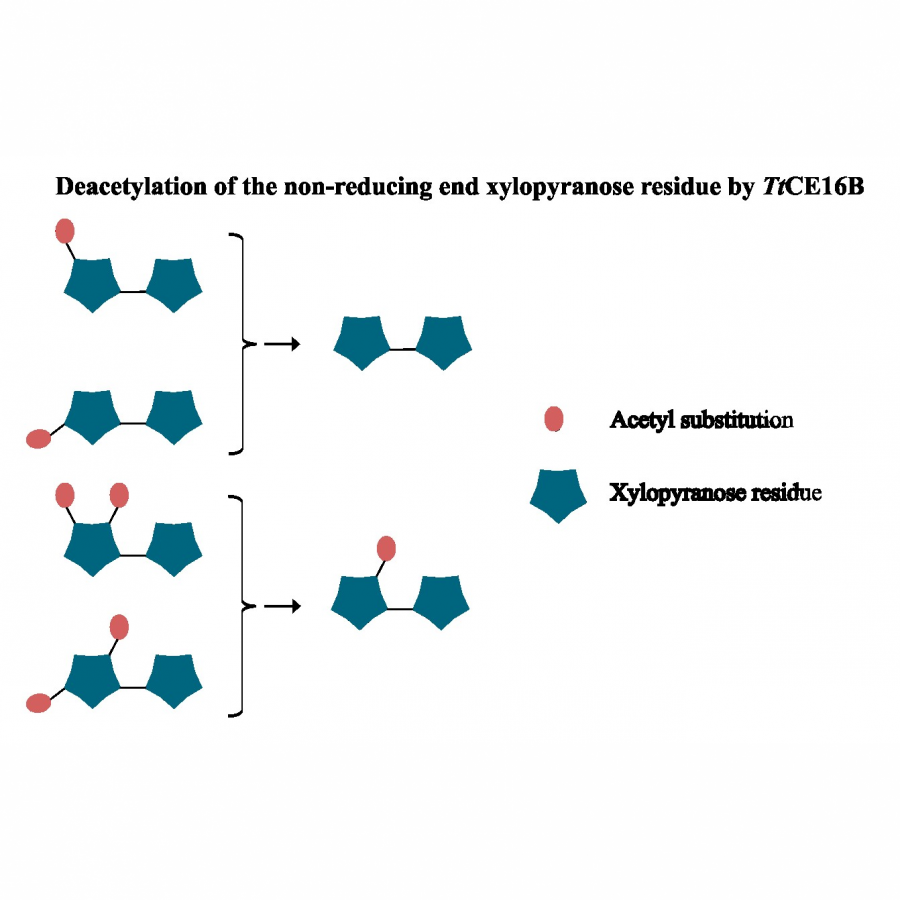 Positional specificity of TtCE16B acetyl esterase TtCE16B preferentially targets acetyl groups from positions O-3 and O-4 of the Xylp residue, given that the other vicinal hydroxyl group is free. |
#1449 | The BioCatHub project, applied research data management in biocatalysis |
|
| Presenting author: | Stephan MALZACHER of FORSCHUNGSZENTRUM JÜLICH |
| Other authors: | Patrizia WARMELINK of FORSCHUNGSZENTRUM JÜLICH Jens LOHMANN of FORSCHUNGSZENTRUM JÜLICH Felix ENGELMANN of FORSCHUNGSZENTRUM JÜLICH William FINNIGAN of UNIVERSITY OF MANCHESTER Nicholas TURNER of UNIVERSITY OF MANCHESTER Alaric PRINS of CAPE PENINSULA UNIVERSITY OF TECHNOLOGY Marilize LE ROES-HILL of CAPE PENINSULA UNIVERSITY OF TECHNOLOGY Jürgen PLEISS of UNIVERSITY OF STUTTGART Jan RANGE of UNIVERSITY OF STUTTGART Dörte ROTHER of RWTH AACHEN UNIVERSITY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Research data management / Automated data collection / Applied biocatalysis / Software development |
| Purpose: | Toxic waste degradation, the (asymmetric) synthesis of active pharmaceutical ingredients or the carbon-neutral synthesis of chemicals are just some examples in which enzymes are expected to play a key role in the years to come. Publicly available experimental data, produced in scientific processes, is one of the most important assets to realise this. It serves as the foundation to reproduce published experimental results, or as the basis for the development and reviewing of theoretical models. However, to be able to fulfil this role, the produced experimental data must be comprehensible, interoperable, and publicly available, which has often not worked in the past, and was even described by its own term: the reproducibility crisis. To counteract the reproducibility crisis, in 2016, the Findable, Accessible, Interoperable and Reusable (FAIR) data principles [1] were published, providing guiding principles in how to collect, document and publish experimental data. Nonetheless, the implementation of research data management (RDM) based on the FAIR data principles is time demanding, requires in-depth technical knowledge, and therefore often exceeds the capacities of the already-busy laboratory staff. For this purpose, we have developed the BioCatHub project. It aims to provide a RDM infrastructure to solve highly independent and individual RDM challenges in biocatalysis. The central component of the BioCatHub project is the BioCatHub platform. It is created to help experimenters to realise standardised data acquisition based on the FAIR data principles in biocatalysis with four central concepts. First, the BioCatHub platform (retrobiohub.org) works as data collection platform and provides a user interface, helping experimenters to describe experimental procedures in a wizard-like structure. Secondly, the BioCatHub platform contains data integration functions to query online databases like SabioRK, Brenda and autocomplete entries for the experimenters during the data collection process. Third, the automated communication between RetroBioCat [2] and the BioCatHub platform enables the planning and seamless integration of data from retrosynthetic reactions. Lastly, the BioCatHub platform writes and reads collected data to and from the machine-readable file format Enzyme markup language (EnzymeML) [3, 4]. This enables the possibility to collect, share, store or publish information about biocatalytic experiments, for example on Zenodo. On top, for data analysis and processing purposes, we have created the BioCatHub data processing repository, enabling the development of customised and reusable data processing workflows, using data previously collected in the BioCatHub platform as EnzymeML documents. For example, the export of the collected data into a spreadsheet for an experimenter friendly data analysis, the automated estimation of Michaelis-Menten parameters or the description of biocatalytic data using a first order reaction were already implemented as workflows. The vision realised with the BioCatHub project is to create an extendable infrastructure for researchers in biocatalysis to perform standardised data collection, storage and publishing based on the FAIR data principles combined with reusable data processing workflows adaptable to the needs of experimenters in the lab. Therefore, it should serve for our community, and should be extended with the needs of our biotransformation community. |
| References: | 1. Wilkinson MD, Dumontier M, Aalbersberg IJ, Appleton G, Axton M, Baak A, et al. Scientific data. 2016;3:18-27. doi:10.1038/sdata.2016.18 2. Finnigan W, Hepworth LJ, Flitsch SL, Turner NJ. Nature Catalysis. 2021:1-7. 3. Range J, Halupczok C, Lohmann J, Swainston N, Kettner C, Bergmann FT, et al. The FEBS Journal 2021. doi:10.1111/febs.16318 4. Lauterbach, S., Dienhart, H., Range, J. et al. Nat Methods 20, 400-402 (2023). https://doi.org/10.1038 |
| Figures: | 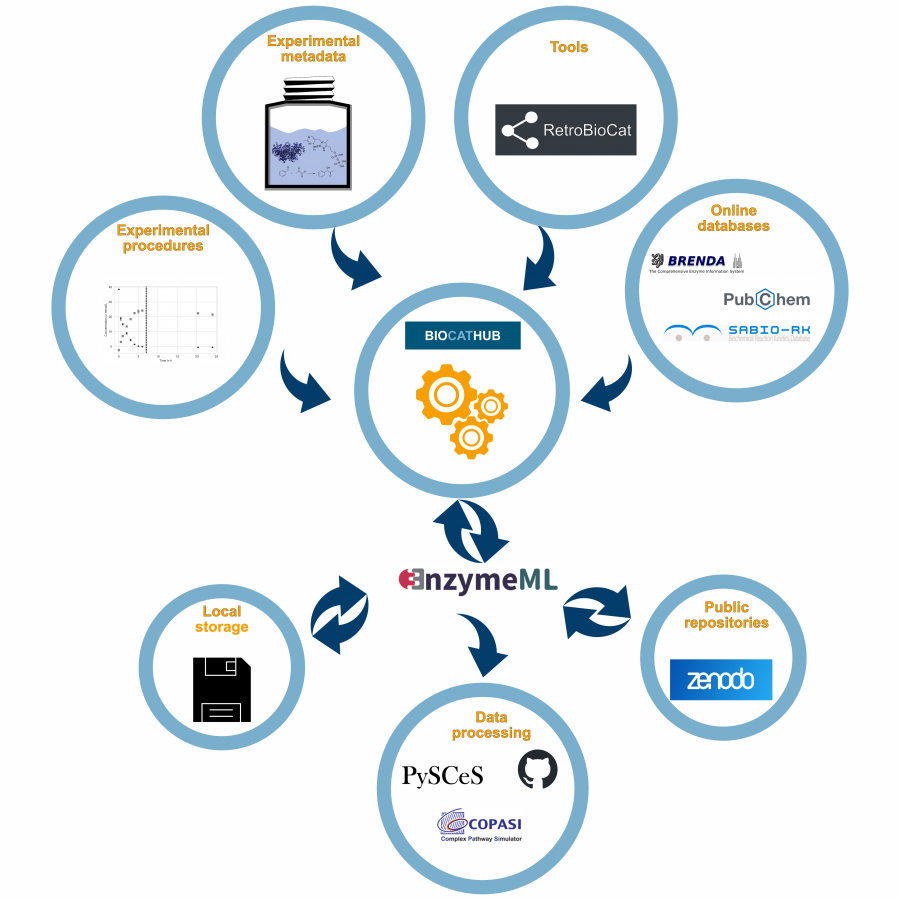 Infrastructure of the BioCatHub project |
#1450 | The LightCas process: A customised solution for a biocatalytic and light regulated three step cascade producing tetrahydroisoquinolines (THIQ) |
|
| Presenting author: | Stephan MALZACHER of FORSCHUNGSZENTRUM JÜLICH |
| Other authors: | Douglas WEBER of FORSCHUNGSZENTRUM JÜLICH Dominik MEISSNER of FORSCHUNGSZENTRUM JÜLICH Doris HAHN of FORSCHUNGSZENTRUM JÜLICH Christiane CLAASSEN of FORSCHUNGSZENTRUM JÜLICH Christian DICK of TH KÖLN Luca FEDE of TH KÖLN Dörte ROTHER of RWTH AACHEN UNIVERSITY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Automation / Synthetic enzyme cascades / NMR |
| Purpose: | Biocatalysis is a potential key technology for the development of carbon neutral and circular economy in the future [1]. One important task for biocatalytic applications is the enzyme catalysed synthesis of stereoisometrically pure active pharmaceutical ingredients (APIs) from renewable sources. An example is the asymmetric synthesis of tetrahydroisoquinolines (THIQ) in a three step enzymatic cascade (shown in Figure 1a) [2]. However, cross reactivities in the first and the third reaction step (shown in Figure 1b) cause a significantly more complicated reaction process and require a separation of the individual reaction step in either time or space. To realise the time separation, the individual enzymes can be fused with photosensitizers, which, upon light exposure produce reactive oxygen species (ROS), inactivating the to the photosensitiser attached enzyme [3]. Nonetheless, even if the light-induced inactivation of the enzymes enables a separation of the reaction steps in time (Figure 1a) and thereby prevents cross reactivities (Figure 1b), the process is still labour intensive for experimenters, as the concentrations of the substrates, products and intermediates of the individual reaction steps have to be measured continuously and the light inactivation as well as the induction of the respective reaction steps need to be performed manually. To solve these challenges, a system is required, which automatically monitors the reaction, performs the light inactivation, and induces the individual reaction steps. The realisation of the just mentioned challenges is the goal of the LightCas process. Using Arduino microcontrollers, we have created a hardware infrastructure, able to irradiate the reaction mixture with light as well as the feeding of the respective enzyme and cosubstrate required for the individual reaction steps. With the automated biocatalysis and benchtop NMR application (ABBA), we have developed a customised software, which automatically monitors the reactions by a benchtop NMR and controls the hardware infrastructure by communicating with the Arduino microcontrollers. With the combination of the Hardware infrastructure and the ABBA software, we have established an automated process (the LightCas process), which handles the complex reaction control, occurring due to the cross reactivities in the synthesis of THIQ (Figure 1a, b). As an additional functionality, a communication with the BioCatHub platform enables the automated storage and publication of the collected data, based on the Findable, Accessible, Interoperable and Reusable guiding principles due to an implemented interface to the BioCatHub platform. |
| References: | 1. Sheldon RA, Brady D. Green Chemistry, Biocatalysis, and the Chemical Industry of the Future. ChemSusChem. 2022;15:e202102628. doi:10.1002/cssc.202102628. 2. Weber D, Souza Bastos L de, Winkler M, Ni Y, Aliev AE, Hailes HC, Rother D. Multi-enzyme catalysed processes using purified and whole-cell biocatalysts towards a 1,3,4-substituted tetrahydroisoquinoline. RSC Advances. 2023;13:10097-109. doi:10.1039/D3RA01210G. 3. Gerlach T, Schain J, Söltl S, van Schie MMCH, Hilgers F, Bitzenhofer NL, et al. Photo-Regulation of Enzyme Activity: The Inactivation of a Carboligase with Genetically Encoded Photosensitizer Fusion Tags. Front. Catal. 2022. doi:10.3389/fctls.2022.835919. |
| Figures: | 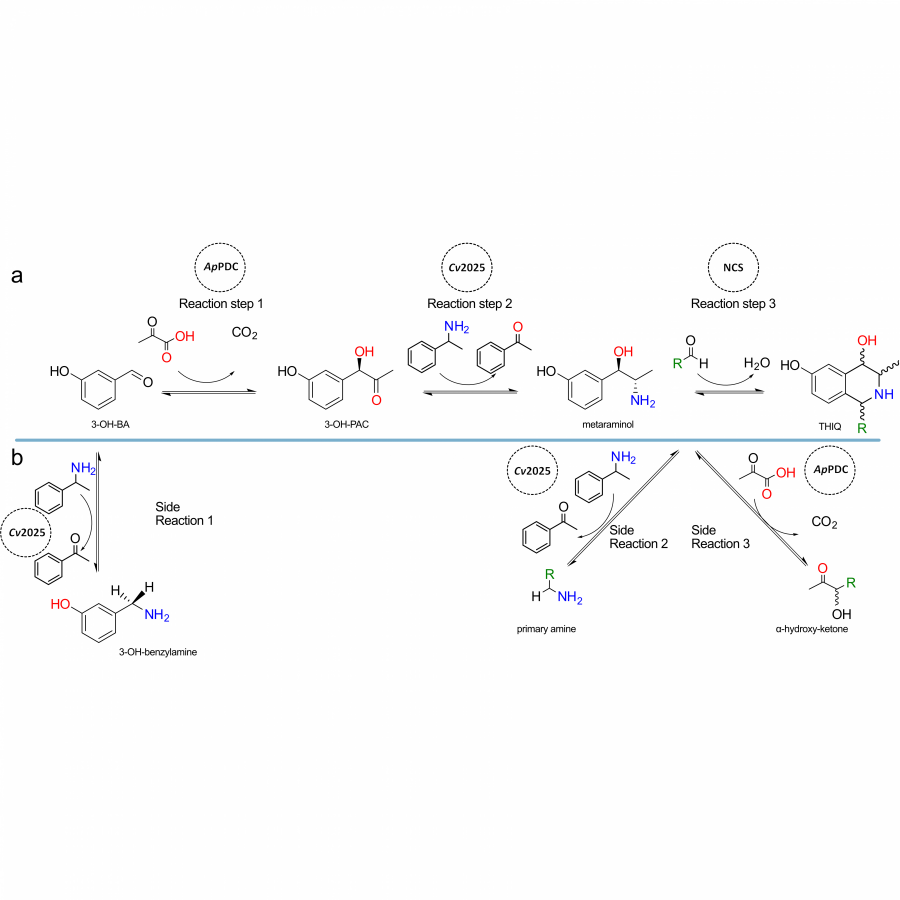 Figure 1 Reaction schema displaying the asymmetric synthesis of THIQ. ApPDC: Acetobacter pasteurianus pyruvate decarboxylase, Cv2025: Chromobacterium violaceum transaminase, NCS: Norcolaurine synthase from Thalictrum flavum. |
#1453 | alpha-L-Fucosidases - a tool for the synthesis of prebiotics |
|
| Presenting author: | Agne KRUPINSKAITE of DEPARTMENT OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY |
| Other authors: | Jonita STANKEVICIUTE of DEPARTMENT OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY Ruta STANISLAUSKIENE of DEPARTMENT OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY Rasa RUTKIENE of DEPARTMENT OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY Rolandas MESKYS of DEPARTMENT OF MOLECULAR MICROBIOLOGY AND BIOTECHNOLOGY, INSTITUTE OF BIOCHEMISTRY, LIFE SCIENCES CENTER, VILNIUS UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | #fucosidases / #fucosylated / #oligosaccharidesynthesis / #humanmilkoligosaccharides |
| Purpose: | α-L-Fucosidases naturally catalyse the hydrolysis reaction of the glycosidic linkage. However, these enzymes are also capable of catalysing glycosidic bond synthesis reactions. This can be achieved by changing the reaction conditions, such as varying substrates, organic compounds, and salt concentrations. This capability of α-L-fucosidases could be used in fucosylated oligosaccharide synthesis since these compounds comprise the most significant part of oligosaccharides found in human milk and are highly beneficial components of infant milk formula and functional foods. Various studies show antiviral, antimicrobial, and immune response-modulating functions of fucosylated oligosaccharides [1]. However, there is still a lack of sustainable methods to synthesise these stereospecific compounds in high yields. Here, we introduce five bacterial α-L-fucosidases selected via metagenomic library screening. Through bioinformatics analysis, we investigate the characteristics of the sequences and structures of these enzymes. Finally, we evaluate the capability of the α-L-fucosidases to perform the trans-fucosylation reaction resulting in oligosaccharide compounds similar to those found in human milk. We also investigate the products of trans-fucosylation reactions using TLC, HPLC-ELSD, and HPLC-MS techniques. Our findings aim to offer valuable insights into utilising α-L-fucosidases for fucosylated oligosaccharides synthesis. Additionally, we aim to identify the structural properties that may affect the efficiency of the trans-fucosylation reaction. |
| References: | [1] Walsh, C., Lane, J. A., van Sinderen, D., & Hickey, R. M. (2020). Human milk oligosaccharides: Shaping the infant gut microbiota and supporting health. Journal of Functional Foods, 72, 104074. https://doi.org/10.1016/j.jff.2020.104074 |
#1455 | A photobiocatalytic one-pot sequential approach to transform aryl diazonium salts into enantioenriched 1-arylpropan-2-ols |
|
| Presenting author: | IVAN LAVANDERA of UNIVERSITY OF OVIEDO |
| Other authors: | LAURA RODRIGUEZ-FERNANDEZ of UNIVERSITY OF OVIEDO JESUS ALBARRAN-VELO of UNIVERSITY OF OVIEDO VICENTE GOTOR-FERNANDEZ of UNIVERSITY OF OVIEDO |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | photocatalysis / alcohol dehydrogenases / diazonium salts / Meerwein arylation |
| Purpose: | Photocatalysis has recently emerged as a powerful tool towards the synthesis of highly valuable (chiral) organic compounds in a straightforward manner and under mild reaction conditions. Interestingly, light-driven chemical transformations can be easily combined with enzymes, giving rise to photobiocatalytic processes that have already been applied for multiple cascades as well as for nicotinamide cofactor recycling purposes.[1] Thus, combining photocatalytic with enzymatic methods allows the creation of molecular complexity from easily accessible starting materials, especially when assembling C–C bond formation and stereoselective processes. Particularly, the Meerwein arylation consists of the coupling of a diazonium salt with an electronically poor alkene using a reducing promoter, typically a copper(I) salt as catalyst, affording the corresponding alkylated arene product.[2] In this context, different groups have exploited the light-driven Meerwein-type α-arylation between aryl diazonium salts and enol acetates using ruthenium complexes[3] or porphyrins[4] as photocatalysts in organic media to produce 1-arylpropan-2-ones. Based on our previous experience in the photobiocatalytic synthesis of optically active 1-arylpropan-2-ols,[5] valuable intermediates of different organic compounds with remarkable biological activities,[6] herein we have envisaged to integrate a Meerwein arylation reaction under light conditions catalyzed by the photosensitizer 9-mesityl-10-methylacridinium perchlorate ([Acr-Mes]ClO4), followed by the action of stereocomplementary alcohol dehydrogenases (Scheme 1). Hence, different enantiopure 1-arylpropan-2-ols have been obtained from simple and easily accessible aryl diazonium salts and isopropenyl acetate. Some of the experiments were easily scaled-up and a one-pot three-step conversion of aniline into enantiopure (R)- or (S)-1-phenylpropan-2-ol has also been demonstrated. |
| References: | [1] L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist, W. Kroutil, ACS Catal. 2019, 9, 4115-4144. [2] S. Kindt, M. R. Heinrich, Synthesis 2016, 48, 1597-1606. [3] T. Hering, D. P. Hari, B. König, J. Org. Chem. 2012, 77, 10347-10352. [4] A. A. N. de Souza, N. S. Silva, A. V. Müller, A. S. Polo, T. J. Brocksom, K. T. de Oliveira, J. Org. Chem. 2018, 83, 15077-15086. [5] J. Albarrán-Velo, V. Gotor-Fernández, I. Lavandera, Adv. Synth. Catal. 2021, 363, 4096-4108. [6] M. Morelli, E. Tognotti, Exp. Neurol. 2021, 342, 113754. |
| Figures: | 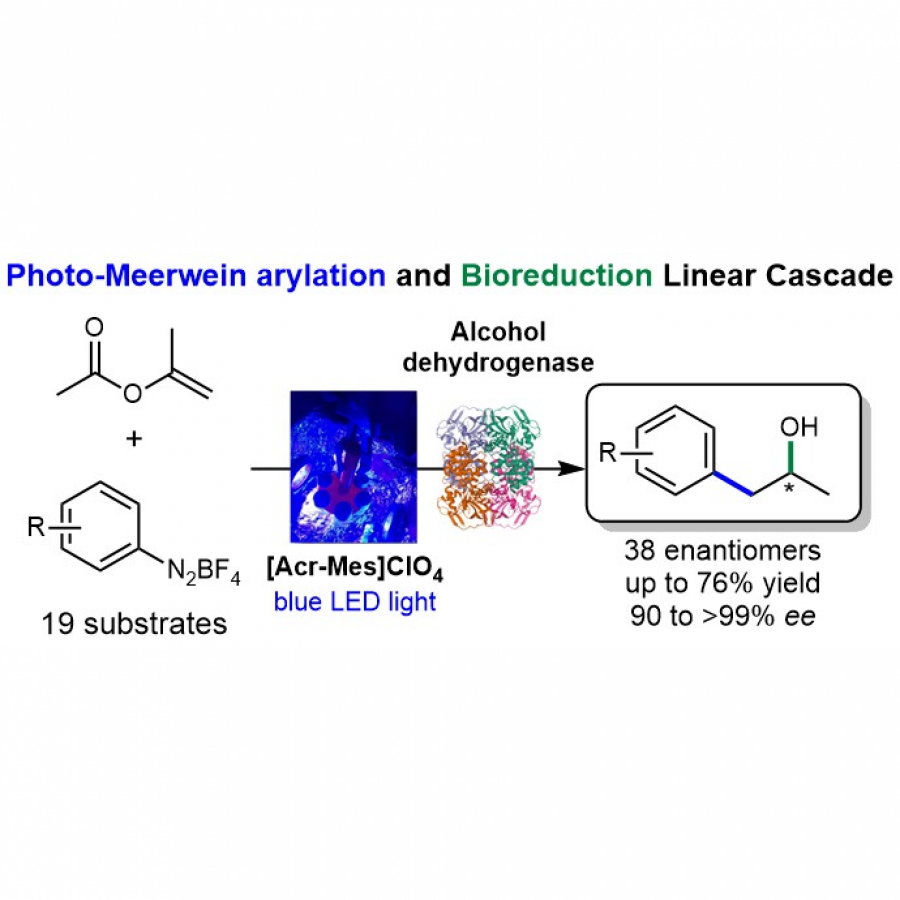 Scheme 1 Photobiocatalytic cascade to obtain enantioenriched 1-arylpropan-2-ols from diazonium salts |
#1457 | Development of a heterogeneous biocatalyst for reduction of aliphatic nitro groups |
|
| Presenting author: | Tara LURSHAY of UNIVERSITY OF OXFORD |
| Other authors: | Daria SOKOLOVA of DEPARTMENT OF CHEMISTRY Tim SUDMEIER of DEPARTMENT OF CHEMISTRY Jack ROWBOTHAM of DEPARTMENT OF CHEMSITRY Sarah CLEARY of HYREGEN LTD. Holly REEVE of HYDREGEN LTD. Georgia STONADGE of DEPARTMENT OF CHEMISTRY Kylie VINCENT of DEPARTMENT OF CHEMISTRY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | #biocatalysis / #nitro-reduction / / |
| Purpose: | Amines are valuable chemical moieties widely used in pharmaceutical synthesis, both as precursors and as drug molecules themselves. They are often produced by late-stage reduction of nitro groups during synthesis.[1] Currently used methods for the nitro reduction reaction usually require unsustainable platinum group metals (e.g., Pt, Pd, Rh) as well as high-intensity conditions including high temperature, pressure and organic solvents.[2] Other hetero- and homo-geneous catalysis methods have been extensively studied, but are not often suited to industrial-scale synthesis.[3] In recent years biocatalysis has generated much interest as a highly efficient route for specific reactions under mild aqueous conditions.[4] The specificity of these reactions also improves atom economy, diminishing waste products from the reaction. During catalyst development, taking a modular approach to the catalyst allows the reaction to be broken-down into its individual components.[5] Current biocatalysts can be limited in substrate scope, and here we show tuning of the biocatalyst components in order to extend this to include a broader range of nitro substrates able to be hydrogenated under mild conditions. |
| References: | [1] Sukhorukov, A. Y., Front. Chem., 2020, 8, 215 [2] S. Sun et al., J Environ Manage, 2022, 305, 114383. [3] D. Formenti et al., Chem Rev, 2019, 119, 2611-2680. [4] M. A. Ramirez, S. J. Srinivasan, S. E. Cleary, Peter. M. T. Todd, H. A. Reeve, K. A. Vincent, Frontiers in Catalysis, 2022, 2, 906694. [5] H. A. Reeve et al., Biochemical Journal, 2017, 474, 215-230. |
| Figures: | 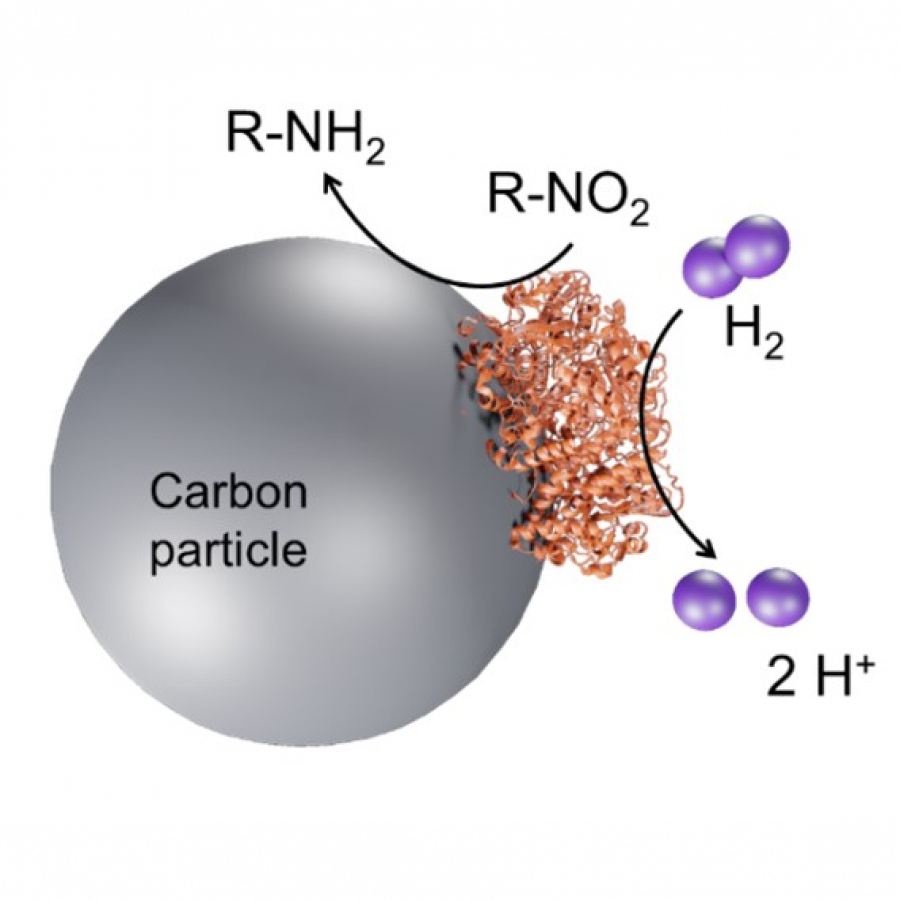 Figure 1 Schematic representation of a modular biocatalyst reducing R-NO2 to R-NH2 with H2 as atom-efficient reductant. |
#1459 | Harnessing the Potential of Fusion Enzymes in Multi-enzymatic Cascades |
|
| Presenting author: | Zinnia DSOUZA of TECHNICAL UNIVERSITY OF MUNICH, CAMPUS STRAUBING |
| Corresponding author: | Volker SIEBER of TECHNICAL UNIVERSITY OF MUNICH, CAMPUS STRAUBING |
| Other authors: | Okke MELSE of TECHNICAL UNIVERSITY OF MUNICH, CAMPUS STRAUBING Gerhard SCHENK of UNIVERSITY OF QUEENSLAND Colin SCOTT of CSIRO |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | The concept of enzyme cascades, where multiple enzymatic reactions are interconnected, has gained substantial interest due to its ability to streamline complex chemical transformations. To mediate redox transformations, Oxidoreductases, a class of enzymes involved in electron transfer reactions, play a crucial role. However, their integration into biocatalytic transformations and multi-enzymatic cascades often presents challenges e.g. the reliance on costly and limited cofactors. To address this issue, fusion enzymes that regenerate their cofactors have emerged as promising tools for cofactor-dependent biocatalysis [1]. In this work, we present a comprehensive overview of the process involved in designing fusion enzymes for cascade applications, namely with the established cascade for the conversion of glucose to isobutanol [2]. In-silico modeling is employed to aid with the creation of fusion enzymes to control the spatial arrangement of catalytic domains and identify cofactor-binding sites. The enzymes are modified to produce chimeric proteins that combine catalytic functions with the capacity to recycle cofactors while retaining the oxidized/reduced cofactor in a confined microenvironment that benefits from the fusion pattern. The approaches used to choose suitable fusion partners are reviewed, and the importance of structural compatibility for preserving catalytic functions is emphasized. Moreover, we want to delve into the challenges and opportunities associated with fusion enzymes, including limitations in in-silico accuracy and the potential for experimental validation. These include optimization of fusion enzyme design, scalability, and compatibility with different cofactors. We aim to highlight possible challenges, concerns, and recent advances in the use of fusion enzymes, as well as to forecast future directions in the field of cofactor regeneration. |
| References: | 1. Hartley, C.J., et al., Engineered Enzymes that Retain and Regenerate their Cofactors Enable Continuous-Flow Biocatalysis. Nature Catalysis 2019. 2(11) 1006-1015. 2. Guterl, J.K., et al., Cell-free metabolic engineering: production of chemicals by minimized reaction cascades. ChemSusChem, 2012. 5(11): p. 2165-72. |
#1462 | Role of a flexible loop in the substrate selectivity of an engineered variant of hyperthermophilic multicopper oxidase McoP |
|
| Presenting author: | Patricia BORGES of ITQB-NOVA |
| Corresponding author: | Lígia MARTINS of ITQB-NOVA |
| Other authors: | Vânia BRISSOS of ITQB-NOVA Carlos FRAZAO of ITQB-NOVA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Multicopper oxidases / Directed evolution / X-ray crystallography / Biotechnology |
| Purpose: | Multicopper oxidases (MCOs) oxidize a range of aromatic substrates such as polyphenols, methoxy-substituted phenols, and amines, concomitantly with reducing dioxygen to water. Some MCOs, denominated metallo-oxidases, exhibit catalytic activity toward metal ions, such as Cu(I) and Fe(II). In this work, we have used directed evolution to improve substrate efficiency of the metalloxidase McoP from the Pyrobaculum aerophilum for organic substrates. Random mutagenesis and DNA shuffling followed by high-throughput screening led to the generation of variant 3F3, showing only six mutations and a 100-fold higher catalytic efficiency for ABTS than wild-type enzyme. The X-ray crystallography was used to solve the crystal structure of the 3F3 variant, using the wild-type structure as a search model (PDB 3AW5, Sakuraba et al). Two mutations (V206I and S331P) are distal to the T1Cu, at ~26 Å and ~33 Å, mutations P292H and F361S are at ~13 Å and ~12 Å, whereas mutations M393V and P390T are at ~ 6 Å to T1Cu; P390T is adjacent to the T1Cu ligand H391, and M393V is adjacent to the T2Cu ligand H394. An analysis of the solvent access in the crystal structures allowed us to define two main pathways from the solvent media to the T1Cu, one tunnel (Path 1) and one cavity (Path 2) parallel to each other. The tunnel is surrounded by a flexible loop (288-310) that contains one of the six mutations, P292H, that together with a short α-helix is covering the T1Cu access in the wild-type. This loop becomes more flexible in the 3F3 variant, resulting in an enlarged diameter (4.0 Å) of the tunnel, when compared with the wild-type (2.8 Å), which is expected to facilitate electron transfer from substrates to T1Cu. The cavity shows the H465 ligand and the nearby residue W355 (at 3 Å) exposed through a pocket (P1). Interestingly, the W355 is approximately three times more solvent-exposed in 3F3 due to the appearance of a new pocket (P2) as a result of the higher flexibility of the loop 288-310. The enlarged tunnel and the extra-pocket P2 in the cavity of 3F3 can explain the significantly higher activity of this variant towards ABTS. This work allows to investigate the molecular mechanisms involved in the evolution of enzyme fitness, which are important for engineering new proteins for biotechnological applications. References: Sakuraba, H., et al, Acta Crystallogr Sect F Struct Biol Cryst Commun, 2011, 67, 753-757. Acknowledgements: This work was supported by the Fundação para a Ciência e Tecnologia, Portugal, by project grants PTDC/BII-BBF/29564/2017, EXPL/BIA-BQM/0473/2021 and 2022.02027.PTDC. B-Ligzymes (GA 824017) from the European Union’s Horizon 2020 Research and Innovation Program is also acknowledged for funding a secondment of P. Borges to Zymvol, Barcelona, SP. |
| References: | Sakuraba, H., et al, Acta Crystallogr Sect F Struct Biol Cryst Commun, 2011, 67, 753-757. |
#1463 | Kinetic evaluation and reaction optimization of a two-step enzymatic cascade for sustainable biosynthesis of coniferol from lignin-derived ferulic acid |
|
| Presenting author: | Sansanee KHIAWJAN of THE ADVANCED CENTRE FOR BIOCHEMICAL ENGINEERING, DEPARTMENT OF BIOCHEMICAL ENGINEERING, UNIVERSITY COLLEGE LONDON |
| Corresponding author: | Gary J. LYE of THE ADVANCED CENTRE FOR BIOCHEMICAL ENGINEERING, DEPARTMENT OF BIOCHEMICAL ENGINEERING, UNIVERSITY COLLEGE LONDON |
| Other authors: | Pinar KARAGOZ of SCHOOL OF ENGINEERING AND INNOVATION, THE OPEN UNIVERSITY Marco P. C. MARQUES of THE ADVANCED CENTRE FOR BIOCHEMICAL ENGINEERING, DEPARTMENT OF BIOCHEMICAL ENGINEERING, UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Coniferyl alcohol / Lignocellulosic biomass / Carboxylic acid reductase (CAR) / Aldo-keto reductase (AKR) |
| Purpose: | Coniferol (CA) is one of promising precursors for varieties of flavor, fragrance, and pharmaceutical chemicals. A two-step enzymatic reaction to convert ferulic acid (FA) into CA via activities of a carboxylic acid reductase from Norcadia iwensis (NiCAR) and an aldo-keto reductase from Coptotermes gestroi (CgAKR-1) has been recently published in literature, mainly focusing on whole cell biocatalysis. FA is a phenolic compound which can be derived from hydrolysis of lignin-containing biomass. Thus, this study focused on evaluation of kinetics of NiCAR and CgAKR-1 and optimization of the enzymatic reaction to outline bioprocess design to improve productivity of CA. Kinetics study of NiCAR towards FA and CgAKR-1 towards CALD pointed out inhibitory effects of both substrates against the enzymes. It suggested that maintaining low concentrations of the substrates might allow greater productivity of CA. Ratio of NiCAR to CgAKR-1 clearly affects productivity of CA which the optimum was found at 1U of NiCAR to 25 U of CgAKR-1 at a certain condition. NADPH regeneration via oxidation of glucose by the glucose dehydrogenase (GDH) was introduced into the system to overcome expensive supplementation of the cofactor. However, development of an effective ATP regeneration is still a challenge. In term of bioprocess design, the enzymes were immobilized on nickel-nitrilotriacetic acid (Ni-NTA) agarose beads. The immobilized enzymes exhibited significantly improved activity and stability without kinetic parameters alteration. As batch, it provided volumetric productivity of CA at 57 mg∙L-1∙h-1. The productivity was clearly improved to 95 mg∙L-1∙h-1 when performed as fed batch. Moreover, the immobilized enzyme can convert FA obtained from an alkaline hydrolysis of sugar beet pulp as batch with productivity of 15 mg∙L-1∙h-1. In summary, the study provided kinetics understandings of NiCAR and CgAKR-1 enzymes to convert FA into CA, contributing to further bioprocess design to intensify the productivity of CA. Moreover, it emphasizes potential of enzymatic cascade reaction, particularly CAR-AKR-GDH, for the synthesis of high value chemicals from lignocellulosic waste stream. |
| Figures: | 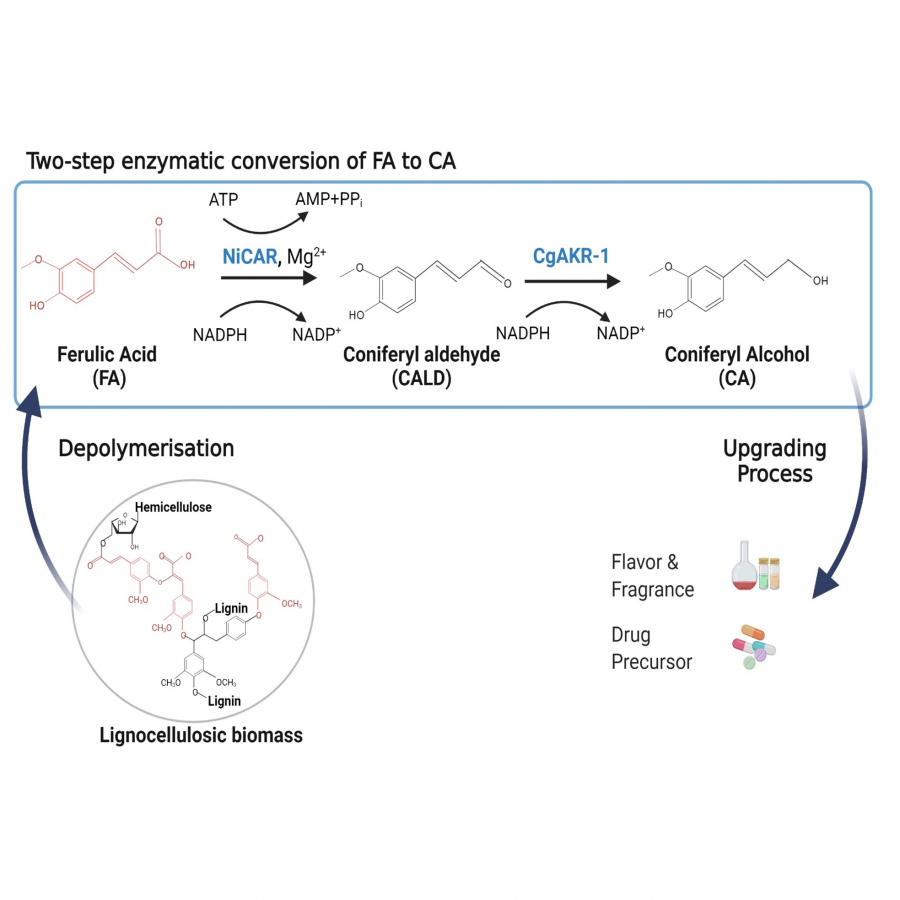 Figure 1 Illustration of two-step enzymatic conversion of FA into CA. The figure shows potential of the two-step enzymatic reaction to convert FA derived from lignocellulosic biomass into CA, potentially serving as a precursor for fragrance and |
#1464 | Exploration of metagenomic sequence space for novel specificities from the PEP-dependent N-acetylneuraminic acid synthase protein family |
|
| Presenting author: | Theofania Pagona ANDREADAKI of PROZOMIX LIMITED |
| Other authors: | Melissa CONTE of TECHNISCHE UNIVERSITÄT DARMSTADT Mehmet Mervan ÇAKAR of UNIVERSITY OF ZAGREB Simon J. CHARNOCK of PROZOMIX LIMITED Wolf-Dieter FESSNER of TECHNISCHE UNIVERSITÄT DARMSTADT Zvjezdana FINDRIK BLAŽEVIĆ of UNIVERSITY OF ZAGREB Aurelio HIDALGO of AUTONOMOUS UNIVERSITY OF MADRID |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | metagenomics / biocatalysis / enzyme discovery / novel activity |
| Purpose: | Carboligases are one of the most desirable enzyme classes in organic synthesis since they form carbon-carbon bonds. PEP lyases (or synthases) are of particular interest as their mechanism of action involves the cleavage of phosphoenolpyruvate with subsequent release of phosphate, thus making their reactions irreversible given the strong driving force for product formation. There are three categories or PEP lyases, (i) N-acetylneuraminic acid synthases (sialic acid synthases), (ii) 2-keto-3-deoxyoetosonic acid 8-phosphate synthases (KDO synthases) and (iii) 3-deoxy-D-arabino-heptulosonic acid 7-phosphate synthases (DAHP synthases)[1]. This project is focused on the ubiquitous, yet largely uncharacterised, sialic acid synthases, where to date research has been limited to genetic, metabolic, biochemical and/or structural studies[2–5]. Thus the fundamental and commercial potential of additional novel specificities harboured by this family are as yet unknown. Towards investigating that end, a screening panel of 112 novel / maximum diversity members of this family was designed through metagenomic mining. All targets were selected using N-acetylneuraminic acid synthase (NeuB) from Neisseria meningitidis (PDB code 1XUZ) as reference sequence, and evenly cover at the phylogenetic level the whole protein family. All target sequences were mined from Prozomix Limited's proprietary metagenomic libraries using "ProzomiGo" software. Preliminary biochemical screening experiments with 58 soluble recombinant products have indicated novel specificities indeed reside within this family and are currently being confirmed. |
| References: | [1] Fessner, W.-D.; Walter, C. Enzymatic C-C Bond Formation in Asymmetric Synthesis; 1996. [2] Gunawan, J.; Simard, D.; Gilbert, M.; Lovering, A. L.; Wakarchuk, W. W.; Tanner, M. E.; Strynadka, N. C. J. Structural and Mechanistic Analysis of Sialic Acid Synthase NeuB from Neisseria Meningitidis in Complex with Mn2+, Phosphoenolpyruvate, and N-Acetylmannosaminitol. Journal of Biological Chemistry 2005, 280 (5), 3555-3563. https://doi.org/10.1074/jbc.M411942200. [3] Joseph, D. D. A.; Jiao, W.; Parker, E. J. Arg314 Is Essential for Catalysis by N -Acetyl Neuraminic Acid Synthase from Neisseria Meningitidis. Biochemistry 2013, 52 (15), 2609-2619. https://doi.org/10.1021/bi400062c. [4] Chen, X.; Varki, A. Advances in the Biology and Chemistry of Sialic Acids. ACS Chemical Biology. February 19, 2010, pp 163-176. https://doi.org/10.1021/cb900266r. [5] Lewis, A. L.; Desa, N.; Hansen, E. E.; Knirel, Y. A.; Gordon, J. I.; Gagneux, P.; Nizet, V.; Varki, A. Innovations in Host and Microbial Sialic Acid Biosynthesis Revealed by Phylogenomic Prediction of Nonulosonic Acid Structure. www.pnas.org/cgi/content/full/. |
#1468 | 1-(4-hydroxyphenyl)-ethanol dehydrogenases from A. aromaticum – mechanism of inactivation |
|
| Presenting author: | Ivana ALEKSIC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHE |
| Other authors: | Mateusz TATARUCH of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY Milan POLAKOVIČ of FACULTY OF CHEMICAL AND FOOD TECHNOLOGY STU Viera ILLEOVÁ of FACULTY OF CHEMICAL AND FOOD TECHNOLOGY STU Anna MIŁACZEWSKA of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY Patrik CABADAJ of FACULTY OF CHEMICAL AND FOOD TECHNOLOGY STU Anna KLUZA of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY Maciej SZALENIEC of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY Tomasz BOROWSKI of JERZY HABER INSTITUTE OF CATALYSIS AND SURFACE CHEMISTRY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | (R)/(S)-1-(4-hydroxyphenyl)-ethanol dehydrogenase / inactivation / aggregation / activity stabilization |
| Purpose: | R- and S-specific 1-(4-hydroxyphenyl)-ethanol dehydrogenases (R-HPED and S-HPED) are enzymes coming from denitrifying bacterium Aromatoleum aromaticum (Azoarcus sp.). They belong to short-chain dehydrogenase/reductase (SDR) family and physiologically catalyze the NAD+-dependent stereospecific oxidation of (R)- or (S)-1-(4-hydroxyphenyl)-ethanol to 4-hydroxyacetophenone [1]. SDRs from A. aromaticum are also capable of catalyzing the reverse reaction (preferentially at slightly acidic pH) and are already used in industry for synthesizing various aromatic and heteroaromatic secondary alcohols with high enantiomeric purity [2, 3]. The presented study focuses on evaluating the enzyme activity and stability as well as investigating the relations between them, both under storage and process condition. The relationship between aggregation dynamics and activity loss under various pHs and in the presence of a glucose stabilizer was analyzed using spectrophotometric techniques and dynamic light scattering. For R-HPED, we found out that at a pH of 8.5, the enzyme exhibits high stability and the highest productivity in the reduction of ketones, despite the relatively low activity shown at basic pH. Based on inactivation experiments, the mechanism of thermal inactivation at pH 8.5 was modelled for R-HPED. The irreversible first-order mechanism of R-HPED inactivation was verified by isothermal and multi-temperature data evaluation, confirming that at the pH of 8.5, aggregation of R-HPED is a secondary process occurring to already inactivated enzyme [4]. The mechanism of S-HPED inactivation was studied at pH 6.5 and 9.0 with and without the addition of glucose as a stabilizer. Analysis of the inactivation curves indicates that S-HPED, like R-HPED, is inactivated according to the "one step – two states" mechanism. The obtained results on the inactivation of HPEDs provide the basis for drawing first general conclusions on the inactivation mechanism of the SDR family. Acknowledgements: This research was funded in part by the statutory research fund of ICSC PAS. This work was supported by grants from the Slovak Research and Development Agency (Grant numbers: APVV-16-0111 and APVV-18-0188). |
| References: | [1] I. Büsing, J. Mol. Microbiol. Biotechnol. 2015, 25, 327-339. [2] P. Borowiecki, Adv. Synth. Catal. 2020, 362, 2012-2029. [3] R. Stürmer, M. Kesseler, B. Hauer, T. Friedrich, M. Breuer, H. Schröder, 2005, EP1745133B1 [4] M. Tataruch, V. Illeova, A. Milaczewska, T. Borowski, M. Mihal', M. Polakovic, Int. J. Biol. Macromol. 2023, 234, 123772. |
#1470 | Designing Genetic Circuits for Laboratory-Adaptive Evolution of Naringenin Production in E. coli |
|
| Presenting author: | Donghyeon KIM of CHEMICAL ENGINEERING / POSTECH |
| Corresponding author: | Jeong Wook LEE of CHEMICAL ENGINEERING / POSTECH |
| Other authors: | Chang Ha WOO of SCHOOL OF INTERDISCIPLINARY BIOSCIENCE & BIOENGINEERING (I-BIO) / POSTECH Da-ae GWON of CHEMICAL ENGINEERING / POSTECH |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | synthetic biology / genetic circuit / laboratory evolution / metabolic engineering |
| Purpose: | Microorganisms have been utilized in various ways to produce desired metabolites through metabolic engineering. However, artificially regulating pathways to increase production can be impractical when the pathways are complicated or not well-known. In such cases, laboratory evolution can be employed to increase the production of desired metabolites. Laboratory evolution involves generating random mutations in the entire genome of microorganisms to obtain strains with new traits, then selecting those that produce a large amount of the target metabolite. This method is very useful in metabolic engineering as it can yield strains that produce large quantities of target metabolites even if the metabolic pathway is complicated or not well-known. However, a method for rapidly generating mutations causing evolution and easily selecting strains of large production is necessary. To address this, we designed genetic circuits for mutation acceleration and strain selection. Various types of mutator devices were tested, each with different characteristics depending on the gene it contains. Each device showed different mutation rates and different levels of leaky expression. Several selection devices were also created, each responsible for different concentrations of target molecules. The naringenin sensor was used as an example. Transcription factor fdeR was utilized to recognize naringenin and activate the fdeA promoter. A UTR region was engineered for wide-range recognition. Antibiotic resistance and fluorescence protein genes were expressed only when the strain produced naringenin. A genetic toggle switch was used to control the operation of mutagenesis and selection. The toggle switch consisted of two cross-regulating transcription factors, lacI, and tetR. We integrated the toggle switch in the E.coli genome, and the switch maintain the state for 72 hours. By combining each device, it was possible to control the mutation state and the selection state repeatedly. In conclusion, genetic circuits were devised for laboratory-adaptive evolution of naringenin. The genetic circuit consisted of three parts, each composed of a mutagenesis device, a selection device, and a toggle switch to control them. Mutagenesis devices were made of two types, considering mutagenesis ability and stability. The selection device consisted of an antibiotic resistance gene and a fluorescence gene based on a transcription factor, and it worked well if naringenin was present. The toggle switch regulated the two states by adding appropriate inducers. The combined genetic circuit can be effectively used during adaptive evolution to produce metabolites on a large scale using E.coli. |
| References: | Lee JW, Gyorgy A, Cameron DE, et al. Creating Single-Copy Genetic Circuits. Mol Cell. 2016;63(2):329-336. |
| Figures: |  Toggle switch test Integrated toggle switch circuit was stable over 72hour. IPTG and ATc was used to change each state. 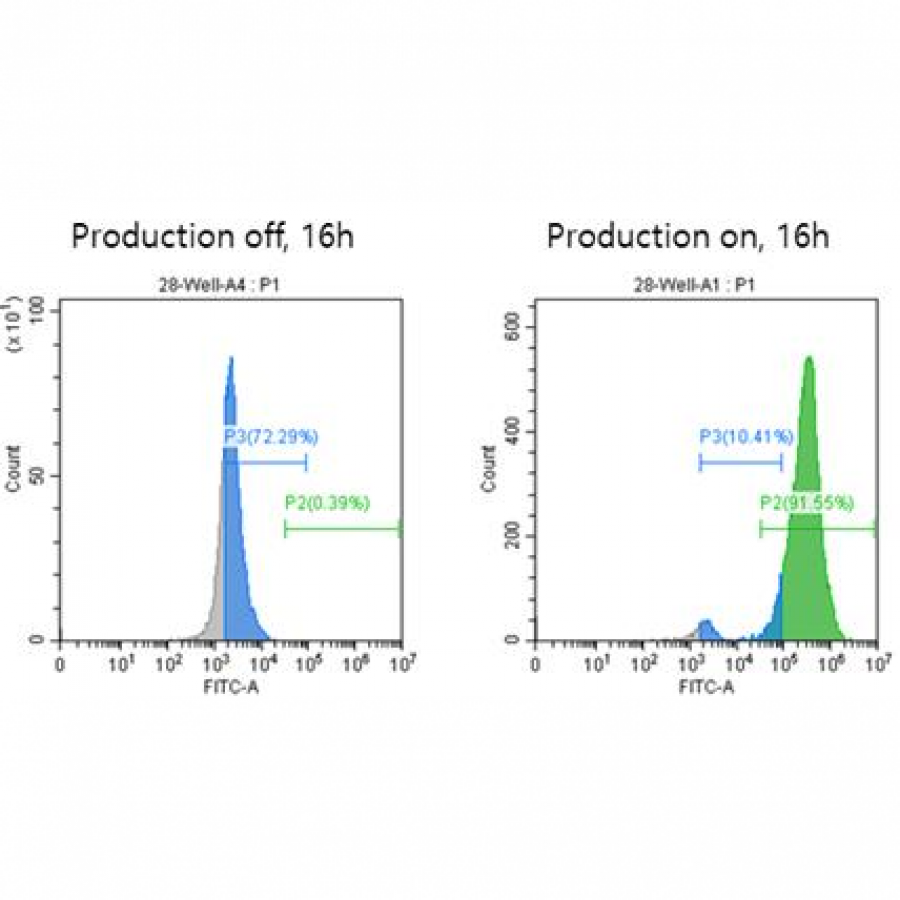 Selector device test Selector device showed fluorescence only when the cell produced naringenin |
#1472 | Lipase from Candida antarctica supported on 3-D printed structured resin packings for reactive distillation |
|
| Presenting author: | FROIDEVAUX Rénato of UMRTBIOECOAGRO - UNIVERSITY OF LILLE |
| Corresponding author: | Cédric DECARPIGNY of UMRT BIOECOAGRO |
| Other authors: | Nicolas CHAUSSARD of CP2M Clémence NIKITINE of CP2M David ROUZINEAU of INSTITUT NATIONAL POLYTECHNIQUE DE TOULOUSE ENSIACET Michel MEYER of INSTITUT NATIONAL POLYTECHNIQUE DE TOULOUSE ENSIACET Pascal FONGARLAND of CP2M Rénato FROIDEVAUX of UMRT BIOECOAGRO |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzymatic reactive distillation / CALB / Structured resin packing / Sol-gel enzyme immobilization |
| Purpose: | Reactive distillation was carried out for the enantioselective transesterification of racemic 2-pentanol with ethylbutyrate using a supported enzymatic catalyst. The reaction was designed to achieve the reaction-separation coupling by using structured resin packings coated with a sol-gel containing the lipase from Candida antarctica (CALB). This study is focus on one hand, on improving the gel properties for the coating process, also to immobilize the lipase without releasing and activity lost, on the other hand, the kinetics and hydrodynamics of the reactive distillation are also studied. Parameters such as catalyst loading, silica precursors, and alcohol were considered to improve the process of dip-coating in terms of gelation time, gel quality, and the resistance and stability of the gel coated on the resin. The design of the interns is established to respond to these problems from a laboratory scale to a pilot scale. |
#1476 | Exploring enzyme biodiversity: paths towards novel biosynthetic routes |
|
| Presenting author: | Damiano BARONI of UNIVERSITY OF GRONINGEN |
| Other authors: | Samantha GITTINGS of PROZOMIX Simon CHARNOCK of PROZOMIX Gerrit POELARENDS of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | #ColorimetricAssay / #Transaminases / #MetagenomicLibrary / #Aminoalcohols |
| Purpose: | Current carboligation methods often make use of hazardous catalysts, work under harsh conditions and generate toxic waste. Therefore, the development of alternative biocatalytic carboligation methodologies is highly desirable. Recent studies highlight the potential of engineered 4-oxalocrotonate tautomerase (4-OT) to efficiently catalyze the aldol addition reaction between acetaldehyde and benzaldehydes yielding various β-hydroxyaldehyde products. In particular, engineered enzymes such as 4-OT(M45T/F50A) or 4-OT homologues from the Prozomix metagenomic libraries, like TAUT015, show a drastically reduced dehydration activity, leading to increased formation of aldol addition products and reduced formation of cinnamaldehydes (Figure 1)1. This desirable aldol addition activity could be improved further by a directed evolution campaign involving a colorimetric prescreening step2 while it has been demonstrated that the evolvability of 4-OT can be enhanced by a gene fusion strategy3. Directed evolution could furthermore be employed in order to expand the substrate scope to benzaldehydes with electron donating substituents. While β-hydroxyaldehydes are useful chiral synthons in their own right, finding applications in the pharmaceutical as well as fine chemical sectors, amination of the carbonyl carbon would lead to even more attractive amino alcohols, which are valuable precursors to blockbuster drugs. In order to synthesize these compounds, transaminases have been identified as ideal candidates for the amination of β-hydroxyaldehydes. We will present recent work that focused on employing a colorimetric screening assay4 in order to discover novel transaminases capable of catalyzing the amination of 3-hydroxy-3-phenylpropanal leading to 3-phenyl-3-hydroxypropylamine, a desirable intermediate for the production of various bioactive compounds and molecules of industrial interest. Several active transaminases have been discovered, which are subjected to further examination to find the best candidate for a one-pot 4-OT/transaminase cascade leading to the production of various amino alcohols under mild conditions from simple biomass-derived starting materials. |
| References: | [1] Saifuddin, M.; Guo, C.; Biewenga, L.; Saravanan, T.; Charnock, S. J.; Poelarends, G. J. Enantioselective Aldol Addition of Acetaldehyde to Aromatic Aldehydes Catalyzed by Proline-Based Carboligases. ACS Catal. 2020, 10 (4), 2522-2527. [2] Biewenga, L.; Crotti, M.; Saifuddin, M.; Poelarends, G. J. Selective Colorimetric “Turn-On” Probe for Efficient Engineering of Iminium Biocatalysis. ACS Omega 2020, 5 (5), 2397-2405. [3] Xu, G.; Kunzendorf, A.; Crotti, M.; Rozeboom, H. J.; Thunnissen, A.-M. W. H.; Poelarends, G. J. Gene Fusion and Directed Evolution to Break Structural Symmetry and Boost Catalysis by an Oligomeric C-C Bond-Forming Enzyme. Angew. Chemie Int. Ed. 2021, e202113970. [4] Baud, D.; Ladkau, N.; Moody, T. S.; Ward, J. M.; Hailes, H. C. A Rapid, Sensitive Colorimetric Assay for the High-Throughput Screening of Transaminases in Liquid or Solid-Phase. Chem. Commun. 2015, 51 (97), 17225-17228. |
| Figures: | 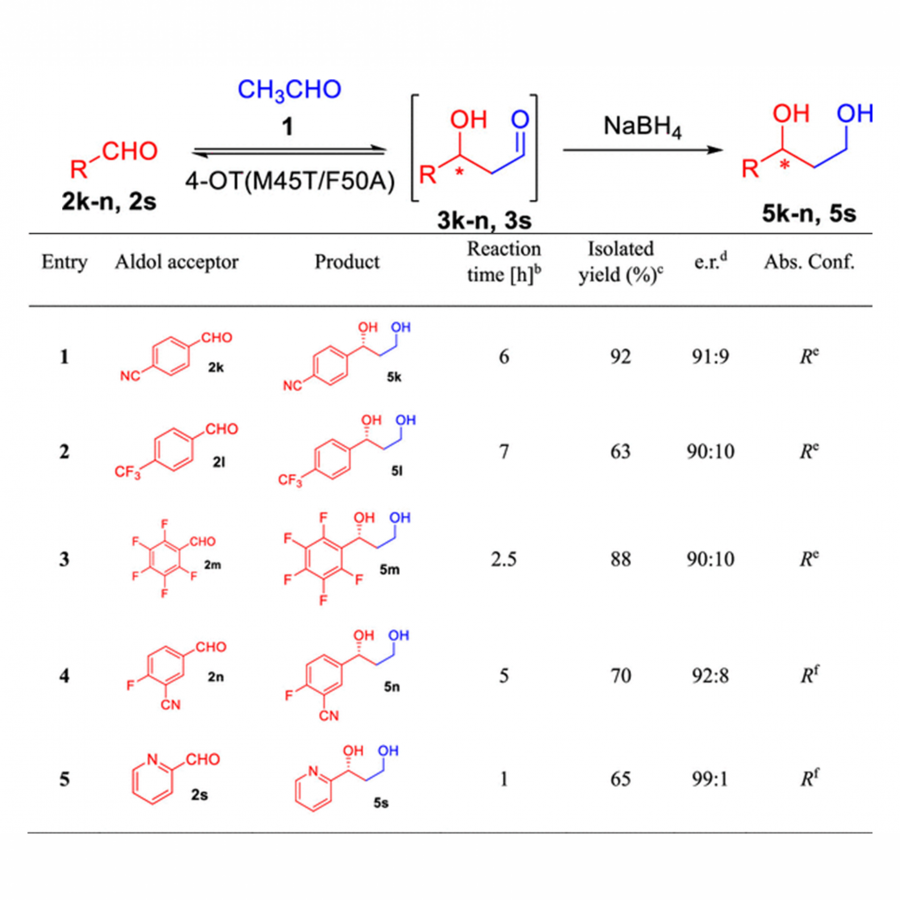 Asymmetric chemoenzymatic synthesis of aromatic 1,3-diols using 4-OT(M45T/F50A) as carboligase a-Conditions (ref. 1)
b-Reaction time of the enzymatic step.
c-Isolated yield of the aromatic product.
d-Determined by HPLC analysis on a chiral stationary phase
e-Configuration determined by chiral HPLC
f-Configuration assigned based on analogy. |
#1478 | Next-generation of enzyme engineering toward improved terminal hydroxylations |
|
| Presenting author: | Jelena SPASIC of GRAZ UNIVERSITY OF TECHNOLOGY |
| Other authors: | Berenika STLOUKALOVÁ of GRAZ UNIVERSITY OF TECHNOLOGY Andrea NIGL of GRAZ UNIVERSITY OF TECHNOLOGY Thomas BAYER of UNIVERSITY OF GREIFSWALD Robert KOURIST of GRAZ UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | protein engineering / hydroxylation / alkane monooxygenase / high-throughput assay |
| Purpose: | Terminal hydroxylation represents an attractive biocatalytic route toward terminal alcohols, aldehydes, and acids, which are of special interest as building blocks for the sustainable production of polymers. To customize biocatalysts for this purpose, several described alkane degradation pathways offer valuable starting points. Particularly interesting are alkane monooxygenases displaying a broad substrate spectrum efficiently ω-oxyfunctionalising alkanes, cycloalkanes, thioesters and fatty acid esters [1, 2]. In-depth knowledge of protein sequence-function relationship would enable the generation of tailored alkane monooxygenases as biocatalysts for stereo- and regioselective biotransformations and further industrial applications. However, due to the lack of crystal structures, enzyme engineering of membrane-bound alkane monooxygenases proved to be challenging. Therefore, we aim to elucidate sequence-function relationships of alkane monooxygenases using simple high-throughput screening (HTS) or selection assays. For the development of the HTS assay, the activity of Pseudomonas oleovorans AlkB alkane hydroxylase system was coupled with alcohol dehydrogenase activity of AlkJ and the LuxAB biosensor system [3]. The activity of AlkB was successfully detected with the established HTS assay starting from different alkane substrates. Additionally, the versatility of the developed HTS assay was corroborated by detecting the activity of four AlkB homologs. The HTS assay will be further applied to obtain datasets for machine learning, predicting mutational hotspots toward improved hydroxylating activities. Overall, this would result in enzyme variants showing desired activities, subsequently, accelerating the engineering of alkane monooxygenases. |
| References: | [1] van Beilen, J. B., Enzyme Microb. Technol. 1994, 16, 904-911. [2] van Nuland, Y. M., Appl. Environ. Microbiol. 2016, 82, 3801-3807. [3] Bayer, T. Catalysts 2021, 11, 953. |
| Figures: | 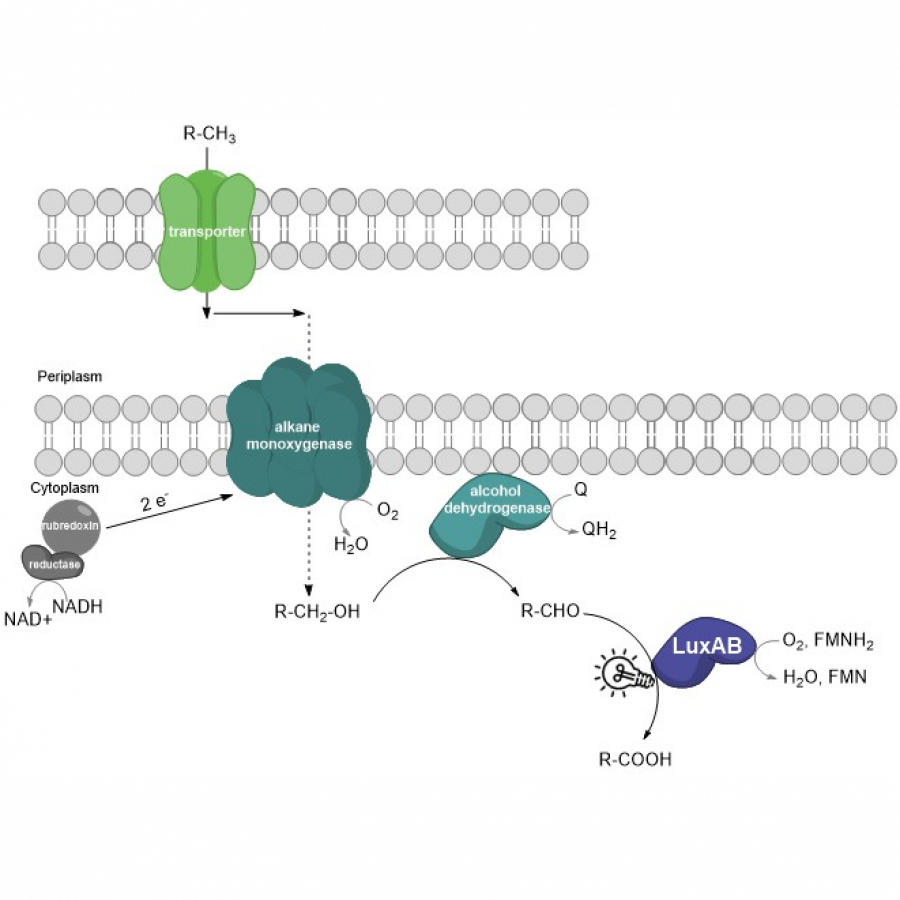 Figure 1. LuxAB-based high-throughput assay for the detection of the activity of alkane hydroxylase system. |
#1479 | Switching regioselectivity in the fructooligosaccharides synthesis by Gluconacetobacter diazotrophicus levansucrase |
|
| Presenting author: | Sandrine MOREL of TBI, UNIVERSITÉ DE TOULOUSE, CNRS, INRAE, INSA |
| Corresponding author: | Lazaro HERNANDEZ of CIGB GRUPO TECNOLOGIA DE ENZIMAS |
| Other authors: | Ana Gabriela MARTINEZ of CIGB, GRUPO TECNOLOGIA DE ENZIMAS Yamira QUINTERO of CIGB, DPT TECNOLOGIA DE ENZIMAS Carmen MENENDEZ of CIGB, GRUPO TECNOLOGIA DE ENZYMAS Alexis MUSACCHIO of CIGB, DEPT BIOLOGIA DE SYSTEMAS Ivan ANDUJAR of CIGB, DPT DE BIOLOGIA DE SISTEMAS Sandra PIZZUT of TBI, UNIVERSITE DE TOULOUSE, CNRS, INRAE, INSA Magali REMAUD-SIMEON of TBI, UNIVERSITE DE TOULOUSE, CNRS, INRAE, INSA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Levansucrase / Fructooligosaccharides / Fructosyltransferase / Fructan |
| Purpose: | Fructooligosaccharides (FOSs) are soluble prebiotic fibbers with proven health-promoting effects in humans and animals. Bacterial levansucrases (EC 2.4.1.10) catalyze fructosyl transfer reactions from the natural substrate sucrose to different acceptors, such as water (sucrose hydrolysis), sucrose (FOSs synthesis), and FOSs (polysaccharide formation and elongation). Gluconacetobacter diazotrophicus levansucrase (Gd_LsdA) is distinguishable for the synthesis and accumulation of the β-(2→1)-linked FOSs 1-kestotriose (1K) and 1,1-kestotetraose (1,1K). The yield of the β-(2→6)-linked polysaccharide levan is rather low due to the minor proportion of the preferred precursor 6-kestotriose (6K). In this work, the amino acids His172 and Asn306 at the active-site cavity of Gd_LsdA were selected as independent targets for saturation mutagenesis aiming to decipher the structural factors involved in the regioselectivity of the fructosyl transfer reaction. His172 binds the fructosyl moiety of the donor sucrose (subsite -1), while Asn306 interacts with the glucosyl moiety of the acceptor sucrose or the second fructosyl moiety of the acceptor FOS (subsite +2). HPAEC-PAD analysis of the time-course transformation of sucrose by the native Gd_LsdA and the mutated variants revealed remarkable differences in the product profiles. As a general behavior, the replacement of His172 failed to shift the FOSs spectrum and caused a drastic reduction in the ratio of the transfructosylation/hydrolysis activities. More interestingly, the substitution of Asn306 by a positively charged amino acid (Arg or Lys but not His) directed the acceptor sucrose molecule in an orientation that favors the synthesis of 6-kestotriose over 1-kestotriose. Contrary to the native enzyme, the N306R and N306K variants mostly yielded β-(2→6)-linked FOSs with degree of polymerization (DP) between 3 and 6 which were no further elongated in prolonged reactions. Our results provide insight into a key structural determinant of the regioselectivity and processivity of the fructosyl transfer reaction in levansucrases. On the other hand, the fully active variant N306R yielding the highest 6K/1K ratio constitutes an attractive catalyst for the production of prebiotic levan-type FOSs, which are currently unavailable in the commercial market. |
#1484 | Enzymatic fructosylation: an eco- friendly tool to enhance the water solubility and stability of the natural aglycon phloretin |
|
| Presenting author: | Sandrine MOREL of TBI, UNIVERSITÉ DE TOULOUSE, CNRS, INRAE, INSA |
| Other authors: | Vincent DULAU of TBI, UNIVERSITE DE TOULOUSE, CNRS, INRAE, INSA Magali REMAUD of TBI, UNIVERSITE DE TOULOUSE, CNRS, INRAE, INSA Lazaro HERNANDEZ of CIGB, GRUPO DE TECNOLOGIA DE ENZIMAS |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Levansucrase / Phenolic compounds / Transfructosylase / |
| Purpose: | Phloretin (2’,4’,6’-trihydroxy-3-(4-hydroxyphenyl)-propiophenone) is a natural dihydrochalcone widely distributed in the leaves, bark and fruit of apple trees. Phloretin exhibits several pharmacological properties, such as antidiabetic, antioxidant, anti-inflammatory, and antitumor activities. As other aglycons, phloretin is poorly soluble in water and consequently its bioavailability is low. In the plant, glucosylation of phloretin at the 2' position is the last step in phlorizin (phloretin-2’-β-D-glucopyranoside) biosynthesis. Phloretin glucosides have also been synthesized in acceptor reactions using sucrose phosphorylase1, amylosucrase2 or UDP-glycosyltransferase3. The in-vivo or in-vitro synthesis of phloretin fructosides has not been reported so far. Microbial and plant enzymes from the glycoside hydrolase (GH) families 32 and 68 (clan GH-J) initiate de novo fructan synthesis by transferring the fructosyl moiety from one sucrose molecule (donor) to another sucrose molecule (acceptor). The fructosyl moiety can be also released to water resulting in substrate hydrolysis. The transfructosylation/hydrolysis (T/H) ratio, the regio-selectivity, and the polymerization capacity are intrinsic properties that vary among clan GH-J enzymes of different origins. In a previous study, enzymatic fructosylation was successfully used to enhance the water solubility and stability of phlorizin4. In this study, five clan GH-J enzymes of different catalytic properties were assayed for transferring the fructosyl residue of the natural substrate sucrose to the aglycon phloretin. The enzymes sucrose:sucrose 1-fructosyltransferase from the plant Schedonorus arundinaceus (Sa_1-SST, EC 2.4.1.99, GH32), and the Lactobacillus reuteri levansucrase (Lr_Lev, EC 2.4.1.10, GH68) and inulosucrase (Lr_Inu, EC 2.4.1.9, GH68) failed to fructosylate the non-sugar acceptor. The Thermotoga maritima β-fructosidase (Tm_BfrA, EC 3.2.1.26, GH32) synthesized a unique phloretin mono-fructoside, but the conversion efficiency was below 10%. More interestingly, the Gluconacetobacter diazotrophicus levansucrase (Gd_LsdA) yielded short-chain series of phloretin fructosides including two positional mono-fructoside isomers with maximal acceptor conversion of 83 %. Due to its high activity and wide acceptor promiscuity, Gd_LsdA is an attractive catalyst for biotech application in the fructosylation of different phenolic compounds, including the apple dihydrochalcones phlorizin and phloretin. |
| References: | [1] J L. Gonzalez-Alfonso et al, Adv. Synth.Catal. 2021, 363, 3079-3089 [2] H. Overwin et al, Journal of biotechnology, 2015, 211, 103-106 [3] R. P. Pandey et al, Applied and environmental microbiology, 2013, 79, 3516-3521 [4] A. Herrera-Gonzalez et al, Enzyme and Microbial Technology, 2021, 147, 109783 |
#1486 | Lights on and action: De novo metalloproteins for photocatalytic diol cleavage |
|
| Presenting author: | Andreas Sebastian KLEIN of TECHNICAL UNIVERSITY OF MUNICH |
| Other authors: | Florian LEISS-MAIER of TECHNICAL UNIVERSITY OF MUNICH Cathleen ZEYMER of TECHNICAL UNIVERSITY OF MUNICH |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | de novo metalloprotein / photocatalysis / C-C bond cleavage / cerium(III) catalysis |
| Purpose: | Enzymes are efficient biocatalysts coupled with tremendous chemo-, regio- and stereoselectivity. Nevertheless, their reaction repertoire is usually confined to the natural chemical space. A large variety of reactions known in organic chemistry therefore remain inaccessible, limiting the applications of these sustainable biocatalysts. With the ongoing advancement of computational methods in the field of protein design, it is now possible to generate desired protein scaffolds de novo and equip them with new catalytic functions.[1–3] Here we demonstrate the development of a photoenzyme based on a de novo protein equipped with a metal-binding site that selectively and with high affinity binds lanthanide ions, such as cerium(III) (A).[4] By irradiation with visible light, this artificial metalloprotein is able to photocatalytically cleave the C–C bond of vicinal diols via a radical mechanism and thus extends the reaction space of natural enzymes by a new-to-nature reaction (B). The enzyme was improved by site saturation mutagenesis and consecutive rational design of the active site. The result of these initial screenings is a variant with higher photostability, faster lanthanide binding kinetics, and improved yield for the C–C bond cleavage of the model substrate hydrobenzoin and derivatives thereof, converting it into the corresponding carbonyl compounds. Future investigations will focus on the stereoselectivity of chiral substrates. |
| References: | [1] A. S. Klein, C. Zeymer, Protein Eng., Des. Sel. 2021, 34; doi: 10.1093/protein/gzab003. [2] J. Wang et al., Science, 2022, 377, 387-394. [3] I. V. Korendovych, W. F. DeGrado, Q. Rev. Biophys. 2020, 53, e3. [4] S. J. Caldwell, I. C. Haydon, N. Piperidou, P.-S. Huang, M. J. Bick, H. S. Sjöström, D. Hilvert, D. Baker, C. Zeymer, Proc. Natl. Acad. Sci. U.S.A. 2020, 117, 30362-30369. |
| Figures: | 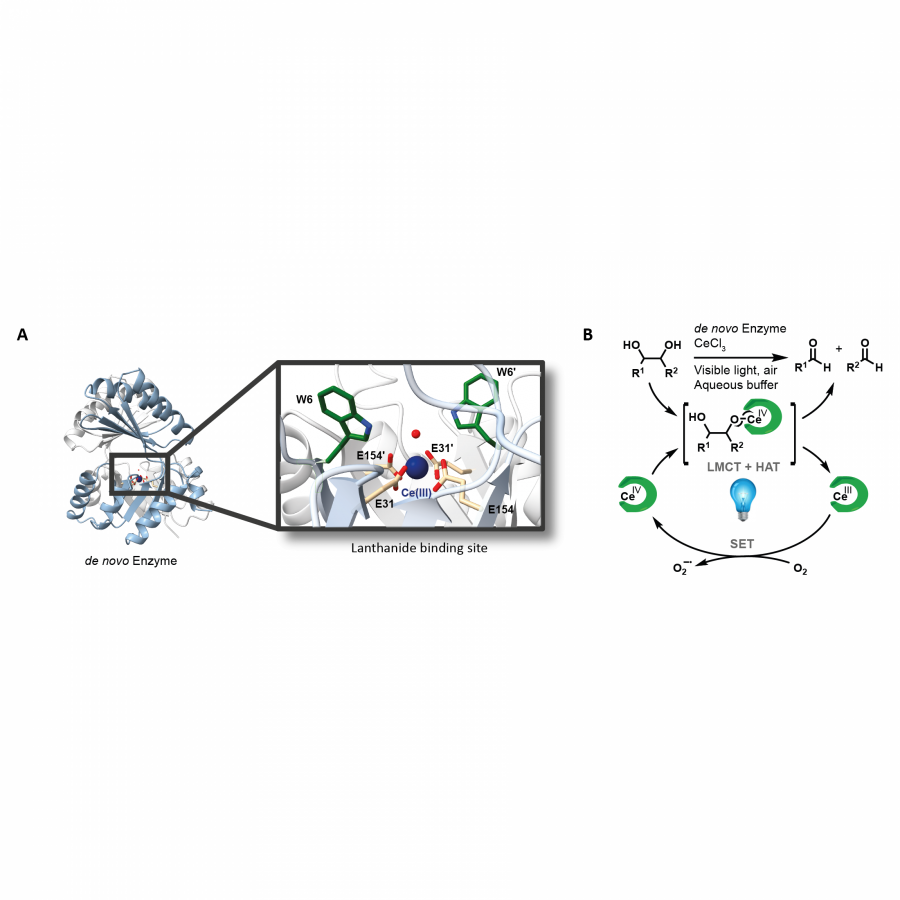 Photocatalytic cleavage of vicinal diols by a de novo metalloprotein A) Cerium (III) bound to the active site of the de novo metalloprotein. B) Proposed radical mechanism for the photocatalytic cleavage of vicinal diols. |
#1488 | Glucosylation of flavonoids by glucosyltransferase from Beauveria bassiana AM278 expressed in E. coli |
|
| Presenting author: | Sandra SORDON of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Karolina LISOWSKA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Agata MATERA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Kinga DULAK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Ewa HUSZCZA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Jarosław POPŁOŃSKI of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | flavonoids / glucosyltransferase / Beauveria bassiana / heterologous expression |
| Purpose: | Flavonoids are a large group of polyphenolic compounds that are present in plants. They exert numerous important physiological functions in plants, having considerable influence on growth and development of plants, protect them from UV radiation, bacterial and fungal infections and provide color to fruits and flowers. Flavonoids found in food have beneficial effects on human health. They exhibit a diverse spectrum of biological activities such as: antioxidant, anticancer, antidiabetic, antibacterial, antiviral and estrogenic [1]. However, many flavonoid compounds very often present a low water solubility and bioavailability. Glycosylation can improve water solubility of natural products, enhance their bioactivity, stability, bioavailability and decrease their toxicity [2]. From the reasons, a lot of effort was put into development of efficient production methods of the glycosylated natural products. Extraction from plants seems to be the easiest method of obtaining natural glycosides, but this approach has many limitations, e.g. low yield as a reason of low concentration in a biomass and strict dependence on seasonal vegetation conditions. The use of organic synthesis for the production of flavonoid glycosides also has many limitations. Disadvantages of chemical glycosylation is use of toxic reagents and the necessity of protection of these hydroxyl groups that are not meant to conjugate with saccharide [3]. Biocatalysts can be utilized to perform eficient glycosylation reaction, additionally resulting in specific products and operating under mild conditions [4]. In our previous studies fungal strains Beauveria bassiana AM278 have been proven to be a useful glycosylation catalysts toward different class of flavonoids. This strain is able to selective introduction of glucose or 4'-O-methylglucose molecule to a hydroxyl group in the flavonoid substrates [5]. Herein, we would like to present that we cloned the dedicated glucosyltransferase from B. bassiana AM278 and heterologously expressed it in E. coli. Sequence identification was based on homology to already described enzymes. The purified enzymatic protein was characterized through in vitro enzymatic reactions as a UDP-glucosyltransferase. In this study we also evaluated biochemical characterisation of this novel flavonoid glucosyltransferase. Basic biochemical parameters, e.g. temp, pH and cofactors were evaluated. This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreements no. 814650 (SynBio4Flav). |
| References: | [1] R.E. Mutha, A.U. Tatiya, S.J. Surana Futur J Pharm Sci 2021, 7, 25. [2] V. Kren, I. Martinkova Curr. Med. Chem. 2001, 8, 1303-1328. [3] Lewis P. S. Kaltia, K. Wähälä J. Chem. Soc. Perkin Trans. 1998, 1, 2481-2484. [4] J. Xiao, T.S. Muzashvili, M.I. Georgiev Biotechnol. Adv. 2014, 32, 1145-1156. [5] S. Sordon, J. Popłoński, T. Tronina, E. Huszcza Bioorg Chem 2019, 93, 102750. |
| Figures: | 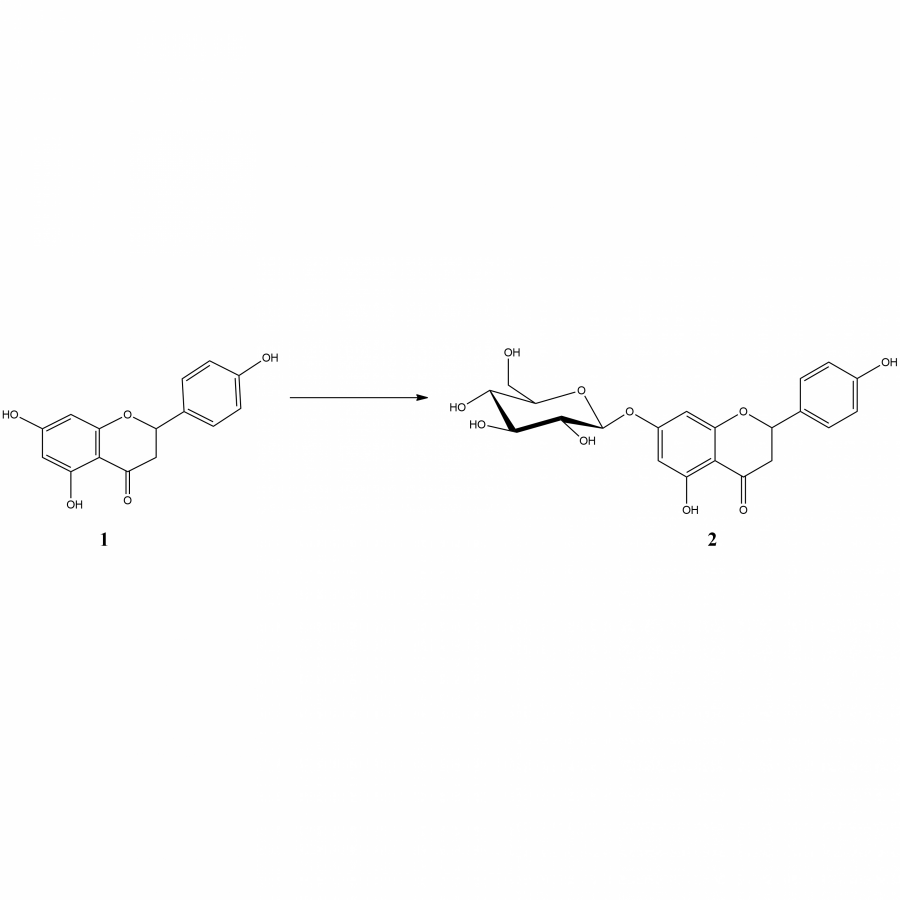 Glucosylation of Naringenin 1 - Naringenin
2 - Naringenin-7-O-β-D-glucopyranoside |
#1490 | Thermostabilizing strategies performed by two computational methods in a DyP-type peroxidase from Pseudomonas putida |
|
| Presenting author: | Constança LORENA of ITQB NOVA |
| Other authors: | Diogo SILVA of ITQB NOVA Carlos FRAZÃO of ITQB NOVA Lígia O. MARTINS of ITQB NOVA Patrícia T. BORGES of ITQB NOVA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | DyP-type peroxidase / X-ray crystallography / Protein interactions / Thermostability |
| Purpose: | Dye-decolorizing peroxidases (DyPs) are enzymes that can oxidize many substrates like anthraquinone and azo dyes, phenolic and nonphenolic lignin units, metals, among others, while reducing hydrogen peroxide to water. This makes DyPs interesting candidates for biotechnological purposes. The computational methods PROSS [1] and FireProt [2] were used to design thermostable DyP variants from Pseudomonas putida (PpDyP) with since this enzyme lacks robust stability properties required for industrial processes and an efficient mechanism to oxidize many substrates with biotechnological interest. The variant PpDyP PR, generated by PROSS, included 29 mutations and exhibited a melting temperature of 88ºC, a 20ºC increase compared to the wild type (WT). On the other hand, the variant PpDyP FP, generated by FireProt, included 21 mutations and exhibited a melting temperature of 75ºC, a 10ºC increase from the WT. Additionally, both variants show approximately 5-fold higher catalytic efficiency (kcat/Km) for hydrogen peroxide and ABTS (unpublished data). The structure of these thermostable variants was determined by X-ray crystallography. Both variants generated thin orange needle-shaped crystals and diffraction data was collected at ALBA Synchrotron Light Facility (Barcelona, Spain) in the BL13-XALOC beamline. Data was processed with a resolution of 2.10 Å for PpDyP PR and 2.35 Å for PpDyP FP. PpDyP PR crystals belong to P 21 21 21 space group and contain two protein molecules per asymmetric unit, whereas PpDyP FP crystals belong to P 32 2 1 space group and show four protein molecules per asymmetric unit. The structures were determined by molecular replacement using PpDyP WT structure (PDB 7QYQ [3]) as search model. The structure of PpDyP PR shows an R-free and R-work values of 23.6% and 19.0%, respectively. Refinement of PpDyP FP is ongoing. Enzymes must have a degree of thermostability to overcome the thermal stress common from industrial processes. To better understand the role of the mutations on the improved thermostability, suggested by PR and FP, we used the webserver Protein Interactions Calculator (PIC; http://pic.mbu.iisc.ernet.in/) to identify interactions in proteins such as hydrophobic interactions, H-bonds and salt bridges, known to contribute for the higher stability in enzymes. Moreover, some other thermostabilizing strategies employed by hyperthermophilic enzymes were reported in the literature and may include structural elements such as shorter loops, maximized protein packing, lower solvent accessible surface, decreased number of internal cavities, among others. Comparative analysis of PpDyP WT and PR variant structures showed, in the PR variant, an increase of the internal packing due to decreased cavities number and volume. The analysis performed at PIC revealed an increase of 14 new hydrophobic interactions in PR relative to the WT, where some involve mutations (6) present in this variant. Moreover, we found that PR has 34 new H-bonds, and 15 mutations are involved in the appearance of these interactions in the variant. Regarding the salt bridges, we did not find any significant differences between the two enzymes. The comparative analysis involving PpDyP FP will also be performed. The structural analysis of these variants will help to understand the variety of stabilization mechanisms that enzymes acquire to become more thermostable which is essential for their application in biotechnology industry. |
| References: | [1] goldenzweig, a., mol. cell 2016, 63, 337-346. [2] musil, m., nucleic acids res. 2017, 45, W393-W399. [3] borges, p. t., comput. struct. biotechnol. j. 2022, 20, 3899-3910. |
#1491 | Development of a high-throughput fluorogenic assay for the detection of promiscuous amidase activity |
|
| Presenting author: | Ina SOMVILLA of UNIVERSITY OF GREIFSWALD |
| Corresponding author: | Uwe T. BORNSCHEUER of UNIVERSITY OF GREIFSWALD |
| Other authors: | Christoffel P. S. BADENHORST of UNIVERSITY OF GREIFSWALD |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | amidase mechanism / high-throughput screening / flow cytometry / |
| Purpose: | Carboxyl esterases are powerful catalysts due to their broad substrate specificity and high regio- and stereoselectivity, as well as their ability to catalyze reactions without cofactors.[1] An esterase from B. subtilis DSM402 (BS2) was found to not only be active towards tertiary alcohols but unlike many carboxyl esterases, shows promiscuous amidase activity.[2] The first steps in unraveling the amidase mechanism of this enzyme have shown that a hydrogen-bond network is essential for stabilizing the transition state.[3] A directed evolution approach resulted in a mutation that improved the amidase activity of BS2 by changing the orientation of the substrate in the active site. This increase was achieved through a π-π stacking network, which enhanced the stability of the tetrahedral intermediate formed during amide bond hydrolysis.[4] We aim to improve the understanding of the amidase mechanism of BS2 to increase its activity further by applying a computational-aided engineering approach using the 3DM tool.[5] For this purpose, we have established a novel whole-cell high-throughput assay based on fluorescence-activated cell sorting (FACS) to screen large mutant libraries and identify variants with improved amidase activity. |
| References: | [1] U. T. Bornscheuer, FEMS Microbiol. Rev. 2002, 26. [2] M. Schmidt, E. Henke, B. Heinze, R. Kourist, A. Hidalgo, U. T. Bornscheuer, Biotechnol. J. 2007, 2, 249. [3] R. Kourist, S. Bartsch, L. Fransson, K. Hult, U. T. Bornscheuer, ChemBioChem 2008, 9, 67. [4] S. Hackenschmidt, E. J. Moldenhauer, G. A. Behrens, M. Gand, I. V. Pavlidis, U. T. Bornscheuer, ChemCatChem 2014, 6, 1015. [5] https://3dm.bio-prodict.com/. |
#1492 | BIOCATALYTIC CASCADES TOWARDS IMINOSUGAR SCAFFOLDS REVEAL PROMISCUOUS ACTIVITY OF SHIKIMATE DEHYDROGENASES |
|
| Presenting author: | Lea GOURBEYRE of UNIVERSITY OF MANCHESTER |
| Corresponding author: | Sabine FLITSCH of UNIVERSITY OF MANCHESTER |
| Other authors: | Christopher SWANSON of UNIVERSITY OF MANCHESTER Grayson FORD of UNIVERSITY OF MANCHESTER Ashley MATTEY of UNIVERSITY OF MANCHESTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | iminosugars / biocatalytic cascades / shikimate dehydrogenase / |
| Purpose: | Iminosugar are highly polar polyhydroxylated N-heterocycles which displays a multitude of bioactivity such as glycosidase inhibitors, antivirals, or pharmaceutical chaperones but their chemical synthesis is lengthy and suffers from poor scalability and purification.1-3 To circumvent all these issues we develop here protecting group-free chemoenzymatic and biocatalytic cascades to synthesize iminosugars from sugar aminopolyols, using Galactose oxidase (GOase) variant F2 followed by a chemical or an enzymatic imine reduction (Figure 1). The cascade has been developed applying biocatalytic retrosynthesis.4,5 The major challenge of this route was to identify a reductase able to reduce the hydroxylated cyclic imine to the iminosugar product for the final step. A first screening on oxidase activity is performed and the GOase variant F2 was identified as the most promising biocatalyst with various substrates. Then putative iminosugar reductases were ‘created’ and developed via Genome Mining. After what a cascade screening was done to identify reductases. Combining GOase F2 with a shikimate dehydrogenase (SDH), provided an efficient one-pot route to various targets, with conversions > 70%. Expanding the reaction system to the whole substrate panel and to circumvent narrow substrate spectrum of SDH, several iminosugar products were obtained using the GOase-NaCNBH3 chemoenzymatic cascade. Iminosugar products were isolated from biotransformations in a single step through development of a gradient-elution ion exchange purification. |
| References: | [1] Alonzi, D. S.; Scott, K. A.; Dwek, R. A.; Zitzmann, N. Biochem. Soc. Trans., 2017, 45 (2), 571-582. [2] Horne, G.; Wilson, F. X.; Tinsley, J.; Williams, D. H.; Storer, R. Drug Discov. Today, 2011, 16 (3-4), 107-118. [3] Nash, R. J.; Kato, A.; Yu, C-Y.; Fleet, G. J. Future Med. Chem., 2011, 3 (12), 1513-1521. [4] De Souza R. O. M. A.; Miranda L. S. M.; Bornscheuer U. T. Chem. Eur. J., 2017, 23, 12040. [5] Finnigan, W.; Hepworth, L. J.; Flitsch, S. L.; Turner N. J. Nat Catal., 2021, 4, 98-104. |
| Figures: | 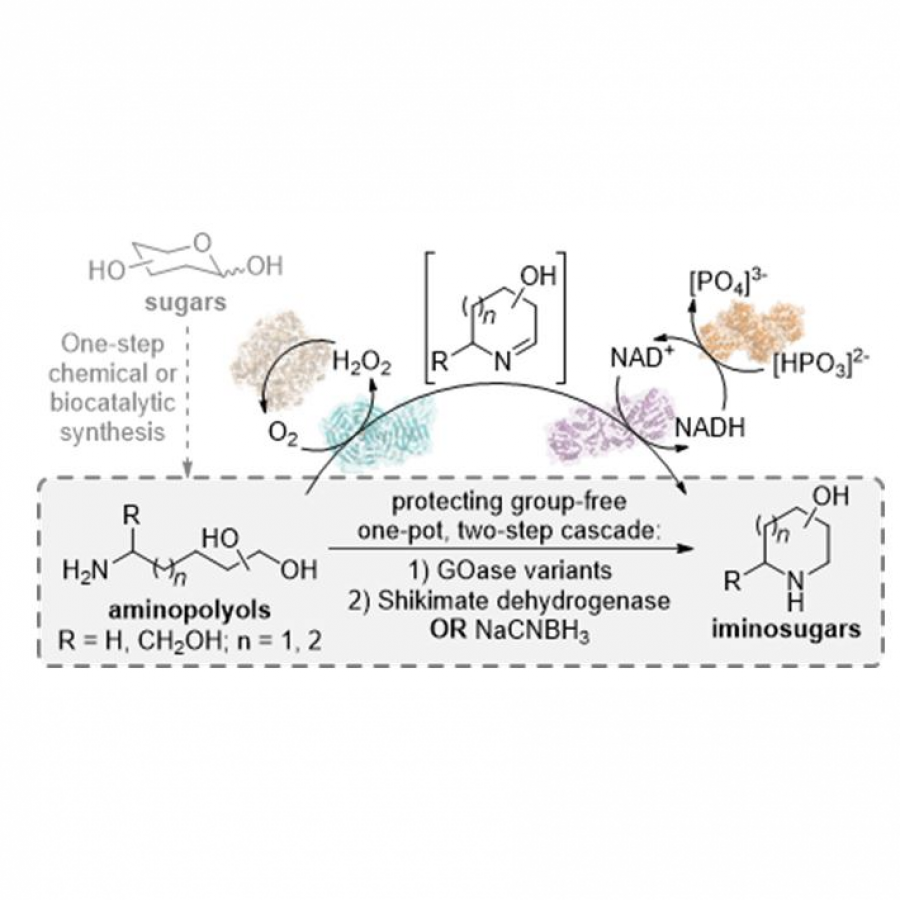 Chemoenzymatic and biocatalytic cascades to synthesize iminosugars from sugar aminopolyols. |
#1494 | Chitosan partial hydrolysis catalyzed by non-glycosidic enzymes |
|
| Presenting author: | ANDRES R. ALCANTARA of COMPLUTENSE UNIVERSITY OF MADRID |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | chitosan / hydrolases / promiscuity / chitooligosaccharides |
| Purpose: | Chitosan is a versatile biopolymer derived from chitin, which is found in the shells of crustaceans such as shrimp, crab, and lobster. It has gained significant attention in recent years due to its numerous potential applications in various fields, including agriculture, medicine, and food industry [1]. Additionally, chitosan has been shown to have antimicrobial, antifungal, and antioxidant properties, making it a potential natural alternative to synthetic preservatives and antibiotics. These properties are related to a defined structure of chitosans, mainly depending on the chain length (MW) and the acetylation degree (AD) [2]. The industrial use of chitosanases for the generation of chitooligosaccharides possessing controlled MWs and ADs, an usual lab protocol, is impeded by the very high price of these glycosidases, which would make the intensified process unaffordable. In this communication, we will present some results obtained upon the use of different commercial hydrolases applied to the partial hydrolysis of chitosan. The standard substrate 4-Nitrophenyl N-acetyl-β-D-glucosaminide, (NP-GlcNAc) was used to test the promiscuous hydrolysis catalyzed by the non-glycosidic hydrolases, and the results were compared with the activity founded in chitosan hydrolysis, followed by GPC using a dextran calibration curve; MW data were compared to the control chitosan. Reactions without enzyme showed that the observed hydrolysis is solely due to the enzyme since no spontaneous depolymerization of the polymer was observed under the experimental conditions. A detailed report of the resulting products shows the different profile of polymeric fractions obtained. |
| References: | [1] Junceda-Mena, I.; García-Junceda, E.; Revuelta, J. From the problem to the solution: Chitosan valorization cycle. Carbohydr. Polym. 2023, 309, doi:10.1016/j.carbpol.2023.120674. [2] Aranaz, I.; Alcántara, A.R.; Civera, M.C.; Arias, C.; Elorza, B.; Heras Caballero, A.; Acosta, N. Chitosan: An Overview of Its Properties and Applications. Polymers 2021, 13, 3256, doi:10.3390/polym13193256 |
| Figures: | 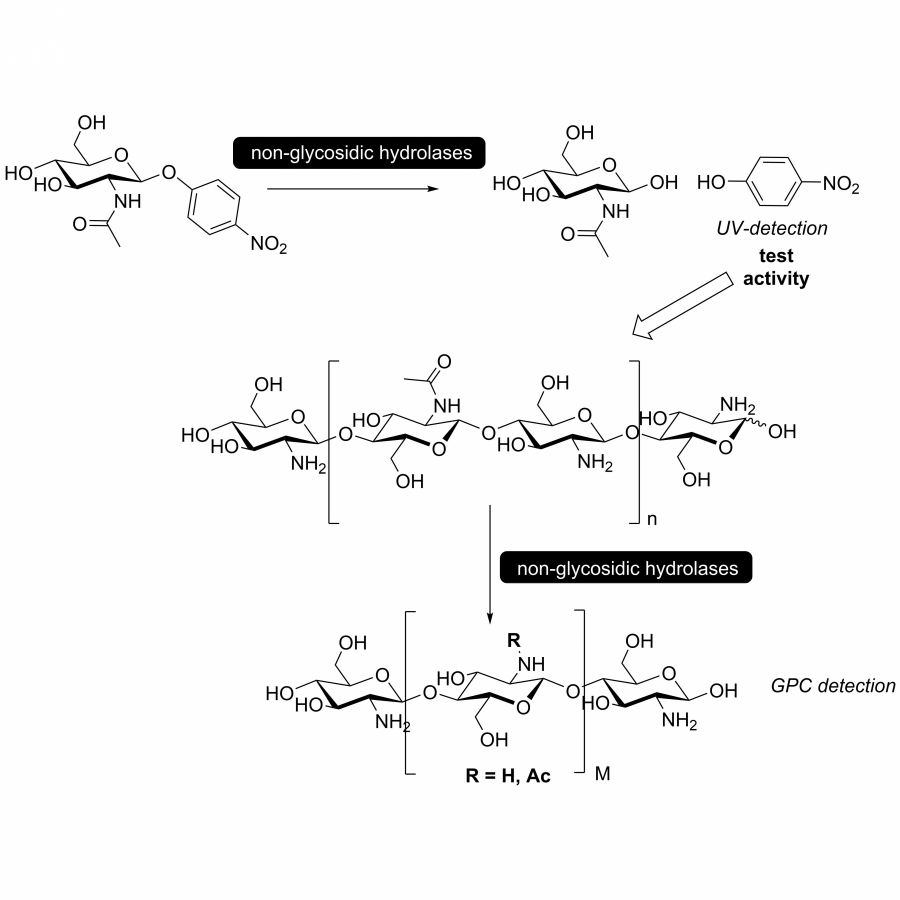 Figure 1 General Scheme |
#1496 | The kinetic characterization of the small laccase from Streptomyces coelicolor using EnzymeML: a technical lesson in reproducibility |
|
| Presenting author: | PLEISS Juergen of UNIVERSITY OF STUTTGART |
| Corresponding author: | Alaric PRINS of CAPE PENINSULA UNIVERSITY OF TECHNOLOGY |
| Other authors: | Max HÄUSSLER of UNIVERSITY OF STUTTGART MARILIZE LE ROES-HILL of CAPE PENINSULA UNIVERSITY OF TECHNOLOGY JÜRGEN PLEISS of UNIVERSITY OF STUTTGART |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | MULTICOPPER OXIDASE / COMPUTATIONAL BIOLOGY / ENZYME CHARACTERISATION / BIOCATALYSIS |
| Purpose: | One of the main challenges in enzyme characterisation is the availability of reliable and reproducible results [1]. Studies have shown that there is a large discrepancy in the ability of researchers to reproduce their own results [2], as well as reproduce the findings of others. The STRENDA commission [3] has outlined a set of guidelines useful for the thorough reporting of enzymology data, including but not limited to protein information, assay conditions, detailed experimental methods as well as processing of results. The small laccase (SLAC) from Streptomyces coelicolor is a two-domain laccase that has been extensively described in both biochemical and structural nature [4, 5, 6], and was selected for use in this study. Using a ThermoScientific Multiskan 1500 microtiter plate reader, the enzymatic oxidation of 2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) (ABTS) was monitored at 340nm (substrate utilisation) between pH values of 3.0 – 5.5, across a substrate concentration range of 0-200µM, and over a temperature gradient of 25-45˚C. Oxidation was monitored for 15 minutes. In total, 30 different assay conditions were performed which yielded more than 100 000 individual reads. In order to process such a large dataset accurately, and subsequently, circumvent the likelihood of erroneous data curation through human error, an EnzymeML [7] workflow was implemented contained in a single Jupyter Notebook. Experimental metadata was captured in an EnzymeML Excel spreadsheet. Photometric measurements for standard curves and oxidation measurements were parsed into individual EnzymeML documents, and the calibration data was used to convert the oxidation measurement into concentration. Michaelis-Menten rate equations, along with different inhibition models were tested to determine which best fits the experimental data, scored based on the lowest Akaike’s Information Criterion (AIC). The kinetic parameters, along with a detailed statistical report are produced. The best catalytic efficiency for SLAC was observed at 45˚C and pH 3.0, at the edge of the parameter space which indicates these are not necessarily the optimal conditions and could therefore be expanded upon. A correlation between the kcat¬ and Km was observed, suggesting too low of an initial substrate concentration was utilised in relation to the true Km of the enzyme. An enzyme half-life of only 10 minutes at the highest catalytic efficiency was observed and further experimentation is required to determine the cause of this inactivation, likely by expanding enzyme concentrations and the use of different substrates. Currently, the SLAC experiment is being expanded to address the problems in the experimental procedure to determine the biochemical characteristics of the enzyme more accurately – namely by increasing enzyme concentration 5- and 10-fold, as well as testing mutants to better understand the nature of the catalytic activity. The robustness of the workflow will also be tested on an expanded set of multicopper oxidases (MCOs), including three-domain MCOs of fungal and bacterial origin, as well as a multicopper polyphenol oxidoreductase laccase (MPOL). This pipeline demonstrated the use of computational biology to describe the kinetics of SLAC, rapidly and accurately, within the limitations of the experimental setup. It is useful for not only describing the biochemical metrics of the enzyme, but also to indicate flaws in the experimental design and allows the end-user to make discriminating choices regarding the technical procedures involved in wet-lab preparation. |
| References: | [1] Begley, C.G. and Ioannidis, J.P., 2015. Reproducibility in science: improving the standard for basic and preclinical research. Circulation research, 116(1), pp.116-126. [2] Baker, M., 2016. Reproducibility crisis. Nature, 533(26), pp.353-66. [3] Tipton, K.F., Armstrong, R.N., Bakker, B.M., Bairoch, A., Cornish-Bowden, A., Halling, P.J., Hofmeyr, J.H., Leyh, T.S., Kettner, C., Raushel, F.M. and Rohwer, J., 2014. Standards for Reporting Enzyme Data: The STRENDA Consortium: What it aims to do and why it should be helpful. Perspectives in Science, 1(1-6), pp.131-137. [4] Machczynski, M.C., Vijgenboom, E., Samyn, B. and Canters, G.W., 2004. Characterization of SLAC: a small laccase from Streptomyces coelicolor with unprecedented activity. Protein science, 13(9), pp.2388-2397. [5] Skálová, T., Dohnálek, J., Østergaard, L.H., Østergaard, P.R., Kolenko, P., Dušková, J., Štěpánková, A. and Hašek, J., 2009. The structure of the small laccase from Streptomyces coelicolor reveals a link between laccases and nitrite reductases. Journal of molecular biology, 385(4), pp.1165-1178. [6] Prins, A., Kleinsmidt, L., Khan, N., Kirby, B., Kudanga, T., Vollmer, J., Pleiss, J., Burton, S. and Le Roes-Hill, M., 2015. The effect of mutations near the T1 copper site on the biochemical characteristics of the small laccase from Streptomyces coelicolor A3 (2). Enzyme and microbial technology, 68, pp.23-32. [7] Pleiss, J., 2021. Standardized data, scalable documentation, sustainable storage-EnzymeML as a basis for FAIR data management in biocatalysis. ChemCatChem, 13(18), pp.3909-3913 |
| Figures: | 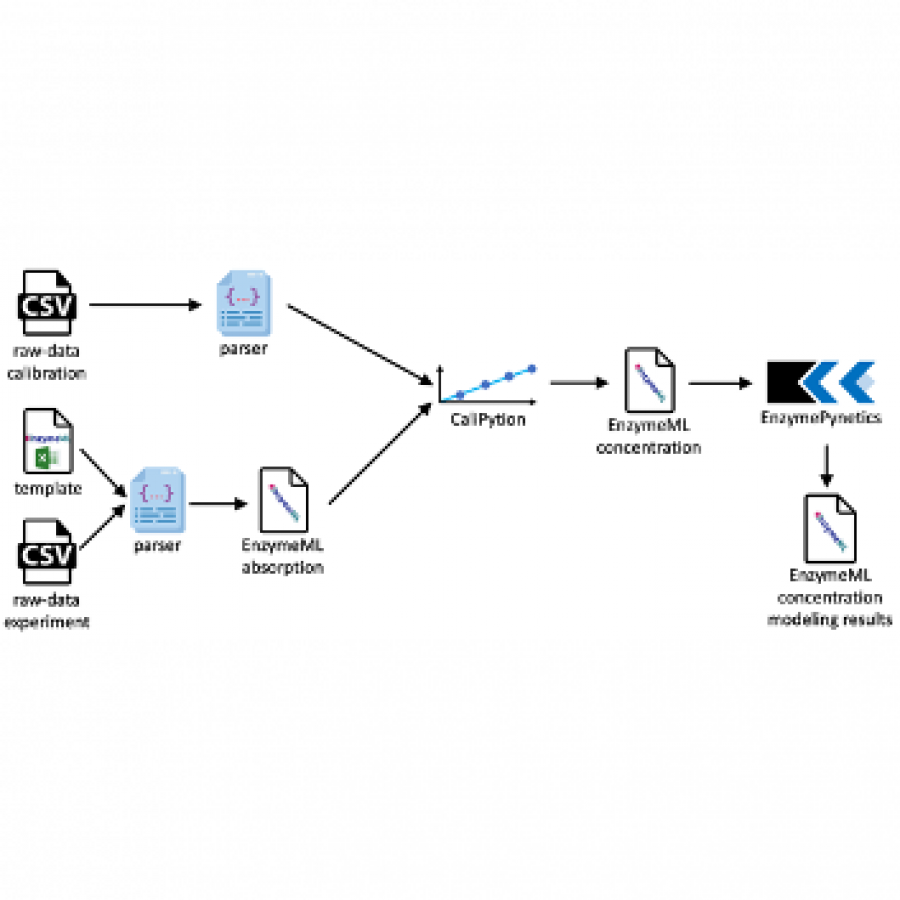 FIGURE: Schematic of the EnzymeML workflow |
#1499 | Structural Characterization and Enzyme Engineering of a Fungal L-Amino Acid Oxidase |
|
| Presenting author: | Simon KOOPMEINERS of BIELEFELD UNIVERSITY |
| Other authors: | Gabriele FISCHER VON MOLLARD of BIELEFELD UNIVERSITY Nils BERELSMANN of BIELEFELD UNIVERSITY Hartmut NIEMANN of BIELEFELD UNIVERSITY Dominic GILZER of BIELEFELD UNIVERSITY Christiane WIDMANN of BIELEFELD UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | L-amino acid oxidase / enzyme engineering / protein crystallization / molecular docking |
| Purpose: | Introduction L-amino acid oxidases (LAAOs, EC 1.4.3.2) catalyze the oxidative deamination of L-amino acids to the corresponding α-keto acids under formation of hydrogen peroxide and ammonium. LAAOs are of high interest for the synthesis of α-keto acids and the deracemization of enantiomeric mixtures of D-amino acids, which are both pharmaceutical building blocks. The fungal HcLAAO4 from Hebeloma cylindrosporum has a broad substrate spectrum, consisting of divergent L-amino acids and derivates, and can be expressed recombinantly with high yield in expression systems like E. coli or Pichia pastoris[1,2]. The HcLAAO4 was already utilized for the production of L-phenylpyruvate[1] and as part of an enzyme cascade with a catalase and transaminase[3]. As optimization by rational enzyme design and directed evolution requires detailed structural information about an enzyme and its active site, we aimed to crystallize the HcLAAO4 and determine its structure. Materials and Methods HcLAAO4 variants were recombinantly expressed in E. coli Arctic Express and purified by affinity chromatography and size exclusion chromatography. The enzyme was crystallized after limited proteolysis with trypsin. Crystals were harvested directly or soaked with different substrates prior to harvesting. Diffraction data were collected at beamlines at the ESRF[4]. The initial model for molecular replacement was created by using the ColabFold-Server[5]. Docking was carried out with AutoDock Tools 1.5.7[6]. The enzyme activity was monitored by a peroxidase coupled photometric assay. Results Crystallization of HcLAAO4 was achieved after removal of unstructured terminal peptides by limited proteolysis with trypsin and by mutating a cluster of hydrophilic residues at the protein surface to alanine, utilizing the surface entropy reduction (SER) strategy. The asymmetric unit (AU) consists of four HcLAAO4 molecules, forming a dimer of dimers. The positioning of the SER-mutation site in the interface between the dimers indicates that the SER-mutations are critical for the formation of this AU. By soaking the crystals with different substrates, we also determined the structure of the substrate-bound enzyme with resolutions of about 2 Å and identified residues involved in substrate binding. Similar to other LAAOs the carboxylate moiety and the amine group of the L-amino acid substrate are coordinated by ionic interactions, hydrogen bridges and a main chain carbonyl group. The active site of HcLAAO4 is overall hydrophobic and mostly consists of aromatic site chains, which stabilize the aliphatic portion of a substrate. The preference for substrates with an aliphatic portion at the γ- and δ-C-atom is also explained by interactions with active site residues. By utilizing a molecular docking model previously unknown, industrially interesting substrates were identified. Finally, we analyzed the role of an active site residue on the substrate scope of HcLAAO4 by generating two point mutants. Both mutants showed an approximately 2-fold increased activity for L tryptophan, which is of high interest for the deracemization of racemic mixtures to the D enantiomer. Conclusion Here we crystallized the HcLAAO4 and determined the structure of the apo enzyme and the substrate bound state. Residues interacting with the substrate in the active site were identified as potential targets for enzyme engineering approaches and by utilizing a molecular docking model previously unknown substrates were found. By site directed mutagenesis we generated mutants with increased activity for industrially interesting substrates, demonstrating the suitability of our structure model for future optimization of the enzyme. |
| References: | [1] Bloess, S., Beuel, T., Krüger, T.et al., Appl. Microbiol. Biotechnol. 2019, 103, 2229. [2] Heß, M. C., Bloess, S., Risse, J. M.et al., MicrobiologyOpen 2020, 9, e1112. [3] Heinks, T., Paulus, J., Koopmeiners, S et al., ChemBioChem 2022, 23, e202200329. [4] Mueller-Dieckmann, C., Bowler, M.W., Carpentier, P. et al., 2015, Eur. Phys. J. Plus 130, 70. [5] Mirdita, M., Schütze, K., Moriwaki, Y. et al., 2022, Nat Methods 19, 679-682. [6] Morris, G. M., Huey, R., Lindstrom, W. et al., 2009, J Comput Chem, 30, 2785-2791. |
| Figures: | 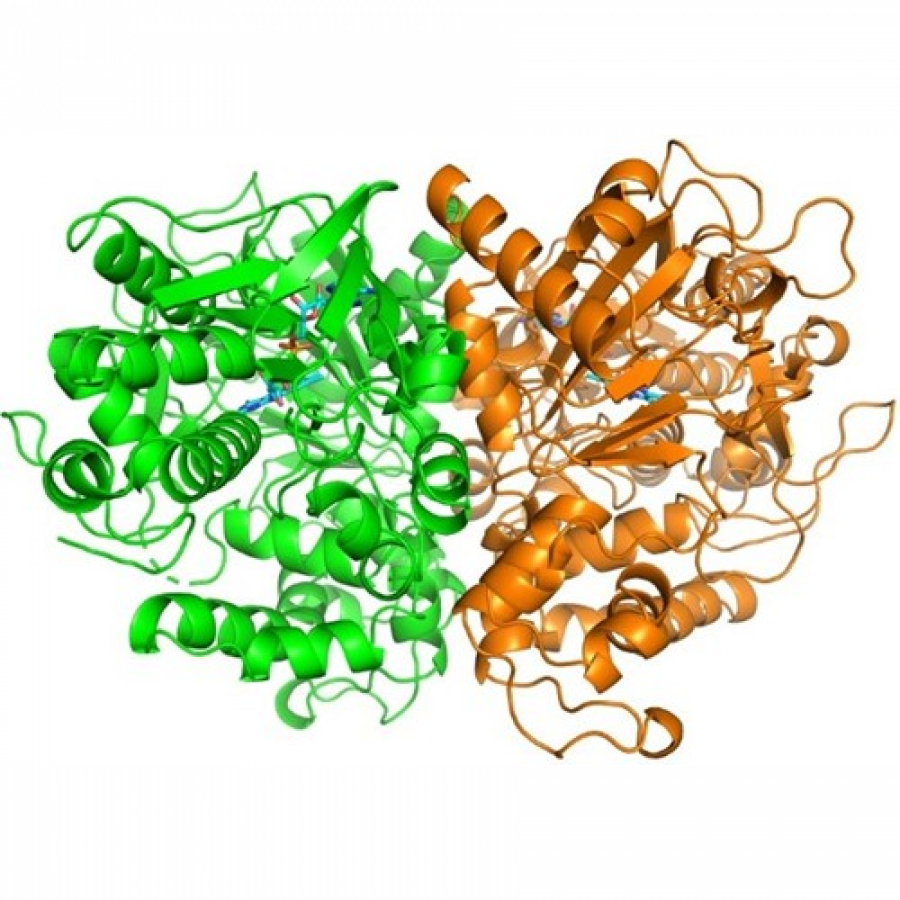 Structure of HcLAAO4. The biological dimer of HcLAAO4 is shown as cartoon with the bound FAD co-factor as sticks. The asymmetric unit contains a second dimer. |
#1501 | Biocatalytic Characterization of an Alcohol Dehydrogenase Variant Deduced from Lactobacillus kefir in Asymmetric Hydrogen Transfer |
|
| Presenting author: | Aleksandra RUDZKA of WARSAW UNIVERSITY OF TECHNOLOGY |
| Other authors: | Beata ZDUN of WARSAW UNIVERSITY OF TECHNOLOGY Natalia ANTOS of WARSAW UNIVERSITY OF TECHNOLOGY Lía MARTÍNEZ MONTERO of UNIVERSITY OF GRAZ Tamara REITER of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ Paweł BOROWIECKI of WARSAW UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Chiral alcohols / Alcohol dehydrogenases / Lactobacillus kefir / Engineered enzyme variant |
| Purpose: | Optically active alcohols are key chiral building blocks supporting the synthesis of high-value-added products. Therefore, it is essential to develop practical, selective, scalable and environmentally friendly methods to obtain them. Unfortunately, most methods for the synthesis of chiral alcohols have significant limitations related mainly to low reaction yields and the high cost of product isolation and purification. Therefore, desymmetrization of prochiral or meso-substrates is considered as one of the most valuable transformations, while the vast majority of efficient and selective biocatalysts capable of reducing carbonyl compounds are alcohol dehydrogenases (E. C. 1.1.1) (ADHs). Hence, hydrogen-transfer biocatalysts for preparing optically pure chiral secondary alcohols are of great interest, especially for sterically demanding ketones. This work reports on the biocatalytic potential of an anti-Prelog (R)-specific ADH variant from Lactobacillus kefir (Lk-ADH-E145F-F147L-Y190C named Lk-ADH Prince) [1] used as an E. coli/ADH whole-cell biocatalyst and its characterization for the stereoselective reduction of prochiral carbonyl substrates. During our studies, crucial parameters of the enzymatic reaction (i.e., reaction medium, cofactor dependence assessment, tolerance to organic co-solvent, and substrate loading) were optimized using pentoxyphylline active agent as a model prochiral ketone. Next, we investigated the substrate scope for Lk-ADH Prince by employing 34 carbonyl derivatives in hydrogen transfer reactions. Our results revealed that E. coli/Lk-ADH Prince is highly stereoselective biocatalyst capable of reducing structurally diverse ketones, providing optically active alcohols with excellent enantiomeric excesses (up to >99%), in high yields (up to 91%) and at relatively high substrate concentration (up to 100 mM) without requiring the addition of costly NADPH cofactor. Acknowledgements: This research was funded by the Warsaw University of Technology within the ‘Excellence Initiative: Research University (IDUB)’ programme in the framework of the ‘Young PW’ starting grant. A.R. and B.Z. are grateful to the IDUB project (’Scholarship Plus’ programme) for providing a research fellowship. The University of Graz and the Field of Excellence BioHealth are acknowledged for financial support. Joerg H. Schrittwieser from the Institute of Chemistry, University of Graz, is thanked for his suggestion to select this enzyme from literature and valuable discussion. |
| References: | [1] Emmanuel, M. A., Greenberg, N. R., Oblinsky, D. G., Hyster, T. K., Nature, 2016, 540, 414-417. |
| Figures: | 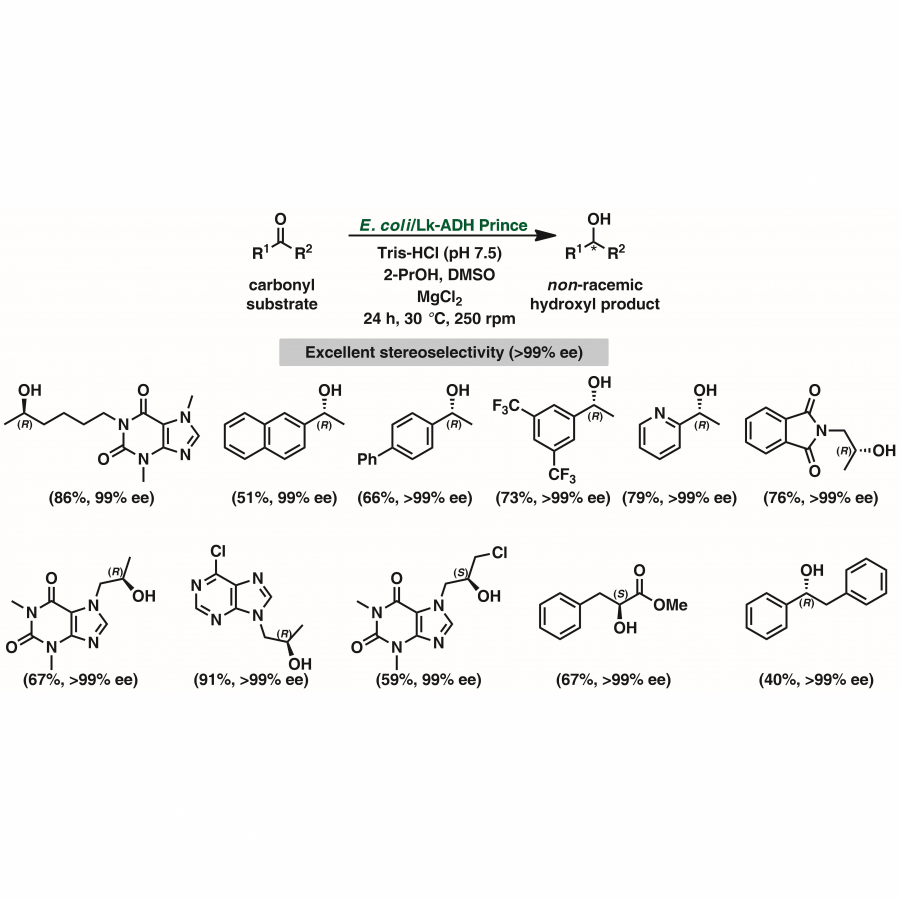 Asymmetric reduction of prochiral ketones using E. coli/Lk-ADH Prince. |
#1502 | The Sugar Methyltransferase Bb278 application in methylated flavonoid glucosides production |
|
| Presenting author: | Jarosław POPŁOŃSKI of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIEN |
| Other authors: | Sandra SORDON of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIEN Agata MATERA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIEN Kinga DULAK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIEN Ewa HUSZCZA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIEN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | methyltransferase / flavonoids / SAM / glycosides |
| Purpose: | Sugar-O-Methyltransferases are a group of relatively small 20-48 kDa monomeric proteins that are known for their specific activity of transferring a methyl group onto sugars or sugar moieties of complex molecules, utilizing S-adenosyl-L-methionine as a methyl donor. Many members of this group have been characterized and are mostly involved in the biosynthesis of specific secondary metabolites, such as macrolides and indolocarbazoles [1-2]. Unfortunately, in most cases, sugar methyltransferases are very selective and specific for their activity, which limits their biocatalytic applications. Recently, a group of enzymes was identified in entomopathogenic filamentous fungi that share a unique ability to transform a variety of natural products to respective 4-O-methylglucosides [3-4]. Our previous work focused on the biochemical characterization of Sugar-O-Methyltransferase from Beauveria bassiana AM278 (Bb278), which revealed a broad acceptor range from small phenolic glucosides to flavonoid glucosides and even bulky steroid glucosides, although limited only to β-glucosides. The application of Bb278 to preparative-scale methylations is mostly limited to in vivo reactions due to the high price of SAM and SAH inhibition, although it suffers from complicated downstream processing. Herein, we would like to present our recent progress on in vitro reaction engineering of Bb278 methylation focused on lowering the SAM consumption and including the SAM regeneration using halide methyltransferases. Acknowledgments: This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreements no. 814650 (SynBio4Flav). www.SynBio4Flav.eu |
| References: | [1]. C. Zhang, C. Albermann, X. Fu, N. R. Peters, J.D. Chisholm, G. Zhang, E. J. Gilbert, P. G. Wang, D. L. Van Vranken, J. S. Thorson, RebG- and RebM-Catalyzed Indolocarbazole Diversification. ChemBioChem, 2006, 7, 795-804. [2]. J. Xiao, Q. Zhang, Y. Zhu, S. Li, G. Zhang, H. Zhang, K. Saurav, C. Zhang, Characterization of the sugar-O-methyltransferase LobS1 in lobophorin biosynthesis. Appl Microbiol Biotechnol, 2013 97, 9043-9053. [3]. L. Xie, L. Zhang, C. Wang, X. Wang, Y.M. Xu, H. Yu, P. Wu, S. Li, L. Han, A.A.L. Gunatilaka, X. Wei, M. Lin, I. Molnár, Y. Xu, Methylglucosylation of aromatic amino and phenolic moieties of drug-like biosynthons by combinatorial biosynthesis, PNAS, 2018, 115, E4980-E4989. [4]. L. Xie, L. Zhang, J. Bai, Q. Yue, M. Zhang, J. Li, C. Wang, Y. Xu, Methylglucosylation of Phenolic Compounds by Fungal Glycosyltransferase-Methyltransferase Functional Modules, J Agr Food Chem, 2019, 67, 8573-8580. |
#1503 | Basecamp Research - Predictive Enzyme Development by Nature-Inspired AI |
|
| Presenting author: | Ahir PUSHPANATH of BASECAMP RESEARCH |
| Other authors: | Molly STEADMAN of BASECAMP RESEARCH Valerio PERENO of BASECAMP RESEARCH |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Generative AI / Protein Design / Metagenomics / Transaminase and Fluorinase |
| Purpose: | Biocatalysis is approaching an inflection point, yet enzyme discovery and evolution still remain bottlenecks for its accelerated adoption across all industries. Directed evolution, the hallmark technique for enhancing natural enzymes to meet industrial process specifications, has provided multiple success stories, but is inherently a resource intensive endeavor. Consequently, there is an increasing need for computational tools to reduce enzyme development times. As the intersection of AI and biology deepens, AI protein design tools are in the spotlight. Despite great advances built on the success of large language models, the protein sequence datasets used for training these algorithms are not diverse or large enough. Therefore, in order to usher in the desired paradigm shift of truly predictive design, a true representation of the protein landscape is required. Basecamp Research's global biodiscovery has yielded hundreds of millions of ethically sourced, novel protein sequences from diverse eco-regions, resulting in a dataset three times larger and four times more diverse than Uniprot. For commonly used biocatalyst classes such as IREDs and KREDS, BaseData™ has over 25 times more than those available on UniProt, many sourced from extreme environments. For every sample collected, extensive environmental metadata is also recorded. Our unique data on nature is not stored in a tabular format but rather as a pre-indexed, interconnected network, incorporating over 3 billion relationships between proteins, genomes, taxonomic communities, and environmental metadata. Using graph deep learning techniques, our BaseGraph™ algorithm can intelligently navigate the protein landscape, using the evolutionary context of a protein to infer complex function and process performance with unparalleled accuracy. Our BaseDesign™ algorithms herald a new vision for enzyme optimisation. By fine tuning protein language models on BaseData, our generated sequences have lower perplexity, higher pLDDT and TM scores compared to other protein design tools trained exclusively on public databases1. The success of our methods are exemplified in two customer case studies. In the first study, Basecamp Research identified a novel broad specificity wildtype transaminase within 1 week using BaseGraph™. The same broad specificity was only achieved after 1.5 years and > £1 million in R&D costs with traditional directed evolution . In the second study, BaseDesign™ was used to generate novel fluorinase enzyme sequences, a rare enzyme class with only 18 representatives in the public database. Some of the generated sequences had 90 mutations over two wildtype literature controls. Out of the 66 novel sequences tested, remarkably, 93% of them were expressed for the first time in the expression host of choice, with 82% of them exhibiting fluorinase activity. Additionally, 64% of the designed sequences outperformed the two literature control sequences in SAM fluorination activity and thermostability. |
| References: | [1] Munsamy, Geraldene, et al. "ZymCTRL: a conditional language model for the controllable generation of artificial enzymes." |
| Figures: | 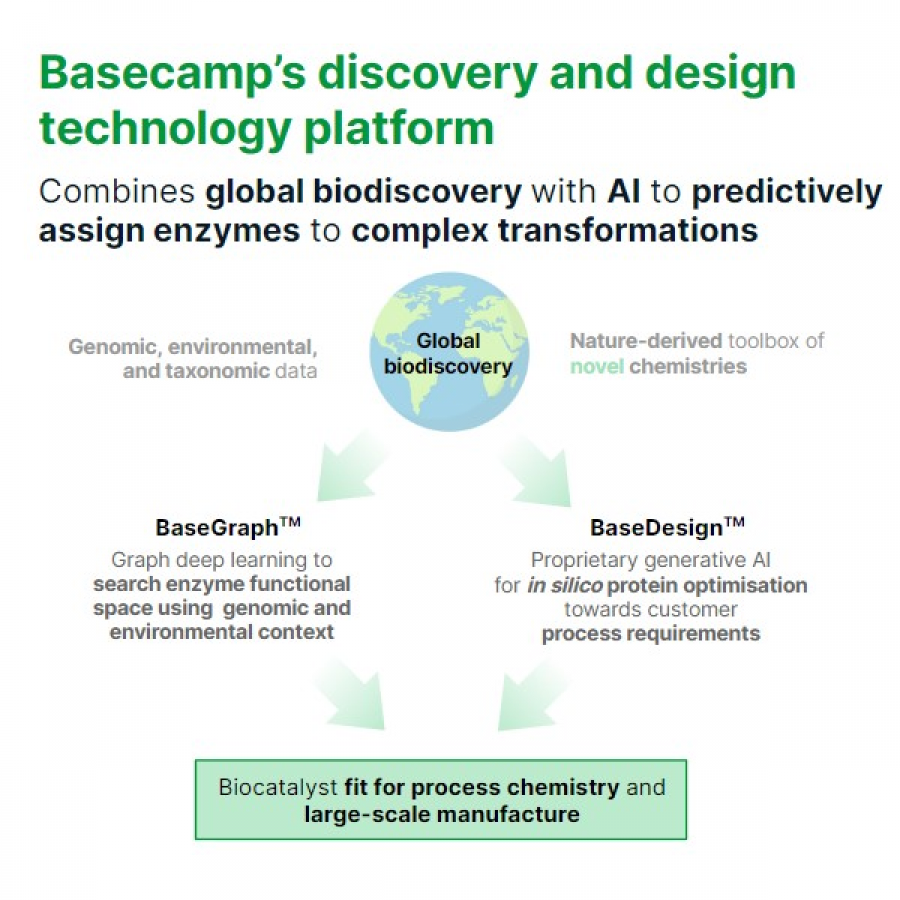 Overview of Technology BaseData™: Unveiling the True Protein Universe.
BaseGraph™: A Search Engine for Proteins, using Evolutionary Context to Infer Complex Function and Performance.
BaseDesign™: Best in Class Protein Design AI. 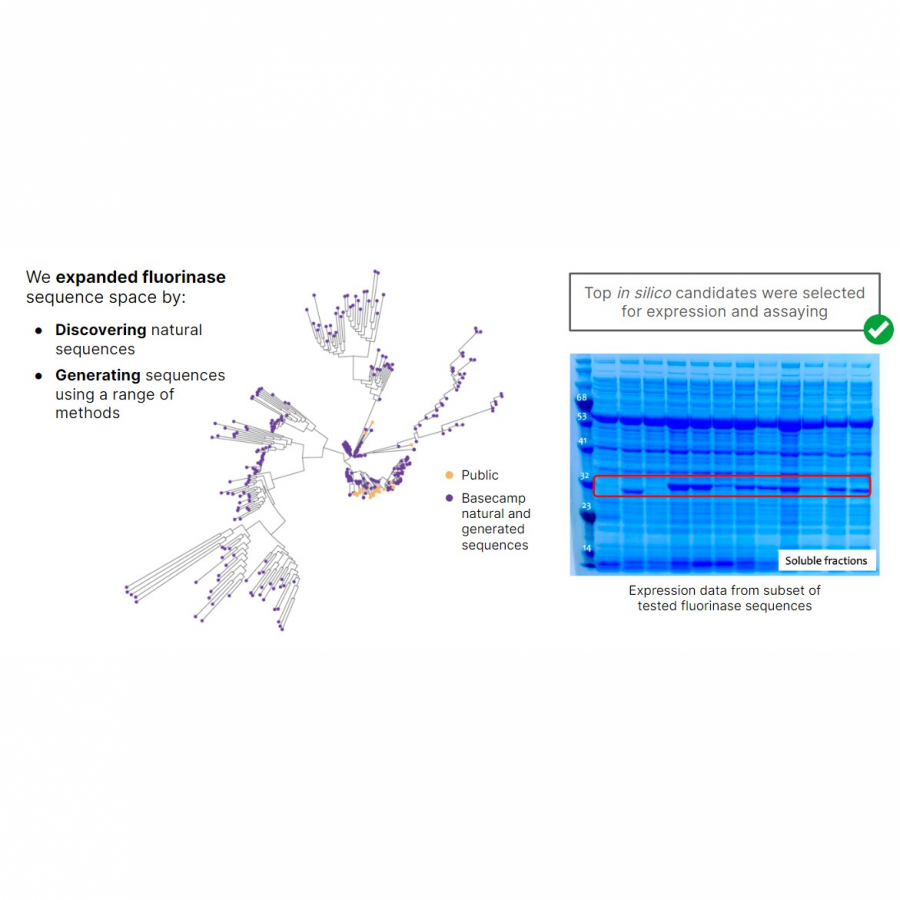 Combination of nature and AI expands the limited sequence pool of a rare biochemistry, fluorination. Expanding Fluorinase Sequence Space with Nature and AI: Basecamp Research Generates Novel Sequences with High Expressibility, Thermostability, and Activity for SAM-Fluorination. |
#1504 | Multienzymatic biotransformation of flavokawain B by entomopathogenic filamentous fungi. |
|
| Presenting author: | Paweł CHLIPAŁA of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Tomasz TRONINA of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Monika URBANIAK of INSTITUTE OF PLANT GENETICS, POLISH ACADEMY OF SCIENCES, Ewa KOZŁOWSKA of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Łukasz STĘPIEŃ of INSTITUTE OF PLANT GENETICS, POLISH ACADEMY OF SCIENCES, Edyta KOSTRZEWA-SUSŁOW of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Tomasz JANECZKO of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biotransformations / entomopathogenic fungi strains / flavokawain B / 4-O-methylglycosylation |
| Purpose: | Flavokawain B is one of the naturally occurring chalcones in the kava plant (Piper methysticum)[1]. It exhibits anticancer[2]–[4], anti-inflammatory[5], [6] and antimalarial properties[7], [8]. Due to its beneficial therapeutic potential, flavokawain B is likely to be used to treat many diseases. However, its poor bioavailability and low aqueous solubility support the view that the application is still limited. The attachment of a sugar unit impacts the stability and solubility of flavonoids and often also determines their bioavailability and bioactivity[9]–[11]. Biotransformation is an environmentally-friendly way to improve the properties of compounds, for example, to increase their hydrophilicity and thus affect their bioavailability[12]. Recent studies proved that entomopathogenic filamentous fungi from the genus Isaria and Beauveria can perform O-methylglycosylation of hydroxyflavonoids or O-demethylation and hydroxylation of selected chalcones[10] and flavones[11]. In the presented study, we examined the ability of entomopathogenic filamentous fungi from Beauveria bassiana, B. caledonica, Isaria farinosa, I. fumosorosea, and I. tenuipes to transform flavokawain B into its glycosylated derivatives. The main process occurring during the reaction is O-demethylation and/or hydroxylation followed by 4-O-methylglycosylation. The substrate used was characterized by low susceptibility to transformations compared to our previously described transformations of flavones and chalcones in the cultures of the tested strains. However, in the culture of the B. bassiana KCh J1.5 and BBT, Metarhizium anisopilae KCh Ma, and I. farinosa KCh It, the expected methylglycosides were obtained with high yields. B. bassiana KCh J1.5 performed either 4’-demethylation, 4’-O-methylglycosylation or 4’-O-methylglycosilation with simultaneous hydroxylation in the meta position in the B ring of the substrate. Also formation of products of 3-O-methylglycosylation and 4-hydroxylation with 3-O-methylglycosylation was observed. In the case of M. anisopilae KCh Ma formation of 4’-O¬-methylglicosides and 4’-O-methylglicosides with concurrent meta or para hydroxylation of B ring of the substrate was noticed. The product of 4’-demethylation of flavokawain B was identified as well. Our results prove that multiple enzymes were involved in forming the products in the entomopathogenic filamentous fungi cultures. |
| References: | [1] K. Zenger, S. Agnolet, B. Schneider, and B. Kraus, ‘Biotransformation of Flavokawains A, B, and C, Chalcones from Kava (Piper methysticum), by Human Liver Microsomes’, J Agric Food Chem, vol. 63, no. 28, pp. 6376-6385, Jul. 2015, doi: 10.1021/acs.jafc.5b01858. [2] J. H. J. A. L. J. Y. K. and J. S. H. Ryu, ‘Flavokawain B and C, Isolated from the Root of Piper methysticum, Inhibit Melanogenesis in Melan-a Cells’, Journal of the Society of Cosmetic Scientists of Korea, vol. 48, no. 1, pp. 11-24, Mar. 2022, doi: 10.1016/j.bmcl.2014.12.082 [3] T. Ji et al., ‘Flavokawain B, a kava chalcone, inhibits growth of human osteosarcoma cells through G2/M cell cycle arrest and apoptosis’, Mol Cancer, vol. 12, no. 1, Jun. 2013, doi: 10.1186/1476-4598-12-55. [4] N. Abu et al., ‘In vivo antitumor and antimetastatic effects of flavokawain B in 4T1 breast cancer cell-challenged mice’, Drug Des Devel Ther, vol. 9, pp. 1401-1417, Mar. 2015, doi: 10.2147/DDDT.S67976. [5] C.-T. Lin et al., ‘Anti-inflammatory Activity of Flavokawain B from Alpinia pricei Hayata’, J Agric Food Chem, vol. 57, no. 14, pp. 6060-6065, Jun. 2009, doi: 10.1021/jf900517d. [6] X. W. Zhang, D. H. Zhao, Y. C. Quan, L. P. Sun, X. M. Yin, and L. P. Guan, ‘Synthesis and evaluation of antiinflammatory activity of substituted chalcone derivatives’, Medicinal Chemistry Research, vol. 19, no. 4, pp. 403-412, May 2010, doi: 10.1007/s00044-009-9202-z. [7] R. H. Hans et al., ‘Synthesis, antimalarial and antitubercular activity of acetylenic chalcones’, Bioorg Med Chem Lett, vol. 20, no. 3, pp. 942-944, Feb. 2010, doi: 10.1016/J.BMCL.2009.12.062. [8] S. K. Awasthi et al., ‘Potent antimalarial activity of newly synthesized substituted chalcone analogs in vitro’, Medicinal Chemistry Research, vol. 18, no. 6, pp. 407-420, Jul. 2009, doi: 10.1007/s00044-008-9137-9. [9] X. Wang, ‘Structure, mechanism and engineering of plant natural product glycosyltransferases’, FEBS Letters, vol. 583, no. 20. pp. 3303-3309, Oct. 20, 2009. doi: 10.1016/j.febslet.2009.09.042. [10] L. Wang et al., ‘Comparing the acceptor promiscuity of a Rosa hybrida glucosyltransferase RhGT1 and an engineered microbial glucosyltransferase OleDPSA toward a small flavonoid library’, Carbohydr Res, vol. 368, pp. 73-77, 2013, doi: https://doi.org/10.1016/j.carres.2012.12.012. [11] R. P. Pandey, R. B. Gurung, P. Parajuli, N. Koirala, L. T. Tuoi, and J. K. Sohng, ‘Assessing acceptor substrate promiscuity of YjiC-mediated glycosylation toward flavonoids’, Carbohydr Res, vol. 393, pp. 26-31, Jun. 2014, doi: 10.1016/j.carres.2014.03.011. [12] M. Dymarska et al., ‘Glycosylation of 6-methylflavone by the strain Isaria fumosorosea KCH J2’, PLoS One, vol. 12, no. 10, Oct. 2017, doi: 10.1371/journal.pone.0184885. |
| Figures: | 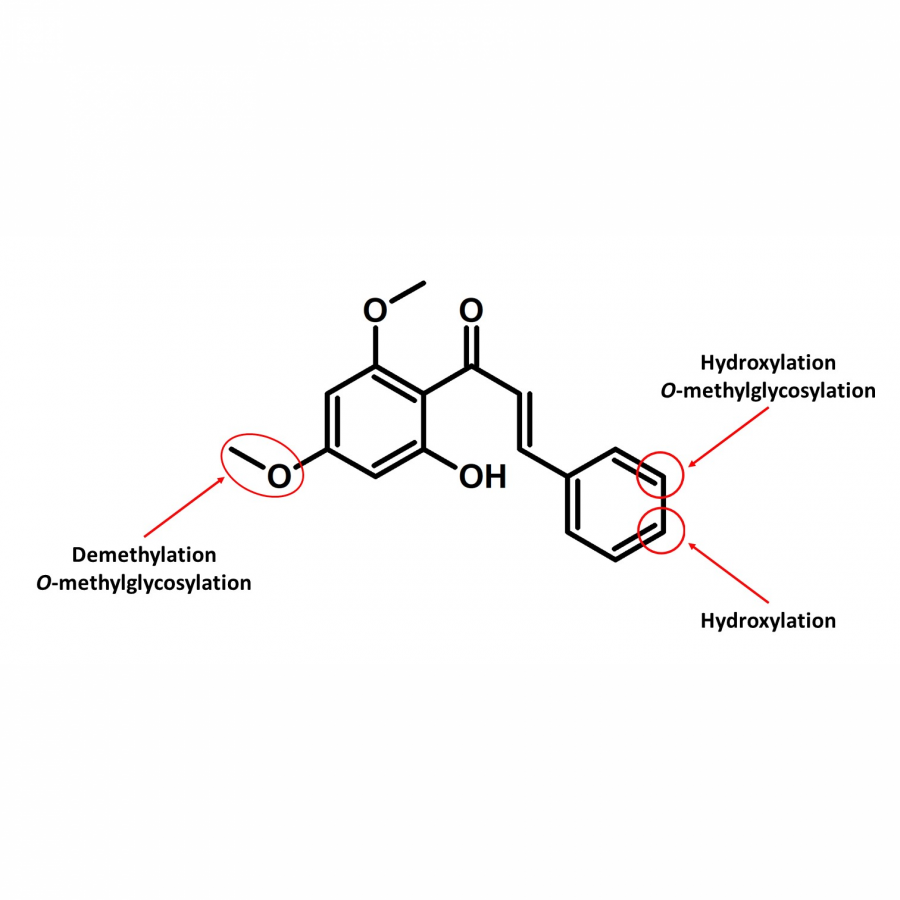 Biotransformation sites in flavokawain B. |
#1505 | The use of tyrosinases in a chemoenzymatic cascade as a peptide ligation strategy |
|
| Presenting author: | Yeke NI of UCL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Pictet-Spengler / tyrosinase / / |
| Purpose: | N-terminal tyrosine-containing peptides play many key roles in biological systems, but the lack of reliable coupling methods under mild conditions limits the generation of natural and unnatural peptieds.[2-7] The Pictet-Spengler reaction (PSR) has been reported to synthsize tetrahydroisoquinolines and tetrahydro--carboline alkaloids via enzymatic process or non-enzymatic method in phosphate buffer.[8-13] In our work, a new N-terminal tyrosine-containing peptide ligation method, with aldehydes, utilising a Pictet-Spengler reaction, has been established. In a key step, tyrosinase enzymes (TYR) have been used to convert L-tyrosine to L-3,4-dihydroxyphenyl alanine (L-DOPA) residues, generating suitable functionality for the Pictet-Spengler coupling. Then, L-DOPA was use in the PSR in phophate buffer with aldehydes to afford tetrahydroisoquinolines. Diverse aldehydes, including benzaldehydes, fluoreseceent aldehydes and peptide aldehydes, were used to study the reacrtion scope. Indicating this new chemoenzymatic coupling strategy can be used for fluorescent-tagging and peptide ligation purposes. 1. Y. Ni, Y. Wang, A. B. Tabor, J. M. Ward and H. C. Hailes, RSC Chem Biol, 2023, 4, 132-137. 2. T. Kimmerlin and D. Seebach, J. Pept. Res., 2005, 65, 229-260. 3. M. Forest, J. C. Martel, S. St-Pierre, R. Quirion and A. Fournier, J. Med. Chem., 1990, 33, 1615-1619. 4. M. Kotani, M. Detheux, A. Vandenbogaerde, D. Communi, J. M. Vanderwinden, E. Le Poul, S. Brezillon, R. Tyldesley, N. Suarez-Huerta, F. Vandeput, C. Blanpain, S. N. Schiffmann, G. Vassart and M. Parmentier, J. Biol. Chem., 2001, 276, 34631-34636. 5. E. J. Mead, J. J. Maguire, R. E. Kuc and A. P. Davenport, Br. J. Pharmacol., 2007, 151, 1143-1153. |
#1506 | Baylis-Hillman cascade reactions |
|
| Presenting author: | Shuyi SHUYI of UCL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Baylis-Hillman reaction / cascade / / |
| Purpose: | Baylis-Hillman cascade reactions Shuyi Zhang The development of enzyme-based sustainable synthetic methods is a key growth area in the field of biotechnology. The Baylis-Hillman (B-H) reaction is a useful synthetic transformation used in C-C bond formation. Earlier work established the use of amine-based organocatalysts[1] and the use of aqueous-based media was found to accelerate the reaction in some cases.[2,3] Recently, an engineered B-Hase has been designed.[4] Also, a paper about the cascade of alcohol dehydrogenase and the engineered B-Hase has already been published.[5] Our aim is to establish if we can develop one-pot chemoenzymatic or enzymatic syntheses using the Baylis-Hillman (B-H) reaction as the first step, and other enzymes in the second step, leading to a stereospecific and effective enzyme cascade. References: 1. V. K. Aggarwal, A. Mereu, Chem Commun., 1999, 2311. 2. A. Lubineeau, J. Auge, Y. Queneau, Synthesis, 1994, 741. 3. J. Auge, N. Lubin, A. Lubineau, Tetrahedron Lett., 1994, 35, 7948. 4. R. Crawshaw, A.E. Crossley, L. Johannissen et al, Nature Chemistry., 2022, 14, 313. 5. Y. Li, C. Bao, Z. Sun et al, ChemCatChem, 2023, e2022016 |
| References: | 1. V. K. Aggarwal, A. Mereu, Chem Commun., 1999, 2311. 2. A. Lubineeau, J. Auge, Y. Queneau, Synthesis, 1994, 741. 3. J. Auge, N. Lubin, A. Lubineau, Tetrahedron Lett., 1994, 35, 7948. 4. R. Crawshaw, A.E. Crossley, L. Johannissen et al, Nature Chemistry., 2022, 14, 313. 5. Y. Li, C. Bao, Z. Sun et al, ChemCatChem, 2023, e2022016 |
#1507 | Dinucleotide-based artificial cofactors: preparation and applications in photobiocatalysis |
|
| Presenting author: | Markus AVERBECK of TECHNISCHE UNIVERSITÄT BERLIN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial cofactors / Photobiocatalysis / New-to-nature functionalities / Protein engineering |
| Purpose: | Nature has generated an impressive array of chemical reactivities in enzymes using only a subset of the 20 proteinogenic amino acids. However, this chemistry is mostly limited to general acid-base catalysis, nucleophilic and electrophilic catalysis. To expand on the chemical reactivities, nature employs cofactors, which are utilised in roughly half of all reactions catalysed by enzymes. Those cofactors can be divided into inorganic, metal-organic, and organic cofactors. The drive to create new-to-nature functions for biocatalysis requires the expansion or modulation of the natural sets of amino acids and cofactors. New-to-nature functions have been implemented by the incorporation of non-canonical amino acids (ncAAs), bioconjugation or supramolecular assembly approaches of cofactors and catalysts. Artificial metalloenzymes are one of the prominent examples. However, artificial organic cofactors have yet seen less attention. Photocatalysis has gained increasing attention to promote otherwise inaccessible reactions by organic synthesis. Photocatalytic reactions often lack stereo- and/or enantioselective control. To overcome this limitation the combination of photocatalysts with the chiral environment of a protein is particularly promising. To date, most photocatalytic approaches have been realised by integrating inorganic or metal-organic photocatalysts either in enzymatic cascade reactions or as cofactors in artificial metalloenzymes. Since these photocatalysts are often based on transition metals, organic photocatalysts could serve as a sustainable alternative. Photobiocatalysis using organic photocatalysts is still underrepresented with only few known examples. We envision to install novel moieties onto known cofactors. Therefore, we have harnessed the base exchange activity of ADP-ribosyl cyclase from Aplysia californica to covalently link organic photocatalytsts to an ADP-ribose backbone. This furnishes an artificial photocatalytic dinucleotide-like cofactor. We aim to utilize the binding characteristics of dinucleotide cofactors, which are mediated to a large extend by the adenosine ring and the phosphodiester group. This allows us to implement a dinucleotide-like cofactor with novel reactivities in given protein scaffolds. This seems particularly promising as many well-known biocatalysts with desirable properties bind dinucleotides. Our setup also allows for easy and direct access of artificial dinucleotide cofactors with modified or improved characteristics not limited to photobiocatalysis. We are currently working on optimizing the performance of the cyclase to establish a versatile tool for the generation of artificial dinucleotide-like cofactors. |
| Figures: | 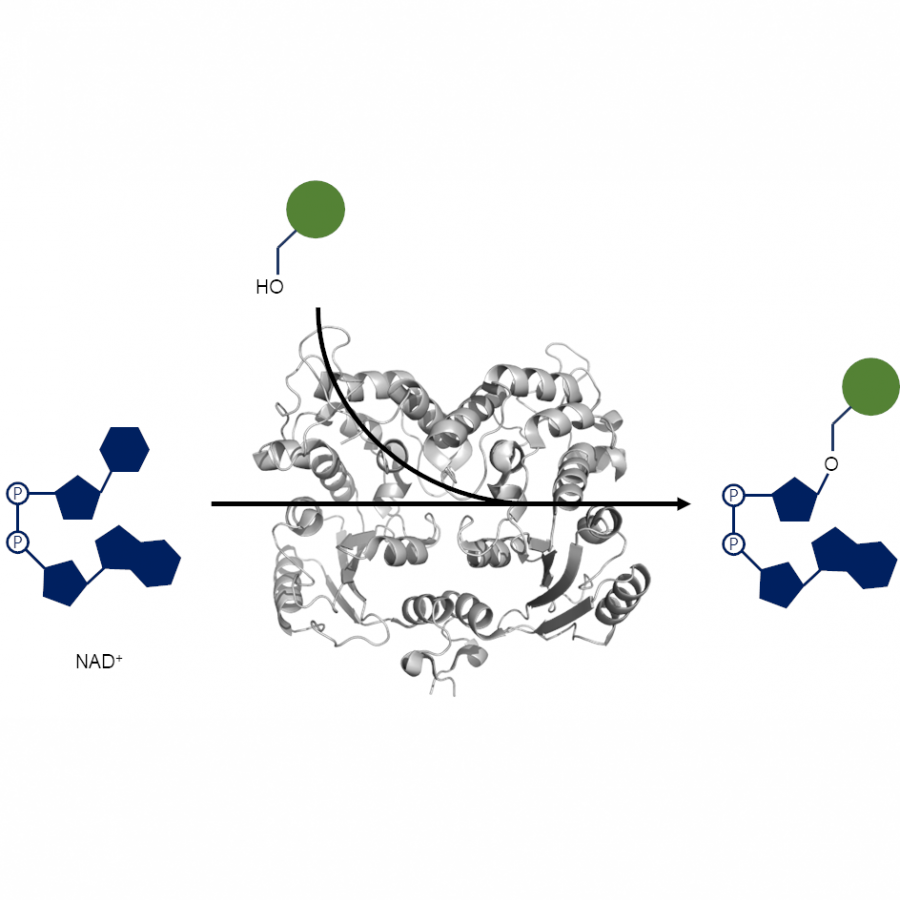 Artificial dinucleotide cofactors by enzymatic reaction General concept of ADPRC-catalysed base exchange reaction with photocatalysts to generate artificial photocatalytic dinculeotide cofactors |
#1510 | Combining enzyme engineering with bioprocess development for a sustainable metaraminol synthesis |
|
| Presenting author: | Berit ROTHKRANZ of FORSCHUNGSZENTRUM JÜLICH GMBH |
| Other authors: | Laura GRABOWSKI of FORSCHUNGSZENTRUM JÜLICH GMBH Nina KLOS of FORSCHUNGSZENTRUM JÜLICH GMBH Sarah SCHMITZ of FORSCHUNGSZENTRUM JÜLICH GMBH Torsten SEHL of FORSCHUNGSZENTRUM JÜLICH GMBH Dörte ROTHER of FORSCHUNGSZENTRUM JÜLICH GMBH |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme engineering / bioprocess engineering / enzymatic cascade / chiral amino alcohols |
| Purpose: | Chiral amino alcohols are subject of interest in the pharmaceutical and fine chemical industries as they play an important role as active pharmaceutical ingredients or precursor molecules for drugs. In recent years, our working group has focused on metaraminol as showcase for the enzymatic production of chiral amino alcohols. An enzymatic two-step reaction was successfully established, starting with the carboligation of 3-hydroxy benzaldehyde and pyruvate towards the intermediate (R)-3-OH-phenylacetylcarbinol ((R)-3-OH-PAC) catalyzed by the pyruvate decarboxylase of Acetobacter pasteurianus (ApPDC). This step is followed by the reductive amination of (R)-3-OH-PAC towards metaraminol using L-alanine as amino donor catalyzed by the amine transaminase of Chromobacterium violaceum (Cv2025) [1,2]. By coupling the enzymatic cascade with an in-situ liquid-liquid extraction, 69 % metaraminol were yielded after three extraction steps [2]. However, to make the process economically feasible, higher product yields and catalyst reuse are envisaged. However, these goals are challenging due to stability issues. Within the scope of our new project MetaProcess, these challenges are targeted by molecular dynamics simulations elucidating the weak points of the applied enzymes. Additionally, the operational stability of the catalysts is to be enhanced by the application of rational design strategies. Besides enzyme engineering, biocatalyst costs can be minimized by optimizing the production process of the catalysts themselves. As whole cell catalysts are applied as a cheap catalyst formulation, high cell densities and simultaneously high specific activities of the heterologous produced enzymes inside the cells are targeted. After optimization of the fed-batch cultivation, first approaches with the amine transaminase of Bacillus megaterium showed a specific activity of 0.81 ± 0.03 U/mg dry cell weight and a cell density of 223 ± 14 g/L wet cell weight. The optimized cultivation protocol led to an overall 45-fold increase of activity compared to shake flask experiments. In the future, this fed-batch protocol can be used to produce the engineered enzymes for the metaraminol synthesis. By combining both strategies, enzyme engineering and bioprocess engineering, the enzymatic production process of chiral amino alcohols can be designed in an economically feasible manner representing a green alternative to classical chemical production routes. |
| References: | [1] K. Mack, M. Doeker, L. Grabowski, A. Jupke, D. Rother, Green Chem., 2021, 23, 4892 [2] M. Doeker, L. Grabowski, D. Rother, A. Jupke, Green Chem., 2022, 24, 295 |
#1511 | Chemoenzymatic synthesis of iminosugars from monosaccharides |
|
| Presenting author: | Marianne Bore HAARR of UNIVERSITY COLLEGE DUBLIN |
| Other authors: | Kathryn YEOW of UNIVERSITY COLLEGE DUBLIN Elaine O'REILLY of UNIVESITY COLLEGE DUBLIN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | iminosugars / transaminase / galactose oxidase / |
| Purpose: | Iminosugars (1-3), are polyhydroxylated alkaloids and sugar mimics, with a nitrogen in the place of the endocyclic oxygen. These naturally occurring compounds are of pharmaceutical importance because they interact with and inhibit carbohydrate processing enzymes, and because of their beneficial drug-like properties.[1] Conventional synthesis of iminosugars from readily available carbohydrates is challenging mainly due to the presence of multiple hydroxyl groups. A key strategy in carbohydrate synthesis has been complex protecting group manipulations in order to perform regioselective functional group manipulations.[2] Inspired by the reported biosynthetic gene cluster for transformation of fructose-6-phosphate into an iminosugar scaffold,[3] we present a biomimetic sequential, three step chemo-enzymatic cascade, whereby minimally protected monosaccharides undergo transamination, selective oxidation, and reduction, via transaminase, oxidoreductase, and catalytic hydrogenation steps, respectively.[4-6] |
| References: | [1] Nash, R. J., Kato, A., Yu, C. Y. and Fleet, G. W., Future Med. Chem., 2011, 3, 1513-1521 [2] Fraňová, P. and Marchalín, Š., Eur. J. Org. Chem., 2022, 35, e202200742 [3] Clark, L. F., Johnson, J. V. and Horenstein, N. A., ChemBioChem, 2011, 12, 2147-2150 [4] Cairns, R., Gomm, A., Ryan, J., Clarke, T., Kulcinskaja, E., Butler, K. and O’Reilly, E., ACS Catal., 2019, 9, 1220-1223 [5] Kuska, J., Taday, F., Yeow, K., Ryan J. and O’Reilly, E., Catal. Sci. Technol., 2021, 11, 4327-4331 [6] Yeow, K., Haarr, M. B., Muldoon, J., O’Reilly, E., Chem. Commun. 2022, 58, 58, 13640-13643 |
| Figures: | 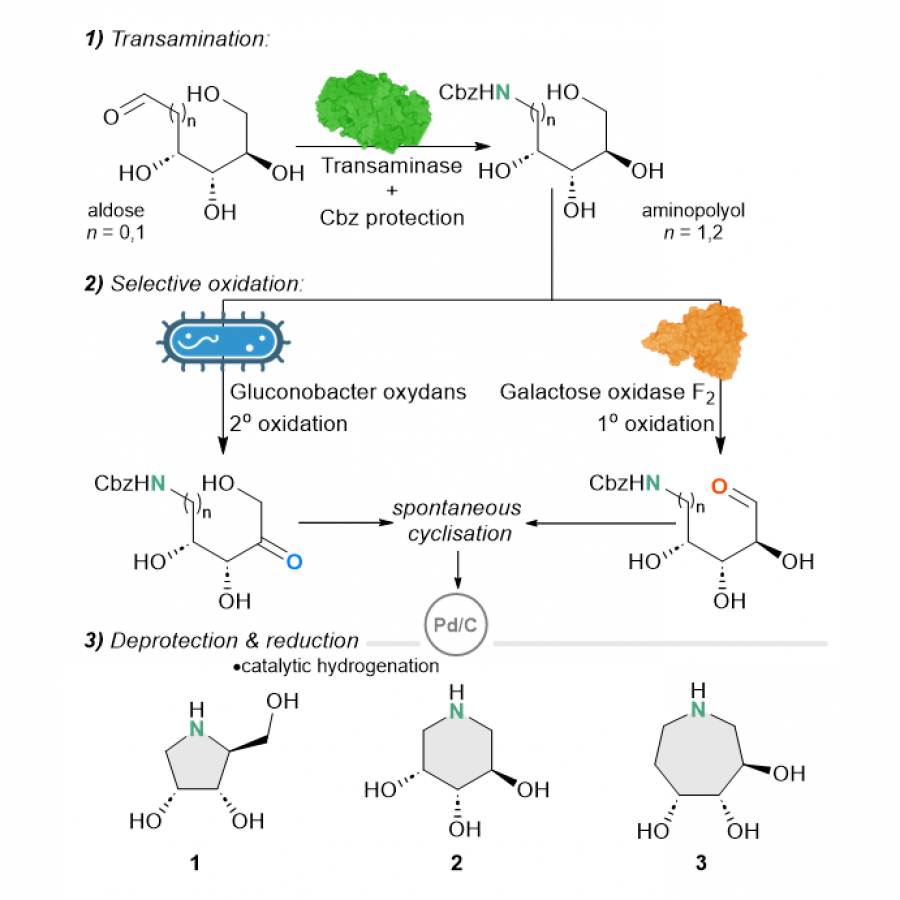 Chemoenzymatic synthesis of iminosugars from monosaccharides Iminosugars of different ring size are accessed from monosaccharides through a four step chemoenzymatic pathway. |
#1512 | The Terpene Mini Path: or how to identify and characterize new terpene synthases ? |
|
| Presenting author: | Agnès AMOURIC of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 |
| Corresponding author: | Katia DUQUESNE of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 |
| Other authors: | Letitia LEYDET of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 Arianna PELISSOU of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 Hugo STEPHANT of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 Gilles IACAZIO of INSTITUT DES SCIENCES MOLÉCULAIRES DE MARSEILLE - UMR7313 - ISM2 Katia DUQUESNE of KATIA.DUQUESNE@UNIV-AMU.FR |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | terpenes / mini-path / biocatalysis / biodiversity |
| Purpose: | Terpenes are very widespread molecules in the world. These natural compounds have a wide range of interests in the flavour industry, insecticides, food and medicine. Although a hundred thousand structures have been solved to date, the world of terpenes leaves a huge field of possibilities to be discovered. Access to terpenes by natural means remains a challenge. Thanks to the terpene mini-pathway (TMP) (see posters and pitch Leydet et al.), we offers a very efficient biotechnological alternative for their access. In only two enzymatic steps we access the universal precursors of all terpenes (IPP and DMAPP) [1]. Then we implemented this simplified pathway with i) a prenyl transferase (FPPS, GPPS, GGPPS) to convert the IPP and DMAPP into C10, C15 or C20 linear diphosphates and ii) a terpene synthase which catalyze the cyclization of these linear molecules into terpenes [2]. Fungi are prolific producers of sesquiterpene compounds with relevant activities. Thanks to a bioinformatic tool recently developed for an accurate identification of fungal sesquiterpene synthases, 18 putative genes have been annotated in the genome of the Polyporales Leiotrametes menziesii [3]. All of these genes have been cloned and overexpressed in order to test in vitro each purified enzyme with the implemented TMP. The characterization of their products by GC/MS in combination with NMR and by biological activities is in progress. Therefore, our biosynthetic pathway offers an easy method to synthesize different terpenes, to discover new terpene synthases, and to determine their substrate promiscuity. |
| References: | [1] J. Couillaud et al. Simplified in Vitro and in Vivo Bioaccess to Prenylated Compounds. ACS Omega 2019, 4, 7838-7849. [2] J. Couillaud et al. In vitro Applications of the Terpene Mini-Path 2.0. Chembiochem 2022, 23(24), e202200595. [3] H. Hage et al. An HMM approach expands the landscape of sesquiterpene cyclases across the kingdom Fungi. Microbial Genomics 2023, 9(4), 000990. |
| Figures: | 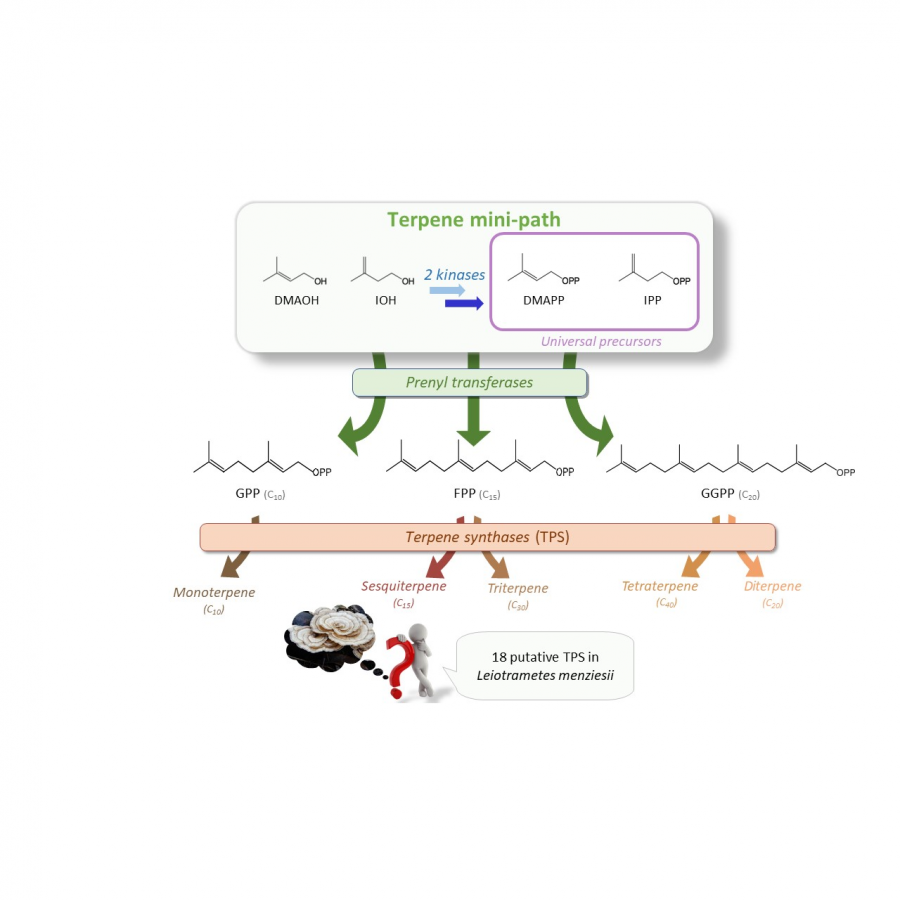 Discovery of the Leiotrametes menziesii sesquiterpene synthases diversity DMAOH : Dimethylallyl-alcohol , IOH : Isopentenol, DMAPP : Dimethylallyl diphosphate, IPP : Isopentenyl diphosphate, GPP : Geranyl diphosphate, FPP : Farnesyl diphosphate, GGPP : Geranylgeranyl diphosphate
|
#1515 | Photoenzymes: method development and applications of non-canonical amino acids as versatile tools in photobiocatalysis |
|
| Presenting author: | Marco SEIFERT of UNIVERSITY OF GREIFSWALD |
| Other authors: | Martin TERMATHE of UNIVERSITY OF GREIFSWALD Matthias HÖHNE of TECHNICAL UNIVERSITY OF BERLIN |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photobiocatalysis / Non-canonical amino acids / Aminoacyl-tRNA synthetase variants / Saturation mutagenesis |
| Purpose: | Enzymes play a fundamental role in catalysing a wide spectrum of biotransformations in living systems. Scientists have long harnessed this natural diversity in various fields and applications. However, natural enzymes often possess limitations like lack of stability or narrow substrate scope, which hamper their utilization in industrial processes. Directed evolution has emerged as a powerful tool to overcome such limitations, by replicating evolutionary processes on a lab scale. In recent years, a variety of methods, such as the development of Artificial Metalloenzymes (ArM), have emerged as innovative approaches to broaden the scope of enzymatic reactions and modulate enzyme activities. Among these techniques, non-canonical amino acids (ncAAs) have been utilized to expand the repertoire of the 20 canonical amino acids with new chemical and physical properties. Thus, ncAAs enable the development of novel enzymes with enhanced or entirely new-to-nature functionalities. This expansion of the genetic code relies on the use of an orthogonal tRNA/aminoacyl-tRNA synthetase (aaRS) pair to incorporate ncAAs at specific and defined positions, by recoding a stop codon. The resulting enzymes possess improved or unique functions, allowing them to catalyze abiological reactions or outperform their natural counterparts. Our research is focused on incorporating photosensitive ncAAs into protein scaffolds to establish a versatile platform for stereo- and enantioselective photobiocatalysis. By taking advantage of light as an abundant and cost-effective energy source, we aim to implement this technology as a sustainable and promising alternative to existing methods in photoredox catalysis. We have assessed the feasibility of our approach using the well-established ncAA Benzoyl-Phenylalanine (Bpa). Following its incorporation into various protein scaffolds, we evaluated their performance in a diverse range of reactions, including radical addition through hydrogen atom transfer (HAT) or decarboxylation and energy transfer reactions (EnT). However, this strategy posed significant challenges due to limited control over the generated radical species during the reactions, as well as the occurrence of undesired side reactions, such as crosslinking. To overcome these limitations, we are focusing on the development of novel ncAAs to expand the range of genetically encoded photosensitizers with improved characteristics, such as thioxanthone and anthraquinone moieties. Here, I will present our efforts on the development and strategies for engineering novel aaRS variants. This includes principles of GoldenGate cloning and different PCR techniques to generate large libraries. With this approach, we enable fast saturation mutagenesis of multiple residues in an aaRS, with the potential for general applicability in various protein scaffolds. In combination with different screening and selection methods, we are able to identify optimized variants in an efficient manner. By elucidating the underlying principles of ncAA incorporation and expanding the toolbox of genetically encoded photosensitizers, we aim to establish light-mediated enzymatic reactions in the field of biocatalysis. |
#1516 | Discovery of the biosynthetic enzymes for the fatty acid-rearranged natural products fischerazoles A-C from a cyanobacterium |
|
| Presenting author: | Kathleen ABT of ICBAS - SCHOOL OF MEDICINE AND BIOMEDICAL SCIENCES/ CIIMAR PORTUGAL |
| Other authors: | Teresa P. MARTINS of CIIMAR, PORTUGAL Sandra A.C. FIGUEIREDO of CIIMAR, PORTUGAL Iñaki LACOMBA of CIIMAR, PORTUGAL Pedro LEÃO of CIIMAR, PORTUGAL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | natural products / cyanobacteria / methyltransferase / P450 |
| Purpose: | Cyanobacteria are prolific producers of secondary metabolites and hold a large amount of unexplored biosynthetic gene clusters (BGCs) encoding enzymes with potential for biotechnological or pharmaceutical application. Using our recently developed metabolite discovery approach that is based on stable isotope-labeled fatty acids and comparative metabolomics, we uncovered novel compounds from the cyanobacterium Fischerella sp. PCC 9431. These new lipopeptides, fischerazoles A-C, had particularly interesting structural features such as extensive chlorination of the fatty acyl chain, a rare gem-dichlorovinylidene moiety and an unusual terminal methylated carboxamide. Most strikingly, they contained a pendant allyl alcohol which, according to our supplementation experiments with stable isotope-labeled precursors, derived from fatty acid rearrangement during the biosynthesis. By mining the genome of PCC 9431 for halogenases, we detected the corresponding BGC (fsh) with three halogenase homologs and a polyketide synthase (PKS)/ non-ribosomal peptide synthetase (NRPS) core. Additional biosynthetic enzymes included a fatty acyl-AMP ligase (FAAL) for initiation of the biosynthesis by fatty acid activation and loading to a carrier protein, a fatty acid desaturase, a SAM-methyltransferase and a cytochrome P450, likely involved in the functionalization of a mid-chain aliphatic carbon to create the pendant allyl alcohol moiety. Although the SAM methyltransferase did not cluster with any characterized enzymes in sequence similarity networks, bioinformatic analysis and in vitro experiments suggest that it creates a cyclopropane on a thioester-tethered palmitoleic acid substrate. We hypothesize that this cyclopropanyl fatty acid is then used as a substrate by the cytochrome P450 to perform a hydroxylation and rearrangement reaction. By uncovering structurally novel natural products, we here discovered several cyanobacterial biosynthetic enzymes that could ultimately expand the biocatalyst’s toolbox. |
#1520 | Developing chassis strains for the sustainable bioconversion of lignocellulose |
|
| Presenting author: | Amias ALSTROM-MOORE of NORTHUMBRIA UNIVERSITY |
| Other authors: | Sonia SANTOS of NORTHUMBRIA UNIVERSITY Warispreet SINGH of NORTHUMBRIA UNIVERSITY Paul JAMES of NORTHUMBRIA UNIVERSITY Jose MUÑOZ-MUÑOZ of NORTHUMBRIA UNIVERSITY Gary BLACK of NORTHUMBRIA UNIVERSITY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Unspecific peroxygenases / Glycoside hydrolases / Lytic polysaccharide monooxygenases / DyP-type peroxidases |
| Purpose: | Eighty million tons of waste lignocellulose biomass is produced each year, and it accounts for 33% of municipal waste worldwide. Therefore, utilising lignocellulose as an abundant and renewable feedstock is appealing. However, lignocellulose is composed of a complex mix of different carbohydrate-based polymers, (cellulose and hemicellulose; xyloglucan, xylan, mannan, and glucomannan) as well as the recalcitrant non-carbohydrate, alkyl-aromatic heteropolymer lignin (Figure 1). Synergistic action of multiple enzyme families, coordinating hydrolytic, non-hydrolytic and oxidative activities, is required for the degradation of these different polymers. Because of the large number of enzymes needed, it is exceedingly rare for any microorganisms to display the simultaneous ability to degrade cellulose, lignin, and hemicellulose. The environment contains organisms that have a rich repository of enzymes with diverse functions. Accordingly, we found a novel isolate cultivated directly from lignocellulose waste, with the ability to degrade cellulose and hemicellulose. This strain had remarkable amenability to genetic manipulation, as well as reasonable metabolic flexibility. We aimed to cultivate this strain into a bona fide chassis. This was achieved by upregulating native glycoside hydrolases, but also adding novel and well-studied enzymes from different sources, aimed at targeting the complex side chains associated with hemicellulose, namely Lytic polysaccharide monooxygenases, acetylxylan, and feruloyl esterases. We have achieved a significant improvement in the strains natural degradation efficiency and increased its substrate range. Furthermore, we have been bio-prospecting for new lignin-degrading enzymes, such as unspecific peroxygenases (UPOs) and DyP-type peroxidases, from meta-genomic databases. After the initial characterization of these enzymes is complete we plan to improve and express them in our host strain. |
| Figures: | 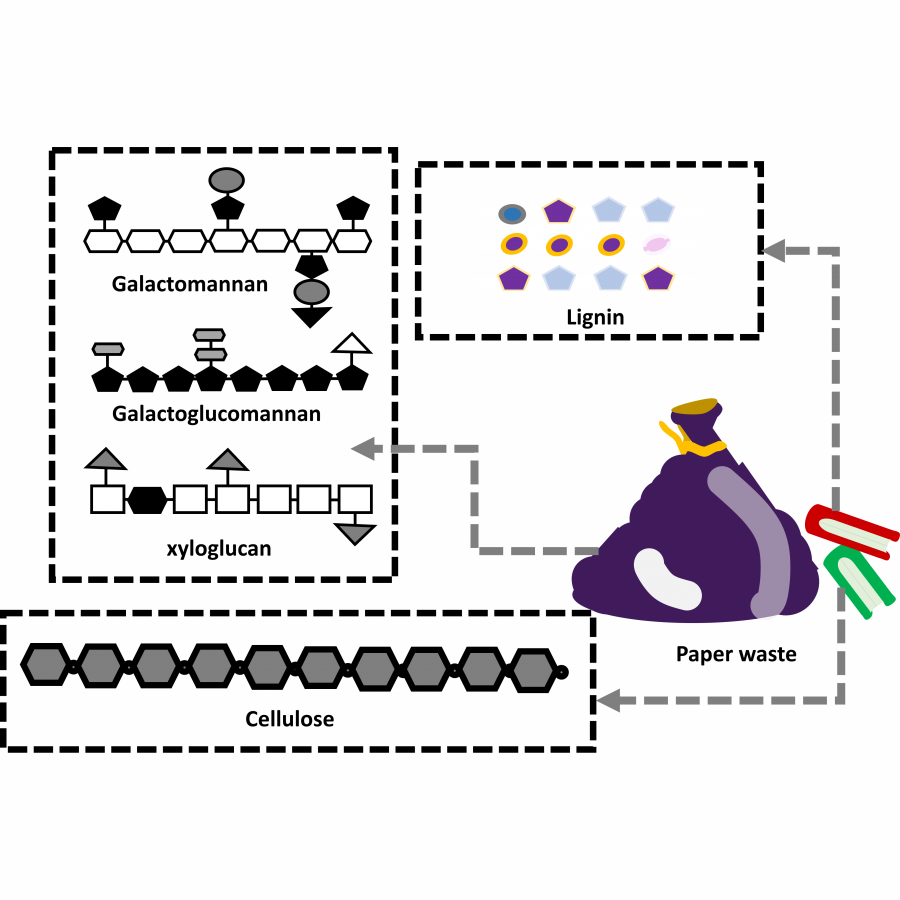 Figure 1 Representation of the complex mix of different polymers that constitute lignosellulosic waste |
#1521 | Extended Biocatalytic Halogenation Cascades Enabled by a Single-Polypeptide Regeneration System for Diffusible FADH2 |
|
| Presenting author: | Nicolai MONTUA of BIELEFELD UNIVERSITY |
| Other authors: | Norbert SEWALD of BIELEFELD UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | One-Pot cascade / Halogenase / Prodrug / Immobilization |
| Purpose: | In recent years flavin dependent halogenases (FHals) have increasingly emerged as attractive biocatalysts for the enzymatic introduction of halogen substituents to unactivated aromatic compounds, even at electronically unfavoured positions. Compared to conventional halogenation procedures, halogenases offer an environmentally friendly and highly regiospecific alternative, requiring only an aqueous buffer, molecular oxygen, and halide salt. While activity and stability of wild type FHals leave much to be desired[1], enzyme engineering and directed evolution campaigns are underway to improve these shortcomings. Tryptophan halogenases in particular have been thoroughly characterized and engineered regarding thermostability[2], regioselectivity[3], and substrate spectrum[4,5]. As most flavin dependent halogenases do not exhibit flavin reductase activity, their application as biocatalysts requires a two-component cascade for the regeneration of diffusible FADH2. Enzymatic cofactor regeneration cascades typically comprising a dehydrogenase enzyme and a separate flavin reductase are most commonly used. This requires multiple cultivations and purification procedures severely limiting scalability. Therefore, we constructed a bifunctional fusion protein for the regeneration of diffusible FADH2 from inexpensive phosphonate as a sacrificial substrate. This fusion protein proved amenable to coexpression with various flavin dependent halogenases and even allowed for the coexpression of an additional dioxygenase enabling the synthesis of L-4-Cl-Kynurenine, a promising prodrug candidate currently undergoing clinical evaluation, at preparative scale in a one-pot reaction directly from inexpensive L-Trp. |
| References: | [1] A. Phintha, K. Prakinee, A. Jaruwat, N. Lawan, S. Visitsatthawong, C. Kantiwiriyawanitch, W. Songsungthong, D. Trisrivirat, P. Chenprakhon, A. Mulholland, K.-H. van Pée, P. Chitnumsub, P. Chaiyen, Journal of Biological Chemistry 2021, 296, 100068. [2] H. Minges, C. Schnepel, D. Boettcher, M. S. Weiss, J. Sproß, U. T. Bornscheuer, N. Sewald, ChemCatChem 2020, 12. [3] A.-C. Moritzer, H. Minges, T. Prior, M. Frese, N. Sewald, H. H. Niemann, Journal of Biological Chemistry 2019, 294, 2529-2542. [4] B. Sana, T. Ho, S. Kannan, D. Ke, E. Li, J. Seayad, C. Verma, H. Duong, F. Ghadessy, ChemBioChem n.d., n/a, DOI 10.1002/cbic.202100210. [5] J. T. Payne, C. B. Poor, J. C. Lewis, Angewandte Chemie International Edition 2015, 54, 4226-4230. |
| Figures: | 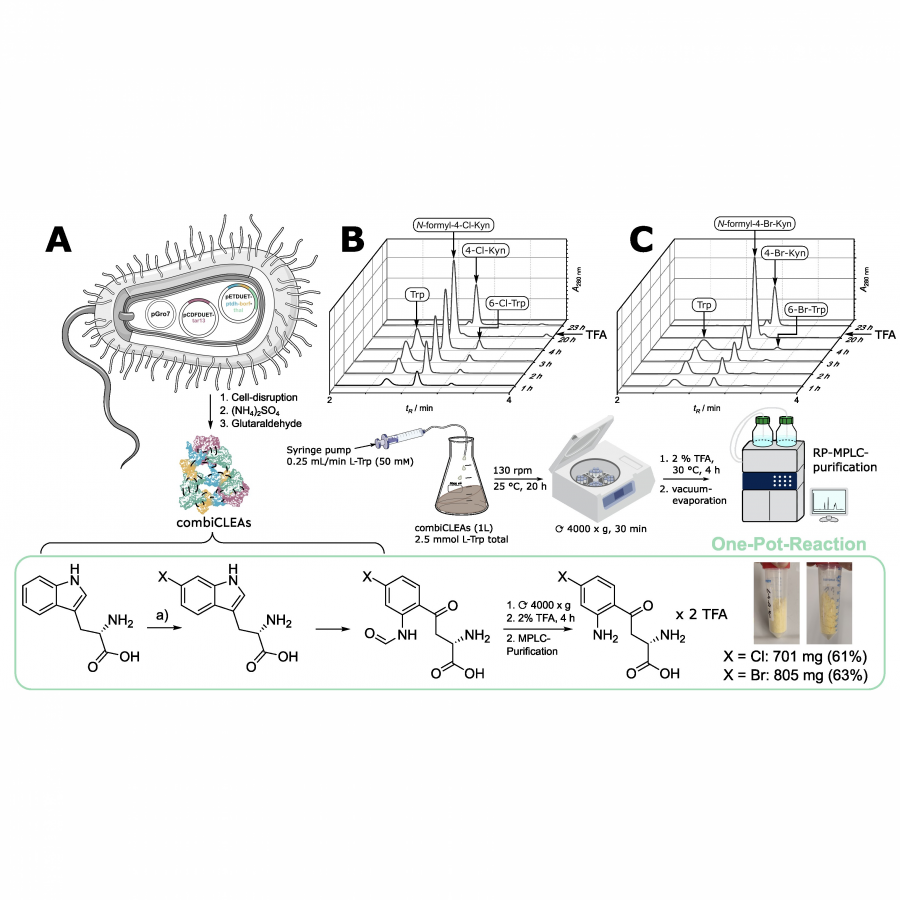 Figure 1 Direct one-pot synthesis of L-4-halo-kynurenine from inexprensive L-Tryptophan using combiCLEAs from lysates of an E. coli BL21 (DE3) derived coexpression strain halogenase and FADH2-regeneration cascade. |
#1524 | Expanding the enzyme universe: Exploration of novel scaffolds for artificial enzymes |
|
| Presenting author: | Alejandro GRAN-SCHEUCH of VRIJE UNIVERSITEIT AMSTERDAM |
| Corresponding author: | Ivana DRIENOVSKÁ of VRIJE UNIVERSITEIT AMSTERDAM |
| Other authors: | Stefanie HANREICH of VRIJE UNIVERSITEIT AMSTERDAM Elisa BONANDI of VRIJE UNIVERSITEIT AMSTERDAM |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Genetic Code Expansion / Non-canonical amino acids / Iminium catalysis |
| Purpose: | Biocatalysis is a sustainable alternative to environmentally damaging and energy-intensive chemical transformations.[1,2] The use of enzymes has become an attractive approach for the chemical and pharmaceutical industries because they can catalyze reactions with exquisite chemo-, regio- and enantioselectivity, along with fulfilling the requirements of green chemistry. However, biocatalytic strategies are often restricted to metabolic reactivities. Whereas, asymmetric chemical approaches, such as the use of organocatalysts; while allowing performing abiological reactions, tend to be less effective exhibiting relatively low turnover numbers.[3,4] These drawbacks can be addressed by combining both systems through the expansion of the genetic code. In the last 20 years, this methodology has emerged as a powerful strategy to incorporate non-canonical amino acids (ncAAs) with new-to-nature chemical groups into proteins (alloproteins/alloenzymes).[5,6] Inspired by well-studied organocatalysts,[3,4] we aimed to expand the repertoire of protein functional groups by incorporating ncAA-bearing secondary amines-based residues. In this work, we (re)designed a set of biomolecular scaffolds for the Michael addition of nitromethane into cinnamaldehyde. This is an abiological reactivity with relevance in the pharmaceutical industry for synthesizing valuable enantiopure γ-nitroaldehydes.[7] First, we proposed a panel of putative alloenzymes by inspecting their natural binding for cinnamaldehyde. Then, we defined a restricted amount of mutations for the amber stop codon (for the cellular incorporation of ncAA). The putative alloenzymes were expressed, purified and the in-house-ncAA cellualr incorporation was confirmed by LC-MS. Finally, the catalytic performance of the alloproteins was investigated upon the iminium catalysis model reaction (Michael addition of nitromethane to cinnamaldehyde). The in vivo incorporation of functional secondary amines showed a biotechnologically attractive potential, expanding the available toolbox for protein engineering and sustainable, abiological biocatalysis. |
| References: | [1] Intasian, P., et al. (2021) Chemical Reviews, 121(17): 10367-10451 [2] Heckmann, C. M., & Paradisi, F. (2020) ChemCatChem, 12(24): 6082-6102 [3] Buckley, B. R. (2009). Annual Reports Section B, 105, 113-128 [4] Bertelsen, S., & Jørgensen, K. A. (2009). Chemical Society Reviews, 38(8), 2178-2189 [5] Agostini, F., et al. (2017). Angewandte Chemie International Edition, 56(33), 9680-9703. [6] Drienovská, I., & Roelfes, G. (2020). Nature Catalysis, 3(3), 193-202. [7] Ordóñez, M. et. al. (2016). Tetrahedron: Asymmetry, 27, 999-1055 |
#1610 | Preparative implementation of in situ-product crystallization in semi-continuous amine transaminase-catalyzed reactions |
|
| Presenting author: | Jan VON LANGERMANN of OTTO-VON-GUERICKE UNIVERSITY MAGDEBURG |
| Other authors: | Sven TIEDEMANN of OTTO-VON-GUERICKE UNIVERSITY MAGDEBURG Feodor BELOV of OTTO-VON-GUERICKE UNIVERSITY MAGDEBURG Alina GAZIZOVA of UNIVERSITY OF ROSTOCK |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transaminase / amine / crystallization / in situ product removal |
| Purpose: | Chiral amines are valuable compounds for the synthesis of various synthetically interesting intermediates and APIs. Here transaminases in particular have established themselves as industrially relevant biocatalysts, which are available with both (R)- and (S)-selectivity. Aside chiral resolution the direct (asymmetric) synthesis of these compounds is often applied due to a theoretic maximum yield of 100%. Such a complete conversion is theoretically possible, but often prevented by an unfavorable reaction equilibrium or other limitations that must be overcome with secondary (bio)chemical reactions, yielding further waste and additional effort during downstream-processing.[1] As a potent alternative we present the preparative implementation of the previously developed integrated use of selective crystallization, which allow an apparent shift of the reaction equilibrium to the product side.[2] The product amine is herein (semi-)continuously removed from the unfavorable biocatalytic reaction equilibrium in the form of a salt, which is precipitated directly from solution (Figure 1). The concept was shown primarily at (S)-3-methoxy-1-phenylethylamine with >1.2 mol/l, which is a valuable intermediate for the synthesis of rivastigmine.[3] The concept was subsequently expanded successfully to a wider range of chiral amines and charcterized regarding choice of counterion, solid phase behavior and synthetic applicability.[3] |
| References: | [1] a) Guo, F.; Berglund, P., Green Chem. 2017, 19, 333-360.; b) Slabu, I.; Galman, J.L., Llyoyd, R.C.; Turner, N., ACS Catal. 2017, 7, 8263-8284. [2] a) Hülsewede, D.; Tänzler, M.; Süss, P.; Mildner, A.; Menyes, U.; von Langermann, J., Eur. J. Org. Chem. 2018, 2018 (18), 2130-2133.; b) Hülsewede, D.; Dohm, J.-N.; von Langermann, J., Adv. Synth. & Catal. 2019, 361, 2727-2733 [3] a) Neuburger, J.; Helmholz, F.; Tiedemann, S.; Lehmann, P.; Süss, P.; Menyes, U.; von Langermann, J., Chem. Eng. Process. Process Intensif. 2021, 168, 108578., b) Tiedemann, S.; Neuburger, J.; Gazizova, A.; von Langermann, J. manuscript in preparation |
| Figures: |  Figure 1 Figure 1: Representation of the integrated selective crystallization of the product (salt) in transaminase-catalyzed reactions |
#1611 | In search of complementary extraction methods for comprehensive coverage of metabolites in Escherichia coli |
|
| Presenting author: | Henry GOULD of NORTHUMBRIA UNIVERSITY |
| Other authors: | William CHEUNG of NORTHUMBRIA UNIVERSITY James FINNIGAN of PROZOMIX LTD Jose MUÑEZ-MUÑEZ of NORTHUMBRIA UNIVERSITY Simon CHARNOCK of PROZOMIX LTD Gary BLACK of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme production / Metabolites / / |
| Purpose: | Escherichia coli is an invaluable research tool for many fields of biology, in particular for its use in expressing recombinant proteins for industrial processes [1]. However, the activity of many such enzymes cannot be determined using standard biochemical assays, as often the relevant substrates are not known, or the products produced are not detectable. Today, the biochemical footprints of such unknown enzyme activities can be revealed via analysis of the metabolomes of the recombinant E. coli clones employed, using sensitive technologies such as mass spectrometry [2]. Before any valuable metabolites can be identified and produced at large scale, it is necessary to achieve as high a coverage of the potential metabolites present within E. coli as possible. Current methods for metabolomics usually involve 1 extraction method and attempting to maximise its efficiency of metabolite recovery [3], [4, 5]. Very commonly these methods also involve a drying step to concentrate the metabolites for better detection, but this removes a large subset of metabolites (see Figure 1). We have therefore analysed a wide range of different extraction methods against the cell free extracts of various recombinant E. coli clones. The results were analysed to determine the minimum number of extractions that achieved high recovery and coverage of metabolites, thus maximising the chance of pinpointing novel metabolites. This revealed that two extraction methods produce significant differences in the metabolomes of the 5 recombinant E. coli clones analysed. These two methods were chosen due to their ability to produce not only high numbers of ions, but wide mass coverage and a high degree of complementarity (Figure 2). One extraction method uses methanol and water, in a 4:1 ratio, which was then dried down and reconstituted in the chromatography running buffer and the other extraction method uses a combination of methanol, water, and chloroform, in a 3:1:1 ratio, that was injected directly onto the chromatography column. |
| References: | 1. Baneyx, F., Recombinant protein expression in Escherichia coli. Current Opinion in Biotechnology, 1999. 10(5): p. 411-421. 2. Gareth A Prosser, G.L.-M., and Luiz Pedro S de Carvalho, Metabolomic strategies for the identification of new enzyme functions and metabolic pathways. EMBO Reports, 2014. 15: p. 657-669. 3. Andresen, C., et al., Comparison of extraction methods for intracellular metabolomics of human tissues. Frontiers in Molecular Biosciences, 2022. 9. 4. Liu, R., et al., Evaluation of two-step liquid-liquid extraction protocol for untargeted metabolic profiling of serum samples to achieve broader metabolome coverage by UPLC-Q-TOF-MS. Analytica Chimica Acta, 2018. 1035: p. 96-107. 5. Prasannan, C.B., et al., An improved method for extraction of polar and charged metabolites from cyanobacteria. PLOS ONE, 2018. 13(10): p. e0204273. |
| Figures: |  Figure 1 (a) Chromatogram for sample extracted using Methanol:Chloroform:Water extraction solvent (no drying step) (red) and Methanol:Water extraction solvent (with a drying step and reconstitution) (Black). (b) Chromatogram for sample extracted using Methanol:Chl  Figure 2 Heat maps showing metabolite abundance for (a) positive mode and (b) negative mode chromatography. For positive mode, 211 features were identified out of 2580 MS2 features < 25% RSD, for negative mode 151 features were identified out of 1864 MS2 features |
#1615 | Novel carbohydrate-active enzymes for improved detergent sustainability |
|
| Presenting author: | Megan GREY of NORTHUMBRIA UNIVERSITY |
| Other authors: | Lily THOMPSON of PROCTER & GAMBLE Nicola BROWN of NORTHUMBRIA UNIVERSITY Hamish YAU of PROTER & GAMBLE Jose MUÑOZ-MUÑOZ of NORTHUMBRIA UNIVERSITY Neil LANT of PROCTER & GAMBLE Gary BLACK of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Carbohydrate-active enzymes / Sustainability / / |
| Purpose: | There is an increasing interest in using enzymes in detergents, attributable to the necessity to discover more sustainable methods for cleaning and breaking down soils. Enzymes are naturally occurring biocatalysts which catalyse reactions with a high rate and efficiency and are better than non-biodegradable surfactants which the industry is striving to phase out. This is largely attributable to the ability of enzymes to catalyze reactions with a higher rate, precise specificity, and greater efficiency [1,3]. Eco-friendly washing conditions are of significant importance to the detergent industry, with many households decreasing temperature used during laundry cycles, this often leads to inefficient stain removal, enzyme technology can aid in overcoming this issue and removing remaining soils whilst encouraging the reduction in washing temperatures and cycle length without compromising cleaning [4]. Enzymes utilized in detergent formulations also bypass the necessity for the use of harsh chemicals and surfactants which are released into the environment and pollute ecosystems [1,2]. Current detergents are formulated with carbohydrate active enzymes (CAZymes) designed to improve cleaning by targeting a variety of distinct polysaccharide-based soil components, current enzymes used for this application include amylase, mannanase, licheninase and cellulase. Recent analysis of residual polysaccharide soils remaining on fabrics following washing cycles suggests potential for other enzyme classes to be used to improve cleaning and bring additional performance benefits by targeting these polysaccharides. This could help pave the route for washing in more environmentally friendly colder and quicker washing conditions and improve the sustainability of detergents by replacing harsh petrochemicals. The aim of the project is to identify and produce novel CAZyme candidates designed to target remaining residual polysaccharides on fabrics, through metagenome mining and by studying the secreted enzymes of microorganisms known to degrade the target polysaccharides, including Bacteroides strains. Work involves collaboration with partners at Newcastle University to study interactions between the enzymes and polysaccharides using molecular probe enabled technology, and visits to Procter & Gamble’s Newcastle Innovation Centre to evaluate the performance of enzymes produced in various tests aimed to evaluate consumer benefits. |
| References: | 1. Al-Ghanayem, A.A. and Joseph, B. (2020) 'Current prospective in using cold-active enzymes as eco-friendly detergent additive', Applied Microbiology and Biotechnology, 104, pp. 2871-2882. Available at: https://doi.org/10.1007/s00253-020-10429-x 2. Dreja, M., Vockenroth, I., Plath, N., Schneider, C. and Martinez, E. (2013) ‘Formulation, Performance, and sustainability aspects of Liquid Laundry Detergents’ Tenside surfactants Detergents, 51(2), pp.108-112. Available at https://doi.org/10.3139/113.110289 3. Victorino da Silva Amatto, I. et al. (2021) “Enzyme Engineering and its industrial applications,” Biotechnology and Applied Biochemistry, 69(2), pp. 389-409. Available at: https://doi.org/10.1002/bab.2117. 4. Kirk, O., Borchert, T.V. and Fuglsang, C.C. (2002) “Industrial enzyme applications,” Current Opinion in Biotechnology, 13(4), pp. 345-351. Available at: https://doi.org/10.1016/s0958-1669(02)00328-2. |
#1616 | Tuning carbon nanomaterials as supports for hydrogenases and other biocatalysts |
|
| Presenting author: | Maya LANDIS of UNIVERSITY OF OXFORD |
| Other authors: | Nicole GROBERT of UNIVERSITY OF OXFORD Kylie VINCENT of UNIVERSITY OF OXFORD |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | The use of hydrogenase enzymes in biotechnology offers possibilities for cleaning up biocatalysis, replacing glucose or formate as reductants with the atom-efficient H2 gas. Biocatalytic hydrogenations represent the equivalent of hydrogenations catalysed by metals such as Pd/C, but without the need for precious metals. Hydrogenases have been employed on carbon supports, together with NAD+ reductase, for heterogeneous catalysis of NADH recycling,1,2 or in solution for recycling of reduced flavins3. This project aims to explore design principles for carbon nanomaterials which are suited to supporting biocatalysts either in batch or continuous flow – more broadly, providing opportunities for making biocatalysis more sustainable and environmentally friendly. Carbon materials are synthesized by aerosol-assisted or floating catalyst chemical vapour deposition and characterized by electron microscopy, x-ray photoelectron spectroscopy, and Raman spectroscopy. Various material qualities are achieved both by targeting diverse intrinsic structural properties and through heteroatom doping. Furthermore, electro-analytical and electro-synthetic methods are employed and the synthesized materials are utilised as heterogeneous supports for redox enzymes. This poster describes new work at the interface between Biocatalysis and Materials Science which will expand the biocatalytic toolbox towards new reactivities, enabling greener chemical synthesis. |
| References: | 1. Reeve, H.A.; Lauterbach, L.; Lenz, O.; Vincent, K.A. Enzyme-Modified Particles for Selective Biocatalytic Hydrogenation by Hydrogen-Driven NADH Recycling. ChemCatChem, 2015, 7, 3480-3487. 2. Zor, C.; Quinson, J.; Thompson, L.A.; Lonsdale, T.H.; Dillon, F.; Grobert, N.; Vincent, K.A. H2-driven Biocatalytic Hydrogenation in Continuous Flow using Enzyme-Modified Carbon Nanotube Columns. Chem. Commun., 2017, 53,9839-9841. 3. Joseph Srinivasan, S.; Cleary, S.E.; Ramirez, M.; Reeve, H.A.; Paul, C.; Vincent, K.A. E. coli Nickel-Iron Hydrogenase 1 Catalyses Non-native Reduction of Flavins: Demonstration for Alkene Hydrogenation by Old Yellow Enzyme Ene-reductases. Ange. Chemie, 2021, 60, 13824-13828 |
#1620 | Expanding the Scope of Transaminase Triggered aza-Michael Chemistry for the Synthesis of High Value Targets |
|
| Presenting author: | Aoife MARTIN of UNIVERSITY COLLEGE DUBLIN |
| Other authors: | Elaine O'REILLY of UNIVERSITY COLLEGE DUBLIN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Transaminase / Enzyme-triggered reaction / Pyrrolidines / Chemoenzymatic |
| Purpose: | The pyrrolidine scaffold is commonly found in natural products,[1] synthetic drugs,[2] and organocatalysts.[1] We propose a transaminase (TA) triggered intramolecular aza-Michael reaction (IMAMR), where a simple prochiral ketoenone undergoes regio- and stereo-selective amination to form a chiral secondary amine that spontaneously cyclises, affording disubstituted pyrrolidines (Figure 1). This work expands the scope of the biocatalytic intramolecular aza-Michael chemistry that has been previously reported by our group to synthesise 2,6-disubstituted piperidines[3]and cyclic β-enaminones.[4] The synthetic route to access the ketoenone substrates has been developed by employing a tandem ozonolysis-Wittig reaction (25 – 49% yields). The small ketoenone panel was converted to their corresponding chiral pyrrolidine products using a commercially available TA (10 – 57% yields). While the transamination reactions are highly selective, the spontaneous IMAMR results in the formation of diastereoisomers that were isolated as inseparable mixtures. Further chemoenzymatic steps are being investigated to isolate an enantio-enriched pyrrolidine product after the transamination reaction. |
| References: | [1.] G. Li Petri, M. V. Raimondi, V. Spanò, R. Holl, P. Barraja and A. Montalbano, Topics in Current Chemistry, 2021, 379. [2.] E. Vitaku, D. T. Smith and J. T. Njardarson, Journal of Medicinal Chemistry, 2014, 57, 10257-10274. [3.] J. Ryan, M. Siauciulis, A. Gomm, B. Macia, E. O'Reilly and V. Caprio, Journal of the American Chemical Society, 2016, 138, 15798-15800. [4.] F. Taday, J. Ryan, S. P. Argent, V. Caprio, B. Maciá and E. O'Reilly, Chemistry - A European Journal, 2020, 26, 3729-3732 |
| Figures: |  Figure 1. The ω-transaminase triggered aza-Michael reaction for the formation of disubstituted pyrrolidines followed by a further chemoenzymatic step to isolate an enantio-enriched product. |
#1621 | Application of SuSy-GT cascade in the production of terpenoid glycosides |
|
| Presenting author: | Oktawia KORCZ of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Maciej SZWECHŁOWICZ of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Paulina CZYRNY of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Paulina PIWOWARCZYK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Agata MATERA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Tomasz JANEK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Jarosław POPŁOŃSKI of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cascade / terpenes / glycosylation / |
| Purpose: | The essential oils of thyme and oregano, members of the Liamiaceae family, contain bioactive isomers known as thymol and carvacrol [1]. These isomers have been found to possess powerful antibacterial properties against both gram-positive and gram-negative bacteria. Their mechanism of action involves destabilizing and disrupting the bacterial cell membrane, which leads to cell lysis and inhibits the activity of membrane pumps, including ATPases. This results in a reduction in bacterial motility and biofilm formation [2]. Animal studies have demonstrated the potential use of these monoterpene and its glycoconjugates as an alternative to antibiotics in animal production. Glucosylation reduces the volatility and pungent taste of the aglycone, making it easier to incorporate into animal feed. However, thymol is rapidly absorbed in the upper gastrointestinal tract, which means that it is challenging to achieve sufficient concentrations of the terpenes in the distal parts of the digestive tract. Additionally, glucosides alter the solubility of secondary plant metabolites, thereby modifying their biological activity. For glucosides to be effective, the active aglycone must be released at the target site by hydrolyzing enzymes [3]. Thymol and carvacrol glucosides are expensive, with a market price of > 1000 EUR per gram of compound. To overcome this challenge, this study aims to use a genetic cascade consisting of YjiC glucosyltransferase from Bacillus licheniformis and sucrose synthase from Glycine max to catalyze the glucosylation reactions of thymol and carvacrol in vitro. Furthermore, this method can reconstitute expensive nucleotide sugars as glucose donors during the reaction, allowing the desired glucosides to be obtained on a preparative scale. The project funded by the programme "The best of the best! 4.0" by the Polish Ministry of Education and Science, organised under the Operational Programme Knowledge - Education - Development co-financed by the European Social Fund POWER programme (No. MEiN/2022/DIR/3529). |
| References: | [1] S. Kokkini, R. Karousou, E. Hanlidou, T. Lanaras, Journal of Essential Oil Research, 2004, 16(4), 334-338. [2] K. Kachur, Z. Suntres, Critical reviews in food science and nutrition, 2020, 60(18), 3042-3053. [3] N. Van Noten, E. Van Liefferinge, J. Degroote, S. De Smet, T. Desmet, J. Michiels, ACS omega, 2020, 5(10), 5241-5248. |
#1622 | Novel enzymatic tools for lignin degradation |
|
| Presenting author: | Davide CARRARETTO of UNIVERSITY OF PAVIA |
| Corresponding author: | Nikola LONČAR of GECCO BIOTECH |
| Other authors: | Lur Alonso COTCHICO of ZYMVOL MARCO FRAAIJE of RUG Andrea MATTEVI of UNIVERSITY OF PAVIA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Green chemistry / Novel enzymes / Protein engineering |
| Purpose: | In Nature, the white-rot basidiomycetes are the most extensively studied natural lignin-degrading microorganisms [1]. The ligninolytic activity of these fungi has been mainly associated with the production of extracellular, non-specific and oxidative ligninolytic enzymes: laccases and high-redox peroxidases [2]. On the other hand, Peroxygenases are largely unexplored and may provide a rich source of new ligninolytic and auxiliary enzymes, given the breadth of different kind of enzymes that belongs to this class, and pathways that functionalise and/or degrade aromatic compounds [3]. Moreover, peroxygenases emerged as a promising group of enzymes that could replace P450s in many reactions; indeed, peroxygenases could step in instead of P450s for biocatalytic transformations and thus enable scale-up of such processes [4]. The utilization of P450s is often complicated due to multi-component systems. Even in the case of BM3-like P450s, the catalytically self-sufficient flavocytochrome fusion proteins, there is still a dependence on NAD(P)H and significant uncoupling rate, which further leads to the enzyme inactivation [5]. This work delves into the substrate scope exploration of the novel bacterial peroxygenases obtained through genome mining. A panel of novel enzymes is efficiently produced in E. coli with good yield (50-300 mg/L). Through screening a set of various substrates and employing available structural data and computational tools the aim is to provide kinetic, stability, spectroscopic, electrochemical, and structural fingerprints of new and engineered ligninolytic enzymes endowed with peroxygenase activities. The identification of the mechanistic and molecular features of wild-type and mutated enzymes will contribute to the construction of a unique toolbox of new ligninolytic enzymes as well as experimental tools for their characterization, which will have a key impact on their further biotechnological exploitation. |
| References: | [1] Biko, O., Viljoen-Bloom, M., & van Zyl, W. H. (2020). Microbial lignin peroxidases: Applications, production challenges and future perspectives. Enzyme and microbial technology, 141, 109669. [2] Martínez AT., Speranza M., Ruiz-Dueñas FJ., Ferreira P., Camarero S., Guillén F. (2005). Biodegradation of lignocellulosics: Microbiological, chemical and enzymatic aspects of fungal attack to lignin. Int Micro-biol, 8, 195-204. [3] Hofrichter M., Ullrich R. (2014) Oxidations catalyzed by fungal peroxygenases. Current Opinion in Chemical Biology, 19, 116-125, 1367-5931, [4] Hofrichter, M., Kellner, H., Pecyna, M.J., Ullrich, R. (2015). Fungal Unspecific Peroxygenases: Heme-Thiolate Proteins That Combine Peroxidase and Cytochrome P450 Properties. In: Hrycay, E., Bandiera, S. (eds) Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450. Advances in Experimental Medicine and Biology, vol 851. Springer, Cham. [5] Thistlethwaite, S., Jeffreys, L. N., Girvan, H. M., McLean, K. J., & Munro, A. W. (2021). A Promiscuous Bacterial P450: The Unparalleled Diversity of BM3 in Pharmaceutical Metabolism. International journal of molecular sciences, 22(21), 11380. |
#1623 | Development of a biological chassis for amine synthesis |
|
| Presenting author: | Josemarco MENDOZA AVILA of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV EVRY, UNIVERSITÉ PARIS-SACLAY |
| Other authors: | Volker DÖRING of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV EVRY, UNIVERSITÉ PARIS-SACLAY Madeleine BOUZON of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV EVRY, UNIVERSITÉ PARIS-SACLAY Louis MOUTERDE of URD AGRO-BIOTECHNOLOGIES INDUSTRIELLES Tanja KNAUS of VAN’T HOFF INSTITUTE FOR MOLECULAR SCIENCES, HIMS-BIOCAT, UNIVERSITY OF AMSTERDAM Francesco G. MUTTI of VAN’T HOFF INSTITUTE FOR MOLECULAR SCIENCES, HIMS-BIOCAT, UNIVERSITY OF AMSTERDAM Carine VERGNE-VAXELAIRE of GÉNOMIQUE MÉTABOLIQUE, GENOSCOPE, INSTITUT FRANÇOIS JACOB, CEA, CNRS, UNIV EVRY, UNIVERSITÉ PARIS-SACLAY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Whole-cell biocatalysis / Amine toxicity / Adaptive evolution / |
| Purpose: | Chiral amines are building blocks for various pharmaceuticals and fine chemicals. Biocatalysis using amine dehydrogenases (AmDHs) can enable the direct synthesis of chiral primary amines from prochiral ketones using ammonia as inexpensive amine donor while generating water as sole by-product (Ducrot et al., 2000; Mutti and Knaus, 2021). These AmDHs are NAD(P)H-dependent enzymes that perform a reductive amination, and can be utilized in the form of purified formenzymes, cell-free extracts, or whole-cells (Houwman et al., 2019). The latter are advantageous in terms of increased enzyme stability, low preparation cost, and direct applicability. However, toxicity of substrates, intermediates, and/or products is, among others, a potential drawback when living cells are used. We therefore aimed at improving the biocatalytic potential of E. coli strains expressing oxidoreductase enzymes for sustainable chiral amine production. In this context, we adapted E. coli BL21 (DE3) cells to grow in the presence of high amine concentrations via adaptive directed evolution using the GM3 technology for automated continuous culture (Mutzel and Mutzel, 2000). The evolved strains displayed up to a five-fold increase in amine tolerance compared to the wild-type strain. Co-expression of genes encoding for AmDH and formate dehydrogenase (FDH) activity in the tested strains, enabled the stereoselective bioamination (ee >99%) of a set of prochiral methyl ketones without any exogenous addition of NAD(P)H. In fact, using a resting cell setup, the adapted cells exhibited superior biocatalytic performance contrasted to the non-adapted ones. Notably, with the adapted cells, some of the tested substrates were converted to the corresponding amine with approximately 80% conversion at high substrate loading (i.e., 200 mM). These results were attributed to an increased survivability of the adapted cells compared to the non-adapted cells during the biotransformation reaction. Future work aims at obtaining a chassis of production by continuing to improve the strains and analyzing the adaptation mechanisms. In conclusion, our E. coli resting cell system represents an important advance towards the development of more robust biocatalysis for amine production with excellent chemical and optical purity in a frame of sustainable chemistry. |
| References: | Ducrot, L.; Bennett, M.; Grogan, G. and Vergne-Vaxelaire, C. et al., Adv. Synth. Catal. 2020, 363, 328-351. Houwman, J. A.; Knaus, T.; Costa, M. and Mutti, F. G., Green Chem., 2019, 21, 3846-3857. Mutti, F. G. and Knaus, T., Biocatalysis for Practitioners, 2021, Eds. Lavandera, I. and De Gonzalo, G., Wiley. Mutzel, R. and Mutzel, P., Patent WO2000034433 A1, 2000. |
#1624 | New sensor for industrial enzymatic catalysis detection |
|
| Presenting author: | Thomas YOUNG of CNRS |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | industrial biocatalysis / phytase / immobilized substrate / hydrolysis |
| Purpose: | The use of enzymes in the industry is in constant growth, reaching a global market worth 7 billion $ in 2022. Industry sectors using enzymes and their applications are numerous such as biofuel production, baking, feed additives or fruit juice treatment, to name only a few. Alongside an increasing use, the necessity of detecting and measuring enzymatic activity represents an important concern. Zymoptiq (https://zymoptiq.com/en/), a French start-up created in 2019, invented an innovative method for measuring hydrolase and lyase enzymatic activities [1]. This method relies on the immobilization of a substrate and its degradation when it is incubated in the presence of enzymatic activity. The immobilized substrate has to be specific of the characterized enzyme. These substrates are usually biopolymers, which can be immobilized using a cross linker. Among the most used enzymes in the industry, some however catalyse the hydrolysis of small molecules. Zymoptiq’s measurement method was not initially designed for the analysis of such enzymes, the limiting step being the substrate immobilization, which usually requires a polymer to work properly. Phytase is one of the most commonly used enzymes in the animal feed industry. With a market size valued at 570 million $ in 2022, it dominates this industry. Phytase is responsible for the step-wise hydrolysis of ester phosphate bonds of phytic acid, leading to the release of inorganic phosphate. It is added in the feed of monogastric animals, which do not express sufficient phytase activity. Besides being the main source of inorganic phosphate, which allows bone growth, in plants (70-80%), phytic acid also acts as an anti-nutrient component by chelating other metallic cations essential to animal growth and can lead to water eutrophication, making its hydrolysis by phytase important for the animal and the environment [2]. This poster will focus on the comprehension of the physico-chemical phenomena allowing the detection of phytase activity based on the progressive destabilization of a spherical shaped deposit (fig 1B) of a suitable substrate. Such structures, which break down when incubated in the presence of a growing phytase activity, are shown in figure 1. This study addresses substrate availability when trapped in a microstructure through a kinetic study using free or trapped phytic acid, we will propose a degradation mechanism of the used immobilized structure and the effect of its conformation on the degradation kinetics. |
| References: | [1] Senez V., Vlandas A., Procédé de détection optique, 2017, FR3067042A1 [2] Namita S., Advances in Animal Biotechnology and its Applications 2018, 269-327 |
| Figures: |  Figure 1 AFM profile (A) and microscope observation (B) of an immobilized substrate incubated in the presence of different phytase activities showing its progressive degradation. |
#1625 | Expanding the amine substrate scope of C-N lyases by homologue discovery |
|
| Presenting author: | LAURA BOTHOF of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | C-N lyases are enzymes that naturally catalyze the cleavage of carbon-nitrogen bonds to yield amines and α,β-unsaturated mono- or dicarboxylic acids. Their ability to reverse this reaction towards C-N bond formation can be a powerful tool for the synthesis of optically pure (un)natural amino acids, which are important synthetic precursors for pharmaceuticals and food additives [1]. The enzyme ethylenediamine-N,N’-disuccinic acid lyase (EDDS lyase), from Chelativorans sp. BNC1, naturally catalyzes a reversible two-step sequential addition of ethylenediamine to two molecules of fumarate, providing (S,S)-EDDS as the final product [2]. This enzyme shows a broad substrate scope, enabling the asymmetric addition of ammonia, various mono- and diamines [2], homo- and hetero cycloalkyl amines [3], arylamines [4,5], arylhydrazines [4], and arylalkylamines [6] to fumarate yielding the corresponding N-functionalized aspartic acids. By means of a one-pot, two-step chemoenzymatic approach, complex heterocycles like pyrazolidinones, dihydrobenzoxazinones and dihydroquinoxalinones could be obtained with high optical purities [4,5]. Furthermore, EDDS lyase can accept a wide variety of (non)natural amino acids with terminal amino groups, supporting the chemoenzymatic synthesis of the fungal natural products aspergillomarasmine A and various related aminocarboxylic acids [7]. Lastly, an engineered variant of EDDS lyase was applied for the enantioselective synthesis of several N-substituted aspartic acids, including precursors to the important dipeptide sweeteners neotame and advantame [8]. The very broad amine substrate scope makes EDDS lyase a promising template for homologue discovery to identify biocatalysts that can convert new unnatural substrates. Here, we used genome mining strategies to select various EDDS lyase homologues from bacteria and explored their biocatalytic applicability. The preliminary results show that some of the homologues are highly promiscuous and can utilize new substrates, expanding the range of C-N bond formation reactions that can be accessed. |
| References: | [1] M. Blaskovich; J. Med. Chem, 2016, 59, 10807-10836. [2] H. Poddar, J. de Villiers, J. Zhang, V. Puthan Veetil, H. Raj, A. Thunnissen, G. Poelarends; Biochemistry, 2018, 57, 3752-3763. [3] J. Zhang, H. Fu, P. Tepper, G. Poelarends; Adv. Synth. Catal. 2019, 361, 2433-2437. [4] H. Fu, A. Prats Luján, L. Bothof, J. Zhang, P. Tepper, G. Poelarends; ACS. Catal, 2019, 9, 7292-7299. [5] M. Bhat, A. Prats Luján, M. Saifuddin, G. Poelarends; ACS Catal. 2022, 12, 11421-11427 [6] M. Abidin, T. Saravanan, L. Bothof, P. Tepper, A. Thunnissen, G. Poelarends; Org. Biomol. Chem, 2021, 19, 6407-6411 [7] H. Fu, J. Zhang, M. Saifuddin, G. Cruiming, P. Tepper, G. Poelarends; Nat. Catal, 2018, 1, 186-191. [8] J. Zhang, E. Grandi, H. Fu, T. Saravanan, L. Bothof, P. Tepper, A. Thunnissen, G. Poelarends; Angew. Chem. 2019, 131, 2-9. |
#1626 | A Magic Piece of Unspecific Peroxigenases? - A Surface Alpha Helix as Key Part for Catalytic Specificity |
|
| Presenting author: | Carsten PICHLER of GRAZ UNIVERSITY OF TECHNOLOGY |
| Other authors: | Vanessa HOLZSCHUSTER of GRAZ UNIVERSITY OF TECHNOLOGY Anton GLIEDER of GRAZ UNIVERSITY OF TECHNOLOGY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | UPO / Pichia pastoris / surface / rational design |
| Purpose: | Unspecific peroxygenases (UPOs, EC 1.11.2.1) are heme thiolate enzymes categorized in two classes - long and short UPOs - that combine peroxidase (one electron transfer) and peroxygenase (two electron transfer) activity [1]. UPOs are capable of catalyzing oxyfunctionalization reactions using hydrogen peroxide as a co-substrate to activate C-H bonds [2] and beyond that they are naturally secreted and stable enzymes. These properties make them interesting for industrial applications, including e.g. the production of agrochemicals, insecticides, dye precursors or pharmaceuticals [3]. But until now these enzymes only have been found in Ascomycota, Basidiomycota and Oomycota [1][4]. Recently, several long and short UPO sequences were successfully expressed by Pichia pastoris and helped expanding the amount of recombinant expressed enzymes but still there is a huge interest to find new or even evolved variants with attractive activities [1][5]. In this study, two novel (Aspergillus brasiliensis, Podospora anserina) and one already described (Hypoxylon sp. E38) [6] short UPO were investigated by random as well as site-directed mutagenesis (SDM). There, a surface located alpha helix close to the heme entrance tunnel turned out to be of particular interest. Replacement of amino acids at certain positions within this alpha helix with polar or nonpolar residues, promote either activity on typical peroxidase (ABTS, 2-6-DMP) or peroxygenase (indole, naphthalene, NBD) substrates. Our results help to expand the repertoire of recombinant expressed UPOs, to clarify mechanistic differences between the two different catalytic reactions (peroxidase vs. peroxygenase), and to support the structure/function relationship in respect to these specific catalytic properties. |
| References: | [1] M. Hofrichter et al., Fungal Peroxygenases: A Phylogenetically Old Superfamily of Heme Enzymes with Promiscuity for Oxygen Transfer Reactions. 2020. [2] J. Martin-Diaz, P. Molina-Espeja, M. Hofrichter, F. Hollmann, and M. Alcalde, “Directed evolution of unspecific peroxygenase in organic solvents,” Biotechnol. Bioeng., vol. 118, no. 8, pp. 3002-3014, 2021. [3] P. Molina-Espeja et al., “Synthesis of 1-Naphthol by a Natural Peroxygenase Engineered by Directed Evolution,” ChemBioChem, vol. 17, no. 4, pp. 341-349, 2016. [4] J. Münch, N. Dietz, S. Barber-Zucker, S. J. Fleishman, and M. J. Weissenborn, “Functional Expression of Novel Unspecific Peroxygenases from Plant Pathogen Fungi Designed Directly From Sequences,” in NextGenBiocat 2023, 2023, p. 22. [5] K. Ebner, L. J. Pfeifenberger, C. Rinnofner, V. Schusterbauer, A. Glieder, and M. Winkler, “Discovery and Heterologous Expression of Unspecific Peroxygenases,” Catalysts, vol. 13, no. 1, 2023. [6] L. Rotilio et al., “Structural and Biochemical Studies Enlighten the Unspecific Peroxygenase from Hypoxylon sp. EC38 as an Efficient Oxidative Biocatalyst,” ACS Catal., vol. 11, no. 18, pp. 11511-11525, 2021. |
#1628 | Artificial metalloenzymes for sustainable chemical synthesis |
|
| Presenting author: | Amanda JARVIS of UNIVERSITY OF EDINBURGH |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Artificial metalloenzymes / Steriod Carrier Protein / late-transition metal catalysis / |
| Purpose: | Artificial metalloenzymes can be prepared using a range of different synthetic techniques from the supramolecular binding of a synthetic metal complex to the incorporation of metal binding amino acids directly in the protein backbone [1]. My group is interested in exploring these different routes to make ArMs based on carrier protein scaffolds. In this poster, I will highlight ongoing projects within the group aimed at making a range of biocatalysts with late-transition metals (Pd, Ir, Ru, Rh) for reactions including Pd-catalysed cross couplings, enantioselective transfer hydrogenation and hydroformylation. |
| References: | [1] Schwizer, F.; Okamoto, Y.; Heinisch, T.; Gu, Y.; Pellizzoni, M. M.; Lebrun, V.; Reuter, R.; Köhler, V.; Lewis, J. C.; Ward, T. R. Chem. Rev. 2018, 118, 142. |
| Figures: | 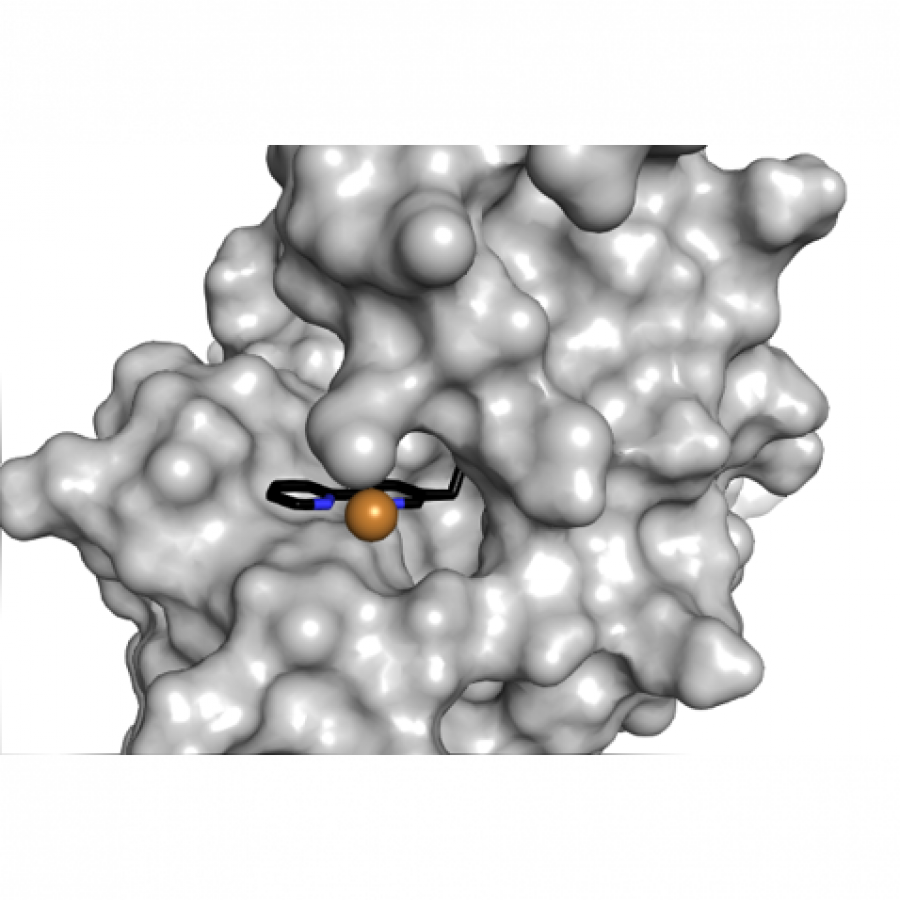 Figure 1 ArM formed from the Steriod Carrier protein including Bipyridylalanine. |
#1629 | Development of a coupled assay for the ultrahigh-throughput screening of 2'-deoxyribosyltransferases using droplets microfluidics |
|
| Presenting author: | Davide Agostino CECCHINI of CENTRO DE BIOLOGÍA MOLECULAR SEVERO OCHOA, UNIVERSIDAD AUTÓNOMA DE MADRID |
| Corresponding author: | Aurelio HIDALGO of CENTRO DE BIOLOGÍA MOLECULAR SEVERO OCHOA, UNIVERSIDAD AUTÓNOMA DE MADRID |
| Other authors: | Jesús FERNÁNDEZ-LUCAS of APPLIED BIOTECHNOLOGY GROUP, UNIVERSIDAD EUROPEA DE MADRID Aurelio HIDALGO of CENTRO DE BIOLOGÍA MOLECULAR SEVERO OCHOA, UNIVERSIDAD AUTÓNOMA DE MADRID |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | nucleosides / microfluidics / coupled assay / |
| Purpose: | Nucleoside analogues are an important class of compound with antiviral, anticancer, or antibacterial activity. Due to the growing environmental concern, nowadays, the enzymatic synthesis of these compounds is gaining ground over the traditional chemical methodologies in the pharma industry. In this contest, one of the enzymes that can be exploited for the synthesis of these non-natural compounds are 2'-deoxyribosyltransferases (NDTs) [1]. Although in recent years many NDTs have been described capable of synthesizing some of these nucleoside analogues, their catalytic efficiencies are far from those shown towards natural substrates, rendering their industrial application not economically attractive. In this regards, new enzymes with improved activities towards these non-natural substrates are needed. Nowadays, there are two main direct route that can be followed to identify candidate proteins with improved activities towards desired substrates: the screening of naturally occurring enzymes by functional metagenomics or the screening of man-made variants by directed evolution. In both cases, the success of screening campaigns relies on the fraction of genetic diversity that can be sampled and analysed. Microfluidics, allows the creation and use of picoliter water-in-oil (w/o) droplets as miniature compartments for enzyme assays that can be screened at ultrahigh-throughput (uHTP) resulting in a concomitant reduction of screening time (>107 per day), reagent consumption (50 μL per library) and costs (10€/library) [2]. In our study we present the development and optimization of a transferase/oxidase/peroxidase fluorogenic coupled assay to perform uHTP screening of 2’-deoxynucleotidyl transferases activity in droplets [3]. This work has received funding from the European Union under grant agreement 685474, 101081957 and the Spanish Ministry of Science and Innovation under grant PID2020-117025RB-I00. |
| References: | [1] Del Arco, J., Acosta, J., Fernández-Lucas, J., Biotechnol. Adv. 2021, 51, 107701. [2] Agresti, J.J., Antipov, E., Abate, A.R., Ahn, K., Rowat, A.C., Baret, J.C., Marquez, M., Klibanov, A.M., Griffiths, A.D., Weitz, D.A., Proc. Natl. Acad. Sci. U S A. 2010, 107, 4004-4009. [3] Cecchini, D.A., Sánchez-Costa, M., Orrego, A.H., Fernández-Lucas, J., Hidalgo, A., Methods. Mol. Biol. 2022, 2397, 19-32. |
#1630 | TOWARDS A TOOLBOX OF OXYDATIVE ENZYMES FOR THE BIOCONVERSION OF PHENOLIC AND ALCOHOLIC COMPOUNDS |
|
| Presenting author: | Teresa Benedetta GUERRIERE of UNIVERSITY OF PAVIA |
| Corresponding author: | Andrea MATTEVI of UNIVERSITY OF PAVIA |
| Other authors: | Marco FRAAIJE of RUG Maria Laura MASCOTTI of RUG |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Green chemistry / / |
| Purpose: | Lignocellulose is a renewable and widely available resource and it is considered one of the best candidates to produce biomaterials, biochemicals, and bioenergy [1]. Since lignocellulosic biomass is composed by cellulose, hemicellulose and lignin, it can be pretreated and fractionated into its main components which can be then converted into biobased products. In particular, one of the main components of the lignocellulose, lignin, is considered one of the largest renewable sources of aromatics on Earth. Several innovative technologies, like the Reductive Catalytic Fractionation (RCF), to extract lignin and convert it into value-added products [2]. Lignin is a biopolymer composed by three main building blocks (p-hydroxyphenyl, guaiacyl, and syringyl units) and the RCF affords the selective formation of these specific lignin monomers. This approach allows the valorization of softwood lignin into 4-n-propylguaiacol, the optimal substrate for enzymatic conversion to vanillin, and of hardwood lignin into syringaresinol, a potential biobased polycarbonate monomer [3]. The overall goal of the project is to generate a set of robust oxidative enzymes and protocols for the biocatalytic usage of aromatic products derived from lignin degradation. These enzymes can yield numerous important molecules that serve for example as pharmaceuticals, precursors for polymers or are employed in personal care products and cosmetics [4]. Specifically, since many enzymes of the VAO/PCMH family are considered valuable biocatalysts to perform the oxidation of phenolic and alcoholic substrates [5], the main challenge is to find some PCMH homologs of biocatalytic interest. p-Cresol methylhydroxylase (PCMH) is a flavocytochrome c found in the periplasm of several types of pseudomonas and it is responsible for the degradation of p-cresol and related phenols by catalyzing the oxidation of p-cresol to p-hydroxybenzyl alcohol [6]. Moreover, another important aim is to develop several promising candidates for the development of oxidative enzymes with boosted activity and stability that can cope with hard-to-modify aromatic compounds that result from lignin degradation. |
| References: | [1] Xiaojun S., Runcang S. (2021). Recent advances in lignocellulose prior-fractionation for biomaterials, biochemicals, and bioenergy; Carbohydrate Polymers, 261, 117884. [2] Van Aelst, K., Van Sinay, E., Vangeel, T., Cooreman, E., Van den Bossche, G., Renders, T.,Van Aelst, J., Van den Bosch, S., Sels, B. F. (2020). Reductive catalytic fractionation of pinewood: elucidating and quantifying the molecular structures in the lignin oil. Chemical science, 11(42), 11498-11508. [3] Galkin M. V., Samec J. S. (2016). Lignin valorization through catalytic lignocellulose fractionation: A fundamental platform for the future biorefinery; ChemSusChem, 9, 1544-1558. [4] De Jong, E., Van Berkel, W. J., Van der Zwan, R. P., De Bont, J. A. (1992). Purification and characterization of vanillyl-alcohol oxidase from Penicillium simplicissimum. A novel aromatic alcohol oxidase containing covalently bound FAD; European journal of biochemistry, 208(3), 651-657. [5] Jin, J., Mazon, H., Van den Heuvel, R. H., Janssen, D. B., Fraaije, M. W. (2007). Discovery of a eugenol oxidase from Rhodococcus sp. strain RHA1; The FEBS journal, 274(9), 2311-2321. [6] Louise M. Cunane, Zhi-Wei Chen, N. Shamala, F. S. Mathews, C. N. Cronin and W. S. McIntire (2000). Structures of the Flavocytochrome p-Cresol Methylhydroxylase and its Enzyme-Substrate Complex: Gated Substrate Entry and Proton Relays Support the Proposed Catalytic Mechanism;J. Mol. Biol. 295, 357±374. |
#1632 | Employment of Zymomonas mobilis as a cellulolytic microbial factory for bioethanol production |
|
| Presenting author: | Melissa POMA of NORTHUMBRIA UNIVERSITY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | bioethanol / cellulose biodegradation / Zymomonas mobilis / molecular toolbox |
| Purpose: | The industrialized world faces progressive depletion of its energetic resources, mainly based on non-renewable fuels.[1] As the world population and its energy demand increases, there is an urgent need to develop environmentally sustainable alternatives. In this context, the employment of microbial cell factories, powered by waste products, is an attractive means for large and sustainable biofuel production.[2] In the present project, we aim at utilizing cellulose waste as feedstock to produce bioethanol in the Gram-negative fermentative bacterium Zymomonas mobilis. Its natural ethanol production coupled to a high sugar uptake make Z. mobilis an appealing platform for a large-scale industrial application. To fully convert Z. mobilis into a cellulolytic microbial factory, it is required the production and secretion of three enzymes: exoglucanases, endoglucanases and β-glucosidases.[3] These enzymes are not present in Z. mobilis; hence, we have developed a tailor-made molecular toolbox to finely tune their heterologous expression. This includes a library of synthetic constitutive promoters and transcriptional terminators. Besides the production of these enzymes, it is fundamental to ensure their secretion extracellularly, where the cellulose biodegradation occurs. Presently, there is a lack of robust secretion tags for use in Z. mobilis and related organisms. To address this, we aim at combining gene knockouts and machine learning to identify which proteins are secreted through the native Type I secretion system and isolate their secretion tags. These can then be repurposed for the secretion of heterologous enzymes. |
| References: | 1. Utama, N. A., Fathoni, A. M., Kristianto, M. A. & McLellan, B. C. The End of Fossil Fuel Era: Supply-demand Measures through Energy Efficiency. Procedia Environ. Sci. 20, 40-45 (2014). 2. Yang, S. et al. Complete genome sequence and the expression pattern of plasmids of the model ethanologen Zymomonas mobilis ZM4 and its xylose-utilizing derivatives 8b and 2032. Biotechnol. Biofuels 11, 1-20 (2018). 3. Dvořák, P., Bayer, E. A. & De Lorenzo, V. Surface Display of Designer Protein Scaffolds on Genome-Reduced Strains of Pseudomonas putida. ACS Synth. Biol. 9, 2749-2764 (2020). |
#1633 | Bioinformatic analysis of immobilized enzymes |
|
| Presenting author: | David ROURA PADROSA of UNIVERSITY OF BERN |
| Other authors: | Francesca PARADISI of UNIVERSITY OF BERN |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme immobilization / Bioinformatics / Protein modelling / Biocatalysis |
| Purpose: | Protein bioinformatics has been applied to a myriad of opportunities in biocatalysis, from enzyme engineering to enzyme discovery, but its application in enzyme immobilization is still very limited. Enzyme immobilization brings clear advantages from the sustainability and cost-efficiency terms but is nowadays still limited in its implementation as it remains tied to a quasi-blind protocol of trial and error. Here, we present the use of a set of bioinformatic tools to rationalize results of protein immobilization that have been previously described. The study of these proteins through new bioinformatic tools, allows the discovery of key driving forces in the process of immobilization that explain the obtained results, moving us a step forward to the final goal: predictable enzyme immobilization protocols. |
| References: | [1] D. Roura Padrosa, V. Marchini, F. Paradisi, Bioinformatics 2021, 37, 2761-2762. [2] R. A. Sheldon, D. Brady, ChemSusChem 2022, 15, DOI 10.1002/cssc.202102628. [3] M. E. Hassan, Q. Yang, Z. Xiao, L. Liu, N. Wang, X. Cui, L. Yang, 3 Biotech 2019, 9, 1-16. |
| Figures: |  Graphical abstract Application of different analysis and bioinformatic tools to rationalize the process of enzyme immobilization. |
#1635 | Manipulation of redox partner protein expression levels in cytochrome P450 systems |
|
| Presenting author: | Kinga NYTKO of UCL & HYPHA DISCOVERY |
| Other authors: | Vincent POON of HYPHA DISCOVERY Jack JEFFRIES of UCL John WARD of UCL |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | P450 / redox partners / oxidation / polycistronic operon |
| Purpose: | Cytochrome P450 monooxygenases (P450s) are one of the most versatile biological catalysts involved in the oxidative transformation of endogenous and exogenous substrates. Mammalian P450s play a major role in the metabolism of xenobiotic compounds and bacterial P450 systems can mimic mammalian biotransformations and convert the drugs to the same metabolites. Microbial P450s are also objects of interest in biotechnology due to their extreme biodiversity and numbers. Hypha Discovery’s PolyCYPs® metabolite screening kit mainly contains recombinant microbial Class I P450 enzymes mined from talented filamentous bacteria called actinomycetes. PolyCYPs® enzymes can be used for the generation of P450s-derived metabolites of drugs, agrochemicals and other small molecules. The Class I microbial P450 systems contain three enzymes: a P450 and two redox partners – ferredoxin and ferredoxin reductase. The redox partners are essential for electron transfer from NAD(P)H to P450’s heme cofactor. However, the selection of suitable redox partners (native or surrogate) and their amount are known to be a bottleneck of Class I P450 activity and their usage in biotechnology. This poster describes efforts to enhance the activity of the 3-component system through modification of the expression level of the three enzymes. In this study, surrogate redox partners from Pseudomonas putida and PolyCYP-6 were used; these redox partners are co-expressed together with P450 within a single polycistronic operon under a T7 promoter. Six plasmids with different modifications within the operon, such as altering gene orders and changing RBS sequences, had a high impact on the production level of proteins and biotransformation activity. Systems with higher amounts of redox partners showed improved biotransformation activity against selected drug compounds. However, the molar ratio between ferredoxin and ferredoxin reductase also plays an important role in biotransformation activity. |
#1637 | Versatile platform for enzymatic polyphenol glucosylation – biocatalyst production and reaction optimisation |
|
| Presenting author: | AGATA MATERA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | KINGA DULAK of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES SANDRA SORDON of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES EWA HUSZCZA of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES JAROSŁAW POPŁOŃSKI of WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sucrose synthase / cascade reaction / flavonoids / expression optimisation |
| Purpose: | Glycosylation is a useful strategy for modifying the chemical and biological properties of natural products. Glycoconjugates exhibit enhanced bioavailability, stability, and water solubility, making them more desirable compounds for industrial applications [1]. As the costs and environmental concerns associated with chemical processes continue to rise, biocatalysis is gaining attention as a more sustainable alternative [2]. In nature, glycosylation processes are largely mediated by glycosyltransferases, a class of enzymes (EC 2.4) that transfer glycosyl moieties from activated (nucleotide of phosphorylated) sugars to nucleophilic glycosyl acceptors [3]. One well-established strategy for highly regio- and stereoselective glucosylation is coupling a nucleotide diphosphate (NDP)-dependent Leloir glycosyltransferase with sucrose synthase in a self-efficient enzymatic cascade [4-6]. With the inner regeneration system of expensive and low-available NDP-glucose, it makes enzymatic glucosylation more profitable and applicable in industrial settings. In the presented study, a collection of polyphenol glucosyltransferases from various sources (bacteria, fungi, and plants) were selected and heterologously co-expressed in Escherichia coli cells with the highly active soybean sucrose synthase (GmSuSy). The resulting library of biocatalysts enabled the stepwise optimization of enzyme production and evaluation of the influence of reaction conditions on enzyme specificity. The collected data allowed for the development of a standardized protocol for the production and application of glucosyltransferases from various sources. Furthermore, the vast substrate promiscuity of the selected biocatalysts created a comprehensive glucosylation platform. This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreements no. 814650 (SynBio4Flav) |
| References: | [1] Sheldon, R. A., Brady, D., Chemsuschem. 2022, 15:e202102628. [2] Mestrom, P., Kowalczykiewicz, P., Kumpf, M., Bento, J., Szymańska, C., Tischler, S., Hanefeld, H., Int. J. Mol. Sci. 2019, 20, 5263. [3] Lairson, L. L., Henrissat, B., Davies, G. J., Withers, S. G., Annu. Rev. Biochem. 2008, 77, 521-555. [4] Rupprath, C., Kopp, M., Hirtz, D., Müller, R., Elling, L., Adv. Synth. Catal. 2007, 349, 1489-1496. [5] Zhang, Y., Xu, S., Jin, Y., Dai, Y., Chen, Y., Wu, X., Sci. Rep. 2020, 10, 6230. [6] Liu, H., Tegl, G., Nidetzky, B., Adv. Synth. Catal. 2021, 363, 2157-2169. |
| Figures: | 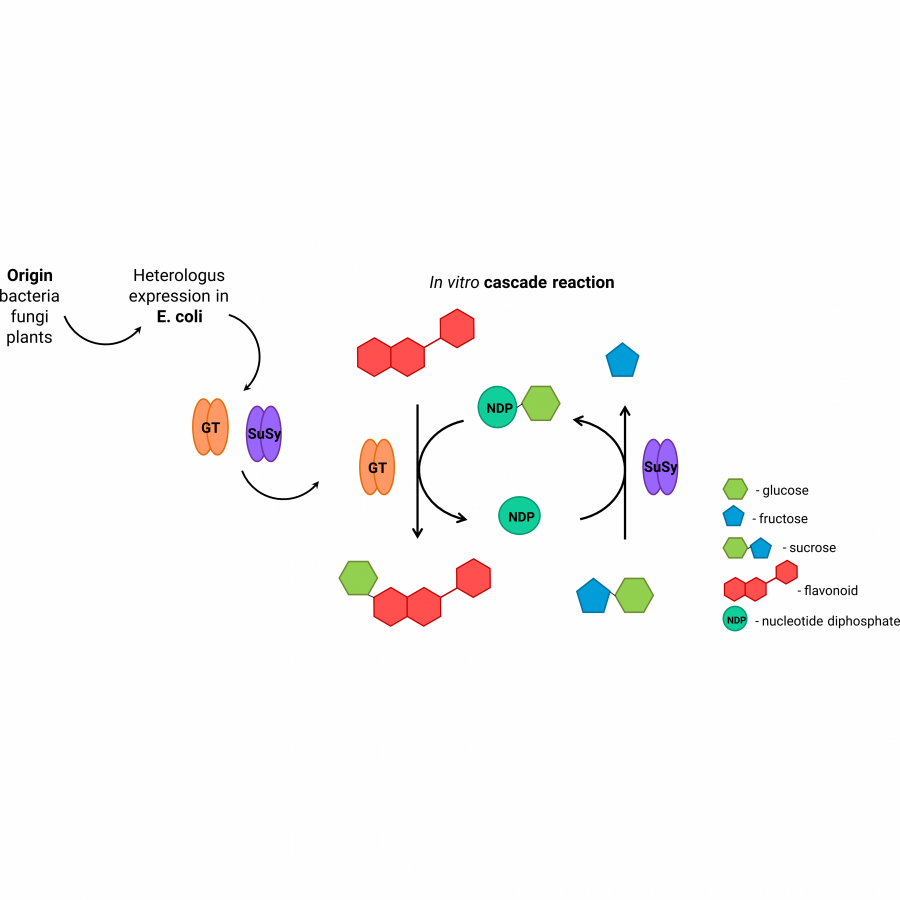 Schematic concept of the research GT - glycosyltransferase
SuSy - sucrose synthase |
#1640 | Reaction development via synthetic aminoacids in LmrR |
|
| Presenting author: | Franco DELLA FELICE of UNIVERSITY OF GRONINGEN |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / Artificial enzyme / Non-canonical aminoacid / new-to-nature reactions |
| Purpose: | Artificial metalloenzymes (ARMs), that is, designed enzymes containing a synthetic metal co-factor within a protein framework, has emerged as a promising area of research for the discovery and development of new synthetic transformations. Recently, the group of Roelfes have successfully exploited the use of the lactococcal multidrug resistant regulator (LmrR) as the host protein in a set of different type of reactions in a selective manner, i.e., Diels-Alder reaction, Friedel-Crafts alkylation and conjugate additions, by the introduction of both non-canonical aminoacids and synthetic transition metal complexes.[1] Intrigued in continuing expanding the synthetic utility of LmrR, here we will present our results on the incorporation via stop codon suppression methodology of synthetic aminoacids bearing a cyclic secondary amine to perform, i.e., enamine catalysis. |
| References: | [1] Roelfes, G. Acc. Chem. Res. 2019, 52, 545. |
| Figures: | 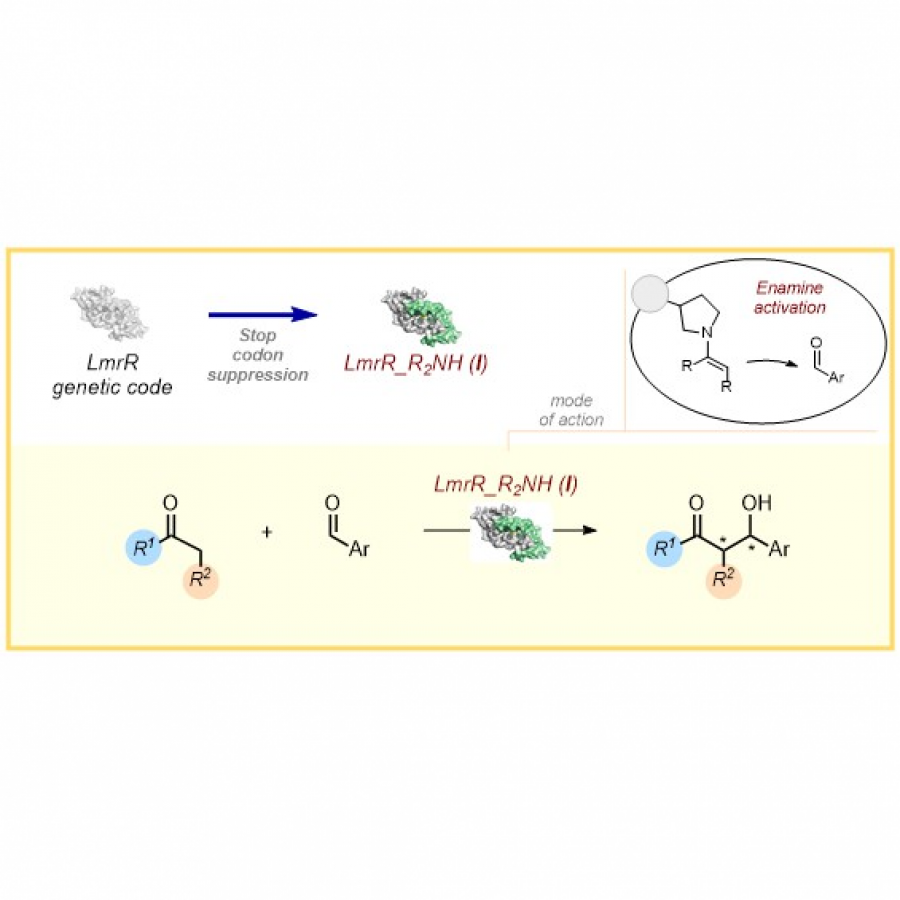 Scheme 1. Expression and validation of LmrR_R2NH (I) for enamine formation |
#1642 | One-Pot Deracemization of sec-Alcohols Combining Photocatalytic Oxidation and Biocatalytic Reduction |
|
| Presenting author: | Paweł BOROWIECKI of WARSAW UNIVERSITY OF TECHNOLOGY |
| Other authors: | Aleksandra RUDZKA of WARSAW UNIVERSITY OF TECHNOLOGY Natalia ANTOS of WARSAW UNIVERSITY OF TECHNOLOGY Tamara REITER of INSTITUTE OF CHEMISTRY Wolfgang KROUTIL of INSTITUTE OF CHEMISTRY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Photoredox catalysis / Biocatalysis / Chemo(enzymatic) cascades / Chiral alcohols |
| Purpose: | Chiral alcohols are versatile building blocks and have gained a particular interest in the asymmetric synthesis of non-racemic active pharmaceutical ingredients, agrochemicals, fragrances, flavors, natural products, etc. Herein we report on a practical and sustainable "one-pot" oxidation-reduction photo-biocatalytic process to synthesize enantiomerically enriched alcohols. In this regard, a relatively new ultra-efficient photocatalytic system, based on irradiation of the reaction mixtures with 440 nm blue LEDs in the presence of inexpensive 9-fluorenone as a metal-free photocatalyst and molecular oxygen as the terminal oxidant in dry DMSO as the solvent and hydrogen peroxide-neutralizing agent at once, was used to oxidize a broad range of (hetero)aromatic-aliphatic racemic alcohols into prochiral ketones quantitively (>99% conv.). The in situ formed carbonyl compounds were subsequently converted into corresponding chiral alcohols via a sequential biotranshydrogenation catalyzed by lyophilized E. coli cells overexpressing highly stereoselective and stereocomplementary recombinant short-chain alcohol dehydrogenases (ADHs) originated from Rhodococcus ruber (E. coli/ADH-A) or Rhodococcus erythropolis (E. coli/ReADH) to obtain (S)-alcohols and Lactobacillus kefir (E. coli/Lk-ADH) to obtain (R)-alcohols, respectively. Overall, the elaborated photo-biocatalytic deracemization of racemic alcohols using 9-fluorenone-O2-blue LED-DMSO-E. coli/ADH system carried out on a semi-preparative scale (0.25 mmol; 63 mM final conc. in 4 mL) yielded non-racemic alcohols with 82–99.9% conv., in up to 92% isolated yield, with high-to-excellent optical purity (97–99.9% ee), and complementary chirality. |
| Figures: | 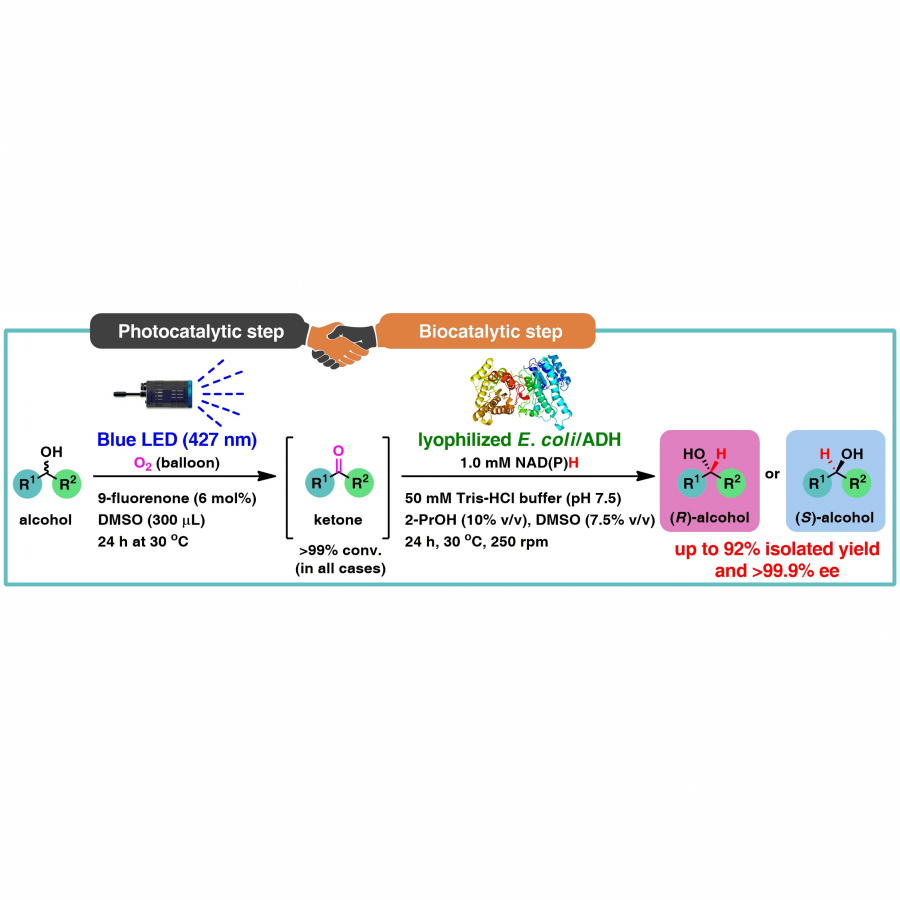 Figure 1. "One-pot" photo-biocatalytic deracemization of sec-alcohols using 9-fluorenone-O2-blue LED-E. coli/ADH system |
#1643 | Chemo-enzymatic Synthesis and Biological Activity Evaluation of Propenylbenzene Derivatives |
|
| Presenting author: | Dawid HERNIK of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Ewa SZCZEPAŃSKA of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Aleksandra WŁOCH of DEPARTMENT OF PHYSICS AND BIOPHYSICS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Hanna PRUCHNIK of DEPARTMENT OF PHYSICS AND BIOPHYSICS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Malwina MULARCZYK of DEPARTMENT OF EXPERIMENTAL BIOLOGY, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Krzysztof MARYCZ of DEPARTMENT OF EXPERIMENTAL BIOLOGY, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Teresa OLEJNICZAK of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Elisabetta BRENNA of DIPARTIMENTO DI CHIMICA, MATERIALI ED INGEGNERIA CHIMICA “GIULIO NATTA”, POLITECNICO DI MILANO Filip BORATYŃSKI of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, WROCLAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biotransformation / fragrances / oxidation / biological activity |
| Purpose: | Propenylbenzenes, such as isosafrole, anethol, isoeugenol, and their derivatives, are widely found in essential oils from plants such as the aniseed tree, liquorice, and the cananga tree [1]. Compounds of this group are economically important and are used in the flavour and fragrance industries as well as the pharmaceutical and cosmetic industries, and as intermediates in the synthesis of more complex products [2]. Propenylbenzenes have broad biological activities, such as antioxidant, antimicrobial, anti-inflammatory, and antiproliferative. In view of the different biological activities of propenylbenzenes and their widespread use in industry, it is worth looking for derivatives of these compounds [3]. As the interest in compounds obtained via environmentally friendly methods is currently growing, the necessity for the development of new methods for producing propenylbenzene derivatives such as diols and hydroxy ketones has increased in recent years. The aim of presented study was to develop an efficient process for synthesising oxygenated derivatives of these compounds and evaluate their potential biological activities. In this research, we propose a two-step biocatalytic method. The first step involves the chemo-enzymatic synthesis of corresponding diols 1b–5b from propenylbenzenes 1a–5a via enzymatic epoxidation followed by epoxide hydrolysis (Figure 1.). The second step involves the microbial oxidation of a diasteroisomeric mixture of diols 1b–5b to yield the corresponding hydroxy ketones 1c–4c, which in this study was carried out on a preparative scale using Dietzia sp. DSM44016, Rhodococcus erythropolis DSM44534, R. erythropolis PCM2150, and Rhodococcus ruber PCM2166. The propenylbenzene derivatives thus obtained and the starting compounds were tested for various biological activities, including antimicrobial, antioxidant, haemolytic, and anticancer activities, and their impact on membrane fluidity. The results indicate the potential utility of these compounds as fungistatics, antioxidants, and proliferation inhibitors of selected cell lines. |
| References: | [1] J. A. M. Lummiss, Journal of the American Chemical Society, 2012, 134, 18889-18891. [2] A. C. Aprotosoaie, Advances in Experimental Medicine and Biology, 2016, 926, 247-267. [3] F. Bruna, Arabian Journal of Chemistry, 2022, 15, 104271. |
| Figures: | 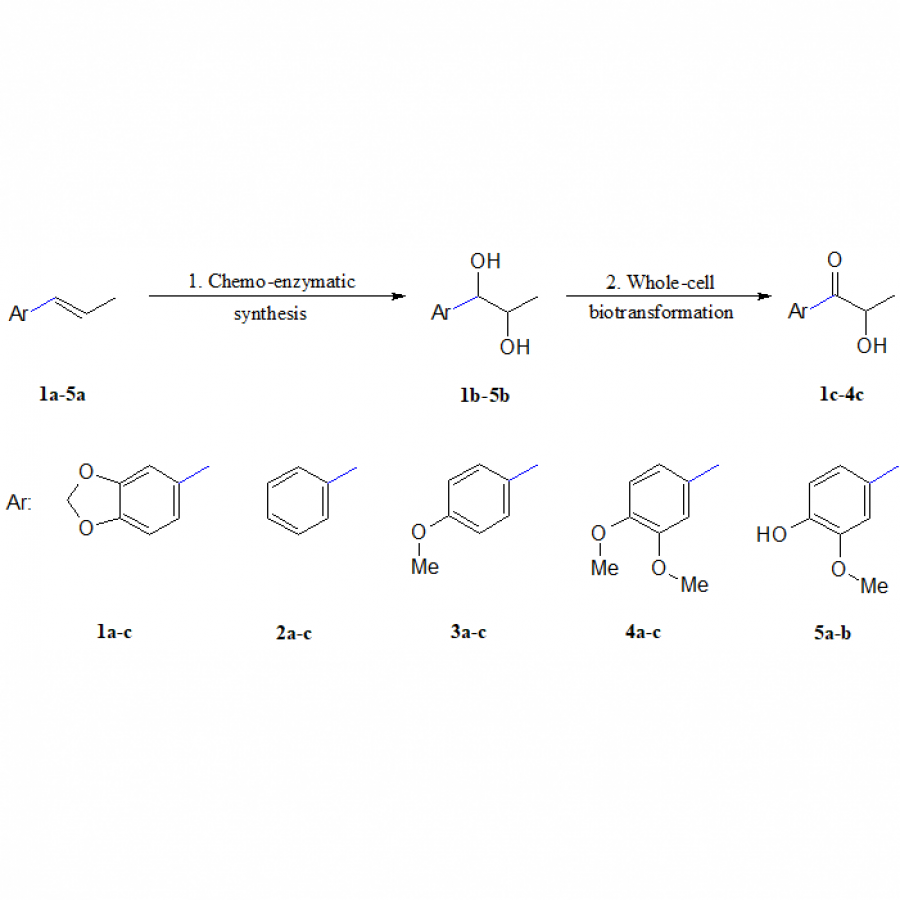 Figure 1. Two-step synthesis of propenylbenzene derivatives: diols 1b–5b and hydroxy ketones 1c–4c. 1. aq. H2O2, Novozym 435, EtOAc, 30 °C, 18 h, KOH, MeOH; 2. PCM medium, 23 °C, 3–11 days. |
#1644 | Enantiopure Sulfoxides via Cyclic Photobiocatalytic Deracemization |
|
| Presenting author: | Silvia GLUECK of ACIB GMBH |
| Other authors: | Sarah BIERBAUMER of UNIVERSITY OF GRAZ Christoph K. WINKLER of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cyclic deracemization / photobiocatalysis / sulfoxides / |
| Purpose: | The combination of biocatalysis and photocatalysis facilitates access to novel synthetic strategies which correspond very well to the requirements of more sustainable and resource-efficient synthesis concepts.[1] In here, we present a cyclic deracemization process combining an enantioselective enzyme-catalyzed sulfoxide reduction with an unselective photocatalyzed sulfide oxidation to yield the desired non-racemic sulfoxide targets.[2] The latter are valuable structural motifs, which occur in many active pharmaceutical ingredients, particularly in proton pump inhibitors such as omeprazole® and its derivatives, as well as in flavor and fragrance compounds.[3] The enantioselective sulfoxide reduction of the racemic model substrate methyl-p-tolyl sulfoxide (MPTSO, R = CH3) was achieved using an (S)-selective methionine sulfoxide reductase from Pseudomonas alcaliphila (paMsr),[4] yielding the corresponding methyl-p-tolyl sulfide, while leaving the unreacted (R)-MPTSO behind. The combination with a subsequent oxidation using various photocatalysts enabled a successful deracemization process towards (R)-MPTSO with enantiomeric excesses (ee) in the range of 56-99%. The biosynthesized photocatalyst protochlorophyllide[5] gave the best results in the deracemization process, yielding optically pure (R)-MPTSO (ee >99%). The concept was further extended by using the commercially available Eosin Y (EY) as photocatalyst for which the deracemization was run in a step-wise fashion in order to overcome incompatible reaction conditions since EY was found to not only degrade dithiothreitol (DTT), but also inactivate paMsr. The generality of the system was demonstrated by applying a small library of biocatalysts as well as structurally diverse substrates to produce the desired optical pure sulfoxides. We acknowledge the Austrian Science Fund (FWF) for funding within the project CATALOX (DOC 46‐B21) and the University of Graz and the Field of Excellence BioHealth for financial support. The COMET center: acib: Next Generation Bioproduction is funded by BMK, BMDW, SFG, Standortagentur Tirol, Government of Lower Austria und Vienna Business Agency in the framework of COMET - Competence Centers for Excellent Technologies. The COMET-Funding Program is managed by the Austrian Research Promotion Agency FFG. |
| References: | [1] a) M. Hall, RSC Chem. Biol. 2021, 2, 958-989 b) C. K. Winkler, J. H. Schrittwieser, W. Kroutil, ACS Cent. Sci. 2021, 7, 55-71 c) L.-E. Meyer, B. E. Eser, S. Kara, Curr. Opin. Green Sustain. Chem. 2021, 31, 100496. [2] a) S. Bierbaumer, L. Schmermund, A. List, C.K. Winkler, S.M. Glueck, W. Kroutil, Angew. Chem. Int. Ed. 2022, 61, e202117103V; b) Nosek, J. Míšek, Angew. Chem. Int. Ed. 2018, 57, 9849-9852. [3] I. Fernández, N. Khiar, Chem. Rev. 2003, 103, 3651-3705. [4] J. Yang, Y. Wen, L. Peng, Y. Chen, X. Cheng, Y. Chen, Org. Biolmol. Chem. 2019, 17, 3381-3388. [5] L. Schmermund, S. Bierbaumer, V.K. Schein, C.K. Winkler, S. Kara, W. Kroutil, ChemCatChem 2020, 12, 4044-4051. |
| Figures: | 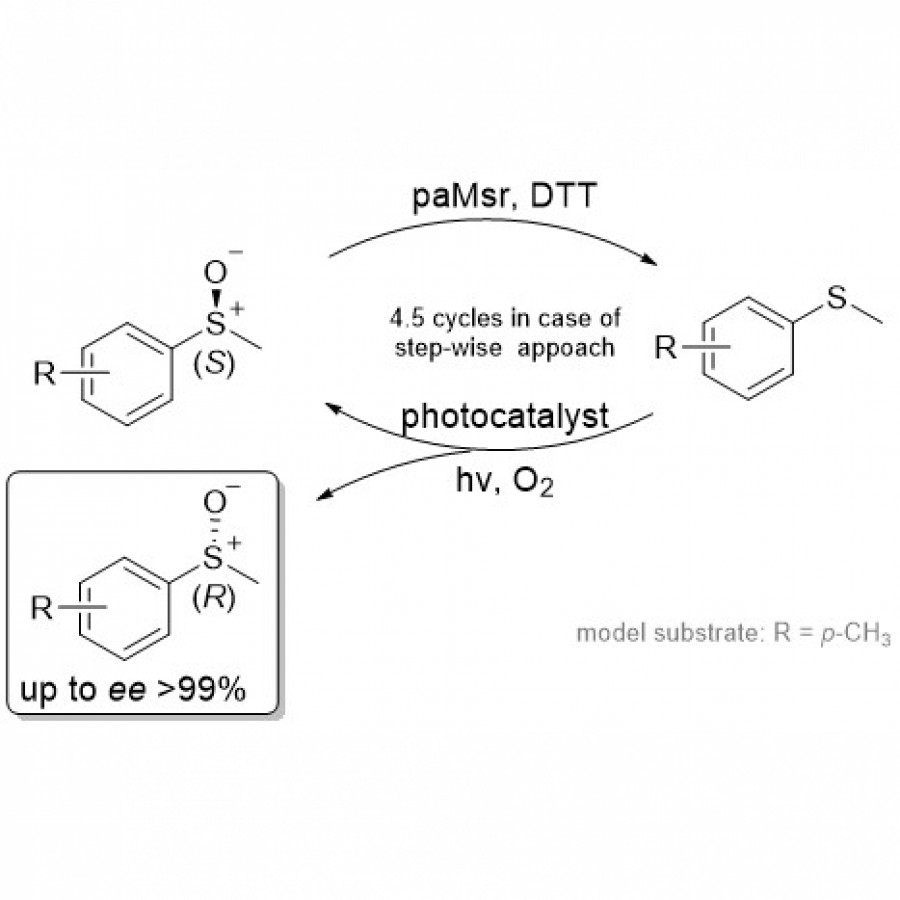 Cyclic Photobiocatalytic Deracemization Figure 1. Cyclic (stepwise) deracemization of rac-sulfoxides via biocatalytic enantioselective reduction and subsequent unselective photocatalyzed oxidation. |
#1645 | Recombinant FAD-dependent monooxygenase used for precise flavonoid functionalization |
|
| Presenting author: | Kinga DULAK of WROCŁAW UNIWERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Agata MATERA of WROCŁAW UNIWERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Sandra SORDON of WROCŁAW UNIWERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Aleksandra WILCZAK of POLISH ACADEMY OF SCIENCES Ewa HUSZCZA of WROCŁAW UNIWERSITY OF ENVIRONMENTAL AND LIFE SCIENCES Jarosław POPŁOŃSKI of WROCŁAW UNIWERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | monooxygenase / recombinant protein / ortho-hydroxylation / enzymatic cascade |
| Purpose: | The potential of natural products, particularly those derived from plants and other biological sources, as a source of new pharmaceuticals is unlimited [1]. Despite technological and societal changes, natural products remain a consistent source of new medicines. In particular, plant secondary metabolites show potent antioxidant activity, which is closely linked to the prevention and treatment of cardiovascular disease and cancer [2]. Their health-promoting properties are mainly related to the presence and position of the hydroxyl group and conjugated double bonds [3]. Although there have been reported advances in methods to obtain hydroxylated polyphenols using hydrogen peroxide and metal catalysts, such as vanadium, palladium, TiO2, or chemical oxidation in supercritical carbon dioxide, these methods are still limited in their selectivity and efficiency. Complex, multi-step processes, such as the Hock process, are still mainly used to obtain hydroxylated polyphenols [4]. Enzyme catalysis is an excellent alternative to these methods. However, most of the identified enzymes for this purpose belong to the cytochrome P-450 monooxygenases, which depend on a compatible reductase [5], and often have very low yields or do not provide sufficient regioselectivity. In contrast, flavoprotein monooxygenases (FMOs), which belong to group B of flavin-dependent monooxygenases, can directly use NADPH as an electron donor [6], and exhibit excellent regioselectivity. Nevertheless, to date, there are limited data on FMOs active against flavonoids [7]. The application of microorganisms or isolated enzymes as biocatalysts is a well-established strategy for the synthesis of high-value natural compounds. This approach provides environmentally friendly and efficient production of compounds that were previously beyond the reach of science [8]. Furthermore, the modification of natural compounds by recombinant protein expands our understanding of their origin, stability, or activity. This offers the potential for their industrial use in the development of new classes of drugs or nutraceuticals that provide social and environmental benefits based on the latest knowledge. The reported here work concerns the heterologous expression and complete characterization of fdeE from H. seropedicae SmR1, the first selective ortho-hydroxylase of isoflavones and flavonols at the C-8 position. Furthermore, a clever maneuver with the reaction environment modification allowed the stable production of hydroxylation products, which exhibited extreme instability in both in vivo and in vitro assays. This approach yielded an 8-hydroxyquercetin titer of 0.16 g/L, which, once optimized, can be widely used for large-scale production of these compounds. This work was supported by the European Union's Horizon 2020 Research and Innovation Programme under Grant Agreements no. 814650 (SynBio4Flav) |
| References: | 1. Schmidt, B. M., Ribnicky, D. M., Lipsky, P. E. & Raskin, I. Revisiting the ancient concept of botanical therapeutics. Nature Chemical Biology 3, 2007, 360-366. 2. Yang, D., Wang, T., Long, M. & Li, P. Quercetin: Its Main Pharmacological Activity and Potential Application in Clinical Medicine. Oxidative Medicine and Cellular Longevity, 2020, 8825387. 3. HemaIswarya, S. & Doble, M. Potential synergism of natural products in the treatment of cancer. Phytotherapy Research 20, 2006, 239-249. 4. Ullrich, R. & Hofrichter, M. Enzymatic hydroxylation of aromatic compounds. Cellular and Molecular Life Sciences: CMLS 64, 2007, 271-293. 5. Ziegler, D. M. Recent Studies on the Structure and Function of Multisubstrate Flavin-Containing Monooxygenases. Annual Review of Pharmacology and Toxicology 33, 1993, 179-199. 6. Huijbersa, M. M. E., Montersinoa, S., Westphal, A. H., Tischler, D. & van Berkel, W. J. H. Flavin dependent monooxygenases. Archives of Biochemistry and Biophysics 544, 2014, 2-17. 7. Hyejin, L., Bong-Gyu, K. & Joong-Hoon, A. Production of bioactive hydroxyflavones by using monooxygenase from Saccharothrix espanaensis. Journal of Biotechnology 176, 2014, 11-17. 8. Baccile, J. A. et al. Plant-like biosynthesis of isoquinoline alkaloids in Aspergillus fumigatus. Nature Chemical Biology 12, 2016, 419-424. |
| Figures: | 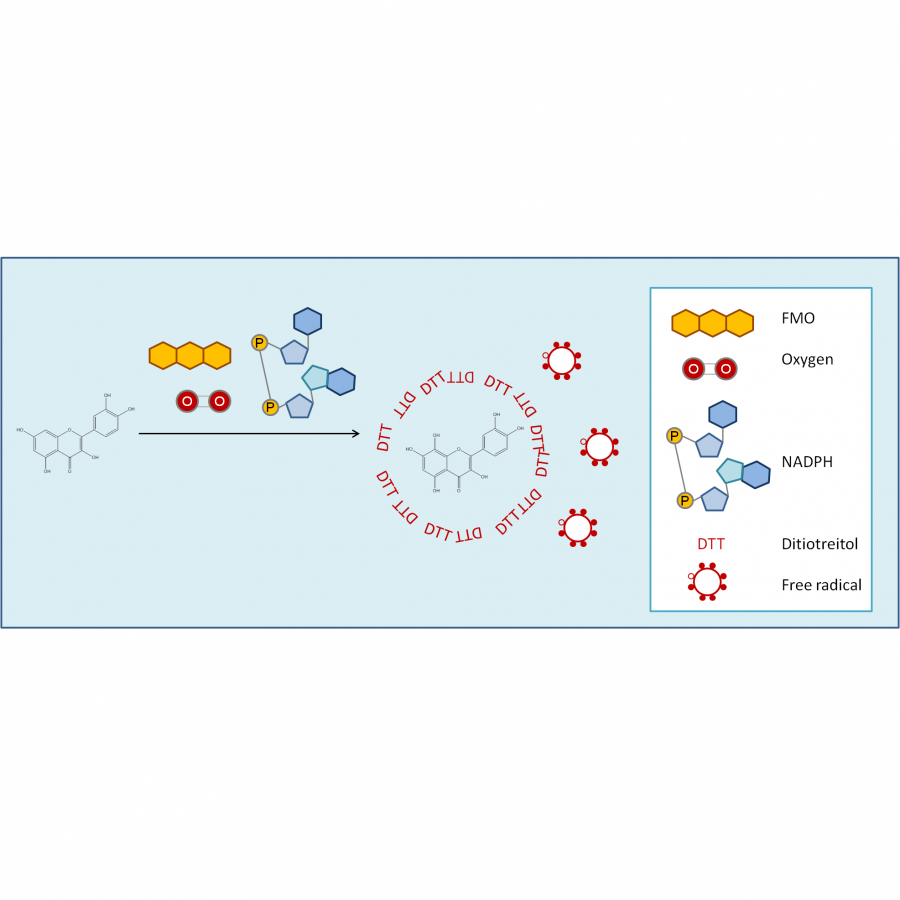 Mechanizm of stable in vitro hydroxylation of flavonoids by FMO. The legend is included in the figure. |
#1647 | Novel activities from an allogenic microbial glycosyltransferase |
|
| Presenting author: | JOSEPH O'SULLIVAN of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | glycosylation / enzymology / glycosyltransferase / glycoconjugates |
| Purpose: | Glycosylation is a function prevalent in eukaryotes, prokaryotes, and archaea, and is required for the formation of important glycoconjugates including cell surface glycans, glycosphingolipids, and glycoproteins.[1] The synthesis of such complexes is possible through the use of glycosyltransferases (GTs), enzymes that are ubiquitous in nature, which catalyse such glycosylations through the transfer of activated sugar donors, frequently nucleotide diphosphates (NDPs), to specific acceptors.[2] GT family classification derives from sequence homology, with each preserving the structural fold and mechanism, and although their activities are substrate specific, families can be utilised with several donor-acceptor substrates.[3] Here, we report studies on a GT from the family 31 (GT31), sourced from the gram-negative gut bacterium Akkermansia muciniphilia. After its expression in Escherichia coli, the enzyme displayed new activities not previously reported for this family. The successful glycosylation of N-acetylglucosamine (GlcNAc) with fucose, and N-acetylgalactosamine (GalNAc) with GlcNAc indicate these new activities and open the question as to the main activity of this expressed GT. Future work will focus on uncovering the predominant activity of this GT, as well as to determine its structure before its classification can be deduced. Verifying this currently unestablished activity for the GT31 suggests many more activities could still be undiscovered in homologues of other families too. The potential to unlock a library of new activities for attainable, soluble GTs presents an alluring opportunity for the facile assembly of desirable enantiomerically pure glycoconjugates. |
| References: | [1] Ito T, et al. Biochemistry. 2010; 49 (11): 2604-2614 [2] Moremen KW, Haltiwanger RS. Nat Chem Biol. 2019; 15 (9): 853-864 [3] Coutinho PM, et al. J Mol Biol. 2003; 328 (2): 307-317 |
| Figures: | 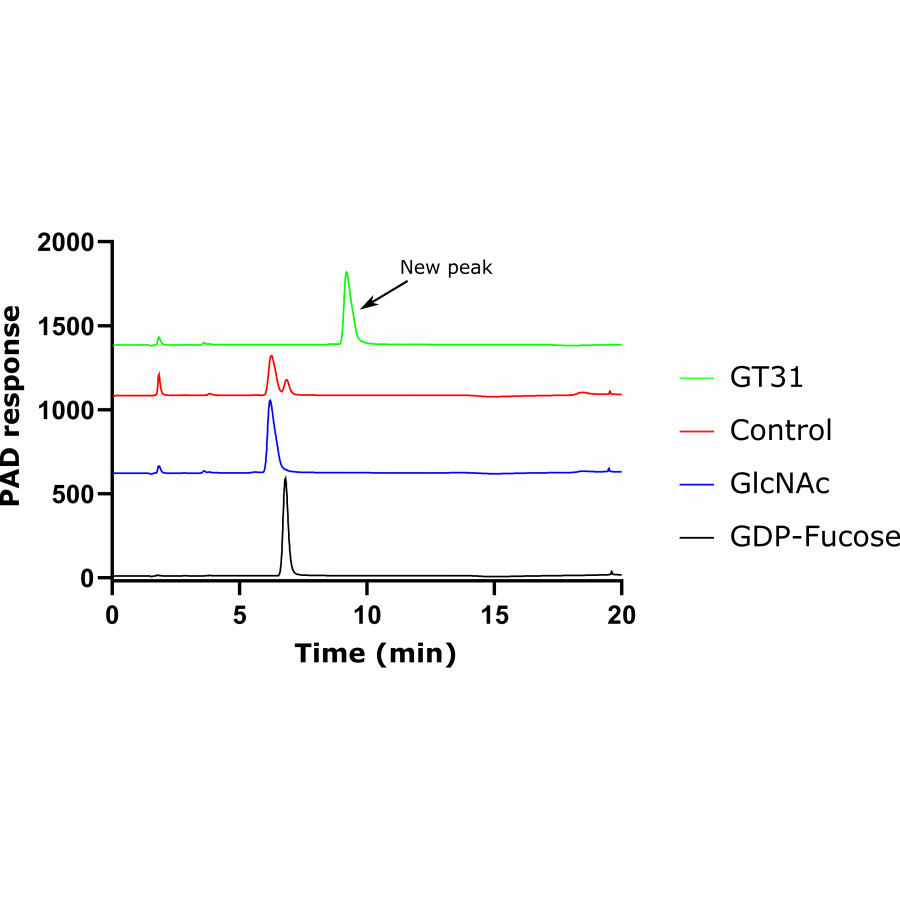 GDP-fucose and GlcNAc enzymatic reactions HPLC chromatograms of enzymatic reactions with a donor control (GDP-fucose) (black), an acceptor control (GlcNAc) (blue), an E. coli control reaction (red) and a GT31 reaction (green). 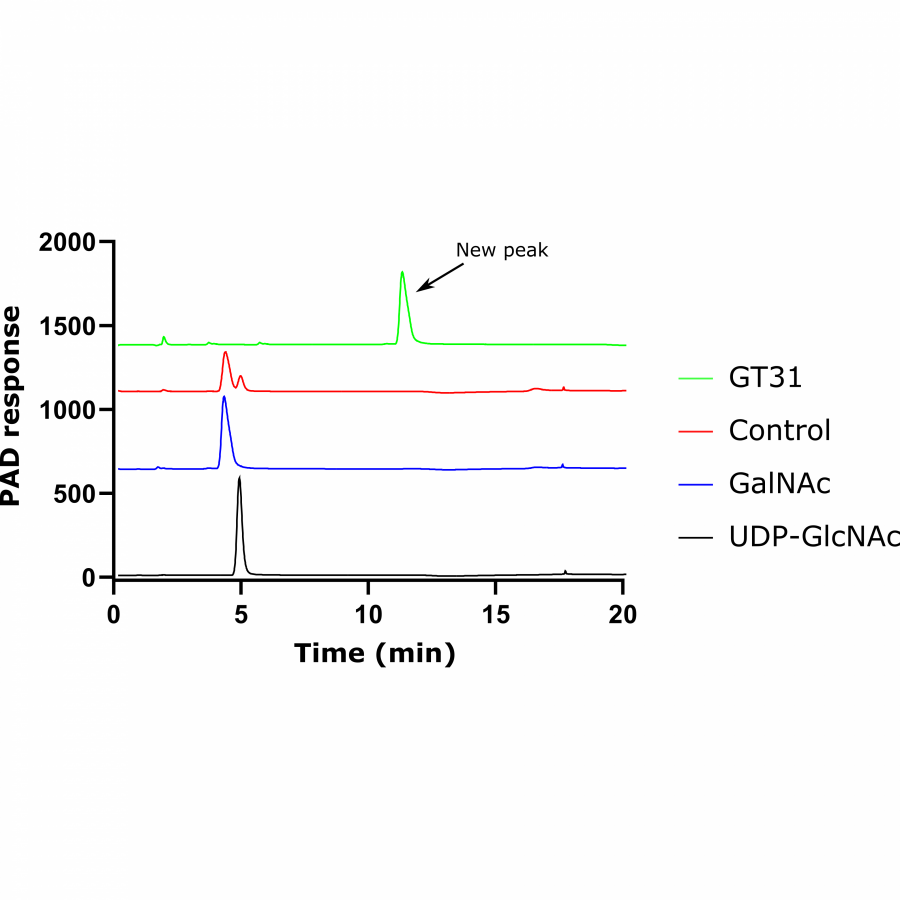 UDP-GlcNAc and GalNAc enzymatic reactions HPLC chromatograms of enzymatic reactions with a donor control (UDP-GlcNAc) (black), an acceptor control (GalNAc) (blue), an E. coli control reaction (red) and a GT31 reaction (green). |
#1648 | Novel Sustainable Approaches for Complex Soil Removal in Cold Water |
|
| Presenting author: | Dragos-Constantine DUMITRESCU of NORTHUMBRIA UNIVERSITY |
| Corresponding author: | Paul JAMES of NORTHUMBRIA UNIVERSITY |
| Other authors: | Steven PATTERSON of PROCTOR AND GAMBLE Gary BLACK of NORTHUMBRIA UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme Discovery / Psychrophile / Biocatalysis / |
| Purpose: | The most significant environmental impact in the process of washing clothes is the energy used to heat the water in a washing machine. Life Cycle Assessment (LCA) shows that the biggest impact on CO2 emission by far is due to the water temperature selected when doing laundry, temperature accounts for up to 90% of the energy needed to do the wash load. Putting this into further perspective, if all wash loads in Europe were washed at 30°C as opposed to 40°C almost 3.5million tonnes of CO2 would be saved per year. Unremoved grease and body soils from fabrics are also a significant factor in customer dissatisfaction. This issue is exacerbated by reduced temperatures during the washing cycle as this contributes to the hardening of lipid-based soils and a drop in the efficiency of surfactants. Additionally, sebum can pose further difficulties due to its reattachment to the fibers during the washing. Improving soil removal at low temperatures without the need of increased surfactant concentrations can help reduce Scope 1 and Scope 3 emissions. Furthermore, preventing the sebum from reattaching to the fibers can also lead to better results from cold washing. Enzymes such as lipases and esterases are known to be used in detergents and have been the subject of much research in the past decades. However, such enzymes have certain limitations in terms of the range of lipids they can target not covering all the substrates present in grease. Targeting relatively minor but rate-limiting grease could provide a breakthrough in terms of the efficiency of cold washing. This project will focus on discovering novel, cold-adapted enzymes that can degrade sebum. Enzymes of interest will be identified from already sequenced psychrophiles and psychrophilic metagenomes. They will be cloned and over-expressed in E. coli and characterised to investigate their potential in a detergent. The structure of these proteins will also be solved to aide potential mutagenic studies to improve stability, activity or change specificity. The project will also explore the use of proteins as a textile surface modification technology in order to achieve soil release benefits on cotton through disrupting the ability of greases to penetrate cotton fibers thus enhancing cold water cleaning. The aim will be to provide surface coating only during the washing cycle after which the proteins would detach from the fabrics. |
#1649 | Valorization of agricultural waste streams by adapting established enzyme cascades using high-throughput screening systems. |
|
| Presenting author: | Dennis ROMEIS of TECHNICAL UNIVERSITY MUNICH |
| Corresponding author: | Volker SIEBER of TECHNICAL UNIVERSITY MUNICH |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Galactosidase / Microfluidics / EnzymeEngineering / EnzymeCascade |
| Purpose: | The utilization of agricultural waste streams as potential feedstock for enzyme cascades in biocatalytic processes has gained significant attention in recent years. These residual streams are typically low-cost or even waste products of agricultural processes and represent an abundant and renewable source of carbon as well as other nutrients that can be utilized in enzyme cascades to yield a variety of valuable products. These cascades involve the sequential action of multiple enzymes to convert various substrates into desired products and offer several advantages over traditional methods of chemical catalysis. In previous studies, our group has established enzyme cascades for the production of various commodity and platform chemicals such as ethanol, isobutanol, butanediol or α-ketoglutarate from glucose or other monosaccharides [1-3]. The aim of the study presented here is to add specific enzyme-catalysed reaction steps to these core cascades in order to incorporate e.g. lactose obtained from agricultural waste streams. To achieve this, enzymes like galactosidase for the hydrolysis of lactose to D-glucose (D-Glu) and D-Galactose (D-Gal) need to be engineered for high catalytic efficiency in the reaction environment of the substrate containing waste streams (salt and substrate concentrations). Microfluidic droplet screening has emerged as a powerful tool for high-throughput screening of enzyme libraries, allowing rapid identification of enzymes with improved catalytic properties for the target application, such as cascade activity, selectivity and stability. In previous studies, our group has successfully applied absorbance and fluorescence-activated droplet sorting systems to improve the catalytic properties of different enzymes [4]. By modifying and optimizing assay setups (Figure 1) and the technical environment of these systems, the enzymes of the extended cascades can be adapted to the above-mentioned reaction conditions using directed evolution techniques. |
| References: | [1] Guterl, J. K., et al., ChemSusChem 2012, 5, 2165-2172 [2] Sutiono, S., et al., Green Chem. 2021, 23, 3656-3663 [3] Beer, B., et al., Metabolic Engineering 2017, 40, 5-13 [4] Zachos, I., et al., ACS Catalysis 2022, 12, 1841-1846 |
| Figures: |  Assay scheme for droplet-based galactosidase screening. Figure 1: Assay scheme for droplet-based galactosidase screening. D-Glu and D-Gal are oxidized to the respective sugar acid by alcohol dehydrogenase, NADH is reducing WST-1 to the absorbant formazan form via the electron mediator mPMS. |
#1650 | Sulfurtransferases as molecular platforms to form C-SH bonds |
|
| Presenting author: | François TALFOURNIER of UNIVERSITÉ DE LORRAINE, UMR 7365 CNRS-UL |
| Other authors: | Anne-Lise CLAUDEL of UNIVERSITÉ DE LORRAINE, UMR 7365 CNRS-UL Erwan GALARDON of UNIVERSITÉ PARIS CITÉ, UMR 8601 CNRS-UP Julien DAIROU of UNIVERSITÉ PARIS CITÉ, UMR 8601 CNRS-UP Sandrine BOSCHI-MULLER of UNIVERSITÉ DE LORRAINE, UMR 7365 CNRS-UL |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Sulfurtransferases / biocatalytic platforms / C-SH bonds / cysteine persulfide |
| Purpose: | The formation of C-S bonds is of utmost importance in material sciences or medicinal chemistry, with for instance approximatively 20% of the approved FDA drugs containing sulfur atoms. While various chemical methods have been developed over the years, essentially based on the nucleophilicity of thiols, a great deal of effort is nowadays directed towards the development of milder and greener methods. Because a versatile methodology allowing for the direct synthesis of thiols from C-H bonds is still lacking, an alternative to the chemical approaches is the growing use of enzymes as biocatalysts. In this context, our project proposes to use sulfurtransferases (STs) as molecular platforms for the challenging formation of C-SH bond by sulfur atom transfer. Indeed, STs, which belong to the rhodanese-fold family, generate a persulfide intermediate on the catalytic cysteine residue in the presence of a suitable donor and the electrophilic distal sulfur atom can then be transferred to various nucleophilic acceptors. Thus, one key challenge relies on the substrate specificity of STs, i.e. on the capacity of nucleophilic compounds to reach their target. However, available structural data indicate that the active sites of some STs are quite solvent exposed therefore supporting the idea that persulfides could react with various nucleophile provided a minimal accessibility. Several STs of various origins were tested for their ability to accommodate unnatural substrates. An original kinetic method based on the fluorescence properties of an unnatural donor substrate was developed. This method was used to screen various STs / model acceptor substrates and the results show that both linear and cyclic acceptor substrates react, at least, with the persulfide intermediate of bacterial STs. Regarding the development of new ecofriendly reactional schemes for C-S bond formation using STs to deliver an activated sulfur atom to various nucleophilic molecules, the proof of concept has been established.with model substrates. |
#1651 | Biochemical and Catalytic Analysis of Computationally Designed Hyperthermostable Bacterial DyP-type Peroxidases |
|
| Presenting author: | Carolina FERRO RODRIGUES of ITQB NOVA, PORTUGAL |
| Other authors: | Diogo SILVA of ITQB NOVA, PORTUGAL Paulo DURAO of ITQB NOVA, PORTUGAL Lígia O. MARTINS of ITQB NOVA, PORTUGAL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Protein Stability / Protein Engineering / Computational Engineering / Biocatalysis |
| Purpose: | DyP-type peroxidases (DyPs) are microbial enzymes that catalyze the oxidation of a wide range of substrates, including lignin-derived compounds and metals, such as Mn2+ and Fe2+ and have enormous biotechnological potential in biorefineries. Nonetheless, several questions on the molecular basis of enzyme function and stability remain unanswered. Protein stability is a complex, multi-variable equation, and tackling it with rational single-point mutations or through iterative rounds of random mutagenesis can be time and energy-consuming. We have applied computational tools to design hyperthermostable variants of PpDyP and have produced and characterized the variants suggested by the web servers PROSS (dsPpDyP_1), with 29 mutations, and FireProt (dsPpDyP_2), with 21 mutations. Both enzymes exhibit remarkable improvements, i) shifting the optimum activity temperature by 35-40 °C and ii) exhibiting a melting temperature of 75-85 °C, circa 10-20 °C higher than wild-type, without jeopardizing the enzyme activity. Moreover, the mutations inserted in the dsPpDyP_1 variant abolished the temperature-induced aggregation of its tertiary structure in a 20 to 100 °C temperature ramp. To assess the mechanisms behind the improved thermostability, DNA shuffling of genes of variants vs. wild-type gene was performed, and libraries of enzyme chimeras were screened to select variants maintaining the hyperthermostable phenotype while showing a reduced mutational load. The biochemical and kinetic analysis of a subset of variants hypothetically containing only neutral or beneficial mutations is presented and discussed. |
#1654 | Biorefinery of Renewable Limonene and Oleic Acid by Designing Multi-Enzymatic Routes |
|
| Presenting author: | Jianhe XU of EAST CHINA UNIVERSITY OF SCIENCE AND TECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | / / / |
| Purpose: | Introduction Based on the design-build-test (DBT) concept of synthetic biology, the natural biosynthesis pathway of l-menthol from limonene in plant was artificially reconstructed in microbial chassis of Escherichia coli by recruiting microbial enzymes to replace some difficult-to-express plant genes encoding membrane proteins. The biosynthesis of isopulegone removes the last technical hurdle for de novo microbial synthesis and large-scale biomanufacturing of l-menthol, one of the most important flavor chemicals with an annual demand of 30,000 metric tons. Green synthesis of eco-friendly chemicals or material monomers from renewable feedstocks is of great significance to alleviate the problems of global warming and white pollutions. We designed and constructed a library of non-natural components/devices for biorefinery of oleic acid into bifunctional chemicals of mid-chain length (C8-C10), such as 8-hydroxyoctanoic acid, 9-aminononanoic acid and 1,10-decanedioic acid. A Baeyer-Villiger monooxygenase with unconventional regio-selectivity was designed, and 6 flexible modules of Plug-and-Play type were constructed for easy assembly into different routes to 7 structurally diverse bifunctional chemicals. Results and Discussion A microbial cell factory was designed and built in E. coli for synthesis of (+)-cis-isopulegone from (−)-limone, removing the last obstacle to (−)-menthol de novo biosynthesis. The production of (+)-cis-isopulegone, up to 281 mg L−1, was improved by 36 times compared with that of the initial strain. A biocatalytic cascade process was designed for the normalization of n-nonanoic acid (NA) into 9-hydroxynonanoic acid (9-HNA) which might be further transformed into other C9 bifunctional chemicals, namely 1,9-nonanedioic acid (1,9-NDA) and 9-aminononanoic acid (9-ANA). More importantly, the cascaded biocatalysis system was operated by using different engineered E. coli consortia composed of the desired cell modules. This represents the first examples for valorizing the by-product (n-nonanoic acid) originated from oleic acid biorefinery, improving the carbon atom economy from 50% to 100%. |
#1669 | RUTHENIUM Metal Complexes for ARTIFICIAL METALLOENZYMES |
|
| Presenting author: | LARA VILLARINO PALMAZ of CENTRO SINGULAR DE INVESTIGACIÓN EN QUÍMICA BIOLÓGICA Y MATERIALES MOLECULARES (CIQUS) /DEPARTAMENTO DE QUÍMICA ORGÁNICA |
| Other authors: | JOSÉ LUIS MASCAREÑAS CID of CENTRO SINGULAR DE INVESTIGACIÓN EN QUÍMICA BIOLÓGICA Y MATERIALES MOLECULARES (CIQUS) /DEPARTAMENTO DE QUÍMICA ORGÁNICA CLARA LÓPEZ DE AGUILETA BUSTERO of CENTRO SINGULAR DE INVESTIGACIÓN EN QUÍMICA BIOLÓGICA Y MATERIALES MOLECULARES (CIQUS) /DEPARTAMENTO DE QUÍMICA ORGÁNICA |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | artificial metalloenzymes / supramolecular assembly / ruthenium catalysis / deallylation reaction |
| Purpose: | Nature employs metalloenzymes to catalyze some of the most remarkable biological processes with high activity and selectivity under mild conditions. In the past decades, much effort has been made to design novel metalloenzymes capable of performing reactions that are not found in nature´s repertoire.[1] In this context, enzyme promiscuity has long been recognized as a critical feature in discovering new activities. Incorporation of an abiotic catalytically active transition metal complex into a host biomacromolecule, also known as artificial metalloenzymes (ArMs), represents an alternative mechanism of enzyme promiscuity. This strategy allows us to combine the broad scope of reactions catalyzed by transition metal catalysts with the high selectivity, biorthogonality, and mild conditions achieved by enzymatic catalysis. Here, we will present a novel artificial metalloenzyme based on the supramolecular anchoring of a series of ruthenium (II) and (IV) piano-stool complexes to the hydrophobic pocket of the promiscuous protein LmrR. [2] These hybrid catalysts will be tested in the uncaging reaction of different prodrug compounds. |
| References: | [1] (a) Zeymer C, Hilvert D. Annu Rev Biochem. 2018; 87, 131. (b) Schwizer, F.; et al. Chem. Rev. 2018, 118 (1), 142-231. [2] (a) P. K. Madoori, H. Agustiandari, A. J. M. Driessen, A.-M. W. H. Thunnissen, EMBO J. 2009, 28, 156-166. (b) J. Bos, W. R. Browne, A. J. M. Driessen, G. Roelfes, J Am Chem Soc. 2015, 137, 9796-9799. (c) L. Villarino, S. Chordia, L. Alonso-Cotchico, E. Reddem, A.-M. W. H. Thunnissen, J.-D. Maréchal, , G. Roelfes, ACS Catalysis 2020, 10, 11783 - 11790. |
| Figures: | 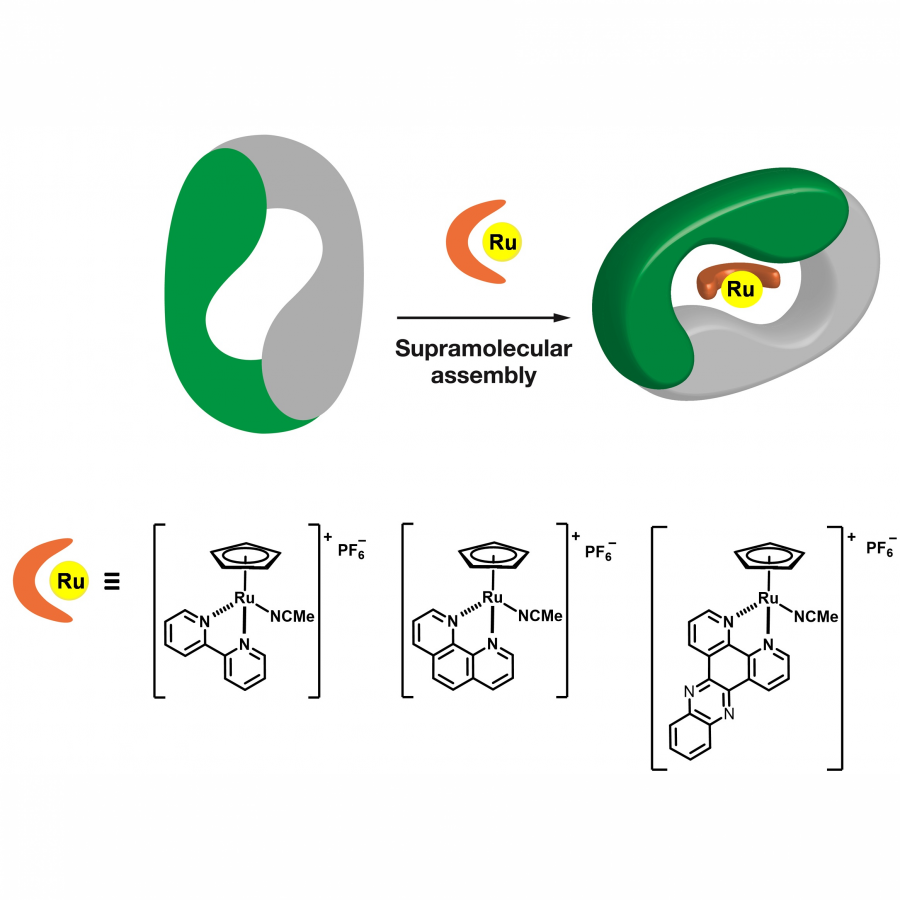 Ruthenium based artificial metalloenzymes Schematic representation of an artificial enzyme based on the supramolecular assembly of different ruthenium metal complexes into the hydrophobic pocket of a protein. |
#1670 | MR YIYU WANG |
|
| Presenting author: | UNIVERSITY LONDON of UNIVERSITY COLLEGE LONDON |
| Other authors: | HELEN HAILES of UNIVERSITY COLLEGE LONDON YU WANG of UNIVERSITY COLLEGE LONDON |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | TRANSAMINASE / BIOCATALYTIC CASCADE / SPONTANEOUS CYCLISATION / AMINE |
| Purpose: | Optically active nitrogen heterocycles are widely found across many living organisms and also play important roles in fine chemical and pharmaceutical industries. The production of a single enantiomers is usually non-sustainable due to the use of toxic solvents and transition metal catalysts. In nature, transaminases catalyse the conversion between carbonyl compounds and amines in a relatively high efficiency and mild conditions. Some approaches have been developed to allow cascade reactions of spontaneous cyclisation to form desired optically pure amine products. |
| References: | 1. Mourelle‐Insua, Ángela, et al. "Conversion of γ‐and δ‐Keto Esters into Optically Active Lactams. Transaminases in Cascade Processes." Advanced Synthesis & Catalysis 360.4 (2018): 686-695. 2. Ryan, James, et al. "Transaminase triggered aza-Michael approach for the enantioselective synthesis of piperidine scaffolds." Journal of the American Chemical Society 138.49 (2016): 15798-15800. |
| Figures: | 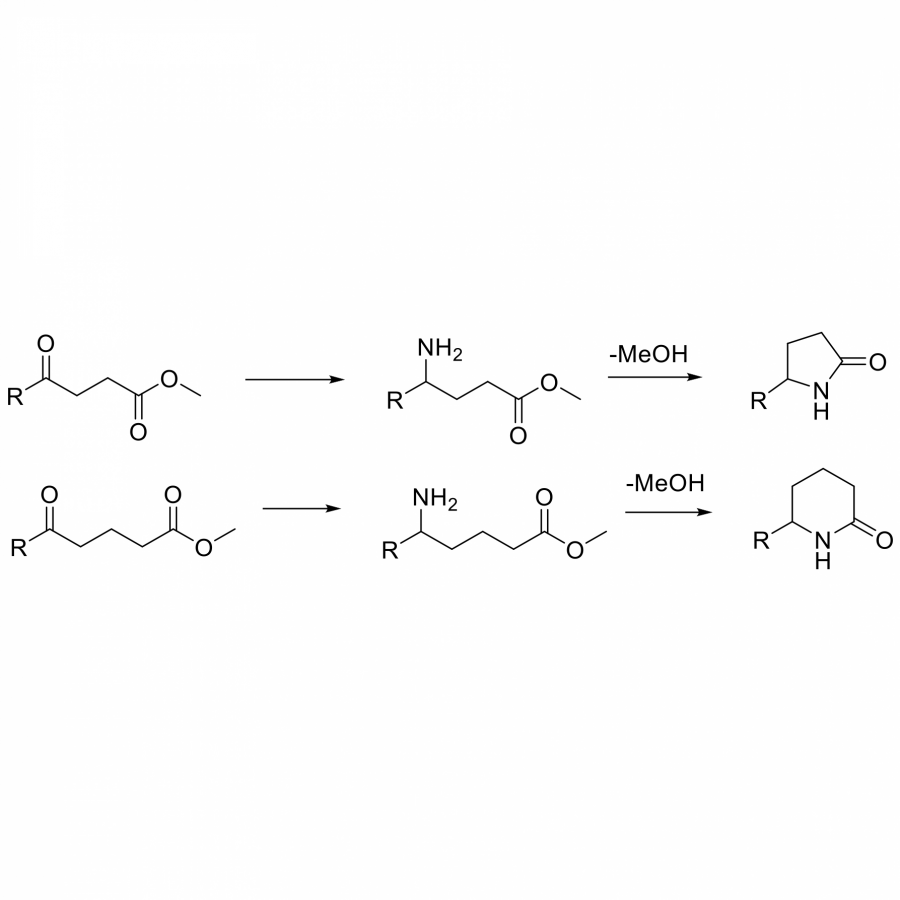 LACTAM FORMATION SUBSTRATES 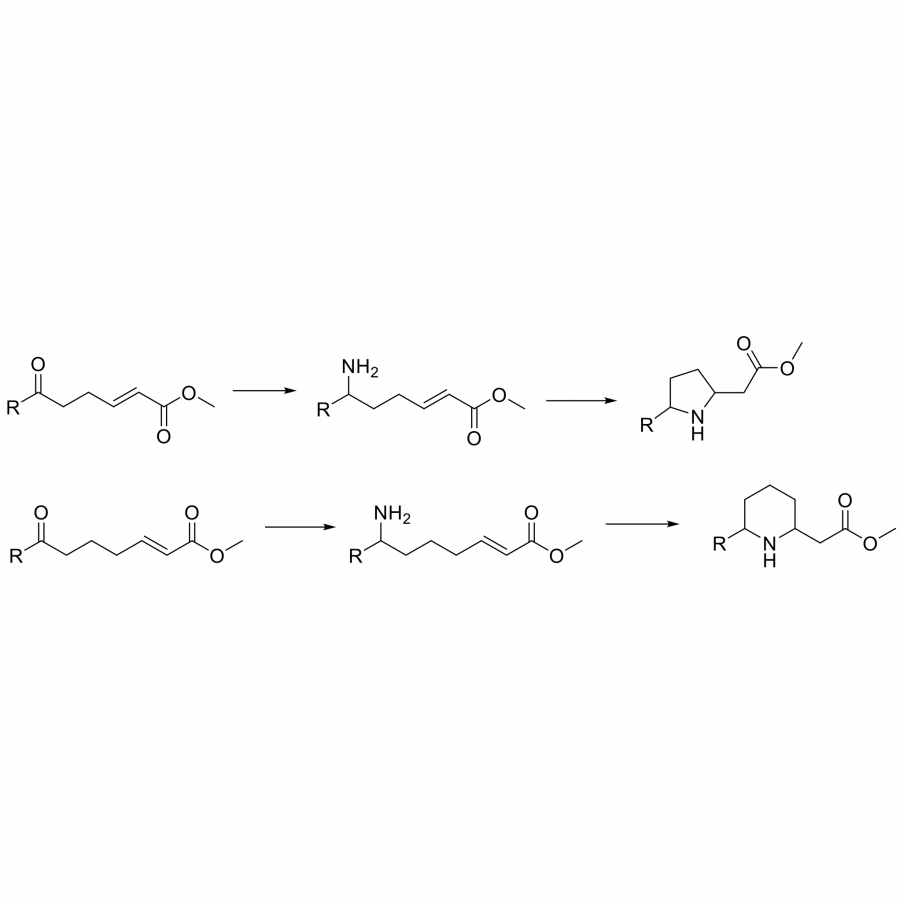 INTRAMOLECULAR MICHAEL ADDITION SUBSTRATES |
#1675 | Biocatalytic Nitro Reduction: From Hit to Process |
|
| Presenting author: | Bruna COSTA of JOHNSON MATTHEY |
| Other authors: | Carmen ARANDA of JOHNSON MATTHEY Peter LINDLEY of JOHNSON MATTHEY Amin BORNADEL of JOHNSON MATTHEY Beatriz DOMINGUEZ of JOHNSON MATTHEY |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | nitro reduction / nitro reductases / / |
| Purpose: | Reduction of nitroaromatic compounds to their corresponding amines is conventionally performed using well-established chemocatalytic methods. However, lack of sufficient activity or chemoselectivity can sometimes limit the effectiveness of those methods. At Johnson Matthey (JM), we have recently developed a biocatalytic approach for the reduction of nitroaromatics combining a nitroreductase (NR) enzyme and vanadium. This technology has shown to overcome a common limitation of NR-catalysed nitro reductions in progressing beyond the formation of the hydroxylamine intermediate. This innovative approach has been demonstrated with a range of functionalised nitroaromatic compounds. By leveraging our diverse portfolio of NRs, we have successfully applied our synergistic NR/vanadium technology to several projects within short timelines. Additionally, the biocatalytic approach developed by our group is complementary to JM’s chemocatalytic solutions. Therefore, JM has a strong position in offering a complete solution for the reduction of nitroaromatics to assist customers and collaborators to rapidly develop cost-effective synthetic routes to aromatic amines. |
| References: | Bisagni et al. Current Research in Chemical Biology 2022, 2, 100026 Bornadel et al. Org. Process Res. Dev. 2021, 25, 648-653 |
#1679 | Directed Evolution of Metalloenzymes through Electrochemical Droplet Microarrays |
|
| Presenting author: | Xie WANG of TECHNISCHE UNIVERSITAT MUNCHEN CAMPUS STRAUBING |
| Corresponding author: | Nicolas PLUMERÉ of TUMCS-ELECTROBIOTECHNOLOGY |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Protein engineering / Surface display / Directed evolution / Electrochemical microarray |
| Purpose: | In recent years, surface-display technology has attracted more and more attention of scientists. It is reported as a leading protein engineering tool to target proteins to the cell surfaces of microorganisms. Because of its correlation between the target proteins and their genetic machineries, it is also considered as an evolution of artificial chemicals (Barderas and Benito-Peña, 2019). It is a perfect tool to adapt different screening methods in directed evolution. With the help of surface display systems, a new electrochemical screening platform is developed for direct enzyme-activity screening and to find out the correlation of enzyme structure and electrode surface. In this way, metalloenzymes could be directed to adapt their application environment, for example, in enzymatic biofuel cells. |
| References: | The 2018 Nobel Prize in Chemistry: phage display of peptides and antibodies. Barderas, R., and Benito-Peña, E. (2019). Anal. Bioanal. Chem. 411, 2475-2479. A dual tag system for facilitated detection of surface expressed proteins in Escherichia coli. Jarmander J, Gustavsson M, Do TH, Samuelson P, Larsson G. Microb Cell Fact. 2012 Sep 3;11:118. doi: 10.1186/1475-2859-11-118. 10.1186/1475-2859-11-118 PubMed 22943700 |
| Figures: | 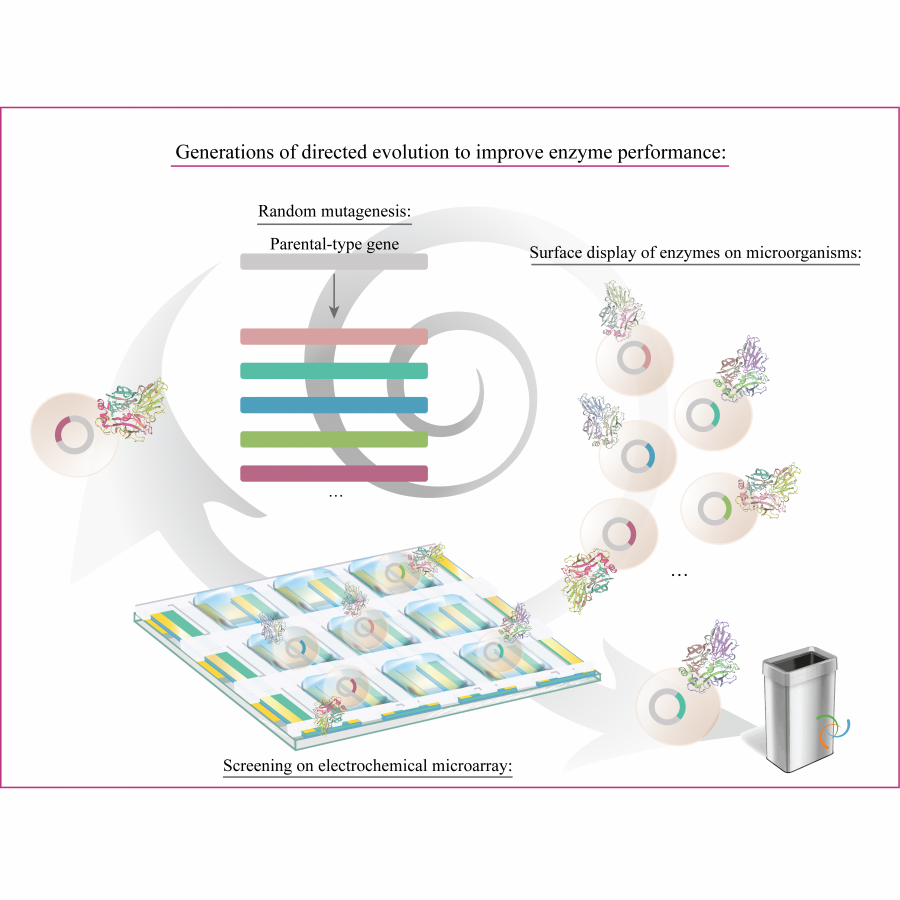 New directed evolution circle The new directed evolution circle is built up by random mutagenesis, surface display systems and electrochemical microarrays. |
#1681 | Baeyer-Villiger monooxygenases from photosynthetic organisms support the production of a multifunctional drug in the cyanobacterium Synechocystis sp. 6803 |
|
| Presenting author: | ELISABETTA BERGANTINO of DEPARTMENT OF BIOLOGY, UNIVERSITY OF PADOVA |
| Other authors: | Giovanni LOPRETE of DEPARTMENT OF BIOLOGY, UNIVERSITY OF PADOVA Giulio CONDOLUCI of DEPARTMENT OF BIOLOGY, UNIVERSITY OF PADOVA |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | BVMO / Synechocystis / whole-cell biotransformation / |
| Purpose: | The cyanobacterium Synechocystis is progressively attracting interest as cell factory for the production of commodity chemicals, fuels and value-added products by essentially consuming carbon dioxide and water, and using light as energy source [1]. It is currently tested in whole-cell biocatalysis by genetic manipulation of its enzymatic content [2]. It is particularly suitable for expressing enzymes candidates for reduction reactions requiring NADPH, being unusually rich in this cofactor that is actively regenerated through photosynthesis [3]. We have produced two Synechocystis strains constitutively expressing two Baeyer-Villiger monooxygenases from eukaryotic photosynthetic organisms that we had described some time ago: CmBVMO and PpBVMO, respectively from the red alga Cyanidioschyzon merolae and the moss Physcomitrella patens [4]. We have examined the two strains in toxicity tests on Petri dishes, by furnishing some molecules that had been identified as preferred substrates upon biocatalytic profiling. Such tests have permitted (i) to verify the permeability of the outer and cytoplasmic cellular membranes to a given molecule, (ii) to evaluate its (approximate) minimal toxic concentration in Synechocystis wild-type cells and (iii) to observe the in vivo activity of the recombinant enzyme, if any, acting on it. After focusing our attention on a substituted aryl chetone that is a natural molecule used as dietary supplement, we moved to biotransformations. By furnishing the molecule to the CmBVMO-expressing strain, we could reveal by HPLC analysis the progressive disappearance of the substrate and the appearance of the expected product (a multifunctional drug) in the growth medium. We will report on this study and its envisaged progresses. |
| References: | [1] Toepel J. et al., Current Opinion in Biotechnology vol. 80 (2023): 102892 [2] Jodlbauer J. et al., Trends in Biotechnology vol. 39 (2021): 875-889 [3] Barone G.D. et al., Biotechnology for Biofuels and Bioproducts vol. 16 (2023): 4 [4] Beneventi E. et al., Journal of Molecular Catalysis B-Enzymatic vol. 98 (2013): 145-154 |
#1683 | Exploration of archaeal nucleotide sugar epimerases unveils a highly promiscuous GDP-Gal4E subgroup |
|
| Presenting author: | Carlos Josué ALVAREZ QUISPE of GENT UNIVERSITY |
| Other authors: | Matthieu DA COSTA of GENT UNIVERSITY Koen BEERENS of GENT UNIVERSITY Tom DESMET of GENT UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Promiscuity / rare sugars / NDP-sugars / enzyme discovery |
| Purpose: | Nucleotide sugar epimerases form a very interesting group of enzymes, as they can invert the configuration of a specific hydroxyl group through a single reaction and without prior activation or protection steps. Within this group, UDP-galactose 4-epimerase (Gal4E, EC 5.1.3.2) is by far one of the best studied members due to its essential role in the Leloir pathway in which it interconverts UDP-galactose and UDP-glucose. Gal4E deficiency is responsible for galactosemia, a hereditary disease, highlighting its vital importance. Although Gal4E was widely studied throughout all domains of life, ranging from eukaryotes to archaea, its biochemical characterization was often limited to UDP-hexoses, neglecting the possibility that Gal4E might be promiscuous towards other NDP-sugars and derivatives thereof. In this study, we identified a novel Gal4E displaying an unprecedented specificity on guanosine diphosphate (GDP) sugars. Indeed, a detailed biochemical investigation performed on Gal4E from Pyrococcus horikoshii (phGal4E_1) revealed that it is a GDP-sugar 4-epimerase. In addition, we confirmed that it accepts a variety of other NDP-sugars including L-sugars moieties, such as GDP-L-Gal/Glc as well as their 6-deoxysugars counterparts GDP-L-fucose and GDP-L-quinovose, respectively. |
| References: | Da Costa et al., Biotechnol. Adv. 2021, 48, 107705. |
#1685 | An Enzyme Cascade Enables Production of Therapeutic Oligonucleotides in a Single Operation |
|
| Presenting author: | Hubert CASAJUS of THE UNIVERSITY OF MANCHESTER |
| Corresponding author: | Sarah LOVELOCK of THE UNIVERSITY OF MANCHESTER |
| Other authors: | Ewan MOODY of THE UNIVERSITY OF MANCHESTER Richard OBEXER of THE UNIVERSITY OF MANCHESTER Florian NICKL of THE UNIVERSITY OF MANCHESTER Reynard SPIESS of THE UNIVERSITY OF MANCHESTER |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Therapeutic oligonucleotides / Enzyme cascade / Biocatalytic oligonucleotides synthesis / |
| Purpose: | Therapeutic oligonucleotides are short DNA analogues that selectively bind to target mRNA through Watson-Crick base pairing and regulate the production of disease related proteins [1]. Following the award of the 2006 Nobel Prize for RNA interfering technology and recent FDA approvals of several RNA-based therapeutics for the treatment of rare diseases, there has been significant investment into therapeutic oligonucleotides as a new drug modality. There are currently more than 160 oligonucleotide products in clinical trials including those for population based indications, in fact Inclisiran a cholesterol-lowering drug was approved in 2020, which has the potential to treat 30 million patients in the USA alone [2]. The increase in the number of potential therapeutics creates a significant manufacturing challenge, as current synthetic approaches are not scalable or sustainable. Existing chemical methods use complex building blocks containing multiple protecting groups (resulting in poor atom economy) and chromatographic purification of the final molecule is required. Syntheses are performed using solid supports which restricts the process to 10 kg batches making it unsuitable for large-scale applications (>100 kg). Moreover, the process uses prohibitively large volumes of acetonitrile (1000 kg per kg of oligonucleotide) which presents a threat to the security of oligonucleotide supply, given recent world shortages of acetonitrile [3,4]. Our group has developed an alternative PCR-based biocatalytic platform for oligonucleotide synthesis that requires no protecting groups, is atom economical and works under aqueous conditions. A combination of polymerases and endonucleases are used to amplify a catalytic template using unprotected deoxynucleotide triphosphate monomers. The target oligonucleotide is produced in a single operation, in high purity and with complete control of stereoselectivity (for molecules containing phosphorothioate linkages). Our methodology is showcased through the synthesis of the phosphorothioate modified therapeutic Vitravene as a single stereoisomer in high yield and purity. |
| References: | [1] khvorova, a. & watts, j. k. nat. biotechnol. 2017, 35, 238-248 [2] kulkarni, j. a. et al. nat. nanotechnol. 2011, 16, 630-643 [3] andrews, b. i. et al. chem. 2021 86, 49-61 [4] tedebark, u., scozzari, a., werbitzky, o., capaldi, d. & holmberg, cell-penetrating peptides: methods and protocols (ed. langel, ü.) 2011 505-524 |
| Figures: | 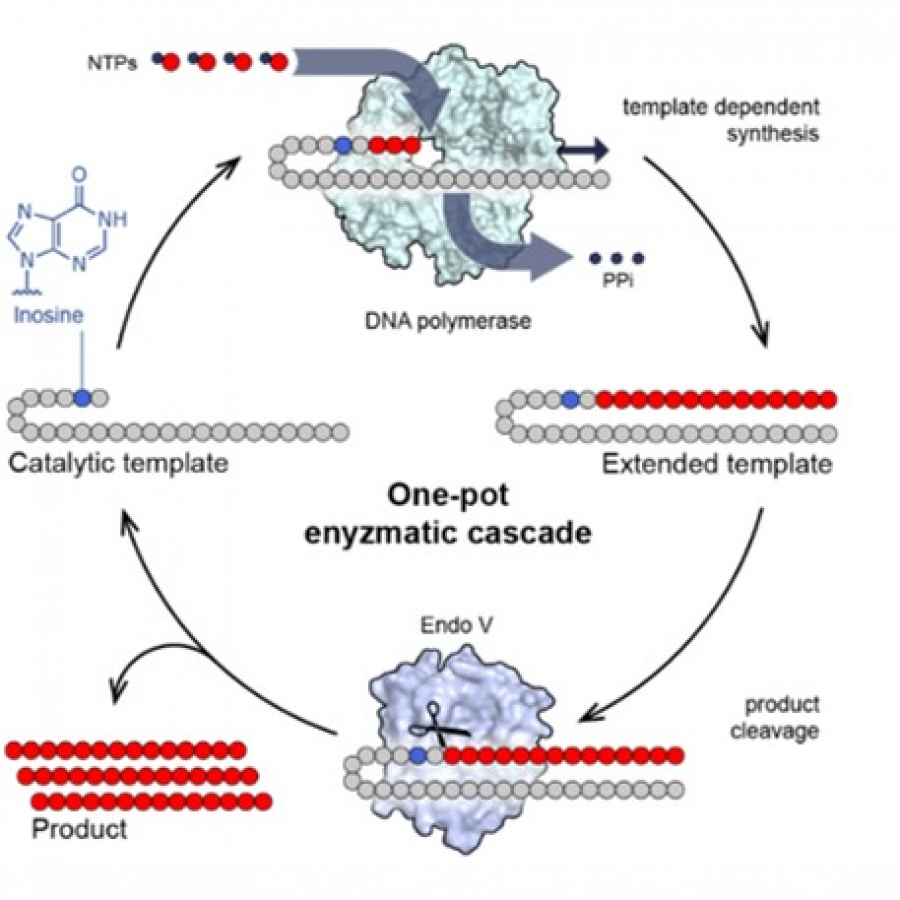 A one-pot enzymatic cascade for oligonucleotide synthesis |
#1689 | Exploration of archaeal nucleotide sugar epimerases unveils a highly promiscuous GDP-Gal4E subgroup |
|
| Presenting author: | Carlos Josué ALVAREZ QUISPE of GHENT UNIVERSITY |
| Other authors: | Matthieu DA COSTA of GHENT UNIVERSITY Koen BEERENS of GHENT UNIVERSITY Tom DESMET of GHENT UNIVERSITY |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Promiscuity / Epimerases / NDP-sugar / Enzyme discovery |
| Purpose: | Nucleotide sugar epimerases form a very interesting group of enzymes, as they can invert the configuration of a specific hydroxyl group through a single reaction and without prior activation or protection steps. Within this group, UDP-galactose 4- epimerase (Gal4E, EC 5.1.3.2) is by far one of the best studied members due to its essential role in the Leloir pathway in which it interconverts UDP-galactose and UDP-glucose. Gal4E deficiency is responsible for galactosemia, a hereditary disease, highlighting its vital importance. Although Gal4E was widely studied throughout all domains of life, ranging from eukaryotes to archaea, its biochemical characterization was often limited to UDP-hexoses, neglecting the possibility that Gal4E might be promiscuous towards other NDP-sugars and derivatives thereof. In this study, we identified a novel Gal4E displaying an unprecedented specificity on guanosine diphosphate (GDP) sugars. Indeed, a detailed biochemical investigation performed on Gal4E from Pyrococcus horikoshii (phGal4E_1) revealed that it is a GDP-sugar 4-epimerase. In addition, we confirmed that it accepts a variety of other NDP-sugars including L-sugars moieties, such as GDP-L-Gal/Glc as well as their 6-deoxysugars counterparts GDP-L-fucose and GDP-L-quinovose, respectively. |
| References: | Biotechnol. Adv., 48 (2021), Article 107705, 10.1016/j.biotechadv.2021.107705 Curr. Opin. Chem. Biol., 61 (2021), pp. 53-62, 10.1016/j.cbpa.2020.09.007 Biotechnol. Adv., 48 (2021), Article 107705, 10.1016/j.biotechadv.2021.107705 Carbohydr. Res., 414 (2015), pp. 8-14, 10.1016/j.carres.2015.06.006 Bioresour. Technol., 110 (2012), pp. 423-429, 10.1016/j.biortech.2012.01.046 |
| Figures: | 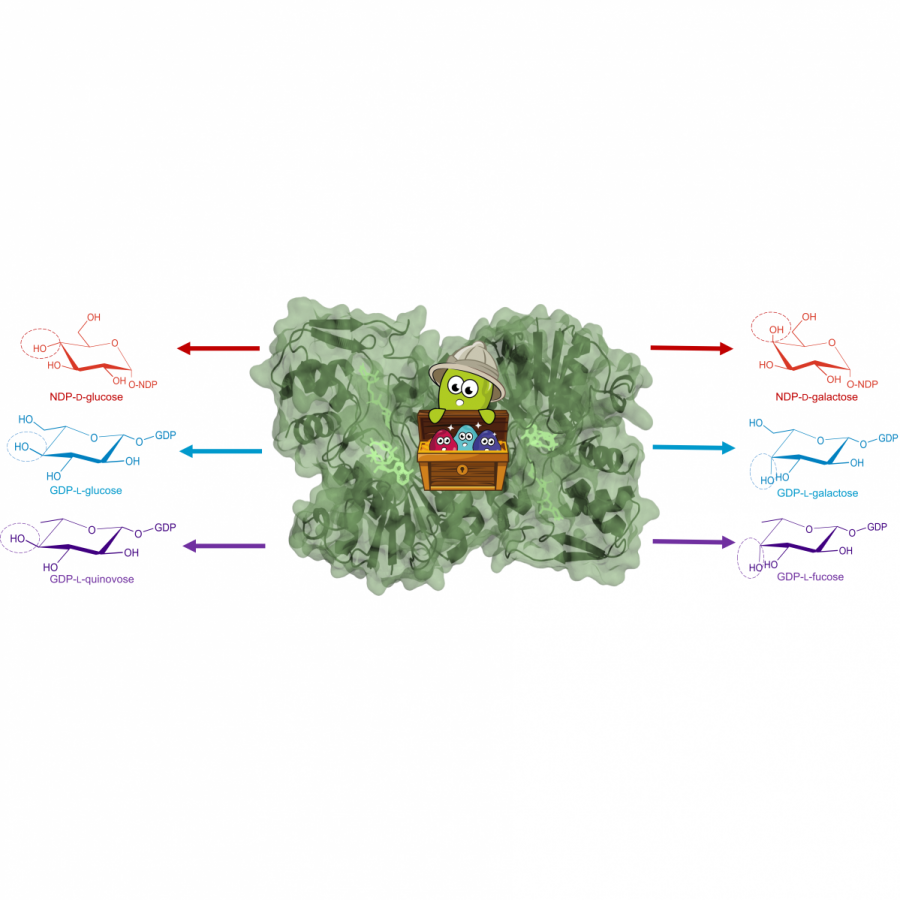 PhGal4E_1 Archaeal epimerase Promiscuity of phGal4E_1 towards different NDP-sugars including L-sugar and deoxy-sugar moieties |
#1693 | Streamlined Chemoenzymatic Synthesis of Cyclic Peptides by Non-ribosomal Peptide Cyclases |
|
| Presenting author: | Kenichi MATSUDA of FACULTY OF PHARMACEUTICAL SCIENCES, HOKKAIDO UNIVE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | cyclic peptide / peptide cyclase / chemoenzymatic synthesis / protein engineering |
| Purpose: | Macrocyclization of peptide improves their pharmaceutical properties such as cell-membrane permeability, target specificity and metabolic stability. However, regio- and chemoselective intramolecular cyclization remain challenging. In the biosynthesis of non-ribosomal cyclic peptides, macrocyclases such as thioesterases (NRPS-TEs) efficiently catalyze the intramolecular peptide cyclization in a regiospecific manner without the use of protecting groups. Thus, NRPS-TEs are promising biocatalysts for the efficient synthesis of macrocyclic peptides. NRPS-TEs generally require the thioester leaving groups on its substrate, which are usually installed by solution-phase coupling reactions during substrate synthesis. However, this step often generates epimerized products which are necessitates the purification of desired peptide from diastereomeric mixture, which is notoriously difficult and time-consuming. In this study, we have developed a streamlined chemoenzymatic approach to synthesize cyclic peptides that bypasses the need for leaving group installation in solution phase[1]. Linear peptides with diol ester functionalities on C-terminus were synthesized on a solid support. Cleavage of the resin-bound peptides yielded the diol esters with sufficient purity, which could be subjected for the subsequent enzymatic cyclization without further purification. The diol-activated peptides were efficiently cyclized in a head-to-tail manner by SurE, a representative penicillin-binding protein-type thioesterase which we previously discovered in biosynthesis of non-ribosomal peptide, surugamides[2-4]. Therefore, we established a streamlined chemoenzymatic approach for cyclic peptides. With this method, we synthesized a library of C-terminally-activated peptides. Using these as substrates, we elucidated the broad substrate specificity of SurE on the substrate N-terminus and the chain length. Notably, the introduction of non-peptidic residues (various length of poly-ethylene glycol) in the middle of the substrates was completely tolerated, suggesting that SurE catalysis could be widely applicable for the cyclization of non-peptidic polymers with the appropriate residues at the ring-closing junction. However, SurE had a limited scope for the substrate C-terminus. Specifically, it was able to cyclize substrates with neutral D-amino acids at the C-terminus, but was less effective with substrates containing acidic and basic residues at the C-terminus. To overcome this limitation, we explored homologous wild type enzymes. SSN analysis of more than 600 homologs as well as the investigating their substrate binding pockets suggested that this peptide cyclase family possess the diverse substrate scope. In particular, WolJ, a homolog of SurE was demonstrated to possess a unique specificity for substrate C-terminus that complements SurE’s specificity. Additionally, we were able to manipulate the specificity of SurE through protein engineering. G235L mutation completely changed SurE’s specificity on C-terminus from D-aa to Gly. These efforts have broadened the scope of the enzymatic macrolactamization[1]. Our study will potentially accelerate the exploitation of NRPS-TEs as biocatalysts. |
| References: | [1] M. Kobayashi, K. Fujita, K. Matsuda*, T. Wakimoto*, J. Am. Chem. Soc. 2023, 145, 3270-3275. [2] T. Kuranaga, K. Matsuda, A. Sano, M. Kobayashi, A. Ninomiya, K. Takada, S. Matsunaga, T. Wakimoto*, Angew. Chem., Int. Ed. 2018, 57, 9447-9451. [3] K. Matsuda, M. Kobayashi, T. Kuranaga, K. Takada, H. Ikeda, S. Matsunaga, T. Wakimoto*, Org. Biomol. Chem. 2019, 17, 1058-1061. [4] K. Matsuda, R. Zhai, T. Mori, M. Kobayashi, A. Sano, I. Abe*, T. Wakimoto*, T. Nat. Catal. 2020, 3, 507-515. |
| Figures: | 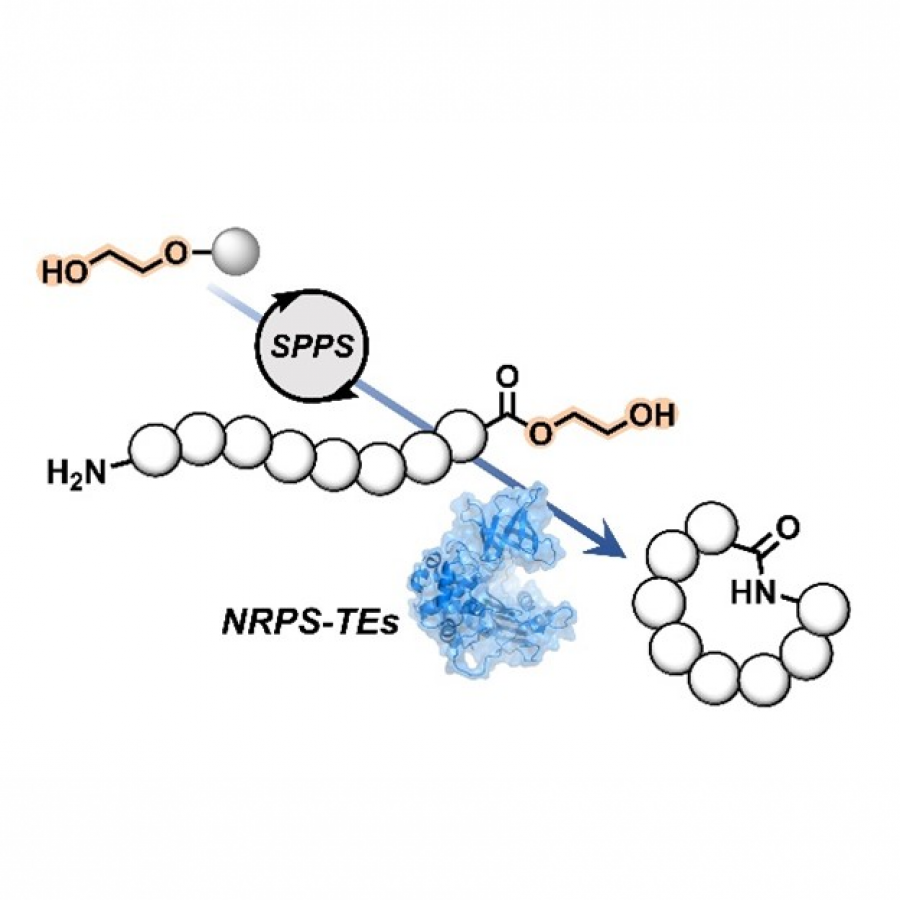 Figure 1 Diol-activated peptides were synthesized by SPPS and cyclized by NRP cyclases. |
#1696 | Combining Bio- and Organocatalysis for the Synthesis Of Piperidine Alkaloids. |
|
| Presenting author: | Adam O'CONNELL of UNIVERSITY COLLEGE DUBLIN |
| Corresponding author: | Elaine O'REILLY of UNIVERSITY COLLEGE DUBLIN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Organocatalysis / Transaminase / Piperidine Alkaloids / Mannich |
| Purpose: | 2-Subtituted piperidine alkaloids are a class of naturally occurring nitrogen-containing heterocyclic compounds that are key components in many traditional medicines, toxins, and modern-day substances of abuse.[1] Due to their low natural abundance and interesting biological properties, development of chemical routes towards the total synthesis of these highly stereospecific compounds has gained much interest. However, these pathways often require the use of expensive metal catalysts and environmentally un-friendly solvents.[2] To circumvent these undesirable components, the introduction of a biocatalytic step would be favorable as it provides a potentially greener way of producing alkaloids and their derivatives, using milder reaction conditions in aqueous solvents. Inspired by the work carried out in the Bella and Turner groups,[3,4] a hybrid bio-organocatalysed approach for the synthesis of 2-subtituted piperidines was developed involving a ω-transaminase enzyme (TA) and the organocatalyst L-proline. The TA mediates the conversion of cadaverine 1 to the reactive intermediate Δ1-piperideine 4, which then undergoes a Mannich-type reaction with an activated methyl ketone (Scheme 1). This novel approach utilises the ketone’s ability to take on a dual role within the cascade, acting as the transaminase acceptor substrate as well as the nucleophile in the Mannich-type reaction. Conversions of up to 75 % were achieved using this methodology and the preparative scale synthesis of the natural product (±)-pelletierine (60 % yield, 85 mg) is reported.[5] |
| References: | [1] B. Debnath, W. S. Singh, M. Das, S. Goswami, M. K. Singh, D. Maiti, K. Manna, Materials Today Chemistry, 2018, 9, 56-72. [2] A. Bari, A. Iqubal, Z. A. Khan, S. A. Shahzad, M. Yar, Syn. Comm., 2020, 50, 2572-2589. [3] M. R. Monaco, P. Renzi, D. M. Scarpino Schietroma, M. Bella, Organic Letters, 2011, 13, 4546-4549. [4] J. L. Galman, I. Shabu, F. Parmeggiani, N. J. Turner, Chem. Comm., 2018, 54, 11316-11319. [5] F. Taday, R. Cairns, A. O’Connell, E. O’Reilly, Chem. Comm., 2022, 58, 1697-1700. |
| Figures: | 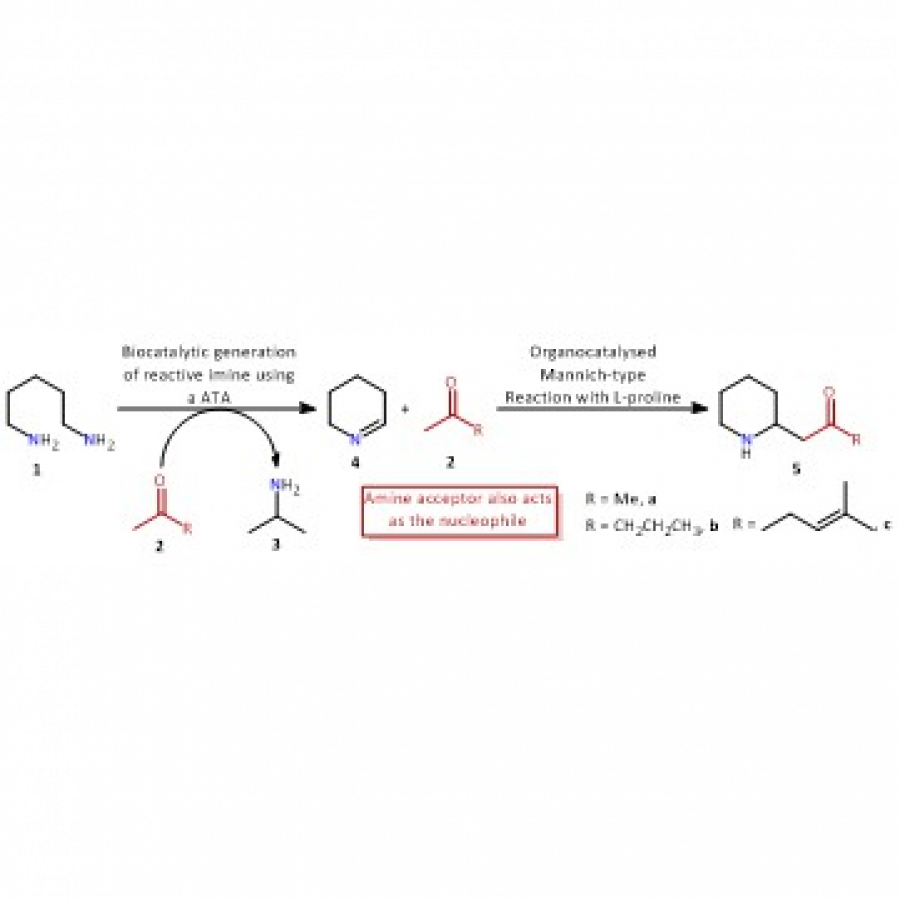 Hybrid bio-organocatalytic approach for the synthesis of 2-subtituted piperidines. |
#1698 | Metagenomic opine dehydrogenases – expanding the enzymatic toolbox for reductive aminations |
|
| Presenting author: | András TELEK of SERVIER |
| Corresponding author: | Gábor TASNÁDI of SERVIER |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / opine dehydrogenases / metagenome mining / reductive amination |
| Purpose: | Opine dehydrogenases (EC 1.5.1.X) are metabolic enzymes found in various species across the tree of life that perform the reductive coupling of pyruvate and amino acids yielding opine products[1]. These chiral secondary amine dicarboxylic acid derivatives represent a valuable class of compounds for the synthesis of bioactive molecules such as peptidomimetics[2]. However, the use of these enzymes as industrial biocatalysts for reductive amination is often restricted by their narrow substrate scope. The only reported example CENDH (N-(1-D-carboxyethyl)-L-norvaline dehydrogenase) was therefore extensively engineered to meet industrial needs [3]. Our study aimed at expanding this underexplored enzyme class by metagenome mining. Metagenomes from extreme environments are often able to provide enzymes with superior properties and altered substrate specificities[4]. Here we report the discovery of 6 novel metagenomic opine dehydrogenases (mODHs). These enzymes exhibit unique substrate specificity and higher thermostability compared to known examples (Figure 1). The feature that they preferably utilize negatively charged polar amino acids is so far unprecedented for opine dehydrogenases. While they still suffer from a relatively narrow substrate scope, their enhanced thermostability and the orthogonality of their substrate preference makes them a valuable addition to the toolbox of enzymes for reductive aminations. In addition, enzymatic reductive aminations with highly polar amines are very rare in the literature, our work contributes to filling that gap. |
| References: | [1] M. Sharma, J. Mangas-Sanchez, N. J. Turner, G. Grogan, Adv. Synth. Catal., 2017, 359, 2011-2025 [2] G. L. Petri, S. Di Martino, and M. De Rosa, J. Med. Chem., 2022, 65, 7438-7475 [3] H. Chen et al., 2013, US Patent 2013/0302859. [4] S. L. Robinson, J. Piel, S. Sunagawa, Nat. Prod. Rep., 2021, 38, 1994-2023 |
| Figures: | 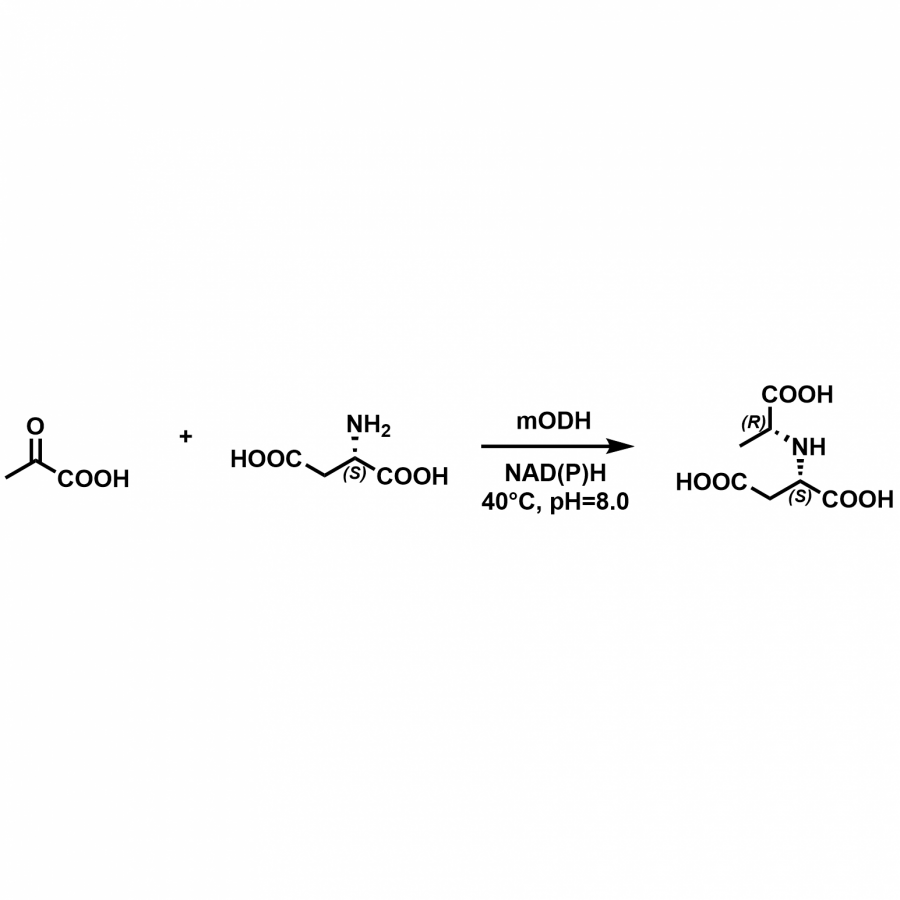 Figure 1 Representative reaction catalysed by the novel metagenomic opine dehydrogenases |
#1700 | Novel Myco-fabrication of Copper and Nickel Nanoparticles and Evaluation of Their Effects Against Antibiotic Resistance Genes in Different Bacterial Strains and Anticancer Potentials |
|
| Presenting author: | EL-SAYED R. EL-SAYED of DEPARTMENT OF FOOD CHEMISTRY AND BIOCATALYSIS, FACULTY OF BIOTECHNOLOGY AND FOOD SCIENCE, WROCŁAW UNIVERSITY OF ENVIRONMENTAL AND LIFE SCIENCES |
| Other authors: | Aisha S. A. AHMED of DEPARTMENT OF CHEMISTRY, FACULTY OF SCIENCE, CAIRO UNIVERSITY Heba K. ABDELHAKIM of DEPAERTMENT OF CHEMISTRY, FACULTY OF SCIENCE, CAIRO UNIVERSITY Zainab ZAKARIA of DEPARTMENT OF CHEMISTRY, FACULTY OF PHARMACY, HELIOPOLIS UNIVERSITY FOR SUSTAINABLE DEVELOPMENT ISMAIL A. ABDELHAMID of DEPAERTMENT OF CHEMISTRY, FACULTY OF SCIENCE, CAIRO UNIVERSITY |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biofabrication / Nanoparticles / Reduction and Transformation / Antibacterial and Anticancer |
| Purpose: | In the current scenario, developing new compounds with innovative modes of action is desperately needed to tackle the increased emergence of drug-resistant microbes [1]. Recently, metallic nanoparticles gained tremendous attention as potential antibacterial agents [2]. Here, we describe the fabrication of copper and nickel nanoparticles by reducing copper sulfate and nickel sulfate using the endophytic fungus Aspergillus terreus, as a potentially simple and eco-friendly method with low cost. Generally, the microbial synthesis of nanomaterials compared to chemical or physical ones is an attractive and emerging prospect for future sustainable industrial production of nanomaterials. copper and nickel nanoparticles were characterized by Fourier transform infrared spectroscopy. X-ray diffraction patterns revealed their crystalline structure. Dynamic light scattering analysis was applied to study the particle size distribution and stability. Transmission electron microscope studies indicated the morphology of the synthesized NPs. The in vitro antibacterial potentials of copper and nickel nanoparticles were evaluated against five MSRA bacterial strains. Additionally, the in vitro anticancer potentials of copper and nickel nanoparticles were assessed against four types of cell lines; Normal human 14 melanocytes (HFB-4), Human breast carcinoma (MCF-7), Hepatocellular carcinoma (HePG-2) and Pulmonary epithelial cell carcinoma (A549). The obtained results confirmed the activity of the two types of nanoparticles against all the tested cell lines. -Acknowledgment: The presented research was supported in part by the BioExplor project No. 2021/43/P/NZ9/02241 co-funded by the National Science Centre and the European Union Framework Programme for Research and Innovation Horizon 2020 under the Marie Skłodowska-Curie grant agreement no. 945339. |
| References: | [1] Giamarellou, H., G. Poulakou, Multidrug-resistant Gram-negative infections: what are the treatment options? Drugs. 2009. 69(14): p. 1879-901. [2] Mousa, S. A., El-Sayed, E. R., Mohamed, S. S., Abo El-Seoud, M. A., Elmehlawy, A. A., Abdou, D. A. M. Novel mycosynthesis of Co3O4, CuO, Fe3O4, NiO, and ZnO nanoparticles by the endophytic aspergillus terreus and evaluation of their antioxidant and antimicrobial activities. Appl. Microbiol. Biotechnol. 2021. 105:741-753. |
| Figures: |  Fabrication of Nanoparticles. 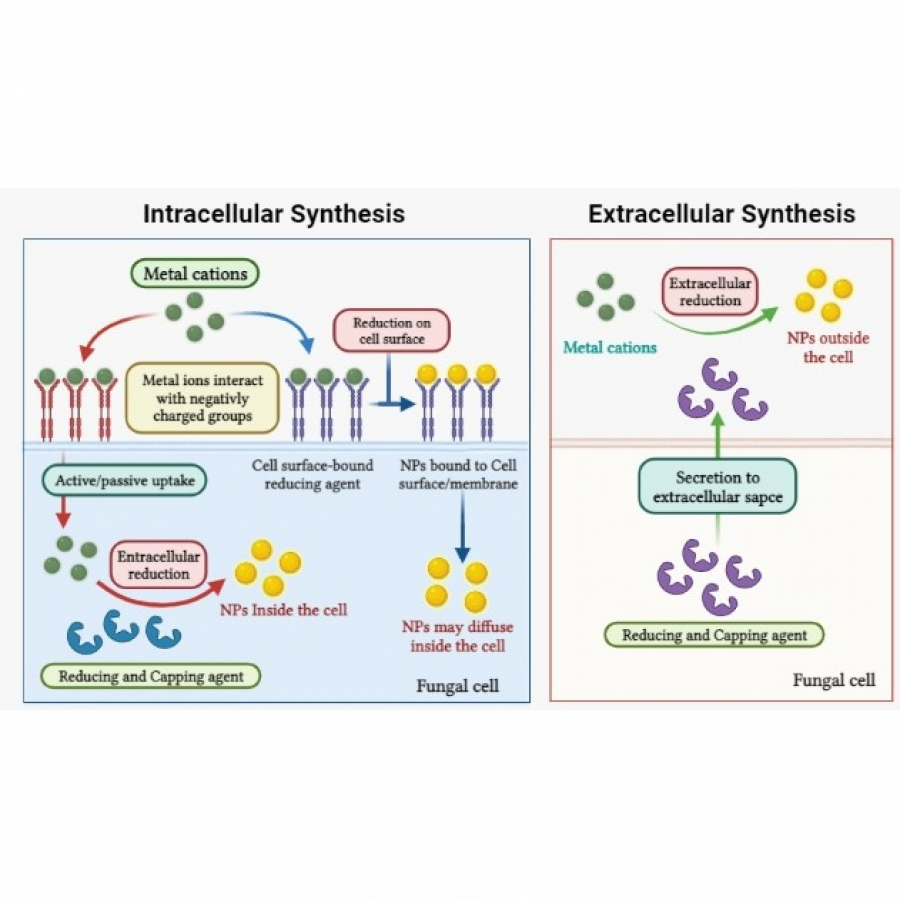 Schematic representation of the cellular process involved in the fabrication of copper and nickel nanoparticles |
#1701 | Identification and Optimization of a Metagenomic Esterase with Hydrolytic Activity towards Polylactic Acid |
|
| Presenting author: | Eva JOSIĆ of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA |
| Corresponding author: | Aleksandra MARŠAVELSKI of THE UNIVERSITY OF ZAGREB, THE FACULTY OF SCIENCE |
| Other authors: | Marko MOČIBOB of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA Ivana KEKEZ of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA Margaux AUBEL of INSTITUTE FOR EVOLUTION AND BIODIVERSITY, UNIVERSITY OF MÜNSTER Erich BORNBERG-BAUER of INSTITUTE FOR EVOLUTION AND BIODIVERSITY, UNIVERSITY OF MÜNSTER Aleksandra MARŠAVELSKI of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | polylactic acid (PLA) / bioplastics / biodegradation / circular economy |
| Purpose: | Polylactic acid (PLA) is a biodegradable polyester with high potential for applications in many fields.[1] The low biodegradability of PLA in natural environments limits its use as a sustainable alternative to conventional plastics. However, microbial enzymes offer a promising strategy to enhance the biodegradation of PLA. Accordingly, we applied a metagenomic approach to identify a novel esterase enzyme, capable of cleaving PLA. A search of metagenomic sequence databases identified an esterase, and its activity towards PLA was demonstrated using emulsified PLA in an agarose test.[2] Subsequent crystallographic studies to obtain the enzyme's structure for further optimization resulted in multiple crystal hits. The structure determination process is ongoing, with initial results indicating the enzyme belongs to the α/β-hydrolase family. To optimize the enzyme's stability, ancestral sequence reconstruction was applied to obtain more robust variants stable at higher temperatures. These variants will be ordered as synthetic genes, expressed, and tested for improved thermostability and activity. (Figure 1). |
| References: | [1] rosenboom, jg., langer, r. and traverso, g. nat. rev. mater. 2022, 7, 117-137. [2] teeraphatpornchai t., nakajima-kambe t., shigeno-akutsu y., nakayama m., nomura n., nakahara t., uchiyama h. biotechnol. Lett. 2003, 25, 23-8. |
| Figures: | 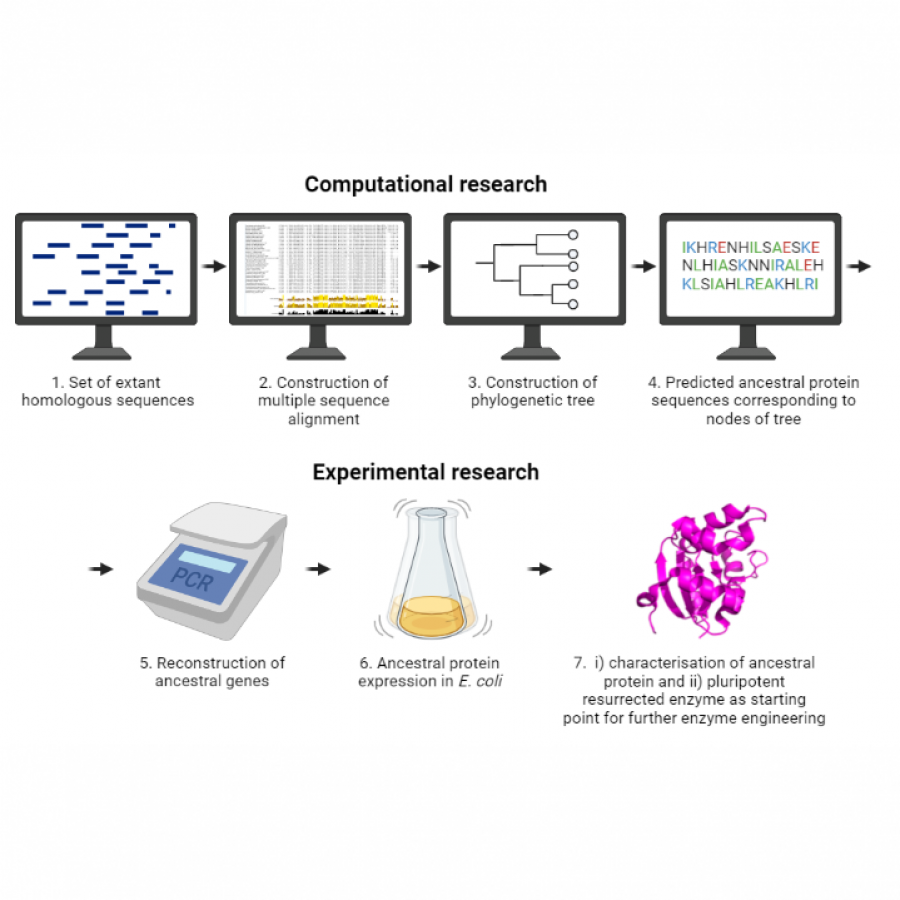 Figure 1 A complete scheme of the conducted research. |
#1702 | Allostery in Purine Nucleoside Phosphorylases Revealed by a Combination of Protein Crystallography and Molecular Dynamics Simulations |
|
| Presenting author: | Aleksandra MARŠAVELSKI of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB, CROATIA |
| Corresponding author: | Zoran ŠTEFANIĆ of RUĐER BOŠKOVIĆ INSTITUTE |
| Other authors: | Boris GOMAZ of RUĐER BOŠKOVIĆ INSTITUTE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | allostery / Purine Nucleoside Phosphorylases / protein crystallography / molecular dynamics simulations |
| Purpose: | Enzymes purine nucleoside phosphorylases (PNPs) [1] have been chosen as a model system for investigating allostery due to several compelling reasons. First, PNPs play a critical role in the purine salvage pathway by catalyzing the synthesis of purine nucleotides. This pathway is essential for maintaining cellular homeostasis and is conserved across various organisms. Second, PNPs exist in different oligomeric forms, such as homohexamers in bacteria and homotrimers in higher organisms, providing an opportunity to study the influence of the oligomeric state on allosteric behavior. Third, the structural and functional diversity observed in PNPs, coupled with the presence of allosteric binding sites, suggests the involvement of intricate communication networks within the protein. Investigating the allosteric mechanisms in PNPs can provide valuable insights into the general principles of allostery and aid in the design of novel strategies for modulating enzyme activity. To fully comprehend the intricacies of allostery, which is fundamentally a dynamic phenomenon, it becomes imperative to move beyond the static structures obtained from X-ray crystallography and delve into the dynamic aspects using molecular dynamics simulations. Allosteric communication within proteins relies on non-covalent interactions between amino acids, and thus, unraveling the allosteric pathways necessitates an examination of the evolving interaction networks over time. Introducing time evolution through molecular simulations adds complexity that can only be managed by programmatic approaches capable of handling the vast quantities of data generated. In this context, machine learning methods emerged as invaluable tools, offering robust capabilities tailored specifically for processing and analyzing such voluminous datasets. By combining molecular dynamics simulations, programmatic data processing, and machine learning techniques, we started to uncover the intricate details of allosteric pathways in PNPs and gain deeper insights into the fundamental principles underlying protein function and regulation. This research is part of the ongoing project Allosteric communication pathways in oligomeric enzymes (ALOKOMP, https://alokomp.irb.hr/, financed by Croatian Science Foundation grant no. IP-2019-04-6764). |
| References: | [1] narczyk m., wojtyś m. i., leščić ašler i., žinić b., luić m., jagusztyn-krynicka e. k., štefanić z., bzowska a. J Enzyme Inhib Med Chem. 2022, 37, 1083-1097. |
| Figures: | 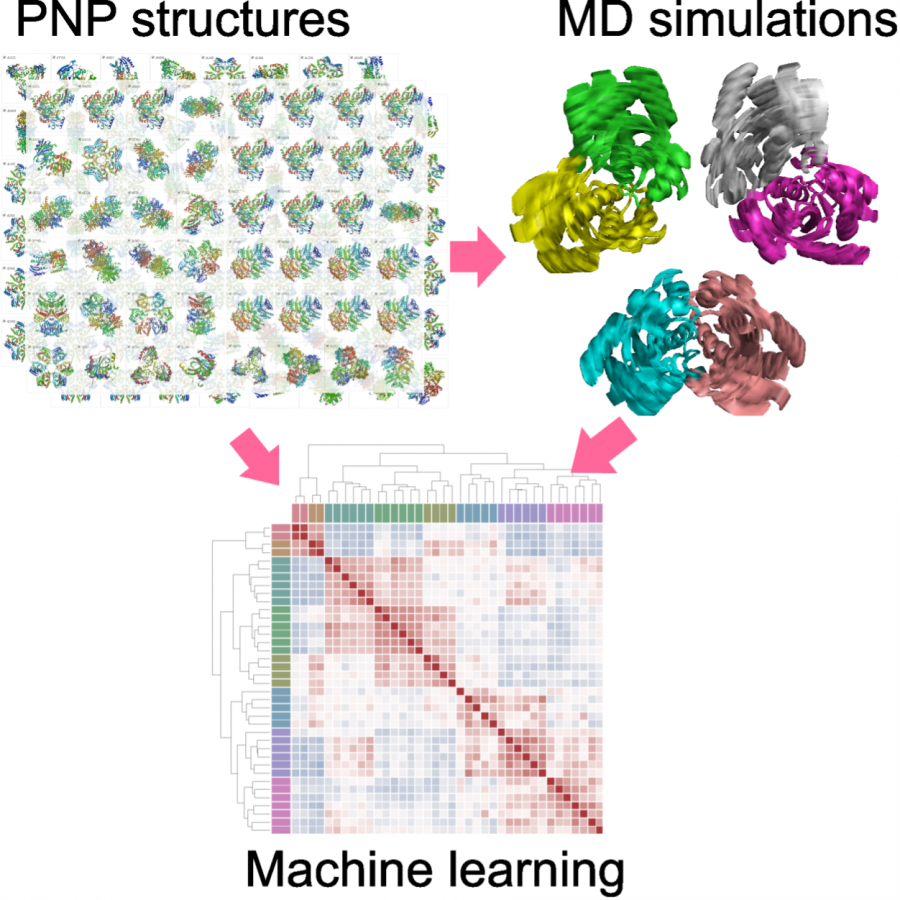 FIGURE 1 A scheme of the conducted research |
#1705 | Iterative metagenome mining leads to expanded substrate scope of the LEH for biocatalytic epoxide opening |
|
| Presenting author: | Bethany HOGG of UNIVERSITY OF MANCHESTER |
| Corresponding author: | Nicholas TURNER of UNIVERSITY OF MANCHESTER |
| Other authors: | Emily KEMPA of ASTRAZENECA Charlotte MORRILL of UNIVERSITY OF MANCHESTER Rhys BARKER of UNIVERSITY OF MANCHESTER James FINNIGAN of PROZOMIX LTD Martin HAYES of ASTRAZENECA Christian SCHNEPEL of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | metagenome mining / biocatalysis / LEH / Epoxide Openeing |
| Purpose: | Recently, the use of metagenome mining to discover novel enzymes has become of increasing interest in biocatalysis. The process has been integral in accelerating the field through identification of enzymes for specific molecule synthesis, particularly drug target synthesis, and exploring sequence space. Interrogating genomic data from environmental samples and comparing these against known, previously characterised sequences through a bioinformatics platform has previously allowed for screening and characterisation of a 384-panel of imine reductases, thus offering a broad screening tool for the synthesis of chiral amines. More recently, this method is being applied to find novelty in well-studied enzyme classes, in the early stage functionalisation of high value intermediates. In this work, we describe the discovery of more than 50 novel limonene epoxide hydrolase (LEH) enzymes from interrogating metagenomic data, adding to this small subset of the epoxide hydrolase (EH) family. Furthermore, we discovered that some candidates demonstrated high activity towards a vastly different substrate scope and displayed good tolerance for more sterically hindered epoxides. These ‘hit’ enzymes introduce novelty to a well-studied but poorly classified enzyme class. Computational studies rationalised the high activity of two both LEHs in particular, which prompted structural analysis by X-ray crystallography revealing substrate and product binding modes which are currently harnessed for further enzyme engineering. A second search of the metagenomic data using the most active LEHs from the first panel as reference points generated a second panel of homologs, which possessed more active and enantioselective candidates for the aforementioned substrate scope. This shows that iterative metagenome mining can be used to walk along protein sequence space and be a powerful, complementary tool in directed evolution and biocatalysis. |
| References: | [1] M. Ferrer, M. Mart?nez-Mart?nez, R. Bargiela, W. R. Streit, O. V. Golyshina, P. N. Golyshin, Microb. Biotechnol. 2016, 9, 22-34. [2] D. Baud, J. W. E. Jeffries, T. S. Moody, J. M. Ward, H. C. Hailes, Green Chem. 2017, 19, 1134-1143. [3] L. Leipold, D. Dobrijevic, J. W. E. Jeffries, M. Bawn, T. S. Moody, J. M. Ward, H. C. Hailes, Green Chem. 2019, 21, 75-86. [4] J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, et al., Nat. Chem. 2021, 13, 140-148. |
| Figures: | 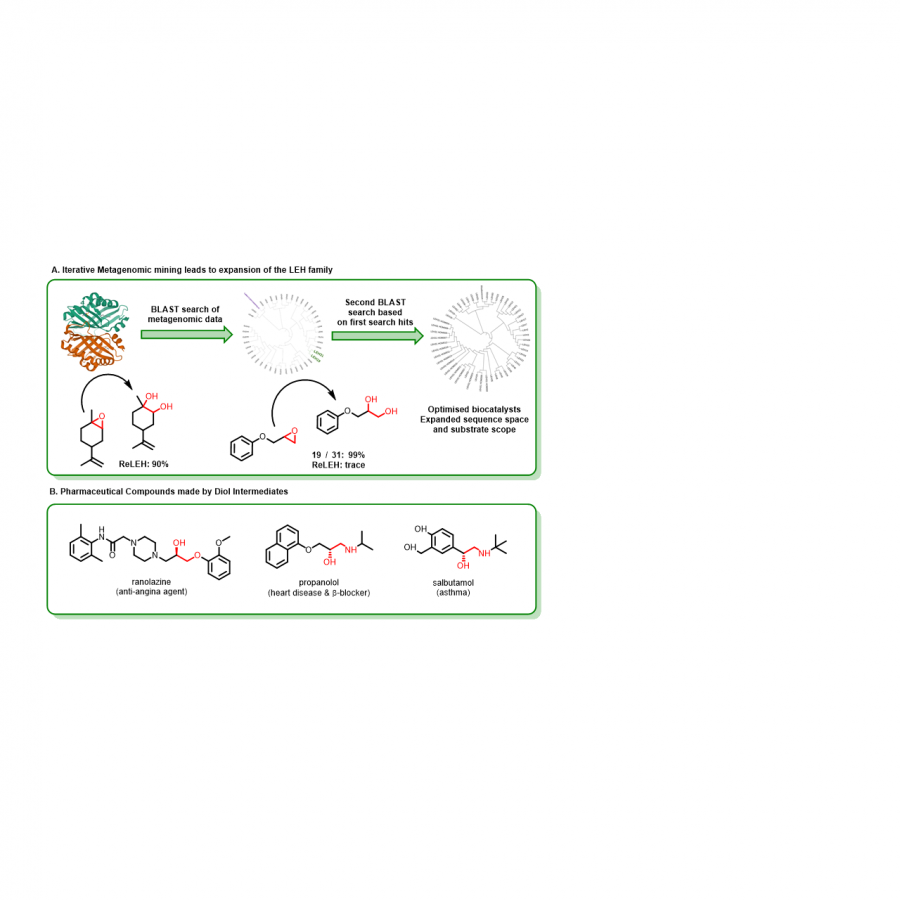 Figure 1 Iterative metagenome mining leads to expanded
enzymes families and novel biocatalysts. 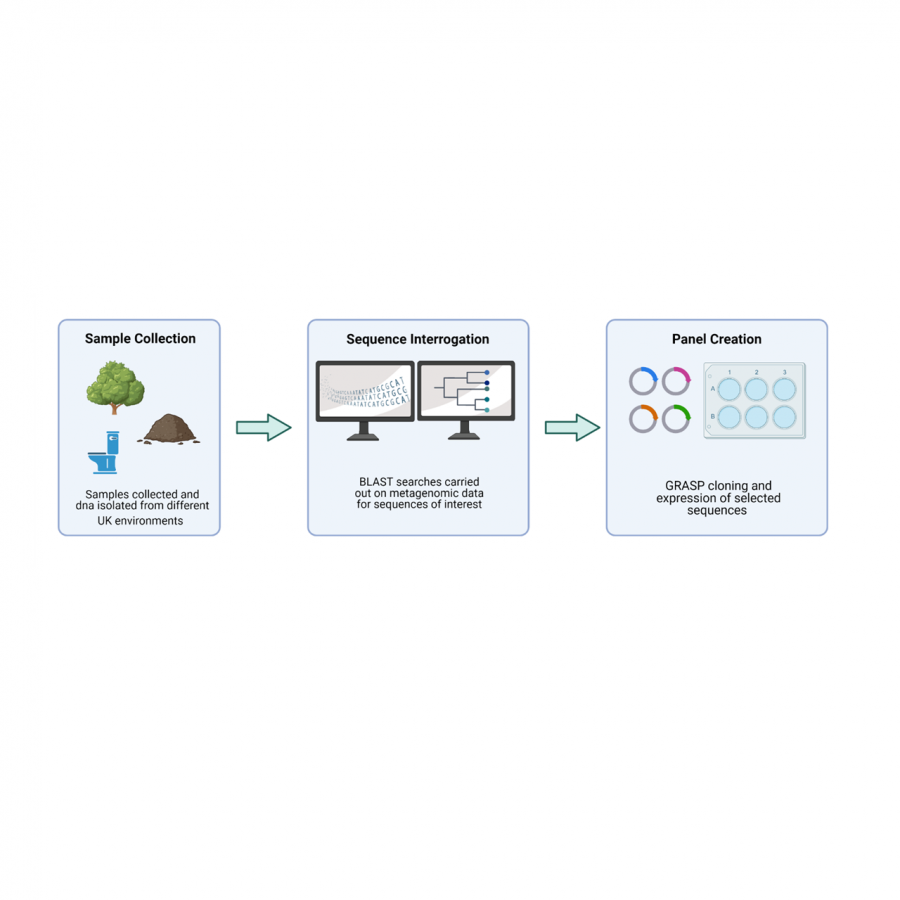 Figure 2 Flowchart showing metagenomic panel creation from
the initial round of mining metagenomic data. |
#1709 | Development of Convergent Biocatalytic Transformations for the Synthesis of Complex Alkaloids |
|
| Presenting author: | Amber BARRY of UNIVERISTY COLLEGE DUBLIN |
| Corresponding author: | Elaine O'REILLY of UNIVERSITY COLLEGE DUBLIN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Bio-organocatalytic / Transaminase / Quinolizidine alkaloid / Cascade |
| Purpose: | Biocatalysis involves the use of catalytic proteins (enzymes) to perform chemical transformations of simple, cheap, achiral materials into high value, chiral products. Nature uses enzymes, often in cascade approaches, to synthesise important building blocks and to assemble complex natural products. The selectivity of enzymes, their tuneability and compatibility with aqueous solvents, have led to their use in the pharmaceutical and fine chemical industry, offering a more attractive alternative to traditional catalysts and as an important synthetic tool in target retrosynthesis. [1] Biocatalysis has been suggested as a greener approach to synthesis, following some of the 12 Principles of Green Chemistry, including mild reaction conditions, production of less waste and the inherent biodegradability of enzymes. Biocatalysts enable more sustainable synthetic routes to access synthetically challenging compounds, offering high levels of chemo-, regio- and stereoselectivity. [2] Alkaloids are a class of naturally occurring nitrogen-containing organic compounds that are known to exhibit many physiological effects, including anticancer and neuroprotective properties. [3,4] Synthetic methodologies accessing natural product N-heterocycles structural motifs and their derivatives is of interest to chemists due to the important therapeutic properties they possess and difficulties in extracting sufficient quantities from natural sources. Previous chemical synthetic routes developed to access such structures often require expensive precious metal catalysts and non-environmentally friendly reaction conditions. The design of a hybrid bio-organocatalytic cascade was envisaged to enable assembly of complex quinolizidine alkaloid structural scaffolds (Figure). Lythraceae alkaloids are a plant derived class of quinolizidine-type alkaloids, [1] containing a 4-arylquinolizidine structural motif 1–6. Alkaloids from this class have shown biological activity, possessing anti-inflammatory, diuretic, sedative, antimalarial, antifungal and antithrombotic properties. The biomimetic cascade approach is initiated by a transaminase catalysed transformation of cadaverine 1 into the naturally occurring reactive cyclic imine, Δ1-piperideine 4, [5] which can subsequently undergo an L-proline 6 facilitated Mannich-aza-Mannich reaction, with an aryl enone 5, forming the 4-arylquinolizidine-2-one scaffold 7, [6] a key structural intermediate in the proposed cascade. The alkaloid 7 has the potential to be further derivatised using additional enzymes, such as a transaminase or alcohol dehydrogenase to generate the amine 8 or alcohol 9 products respectively, providing a chemical handle to further react. The cascade has the potential to be significantly expanded by using alternative enones and incorporating additional enzymes to further functionalise the products, enabling access to a panel of natural products. The stereoselectivity and chemoselectivity of enzymes can enable a one-pot approach under mild conditions, forming complex alkaloid structures. BiOrbic is funded under the Science Foundation Ireland Research Centres Programme and is co-funded under the European Regional Development Fund. The A2P CDT is supported by Science Foundation Ireland (SFI) and the Engineering and Physical Sciences Research Council (EPSRC) under the Grant No. 18/EPSRC-CDT/3582 |
| References: | [1] E. L. Bell et al., Nat. Rev. Methods Primers, 2021, 1, 1, 46. [2] C. M. Heckmann and F. Paradisi, ChemCatChem, 2020, 12, 6082-6102. [3] B. R. Lichman, Nat. Prod. Rep., 2021, 38, 103-129. [4] J. Kurek, ed. Alkaloids: their importance in nature and human life. BoD, 2019, IntechOpen [5] F. Taday et al., Chem. Commun., 2022, 58, 1697-1700. [6] S. Virk and S. V. Pansare, Org. Lett., 2019, 21, 14, 5524-5528. |
| Figures: | 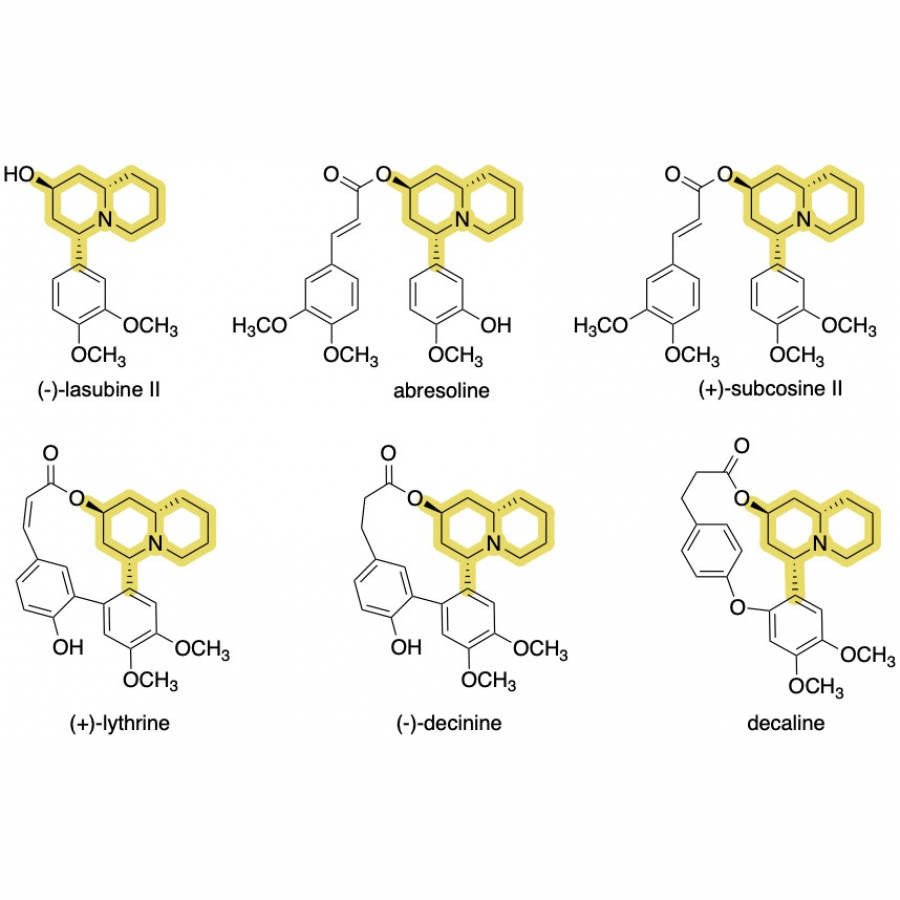 Plant derived Lythraceae alkaloids containing 4-arylquinolizidine ring structure motif. Plant derived Lythraceae alkaloids containing 4-arylquinolizidine ring structure motif. 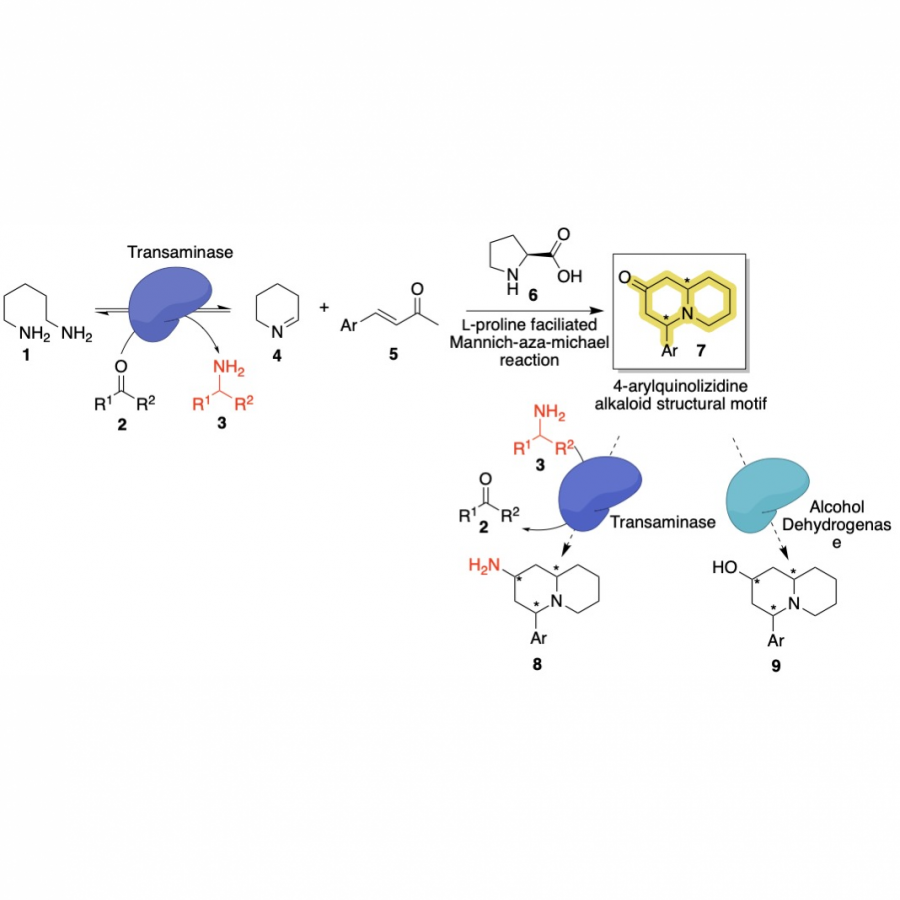 Proposed hybrid bio-organocatalytic route for the synthesis of quinolizidine alkaloids. Transformation of cadaverine 1 to Δ1-piperideine 4, to react with aryl enone 5, forming the key intermediate 7, with further derivatization |
#1712 | Reversal of stereoselectivity in ERED-catalyzed reduction through a smart positioning of double bond |
|
| Presenting author: | Laura LEEMANS MARTIN of NOVARTIS PHARMA AG |
| Corresponding author: | Frederic STANGER of NOVARTIS PHARMA AG |
| Other authors: | David ENTWISTLE of CODEXIS INC Kirsten SCHROER of NOVARTIS PHARMA AG Theo PESCHKE of NOVARTIS PHARMA AG Greg MANN of NOVARTIS PHARMA AG Nhat Quang NGUYEN of NOVARTIS PHARMA AG Oscar ALVIZO of CODEXIS INC Thierry SCHLAMA of NOVARTIS PHARMA AG Herve KOEHL of NOVARTIS PHARMA AG Franziska GRASSINGER of NOVARTIS PHARMA AG Philippe DREIER of NOVARTIS PHARMA AG Oemer ERDOGAN of NOVARTIS PHARMA AG Nathalie BURRER of NOVARTIS PHARMA AG Lorenzo PICCIONI of NOVARTIS PHARMA AG Leann TEADT of CODEXIS INC |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Ene-reductase / Protein engineering / Process development / |
| Purpose: | Biocatalysis allows the environmental-friendly and cost-effective synthesis of intermediates or pharmacologically relevant compounds and is of particular interest for chiral molecules. Ene-reductases (EREDs) catalyze the asymmetric hydrogenation of alkenes in a highly stereoselective manner. We screened a large enzyme panel for the selective reduction of several key intermediates. Depending on the localization of the unsaturated C=C bond (internal vs external), we identified EREDs able to catalyze the formation of either the (R) or the (S) enantiomer with high enantiomeric excess (ee). Combining protein engineering and process development, we obtained an ERED able to fully convert the substrate to the desired product with great selectivity (> 98% ee) at lab scale. |
| References: | Laura LEEMANS MARTIN / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Frederic STANGER / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Nhat Quang NGUYEN / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Greg MANN / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Theo PESCHKE / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Kirsten SCHROER / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Nathalie BURRER / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Franziska GRASSINGER / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Lorenzo PICCIONI / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Oemer ERDOGAN / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Herve KOEHL / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Philippe DREIER / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Thierry SCHLAMA / NOVARTIS PHARMA AG, CHEMICAL & ANALYTICAL DEVELOPMENT, BASEL Oscar ALVIZO / CODEXIS INC, REDWOOD CITY, REDWOOD CITY David ENTWISTLE / CODEXIS INC, REDWOOD CITY, REDWOOD CITY Leann TEADT / CODEXIS INC, REDWOOD CITY, REDWOOD CITY |
#1718 | Regio- and Stereo-Selective Amination of Fatty Acids to D-Amino Acids by a Three-Step One-Pot Cascade |
|
| Presenting author: | Zhijun ZHANG of EAST CHINA UNIVERSITY OF SCIENCE AND TECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Fatty acid / C-H bond functionalization / Enzymatic cascade reaction / D-amino acids |
| Purpose: | Biocatalytic regio- and stereo-selective functionalization of renewable fatty acids for sustainable synthesis of value-added chiral chemicals is highly desirable but remains a great challenge. Herein, a three-step one-pot multienzyme cascade for the asymmetric synthesis of D-amino acids from renewable fatty acids was developed. Combination of P450 peroxygenase with two enantiocomplementary hydroxyacid oxidase(s) enabled the regioselective oxyfunctionalization of fatty acids into prochiral α-ketoacids with internal H2O2 recycling. An engineered D-amino acid dehydrogenase with formate dehydrogenase for self-recycling of the expensive cofactor NADPH was adopted for the reductive amination of α-ketoacids to D-amino acids. Various fatty acids (C6-C10) with different chain length can be efficiently converted into the corresponding D-amino acids with high yield (up to 99%) and excellent ee value (>99%). This study exploits the advantage of cascades and showcases the potential for synthesizing valuable chiral chemicals from inexpensive renewable feedstocks. |
| References: | Xing Yu, Xin-Yi Chen, Hui-Lei Yu, Jian-He Xu, Zhi-Jun Zhang*. Regio- and stereo-selective amination of fatty acids to D-amino acids by a three-step one-pot cascade. Green Chem. 2023, 25: 3469-3474. |
#1719 | Rational Design of Carbonyl Reductase CgKR1 Based on Molecular Simulation Methods |
|
| Presenting author: | Qi CHEN of EAST CHINA UNIVERSITY OF SCIENCE AND TECHNOLOGY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | ketoreductase / residue interaction networks / rational design / thermal stability |
| Purpose: | Chiral alcohols are important chiral blocks for the synthesis of chiral drugs, fine chemicals, etc. and have high application value. Carbonyl asymmetric reduction is an important method for the biocatalytic synthesis of chiral alcohols, which has the advantages of mild reaction conditions, environmental friendliness and better stereoselectivity. In our previous work, the carbonyl reductase CgKR1-F92C/F94W from Candida glabrata (as the starting parent M0) was discovered with high catalytic activity and stereoselectivity. However the unsatisfying thermal stability has become the bottleneck for its practical application. Here in this work, we focused on computer-based rational design of the enzyme to address the problem of its low thermal stability. Since the interactions between residues in the protein structure play an important role in maintaining the thermal stability of proteins. The RINVES (Residue Interaction Network-based Virtual Design for Enzyme Stability) strategy based on residue interaction networks was constructed. Firstly, three-dimensional structure of proteins was converted into a two-dimensional network with residues as nodes and inter-residue interaction forces as edges. A virtual mutation library with site-saturated mutations was then established, and the changes in the topology and other network parameters were calculated and investigated to select mutations that were beneficial to the stability improvement. Using this virtual screening strategy, the best mutation CgKR1-M3 with significantly improved thermal stability was obtained, representing a 62.3-fold increase in t1/2 at 50 °C and a 13 °C increase in T15 50 compared to the parent, and maintained a high viability. The structural mechanism of the enhanced thermal stability of the mutant was further investigated based on molecular dynamics simulation analysis. Subsequently, the optimal mutant CgKR1-M3 were coupled with glucose dehydrogenase to achieve a coenzyme cycle, resulting in the asymmetric reduction of 100 g L-1 of N-Boc-3-piperidone. 95% conversion of the mutant CgKR1-M3 was achieved in about 4 h, whereas the parent M0 was only converted to 56% in the end due to rapid inactivation. Validation of the reaction showed that the resulting dominant mutant strain was able to efficiently synthesize (S)-N-Boc-3-hydroxypiperidine, a key chiral intermediate of the anticancer drug ibrutinib, with promising applications. |
| References: | [1] Ma H, Yang L, Ni Y, Zhang J, Li CX, Zheng GW, Yang H, Xu JH. Stereospecific reduction of methyl o-chlorobenzoylformate at 300 g⋅L-1 without additional cofactor using a carbonyl reductase mined from Candida glabrata. Advanced Synthesis & Catalysis, 2012, 354(9): 1765-1772. [2] Huang L, Xu JH, Yu HL. Significantly improved thermostability of a reductase CgKR1 from Candida glabrata with a key mutation at Asp 138 for enhancing bioreduction of aromatic α-keto esters. Journal of Biotechnology, 2015, 203: 54-61. [3] Zheng GW, Liu YY, Chen Q, Huang L, Yu HL, Lou WY, Li CX, Bai YP, Li AT, Xu JH. Preparation of structurally diverse chiral alcohols by engineering ketoreductase CgKR1. ACS Catalysis, 2017, 7(10): 7174-7181. [4] Gong XM, Qin Z, Li FL, Zeng BB, Zheng GW, Xu JH. Development of an engineered ketoreductase with simultaneously improved thermostability and activity for making a bulky atorvastatin precursor. ACS Catalysis, 2019, 9(1): 147-153. |
#1721 | De novo design of a multifunctional thermostable protein catalyst with a novel fold |
|
| Presenting author: | Wael ELAILY of INSTITUTE OF BIOCHEMISTRY, TU GRAZ |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | De novo enzyme design / Thermostable protein / Multifunctional enzyme / Rosetta software |
| Purpose: | The design of a stable de novo protein scaffold that can be functionalized to catalyze a wide range of reactions is a promising but challenging task. This study aims to use the Rosetta software to parametrically design a novel thermostable helix bundle with the ability to accommodate an active site. Our initial design (6H5L) forms a helical barrel structure with a hydrophobic channel and is comprised of six straight antiparallel helices, which are connected by loops (Figure 1). As a first reaction model, we designed several variants to catalyze the retro-aldol reaction, which has extensive application in biocatalysis and could be extended to perform various non-native carboligation reactions. All variants are readily produced in E. coli and characterized using biochemical and biophysical methods including circular dichroism spectroscopy (CD), UV-Vis and fluorescence spectroscopy, SAXS analysis, and X-ray crystallography studies. The CD spectra of the designs confirm their alpha-helical fold and, moreover, a thermal stability up to 95°C with only minimal loss of signal at high temperatures and complete refolding after cooling back to 20°C. To improve protein production, we redesigned the protein’s surface using ProteinMPNN, which increased protein production 10-fold. Alphafold2 showed a high predicted IDDT score of over 90 for the originally designed sequence and most variants. To test retro-aldolase activity, we performed an inhibition reaction assay with a naphthalene diketone derivative which reacts with a nucleophilic lysine residue and forms an imine intermediate that can be monitored photometrically. The retro-aldol reaction of our designs with 4-hydroxy-4-(6-methoxy-2-naphthyl)-2-butanone (methodol) as a substrate was followed by measuring the increase of fluorescence upon product formation. 6H5L and all its variants showed noticeable retro-aldolase activity. To increase the activity, we used rational and computational design approaches. The best variant showed a 10-fold activity increase compared to the initial design (Figure 2). SAXS measurements of all variants gave an overall good fit between the measured and the calculated scattering profiles with low chi² values. We were able to determine the crystal structures of the apo design (2.2 Å) and a variant with the covalently bound ligand (naphthalene diketone derivative, 3.0 Å). To demonstrate the multifunctional use of our designs, we assayed them for other reactions and found some variants which showed notable Michael-addition activity and good PLP binding affinity. We are currently working on improving all catalytic activities. |
| Figures: |  6H5L design Structure representation of 6H5L in side view. 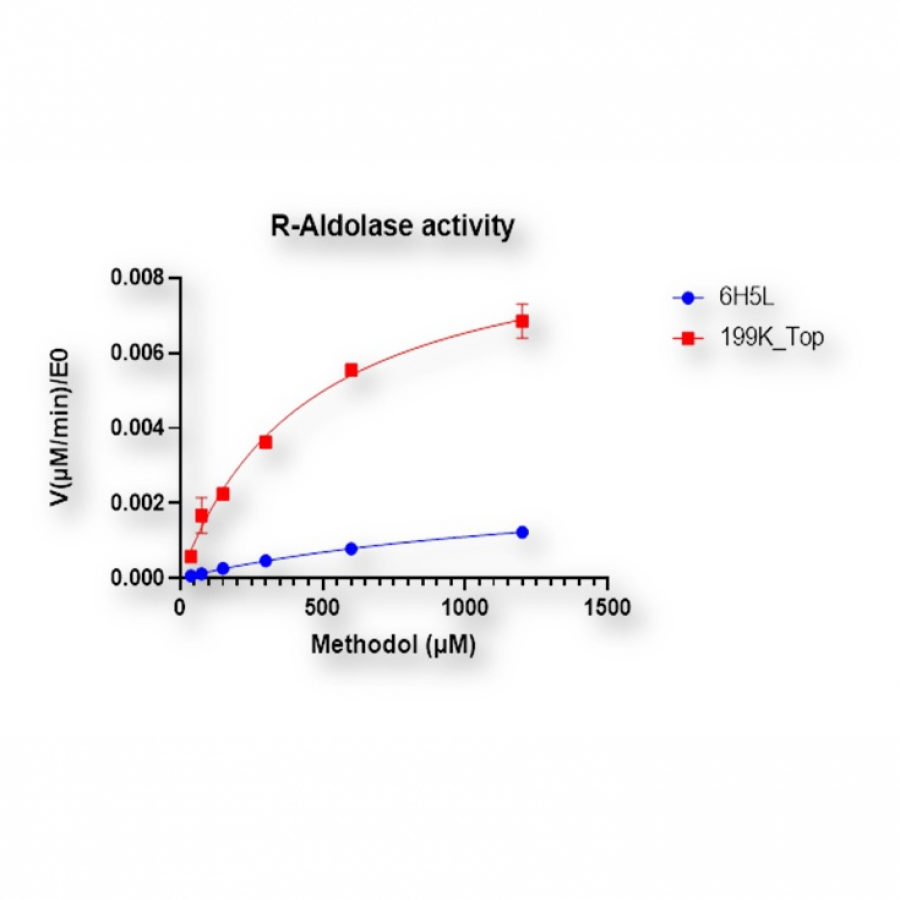 Retro-aldolase activity Michaelis Menten plots of 6H5L and 199K_Top variant with Methodol as a substrate. E0, total enzyme concentration; V, initial reaction velocity. |
#1724 | Regioselective Ring-Opening Reactions with Non-Natural Substrates using Engineered Halohydrin Dehalogenase |
|
| Presenting author: | Xinhang YANG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO |
| Corresponding author: | Haibin CHEN of ENZYMASTER (NINGBO) BIO-ENGINEERING CO |
| Other authors: | Xiao LUO of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Fenshuai SUN of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Junxia ZHAO of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Kuifang HE of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Qinli PENG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Ruimei HONG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Marco BOCOLA of ENZYMASTER DEUTSCHLAND GMBH Neha VERMA of ENZYMASTER DEUTSCHLAND GMBH Hao YANG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Thomas DAUSSMANN of ENZYMASTER DEUTSCHLAND GMBH Osama MAHMOUD of ENZYMASTER DEUTSCHLAND GMBH |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme Evolutions / BioEngine / Halohydrin Dehalogenase / Styrene Oxide |
| Purpose: | Epoxides are important building blocks for chemical and pharmaceutical synthesis[1], and many of their ring-opening derivatives have been used as chiral building blocks or auxiliary agents such as chiral diols[2], halohydrins, glycinols[3], and Evans-type auxiliaries[4]. The conventional methods for epoxide ring-opening suffers from poor regioselectivity, which poses a challenge for the direct utilization of epoxide derivatives. Halohydrin dehalogenases (HHDH) are industrially relevant enzymes that catalyze the reversible dehalogenation of vicinal haloalcohols, yielding the corresponding epoxides[5]. In the reverse reaction, non-native nucleophiles like NaOCN[6] and NaSCN[7] may lead to the enzymatic SN2 ring-opening and spontaneous ring re-closing processes for desired products such as oxazolidines. As a result, such non-native reactions would give 100% conversion. BioEngine® is an integrated directed enzyme evolution platform that offers the full-spectrum solution from enzyme discovery, enzyme engineering, process development, all the way to qualified product manufacturing. Powered with proprietary data collection as well as BioNavigator® toolbox and EM2L toolbox, Enzymaster’s BioEngine® platform delivers biocatalytic solution effectively and efficiently. From our research, a novel HHDH has been engineered by computer-aided direct enzyme evolution in our enzyme engineering lab which enables highly regio- and stereo-selective synthesis of Evans-type auxiliary reagents and other chiral glycinols at high substrate loading. For our engineered HHDH variants, their substrate tolerance has been improved by more than 100 fold for full conversion , while the alpha ring-opening products have excellent chirality with minimum by-products. A panel of HHDH variants was developed to enable chemoenzymatic synthesis of a series of high-value products w/o assistance of epoxide hydrolase(EH), including chiral glycinols, halohydrins, epichlorohydrins and mandelic acid. These enzymatic synthesis routes will provide more economic and environmental-friendly technologies to the chemical industry. |
| References: | (1) Moschona, F.; Savvopoulou, I.; Tsitopoulou, M.; Tataraki, D.; Rassias, G. Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts 2020, 10 (10), 1117. https://doi.org/10.3390/catal10101117. (2) Kamble, M. P.; Yadav, G. D. Biocatalytic Resolution of (R,S)-Styrene Oxide Using a Novel Epoxide Hydrolase from Red Mung Beans. Catalysis Today 2018, 309, 236-241. https://doi.org/10.1016/j.cattod.2017.06.013. (3) Wang, H.-H.; Wan, N.-W.; Miao, R.-P.; He, C.-L.; Chen, Y.-Z.; Liu, Z.-Q.; Zheng, Y.-G. Identification and Structure Analysis of an Unusual Halohydrin Dehalogenase for Highly Chemo-, Regio- and Enantioselective Bio-Nitration of Epoxides. Angewandte Chemie International Edition 2022, 61 (37), e202205790. https://doi.org/10.1002/anie.202205790. Heravi, M. M.; Zadsirjan, V.; Farajpour, B. Applications of Oxazolidinones as Chiral Auxiliaries in the Asymmetric Alkylation Reaction Applied to Total Synthesis. RSC Adv. 2016, 6 (36), 30498-30551. https://doi.org/10.1039/C6RA00653A. Fauzi, A. M.; Hardman, D. J.; Bull, A. T. Biodehalogenation of Low Concentrations of 1,3-Dichloropropanol by Mono- and Mixed Cultures of Bacteria. Appl Microbiol Biotechnol 1996, 46 (5), 660-666. https://doi.org/10.1007/s002530050877 (6) Wan, N.; Tian, J.; Zhou, X.; Wang, H.; Cui, B.; Han, W.; Chen, Y. Regioselective Ring-Opening of Styrene Oxide Derivatives Using Halohydrin Dehalogenase for Synthesis of 4-Aryloxazolidinones. Advanced Synthesis & Catalysis 2019, 361 (20), 4651-4655. https://doi.org/10.1002/adsc.201900786. (7) Ma, R.; Hua, X.; He, C.-L.; Wang, H.-H.; Wang, Z.-X.; Cui, B.-D.; Han, W.-Y.; Chen, Y.-Z.; Wan, N.-W. Biocatalytic Thionation of Epoxides for Enantioselective Synthesis of Thiiranes. Angewandte Chemie International Edition 2022, 61 (52), e202212589. |
| Figures: | 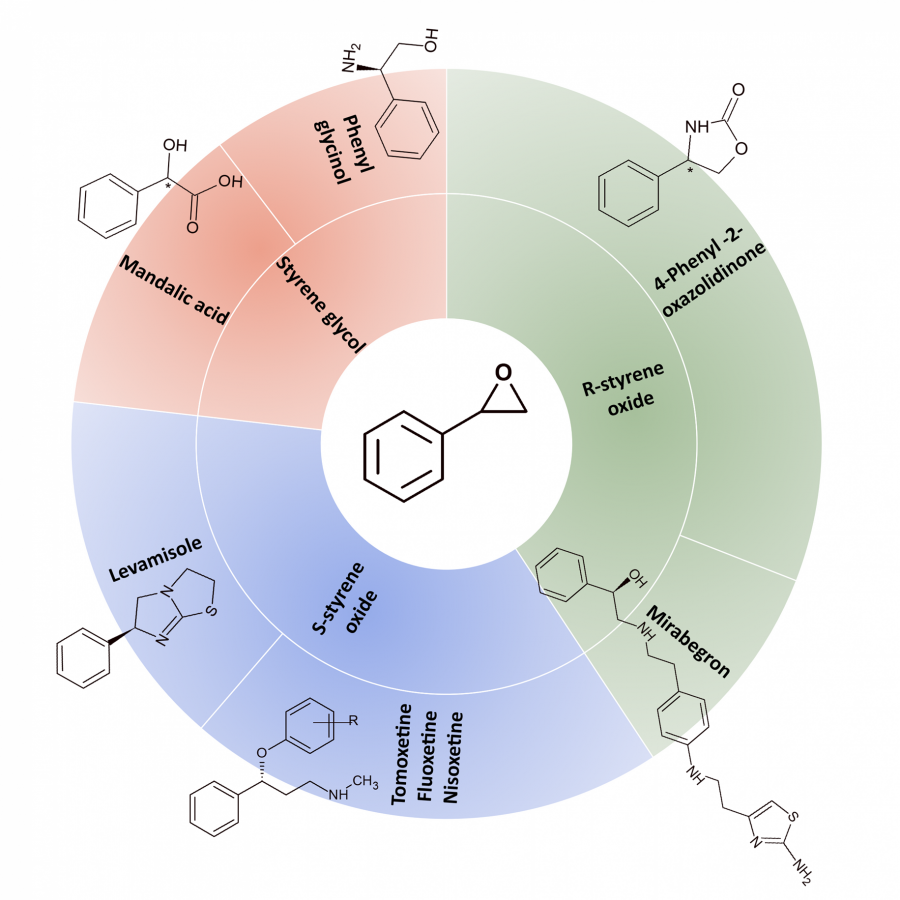 Product tree of styrene oxide All potential medical intermediates and potential API products from styrene oxide are listed 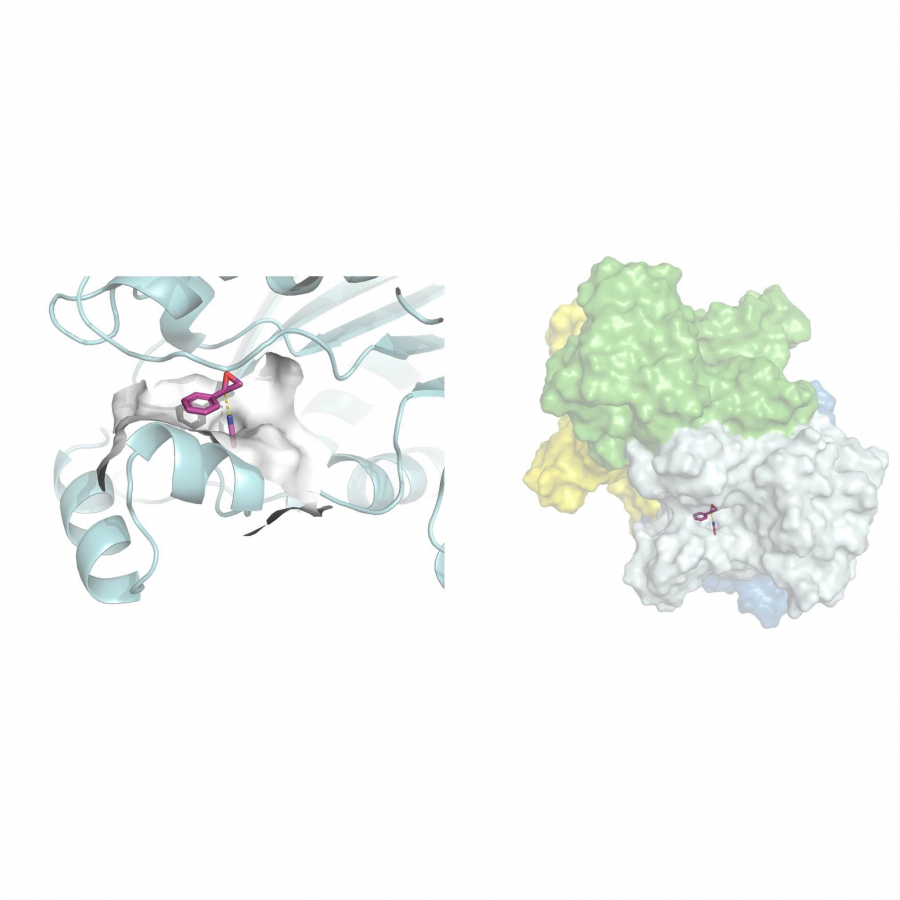 Substrate docking results The docking results from our BioEngine platform, leading to the library designs |
#1725 | Discovery of a novel thermophilic laccase for low-density polyethylene degradation |
|
| Presenting author: | Yan ZHANG of AARHUS UNIVERSITET |
| Corresponding author: | Zheng GUO of AARHUS UNIVERSITET |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme discovery / Laccase / PE degradation / Dye decolorization |
| Purpose: | Petroleum-derived plastics and synthetic dyes are hazardous pollutants that threaten human health and the planet's ecosystems. PE is exclusively made up by a C-C backbone without any hydrolysable groups while industrial dyes have complex aromatic structures, making both pollutants highly recalcitrant to biodegradation. [1] Biodegradation seems to be the most attractive and eco-friendly method to combat this growing environmental problem. [2] For instance, by secreting extracellular enzymes, microbes have been identified to facilitate the functionalization and further depolymerization of PE polymers into shorter oligomers. The modified oligomers can enter the cells and can eventually be assimilated in the metabolic pathways. Enzymes involved in lignin degradation pathways, specifically laccases, have been proven to possess PE-degrading and dye-decolorizing abilities. [3] However, the discovery of new laccases to enrich the enzyme toolbox for PE and dye degradation is still valuable and should be explored further. In the presented work, we designed a screening strategy for discovery of new enzymes that were possibly involved in pollutant-degradation pathways from reported PE-degrading microorganisms (Fig. 1). First, reported PE-eating bacteria were collected from German Collection of Microorganisms and Cell Cultures (DSMZ) and cultivated in our lab for obtaining their genomic DNA. In addition, we hypothesize that enzymes that can facilitate the functionalization and further depolymerization of PE polymers should have: (1) An open substrate-binding pocket; (2) Metal catalytic centers. According to the hypothesis, the computer-aided structure analysis was performed to identify the potential enzyme candidates. From the structural view, laccases with a relatively open catalytic pocket and copper centers are expected candidates for further investigations. After an activity-based screening of selected enzyme candidates, a novel thermophilic laccase-LfLAC3 was discovered which is applicable for the decolorization of various commercial dyes, as well as the degradation of unpretreated low-density polyethylene films. |
| References: | [1] C. Jonsson, R. Wei, A. Biundo, J. Landberg, L. Schwarz Bour, F. Pezzotti, A. Toca, M.J. L, U.T. Bornscheuer, P.O. Syren, ChemSusChem 14 (2021) 4028. [2] Y. Zhang, J.N. Pedersen, B.E. Eser, Z. Guo, Biotechnol. Adv. 60 (2022) 107991. [3] R. Wei, W. Zimmermann, Microb. Biotechnol. 10 (2017) 1308-1322. |
| Figures: | 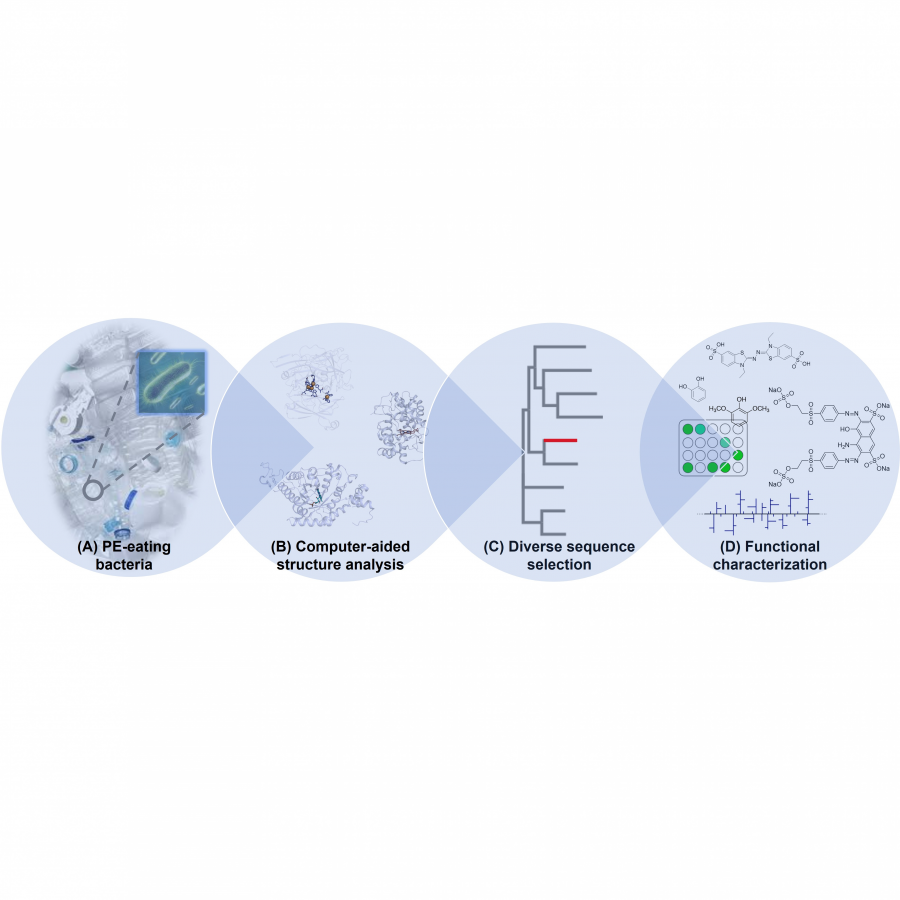 Figure 1 Strategy for discovery of new enzymes that potentially have pollutant-degrading properties. |
#1732 | Production of furan-based renewable monomers by employing novel enzymes of fungal origin |
|
| Presenting author: | Evangelos TOPAKAS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | Anthi KARNAOURI of AGRICULTURAL UNIVERSITY OF ATHENS |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biotransformation / Enzyme catalysis / Oxidases / CAZymes |
| Purpose: | Plastics have become ubiquitous materials, present in almost every aspect of modern life, however their dependence on dwindling fossil fuel sources and their durability has raised concerns about their sustainability and the impact of their use in the environment. The development of biobased polymers, including biobased and/or biodegradable plastics, addresses the need for greener, environmentally responsible materials. Furans, such as 5-(hydroxymethyl)furfural (HMF) and furfural (FA), that can be obtained by chemical dehydration of sugars, have gained significant attention due to their chemical attributes as platform chemicals for the production of bio-polyester materials [1]. HMF is the precursor of 2,5-furandicarboxylic acid (FDCA), a monomer of polyethylene 2,5-furandicarboxylate (PEF) which is an alternative to polyethylene terephthalate (PET) produced from fossil fuel, while 2-furancarboxylic acid (FCA), produced by FA oxidation, also possesses significant potential for use as a monomer in polyesters. Biocatalytic oxidation of furans with redox enzymes offers a facile and regioselective route of reaction under mild conditions [2]. Τhe current study targeted at the enzymatic biotransformation of HMF and FA using novel fungal biocatalysts from the Auxiliary Activity AA3 and AA5 families of CAZy database [3]. Through intelligent exploration of Ganoderma lucidum genome, it was possible to retrieve one sequence with putative glyoxal oxidase activity (GlGlyOx1) and one with aryl-alcohol oxidase (GlAAOx1) activity based on their homology with known furan-transforming fungal catalytic activities. The genes were heterologously expressed in yeast Pichia pastoris, the respective enzymes were purified to their homogeneity and biochemically characterized. GlGlyOx1 and GlAAOx1 were evaluated, both individually and synergistically (along with the presence of in-house produced galactose oxidase FoGalOx from Fusarium oxysporum [4] and a commercially available horseradish peroxidase HRP), for their ability to act on furans and produce value-added oxidized derivatives. Our results demonstrate the potential of G. lucidum enzymes for obtaining furan-based monomers from lignocellulosic biomass residues, which can be used as building blocks for the production of biobased polymers. Acknowledgements The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the “3rd Call for H.F.R.I. Research Projects to support Post-Doctoral Researchers” (Project Number: 7315). |
| References: | [1] van Putten R-J, et al. 2013, Chem. Rev. 113, 3, 1499-1597 [2] Cajnko M. et al. 2020, Biotechnol Biofuels 13, 66 [3] Drula E. et al. 2022, Nucleic Acids Res 50: D571-D577 [4] Dedes G., Karnaouri A., Topakas E. 2022, P-234, 13th Panhellenic Scientific Conference of Chemical Engineering (PESXM), Patras, Greece |
#1734 | Identification and Optimization of a Metagenomic Esterase with Hydrolytic Activity towards Polylactic Acid |
|
| Presenting author: | Eva JOSIC of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB |
| Corresponding author: | Aleksandra MARSAVELSKI of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB |
| Other authors: | Marko MOCIBOB of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB Ivana KEKEZ of THE FACULTY OF SCIENCE, THE UNIVERSITY OF ZAGREB Margaux AUBEL of INSTITUTE FOR EVOLUTION AND BIODIVERSITY, UNIVERSITY OF MÜNSTER Erich BORNBERG-BAUER of INSTITUTE FOR EVOLUTION AND BIODIVERSITY, UNIVERSITY OF MÜNSTER Aleksandra MARSAVELSKI of THE UNIVERSITY OF ZAGREB, THE FACULTY OF SCIENCE |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | polylactic acid (PLA) / bioplastics / biodegradation / circular economy |
| Purpose: | Polylactic acid (PLA) is a biodegradable polyester with high potential for applications in many fields.[1] The low biodegradability of PLA in natural environments limits its use as a sustainable alternative to conventional plastics. However, microbial enzymes offer a promising strategy to enhance the biodegradation of PLA. Accordingly, we applied a metagenomic approach to identify a novel esterase enzyme, capable of cleaving PLA. A search of metagenomic sequence databases identified an esterase, and its activity towards PLA was demonstrated using emulsified PLA in an agarose test.[2] Subsequent crystallographic studies to obtain the enzyme's structure for further optimization resulted in multiple crystal hits. The structure determination process is ongoing, with initial results indicating the enzyme belongs to the α/β-hydrolase family. To optimize the enzyme's stability, ancestral sequence reconstruction was applied to obtain more robust variants stable at higher temperatures. These variants will be ordered as synthetic genes, expressed, and tested for improved thermostability and activity. (Figure 1). |
| References: | [1] rosenboom, jg., langer, r. and traverso, g. nat. rev. mater. 2022, 7, 117-137. [2] teeraphatpornchai t., nakajima-kambe t., shigeno-akutsu y., nakayama m., nomura n., nakahara t., uchiyama h. biotechnol. Lett. 2003, 25, 23-8. |
| Figures: | 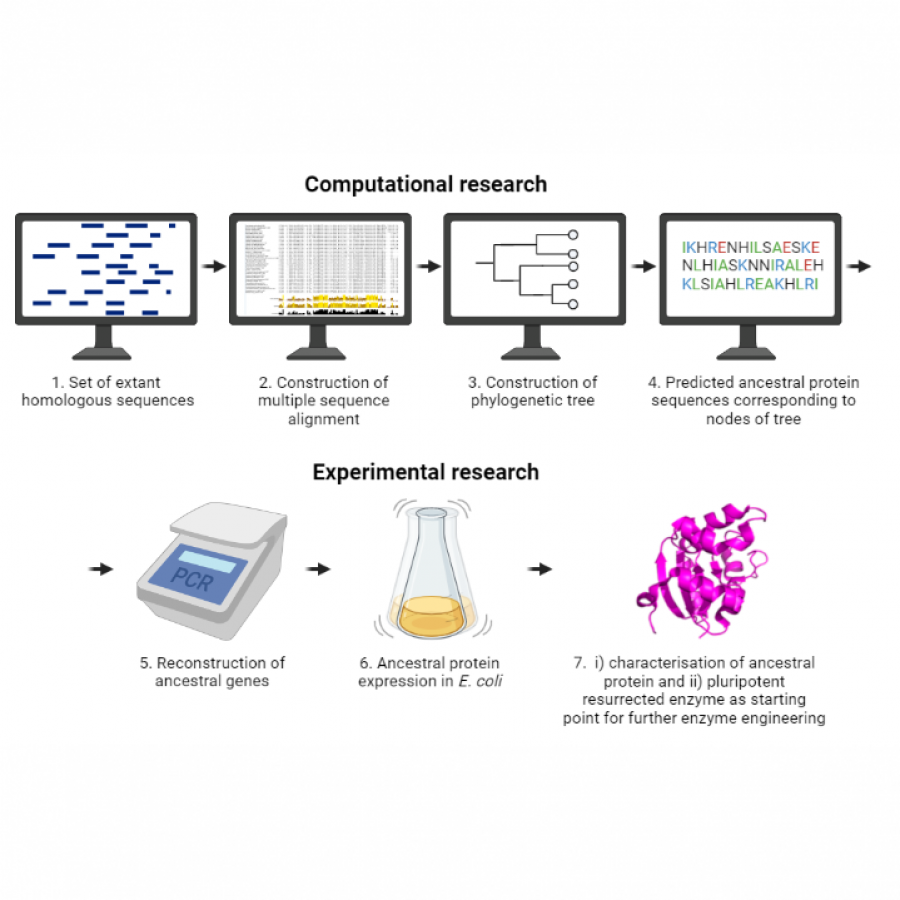 FIGURE 1 A complete scheme of the conducted research. |
#1737 | PC |
|
| Presenting author: | Nikolaos KALOUDIS of UNIVERSITY OF CRETE |
| Other authors: | FOTEINI-ILEKTRA MARIDAKI of UNIVERSITY OF CRETE IOANNIS V. PAVLIDIS of UNIVERSITY OF CRETE |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | transaminases / immobilization / asymmetric synthesis / |
| Purpose: | The (S)-selective amine transaminases (EC 2.6.1.X) belong to fold type I, and they have been demonstrated to have great potential for biotechnological applications due to their ability to catalyze the stereoselective amination of prochiral ketones. However, their industrial use is still limited due to issues such as stability and recycling. Covalent immobilization is a promising solution to these drawbacks, allowing biocatalyst reuse and providing higher stability. In this work, three (S)-selective amine transaminases, namely (PDB entries) 3HMU, 3FCR_4M_I234M, and 3FCR_4M_L382M_G429A, were covalently immobilized on amino-functionalized beads using glutaraldehyde as a crosslinker. The resultant immobilized biocatalysts were characterized in terms of residual activity, thermal stability and reusability. The immobilization efficiency was higher than 95% in every case, and the immobilization time was approximately 3 hours. Also, despite the fact that residual activity was less than 10%, the biocatalysts could be successfully reused for at least 5 cycles retaining at least 50 % of their initial activity. In addition, 3HMU was also evaluated in terms of co-solvent (DMSO) stability where immobilized catalyst didn't show any improvement when compared to the free. Furthermore, both immobilized 3FCR mutants were improved in terms of storage stability. Finally, the immobilized biocatalysts were applied to the asymmetric synthesis of chiral amines using either sopropylamine or alanine as the amino donor. This study provides new insights into the development of efficient immobilized biocatalysts for the production of chiral amines with industrial relevance. Acknowledgements: The research project was supported by the Hellenic Foundation for Research and Innovation (H.F.R.I.) under the "1st Call for H.F.R.I. Research Projects to support Faculty Members & Researchers and the Procurement of High-and the procurement of high-cost research equipment grant" (Project Number:664). |
#1738 | Chemoenzymatic synthesis of Tenofovir |
|
| Presenting author: | Beata ZDUN of WARSAW UNIVERSITY OF TECHNOLOGY, FACULTY OF CHEMISTRY |
| Other authors: | Wolfgang KROUTIL of INSTITUTE OF CHEMISTRY, UNIVERSITY OF GRAZ, GRAZ, AUSTRIA Tamara REITER of INSTITUTE OF CHEMISTRY, UNIVERSITY OF GRAZ, GRAZ, AUSTRIA Paweł BOROWIECKI of WARSAW UNIVERSITY OF TECHNOLOGY, FACULTY OF CHEMISTRY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Tenofovir / chemoenzymatic synthesis / lipase / alcohol dehydrogenase |
| Purpose: | Tenofovir, and its lipophilic prodrugs [i.e., tenofovir disoproxil fumerate (TDF) and tenofovir alafenamide (TAF)] are powerful antiretrioviral agents that have already gained the status of the ‘frontline drugs’ for the treatment of Human Immunodeficiency Virus (HIV) infection and chronic hepatitis B caused by HBV [1-2]. The anti-HIV activity of parent tenofovir, resulting from competitive inhibition of HIV reverse transcriptase inhibitor concerning for to dATP, is strictly related to the absolute configuration of its stereogenic center. In this context the (R)-configurated Tenofovir is ca. 100-fold more active as a nucleoside reverse transcriptase inhibitor (NRTI) than its enantiomeric counterpart [3]. In this study, optimization of the reaction conditions for lipase-catalyzed kinetic resolution of a racemic 1-(6-chloro-9H-purin-9-yl)-propan-2-ol was performed. This task allowed us to select the most efficient biocatalytic system, which consisted of immobilized lipase from Burkholderia cepacia (Amano PS-IM) suspended in toluene and vinyl acetate as an acyl donor. The optically pure (R)-ester (>99% ee) was obtained on a 500 mg-scale (60mM conc.) in 47% yield when employing preparative silica-gel column chromatography at a purification step or in 31% yield when using liquid-liquid extractive workup. Alternatively, stereoselective bioreduction of the corresponding prochiral ketone, namely 1-(6-chloro-9H-purin-9-yl)-propan-2-one was performed. The most satisfactory result in terms of enantiomeric purity of the product was obtained in the reaction catalyzed by lyophilized E. coli cells harboring recombinantly overexpressed variant of alcohol dehydrogenase (ADH) from Lactobacillus kefir (E.coli/Lk-ADH-Prince). In this case, the desired (R)-alcohol was isolated with 86% yield and excellent optical purity (>99% ee). The key-(R)-intermediate obtained in this way was further functionalized toward a tenofovir in a three-step reaction sequence consisting of “one-pot” aminolysis-hydrolysis or (R)-acetate in NH3-saturated methanol, alkylation of the resulting (R)-alcohol with tosylated diethyl (hydroxymethyl)phosphonate, and bromotrimethylsilane (TMSBr)-mediated cleavage of the formed phosphonate ester into the free phosphonic acid. The overall isolated yield of enantiomerically enriched tenofovir (99% ee) prepared in this manner reached 72% after four steps. The elaborated enzymatic strategy could be applicable in the asymmetric synthesis of two other blocbuster antiretroviral tenofovir prodrug derivatives, including 5’-disoproxil fumerate (TDF, Viread ®) and 5’alafenamide (TAF, Vemlidy ®), respectively. Acknowledgments: This research was funded by the National Science Center (NCN) of Poland grant SONATA 15 (No: 2019/35/D/ST4/01556). B.Z. is grateful to the IDUB project (Scholarship Plus Programme) for providing a research fellowship. |
| References: | [1] a.s. ray, m.w. fordyce, m. j. hitchcock, antivir. res. 2016, 125, 63-70. [2] f.amblard, d. patel, e. michailidis, s.j. coats, m.kasthuri, n, biteau, z. tber, m. ehteshami, r.f. schinazi, eur. j. med. chem. 2022, 114554. [3] j. balzarini, a. holy, j. jindrich, l. naesens, r. snoeck, d. schols, e. de clereq, antimicrob. agents chemother. 1993, 37(2), 332-338. |
| Figures: | 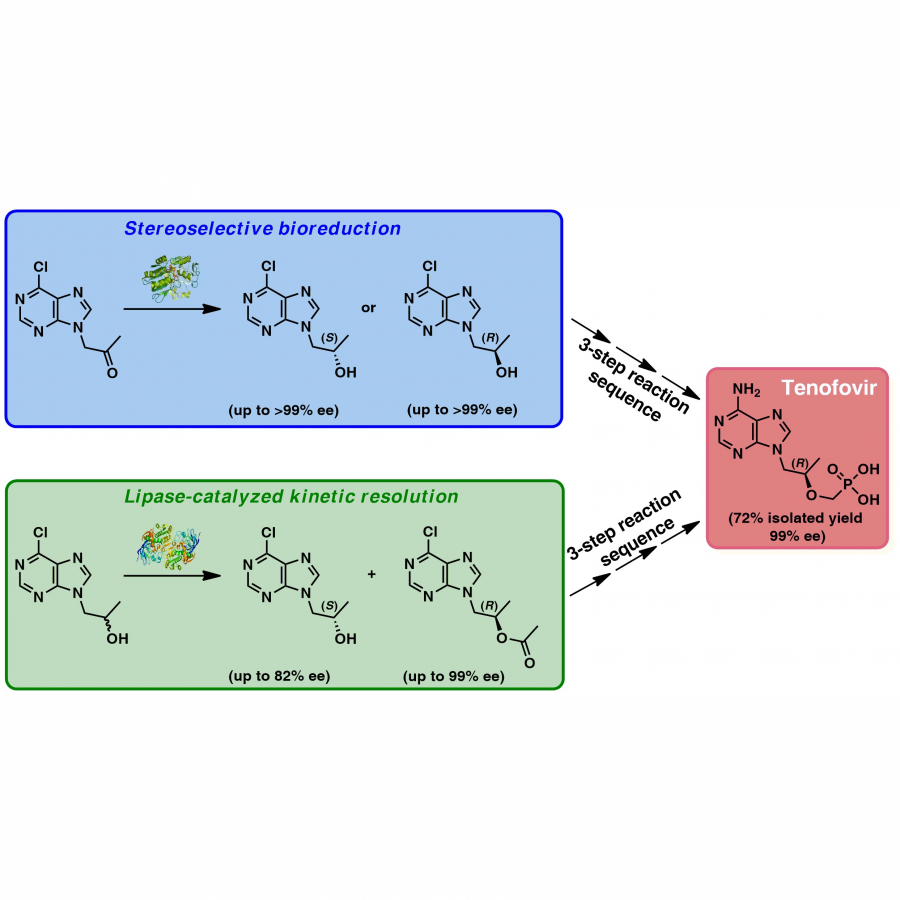 Chemoenzymatic Synthesis of Tenofovir. |
#1741 | Deep eutectic solvents enable robust hydroxylation of fatty acids: A correlation between water activity and thermostability of FA-HY1 |
|
| Presenting author: | Pengfei ZHOU of SERICULTURAL & AGRI-FOOD RESEARCH INSTITUTE GUANGDONG ACADEMY OF AGRICULTURAL SCIENCES |
| Corresponding author: | Mingwei ZHANG of SERICULTURAL & AGRI-FOOD RESEARCH INSTITUTE GUANGDONG ACADEMY OF AGRICULTURAL SCIENCES |
| Topic: | Reaction design |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Deep eutectic solvents / Fatty acid hydroxylation / Hydratase / Thermostability |
| Purpose: | This work aims to explore the performance and application of fatty acid hydratases in green and sustainable deep eutectic solvents (DESs). As the first case, a robust system with the combination of fatty acid hydratase-1 (FA-HY1) from Lactobacillus Acidophilus and DESs for biocatalytic hydroxylation of fatty acids was developed. The results indicated that the catalytic performance of FA-HY1 was mainly determined by the composition of DESs. Especially water molecules, as the co-substrate of hydroxylation reactions, have been proven as the key factor of the thermostability of FA-HY1 in DESs systems as well. By varying the water activity of DESs, we found that FA-HY1 displayed improved thermostability at lower water activity. In particular, the half-life time of FA-HY1 increased 6-fold in choline chloride/sorbitol system with water activity (aw = 0.84) compared to aqueous (Kpi) system. Moreover, the thresholds of water activity in betaine/glycerol and choline chloride/sorbitol systems were determined as 0.64 and 0.75, respectively. We further investigated the recyclability of FA-HY1 in choline chloride/sorbitol system, after three rounds of recycling, the hydroxylation efficiency of 83.2% was still observed. However, the conversion dropped to only 8.3% in aqueous system. In addition, molecular dynamics simulation was performed in DESs and aqueous systems by determining the RMSD, RMSF, Rg, and RDF values, which revealed the mechanistic features of the thermostability of FA-HY1 in DESs. |
| Figures: |  Figure 1 Graphic Abstract |
#1745 | Concurrent Hybrid Catalysis at acidic pH for Rare Monosaccharide Production by Combining Aldolase and N-Heterocyclic Carbene Gold Catalysts |
|
| Presenting author: | Cédric GASTALDI of UCL |
| Other authors: | Christine GUERARD-HELAINE of ICCF Virgil HELAINE of ICCF Claude FORANO of ICCF Arnaud GAUTIER of ICCF Muriel JOLY of ICCF |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Hybrid catalysis / Layered double hydroxide / Organometallic catalyst / enzyme |
| Purpose: | Hybrid catalysis has been increasingly developed in recent years. Indeed, it takes advantage of the robustness and the broad range of applications of the chemical catalysts combined with the high regio, chemo and/or stereoselectivity of the enzymes. Among chemical catalysts, transition-metal complexes are considered as one of the most powerful. Furthermore, metal catalysts working in water are recently emerging, giving opportunities to couple them with enzymes whose natural medium is aqueous. Thus, various examples of combination of biocatalysts and metal-catalysts for linear cascades have been reported, but often in a sequential mode, because of various incompatibilities that could not be solved (for instance: pH, temperature, substrate concentrations, inhibition problems, etc…). Indeed, few one-pot one-step processes, i.e., involving the addition of all the reagents and catalysts from the beginning, without any changes in the operating conditions till the end, are described. The reason is often an incompatibility of the catalysts with each other, or with the operating conditions. Several techniques have overcome these difficulties by compartmentalization using either cells, biphasic systems, artificial metallo-enzymes or supramolecular hosts. Combined with techniques of enzymes evolution, in order to make biocatalysts more robust, in particular with respect to the temperature of the medium or the concentrations of substrates, these methods have made it possible to solve almost all the reasons of incompatibilities of the catalysts between them, except, to our knowledge, when the incompatibility comes from the pH. In our work, a chemical catalyst, a N-heterocyclic carbene gold complex, only capable of operating at pH 3, was successfully coupled to an aldolase, the Fructose-6-phosphate Aldolase, previously confined in cells to protect it from this extreme pH. The catalyst working better at 60°C, cells were in turn immobilized on Layered Double Hydroxides (LDH), specially designed to resist to pH 3, so as not to degrade cell membranes at this temperature. Finally, to avoid any mutual deleterious interactions between chemical catalyst and the immobilized cells harboring the aldolase, a compartmentalization has been set up thanks to a semi-permeable membrane. Thus, as proof of principle, propargyl alcohol, a cheap and achiral compound, could be hydrated, using a fair range of substrates concentrations (200-500mM) for easier scale-up, and subsequently converted to the corresponding aldol, a high added-value compound with one asymmetric center with fixed configuration, at pH 3 and 60°C, in 70% isolated yield. Interestingly, this process could be exemplified to another concurrent reaction involving an acidic resin catalyzed acetal hydrolysis to the corresponding aldehyde, as aldolase electrophilic substrate, particularly useful in the case of unstable aldehydes. This latter system, even simpler than the previous one, gave the corresponding aldol in 98% yield, with two asymmetric centers with fixed configurations. |
| References: | [1] Huang, X.; Cao, M.; Zhao, H. Curr. Opin. Chem. Biol. 2020, 55, 161-170. [2] a) Simons, C.; Hanefeld, U.; Arends, I. W. C. E.; Maschmeyer, T.; Sheldon, R. A. Top. Catal. 2006, 40 (1-4), 35-44. b) Prastaro, A.; Ceci, P.; Chiancone, E.; Boffi, A.; Cirilli, R.; Colone, M.; Fabrizi, G.; Stringaro, A.; Cacchi, S. Green Chem. 2009, 11 (12), 1929-1932. c) Ríos-Lombardía, N.; Vidal, C.; Cocina, M.; Morís, F.; García-Álvarez, J.; González-Sabín, J. Chem. Commun. 2015, 51 (54), 10937-10940. d) Burda, E.; Hummel, W.; Gröger, H. Angew. Chem. Int. Ed. 2008, 47 (49), 9551-9554. [3] a) Foulkes, J. M.; Malone, K. J.; Coker, V. S.; Turner, N. J.; Lloyd, J. R. E. ACS Catal. 2011, 1 (11), 1589-1594. b) Denard, C. A.; Huang, H.; Bartlett, M. J.; Lu, L.; Tan, Y.; Zhao, H.; Hartwig, J. F. Angew. Chem. Int. Ed. 2014, 53 (2), 465-469. c) Köhler, V.; Wilson, Y. M.; Dürrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.; Knörr, L.; Häussinger, D.; Hollmann, F.; Turner, N. J.; Ward, T. R. Nat. Chem. 2013, 5 (2), 93-99. d) Wang, Z. J.; Clary, K. N.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. A. Nat. Chem. 2013, 5 (2), 100-103. [4] Gastaldi, C.; Mekhloufi, G.; Forano, C.; Gautier, A.; Guérard-Hélaine, C. Green Chem. 2022, 24 (9), 3634-3639. [5] Gastaldi, C.; Hélaine, V.; Joly, M.; Gautier, A.; Forano, C.; Guérard-Hélaine, C. Catal. Sci. Technol., 2023, Accepted Manuscript |
| Figures: | 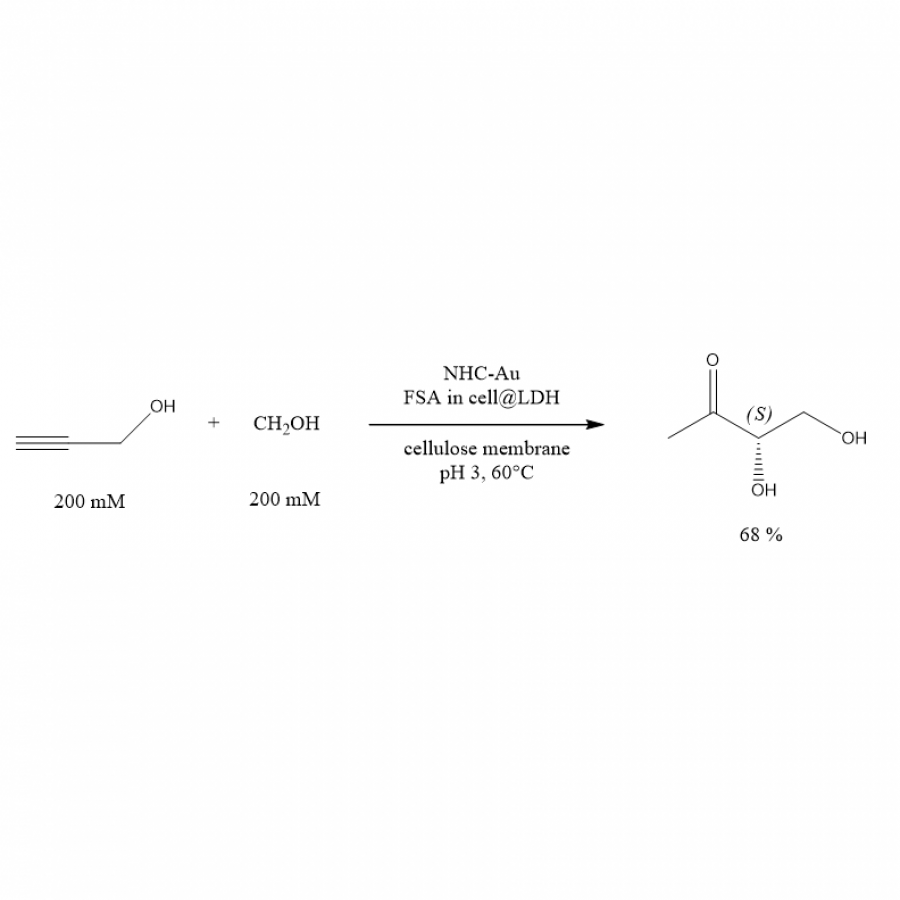 Hybrid system optimized conditions for the one-pot concurrent hydration-aldolisation reaction |
#1747 | Enzyme engineering approaches on a stereospecific indole C-methyltransferase – challenges, new functions and solutions for practical applications |
|
| Presenting author: | Diana-Alexandra AMARIEI of HEINRICH-HEINE UNIVERSITY, DÜSSELDORF |
| Other authors: | Julia TENHAEF of IBG-1, FORSCHUNGSZENTRUM JÜLICH Tobias ROSCH of IBG-1, FORSCHUNGSZENTRUM JÜLICH Mona HAASE of HEINRICH-HEINE UNIVERSITY, DÜSSELDORF Pascal SCHNEIDER of HEINRICH-HEINE UNIVERSITY, DÜSSELDORF Stephan NOACK of IBG-1, FORSCHUNGSZENTRUM JÜLICH Jörg PIETRUSZKA of HEINRICH-HEINE UNIVERSITY, DÜSSELDORF, IBG-1, FORSCHUNGSZENTRUM JÜLICH |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | methyltransferase / enzyme engineering / indole methylation / library screening |
| Purpose: | The indole and pyrroloindoline structural motifs are common among bioactive natural product alkaloids with various biological activities.[1] AChE inhibitor physostigmine was the inspiration for the development of a stereoselective enzymatic methylation platform for these scaffolds. A key step in the biosynthesis of physostigmine is the enzymatic C-methylation of an indole derivative, which drives the subsequent intramolecular cyclization, forming a stereogenic centre.[2] By using a chemo-enzymatic approach, we were able to produce physostigmine analogs in an enantiopure fashion. However, the limits of the enzyme are preventing application on a broader scope.[3] Site-specific and saturation mutagenesis approaches allowed the modification of the catalytic site, with implications on the product size and chemistry. Our mechanistic study, as well as an analysis of known methyltransferases containing a Rossmann-fold motif are a source of inspiration for targeted modifications, while directed evolution techniques were able to fill in the gaps towards a substrate promiscuous – but stereoselective engineered catalyst. Further enzymatic alkylations of the indole scaffold were also enabled by this approach, using SAM analogs. In order to support the screening effort, we developed an automated process for the cultivation, expression and activity testing of the resulting mutant libraries. Our results lead us to an enhanced biocatalytic approach for the production of physostigmine analogs. |
| References: | [1] de Sa Alves, F. R.; Barreiro, E. J.; Manssour Fraga, C. A. Mini Rev. Med. Chem. 2009, 9, 782-793. [2] Liu, J.; Tailun, Ng.; Rui, Z.; Ad, O.; Zhang, W. Angew. Chem. Int. Ed. 2014, 53, 136-139. [3] Amariei, D. A.; Pozhydaieva, N.; David, B.; Schneider, P.; Classen, T.; Gohlke, H.; Weiergräber, O. H; Pietruszka, J. ACS Catal. 2022, 12(22), 14130-14139. |
| Figures: | 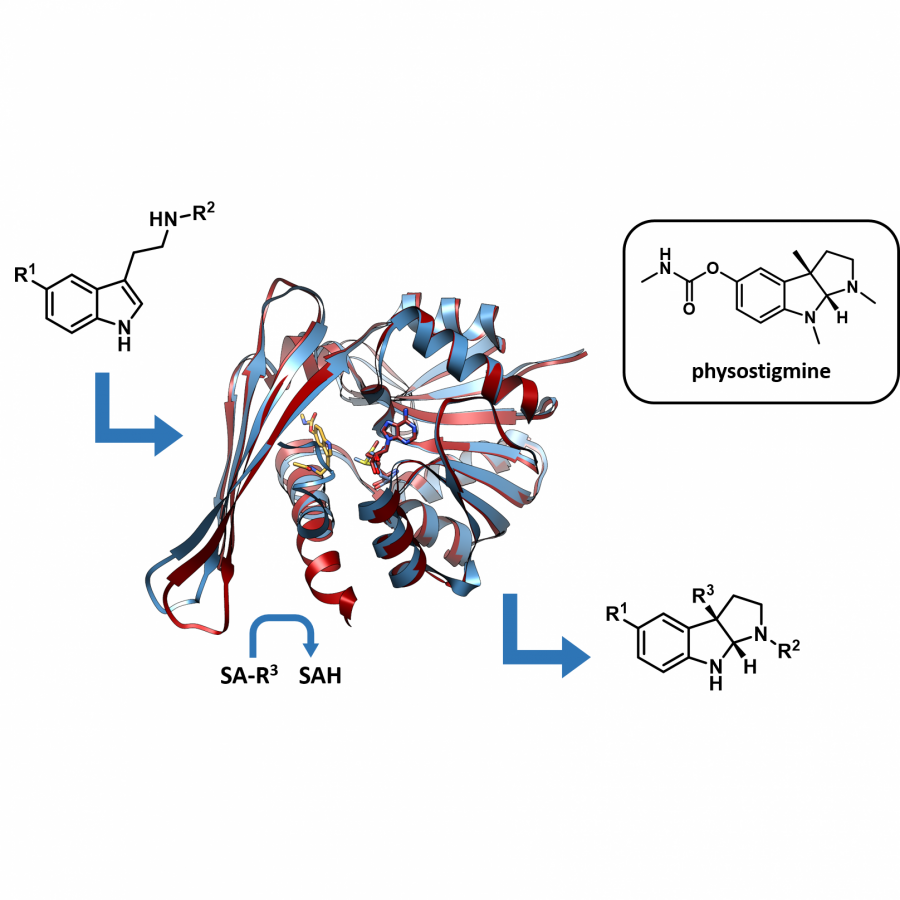 General process catalyzed by the engineered methyltransferase 3 points of interest were selected for the structural diversification of the products. SAM analogs were used for alkylation in the 3 possition (ex. R3=Me for methylation) |
#1748 | A microfluidics-enabled workflow for rapid large-scale fitness data generation informs imine reductase engineering |
|
| Presenting author: | Maximilian GANTZ of UNIVERSITY OF CAMBRIDGE |
| Corresponding author: | Florian HOLLFELDER of UNIVERSITY OF CAMBRIDGE |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Protein engineering / Microfluidics / Imine reductase / Deep Mutational Scanning |
| Purpose: | Biocatalysis is emerging as an environmentally friendly and cost-effective alternative to the chemocatalytic synthesis of chiral amines. Directed evolution is the method of choice for optimizing biocatalysts but is often time consuming and its success is unpredictable. To meet the fast pace of product development in industry the turnaround time of engineering enzymes needs to become shorter. Here we present a workflow for rapid large-scale sequence–function data generation and show that this data can guide and thereby accelerate enzyme engineering. As a model system we chose an imine reductase (IRED) from S. roseum (SrIRED) catalysing reductive amination. To rapidly assess the fitness of >10000 IRED variants, we developed a cheap and versatile absorbance-activated droplet sorting-based ultra-high throughput screening setup. Coupled to a novel deep sequencing protocol combining next generation- and long-read-sequencing, this workflow allows the generation of large-scale fitness data using unbiased mutagenesis by error-prone PCR and the resolution of epistasis in less than 2 weeks. Using this novel workflow, we identified hot spots for engineering substrate specificity in the active site and at the periphery of the enzyme. We used higher-order mutation data to score the combinability of individual improving mutations and rationally engineered highly active variants (10-fold improved kcat/KM) displaying positive epistasis showing that our workflow can give rapid access to highly improving variants. Due to its versatility and broad applicability, we envision that this workflow will become a valuable part of the protein engineer’s toolkit for rapid enzyme engineering. |
#1753 | Engineering 4-oxalocrotonate tautomerase for Michael-type addition reactions |
|
| Presenting author: | Veronika CHADIMOVÁ of UNIVERSITY OF GRONINGEN |
| Other authors: | Gerrit POELARENDS of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | 4-oxalocrotonate tautomerase / carboligase / Michael-type addition / |
| Purpose: | Carbon-carbon bond formation is a critical step in the synthesis of many compounds for industrial application. The traditional synthetic strategies often involve the use of toxic reagents, solvents and extreme reaction conditions, resulting in increased waste generation and energy consumption. Biocatalysis is a promising alternative method employing enzymes that yield carbon-carbon bonds with high selectivity, in an efficient and biosustainable manner, however the carboligase toolbox is still rather limited. In this work, we present an evolved variant of 4-oxalocrotonate tautomerase (4-OT), a highly promiscuous biocatalyst mimicking widely known organocatalysts. It utilizes an N-terminal proline as catalytic residue allowing the formation of both enamine and iminium intermediates. 4-OT has been applied in diverse biotransformations involving aminocatalytic modes, including reactions forming C-C and C-O bonds (1,2). Applying the gene fusion strategy to 4-OT expands the available sequence space to allow for further exploration of reaction promiscuity within the 4-OT scaffold, while keeping a high regio- and stereochemical control typical for biocatalysis(3). Our study highlights the potential of 4-OT as a promising biocatalyst for the production of valuable pharmaceutical precursors. We show that the tandem-fused 4-OT variant evolved for Michael-type addition of nitromethane to cinnamaldehyde(3), has a broader catalytic scope that includes other addition and cycloaddition reactions. We also report a scalable and efficient purification strategy to improve the enzyme production and support the ongoing engineering efforts. This project is funded by the European Union's Horizon 2020 Research and Innovation Programme under the Marie Sklodowska-Curie Grant Agreement no. 956631 — CC-TOP |
| References: | (1) Kunzendorf, A.; Xu, G.; Saifuddin, M.; Saravanan, T.; Poelarends, G. J. Biocatalytic Asymmetric Cyclopropanations via Enzyme-Bound Iminium Ion Intermediates. Angewandte Chemie - International Edition 2021, 24059-24063. https://doi.org/10.1002/anie.202110719. (2) Xu, G.; Crotti, M.; Saravanan, T.; Kataja, K. M.; Poelarends, G. J. Enantiocomplementary Epoxidation Reactions Catalyzed by an Engineered Cofactor-Independent Non-Natural Peroxygenase. Angewandte Chemie - International Edition 2020, 59 (26), 10374-10378. https://doi.org/10.1002/anie.202001373. (3) Xu, G.; Kunzendorf, A.; Crotti, M.; Rozeboom, H. J.; Thunnissen, A. M. W. H.; Poelarends, G. J. Gene Fusion and Directed Evolution to Break Structural Symmetry and Boost Catalysis by an Oligomeric C-C Bond-Forming Enzyme. Angewandte Chemie - International Edition 2022, 61 (8). https://doi.org/10.1002/anie.202113970. |
#1754 | Pichia pastoris - Novel strategies for enzyme production |
|
| Presenting author: | Claudia RINNOFNER of MYBIOS GMBH |
| Topic: | Enzyme production, immobilization |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Enzyme / Pichia pastoris / Methanol-free / |
| Purpose: | Nature provides us with an inspiring wealth of different proteins. Protein-based (biological) molecules have unique physical and biochemical properties and are biodegradable. Our mission is to provide solutions for the recombinant production of natural biomaterials and enzymes. Biopolymers have repetitive amino acid sequences, which give them special properties such as strength or elasticity. Others, such as transmembrane glycoproteins of viruses, may be used in diagnostics or as vaccines. Yet others can be applied as catalysts in drug design. Recombinant enzyme production allows us to create smart materials and catalysts. To produce our enzymes we build on yeasts such as Pichia pastoris, which is known for strong gene expression, secretion, high yields, high cell densities and good scalability. Building on license-free tools and own developments we have established or own toolbox for methanol-induced and -free protein production. |
#1756 | Biocatalytic alkene isomerization for asymmetric synthesis |
|
| Presenting author: | Federico ROSSI of UNIVERSITY OF GRAZ |
| Other authors: | Marina TOPLAK of UNIVERSITY OF GRAZ Mélanie HALL of UNIVERSITY OF GRAZ |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Biocatalysis / One-pot enzymatic cascade / Flavin-dependent Old Yellow Enzymes (OYEs) / Stereocomplementary cascade |
| Purpose: | Nowadays, the pharma and chemical industries are increasingly looking for cleaner routes to accessing chiral synthons in enantiopure form. For this reason, it is essential to control the stereochemistry of a chemical reaction while maximising the performance of the process and minimising waste[1]. The angelica lactones and gamma-valerolactones are versatile bio-based building blocks derived from glucose and represent platform chemicals with broad industrial applications, such as solvent production, fuel additives, precursors of bio-based polymers, building blocks of natural products and drugs, and fragrance/flavouring agents[2]. Unfortunately, most chemical methods available to isomerise the prochiral alpha-angelica lactone into the chiral beta-angelica lactone and, subsequently, access the reduced chiral gamma-valerolactone require harsh acid and base conditions and produce racemic products with low yields. Furthermore, only a few organocatalytic protocols can isomerise stereoselectively beta,gamma-unsaturated butenolides into the corresponding chiral gamma-substituted alpha,beta-unsaturated butenolides[3]. Recently, the promiscuous behaviour of flavin-dependent Old Yellow Enzymes (OYEs) able to isomerise asymmetrically alpha-angelica lactone in the absence of nicotinamide was identified[4]. We coupled this redox-neutral step with a nicotinamide-dependent bioreduction using originally two separate OYEs. The system was improved by designing a fusion bi-molecular protein to generate a one-pot enzymatic cascade for the formal stereo-divergent reduction of alpha-angelica lactone[2]. We are now exploring this synthetic platform on a range of non-activated C=C bonds to provide access to various chiral molecules in enantiopure form and with possible additional chiral centres. Acknowledgements Acknowledgement is made to the FWF (Austrian Science Fund) P36538-N for supporting this research. In addition, Alessia Tonoli, Karla Wagner, Arianna Bacchin, Marina Robescu and Elisabetta Bergantino are thanked for their past contributions to the project. |
| References: | [1] V. Farina, J. T. Reeves, C. H. Senanayake, J. J. Song, Chem. Rev. 2006, 106, 2734-2793. [2] A. Tonoli, K. Wagner, A. Bacchin, T. Reiter, E. Bergantino, M. S. Robescu, M. Hall, ChemBioChem 2023, e202300146. [3] Y. Wu, R. P. Singh, L. Deng, J. Am. Chem. Soc. 2011, 133, 12458-12461. [4] M. S. Robescu, L. Cendron, A. Bacchin, K. Wagner, T. Reiter, I. Janicki, K. Merusic, M. Illek, M. Aleotti, E. Bergantino, M. Hall, ACS Catal. 2022, 12, 7396-7405. |
| Figures: | 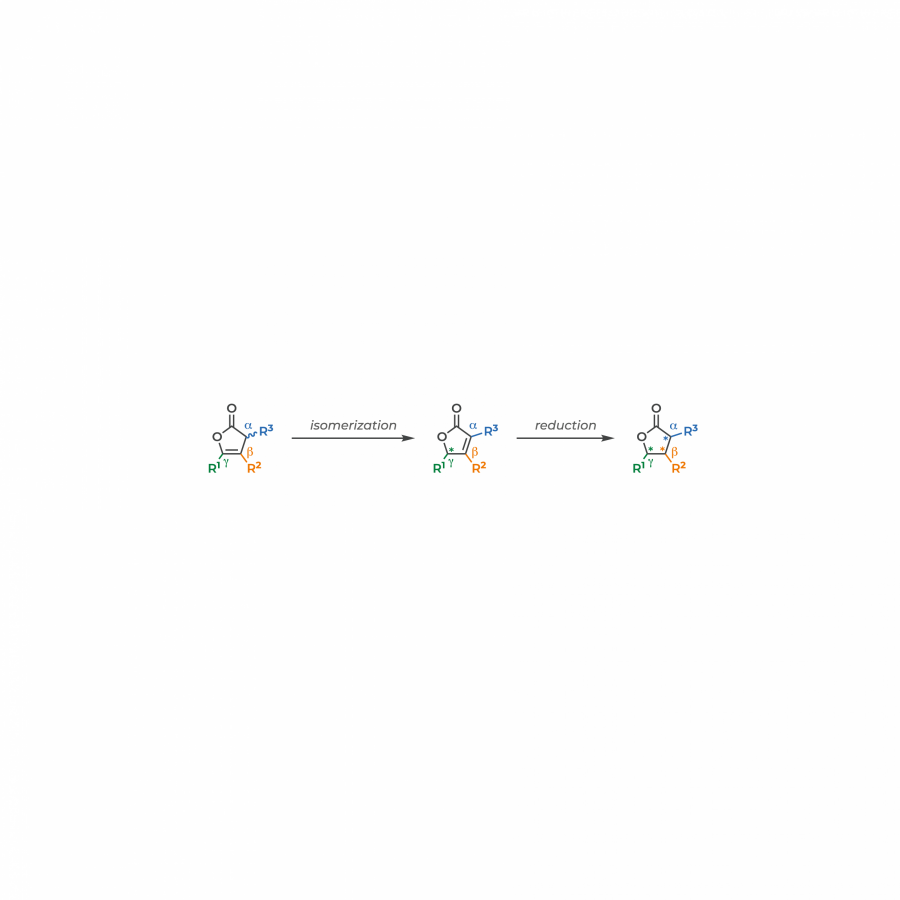 General approach to generate chiral lactones in a one-pot cascade. 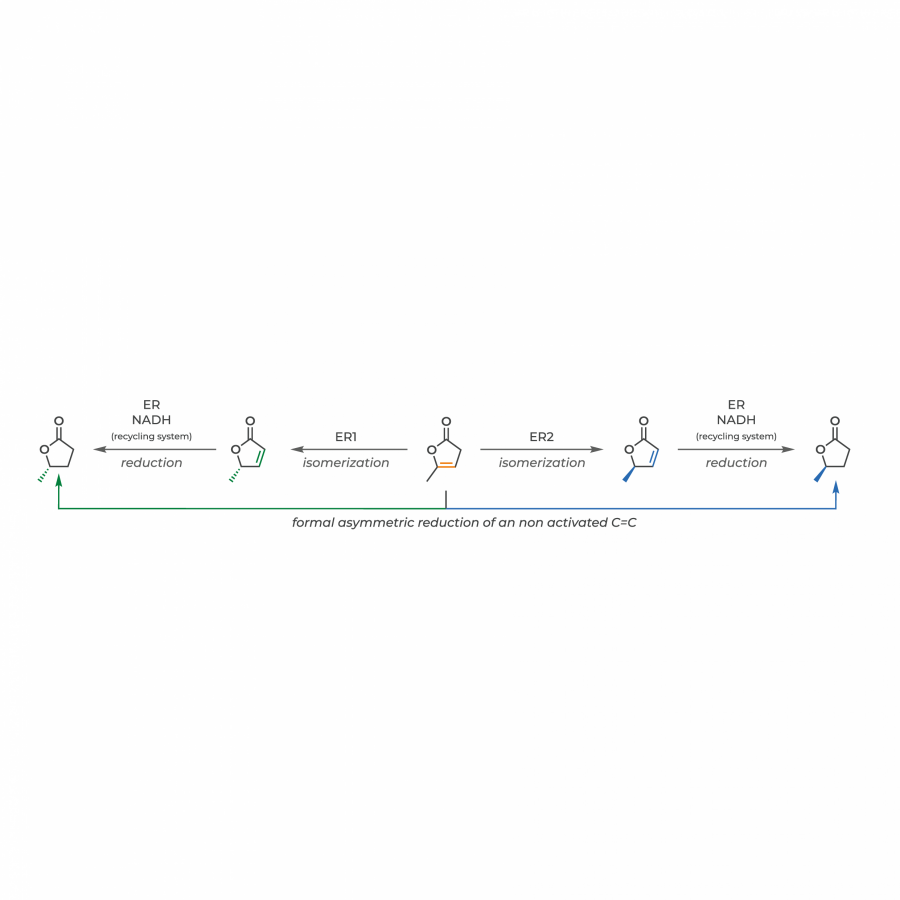 Stereocomplementary isomerisation-reduction cascade catalysed by ene-reductases (ER) applied to alpha-angelica lactone. |
#1759 | Controlled Biocatalytic Synthesis of Metal Nanoparticle-Enzyme Hybrids: Demonstration for Catalytic Hydrogen-Driven NADH or Flavin Recycling |
|
| Presenting author: | Lucy BROWNE of UNIVERSITY OF OXFORD, INORGANIC CHEMISTRY LABORATORY |
| Other authors: | Kylie VINCENT of UNIVERSITY OF OXFORD, INORGANIC CHEMISTRY LABORATORY |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | biocatalysis / biohybrid metal nanoparticles / chemoenzymatic / cofactor regeneration |
| Purpose: | Having control over the shape and size of metal nanoparticles (NPs) is key for making them suitable for specific applications: ranging from drug delivery, biosensing to catalysis. This has given rise to substantial effort into exploring different synthesis routes for improving the control over the NPs polydispersity as well as the sustainability of the process.[1] Here, we demonstrate the controlled synthesis of metal NPs under mild conditions. An isolated enzyme, an NAD+ reductase, oxidizes a nicotinamide cofactor (either the native NADH or the cheaper, synthetic cofactors: BNAH or AmNAH)[2] which provides electrons for metal reduction (Fig. 1A). These metal NPs can subsequently be used as H2 oxidation catalysts, supplying electrons back to the enzyme, which can then selectively reduce NAD+ to the bioactive cofactor 1,4-NADH (Fig. 1B). This biohybrid was then utilized as a H2-driven NADH recycling catalyst for an alcohol dehydrogenase to carry out an enantioselective ketone reduction. We have also explored the synthesis of another metal NP-enzyme hybrid, which uses H2 as the reductant and therefore leads to no carbon-containing by-products. Here, the hydrogenase enzyme reduces metal salts while oxidising H2 (Fig. 2A). Initial studies have shown these metal NP-enzyme hybrids can give increased activities in comparison to the enzyme alone, likely due to the metal NP increasing the electronic surface area between the hybrid catalyst and the substrate (Fig. 2B). In both systems we have observed the enzymes’ compatibility with the metal NPs; we hope this is an area which can help bring about new reactivities for more sustainable processes by using both the enzyme and metal NP properties either in a cascade reaction or synergistically. |
| References: | [1]J. E. Ortiz-Castillo, R. C. Gallo-Villanueva, M. J. Madou, V. H. Perez-Gonzalez, Coord. Chem. Rev. 2020, 425, 213489. [2]H. A. Reeve, J. Nicholson, F. Altaf, T. H. Lonsdale, J. Preissler, L. Lauterbach, O. Lenz, S. Leimkühler, F. Hollmann, C. E. Paul, K. A. Vincent, Chem. Commun. 2022, 58, 10540-10543. |
| Figures: | 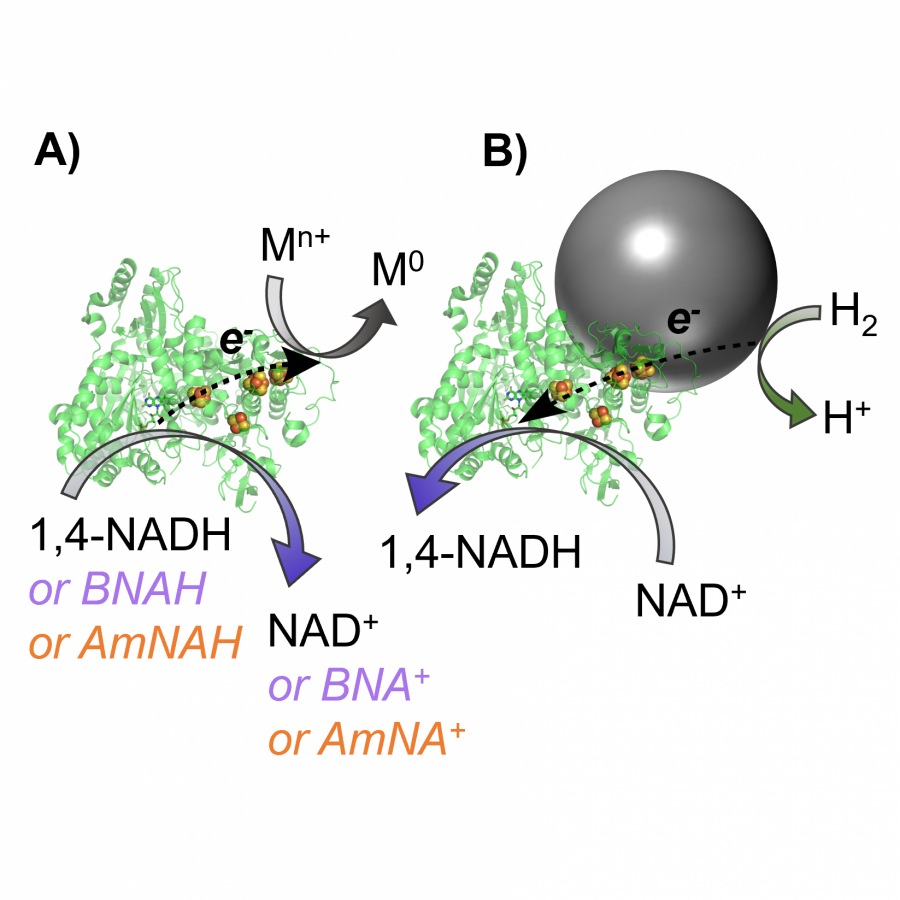 NAD+ reductase used to make metal NPs, subsequently creating a metal biohybrid catalyst. Using NAD+ reductase enzyme to reduce metal salts, leading to the controlled synthesis of metal NPs and utilising the resulting metal NP-enzyme hybrids as catalysts for selective NAD+ reduction. 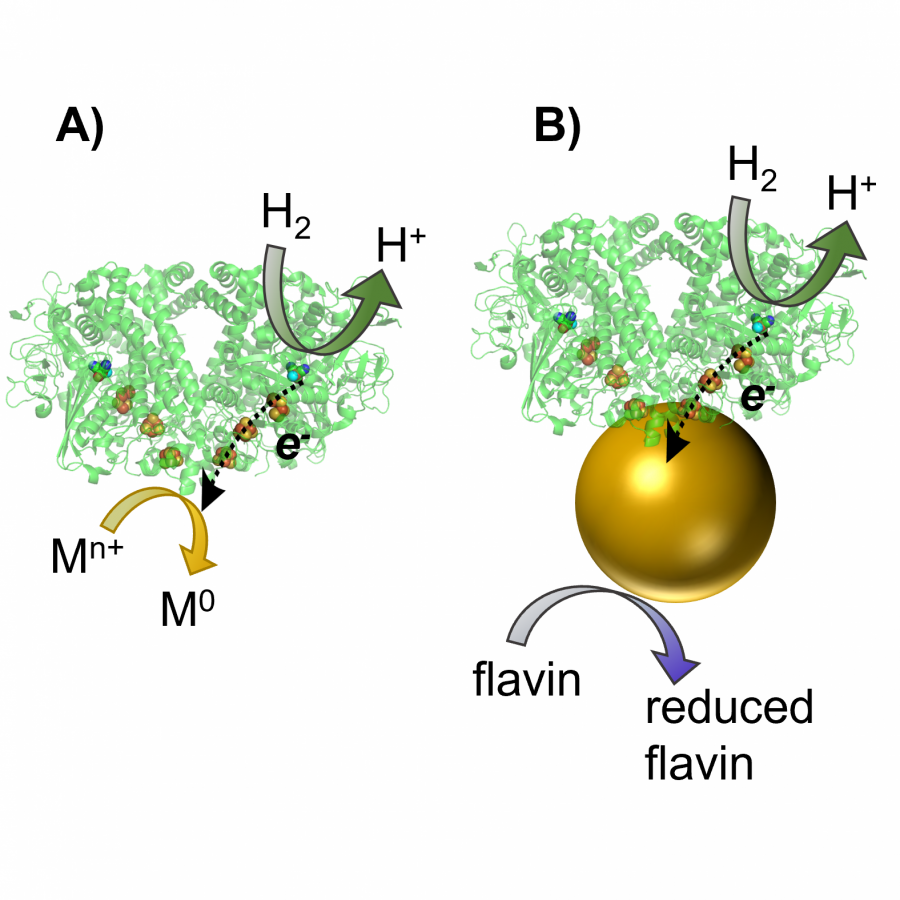 Using hydrogenase for H2-driven metal NP synthesis and flavin recycling. Hydrogenase oxidises H2, releasing electrons for metal reduction. The resulting metal NP-enzyme hybrid can then be used for catalytic applications such as flavin recycling. |
#1761 | Asymmetric mono-reduction of 1,2-dicarbonyls by Old Yellow Enzyme and glucose dehydrogenase |
|
| Presenting author: | Allison WOLDER of TU DELFT |
| Other authors: | Caroline PAUL of TU DELFT |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | asymmetric reduction / ene reductases / hydroxy ketones / dicarbonyls |
| Purpose: | Chiral α-hydroxy ketones are valuable molecules that can be used as fine chemicals or as building blocks for asymmetric synthesis to obtain bioactive compounds [1]. These enantiopure molecules can be obtained via asymmetric reduction of one carbonyl from a vicinal diketone or other (enzymatic) approaches [2]. Alcohol dehydrogenases typically catalyse the double reduction of diketones to the corresponding diols, with some exception for the mono-reduction of certain aliphatic and aromatic 1,2-diketones [3]. Recent studies on flavin-dependent ene reductases from the Old Yellow Enzyme family (OYE) have revealed compelling non-conventional activity without illumination, in particular oxime reduction [4] and C-C bond formation [5], that are distinct from the typical asymmetric reduction of α,β-unsaturated compounds. Evidence alludes to yet another variant reaction, where OYEs might catalyse the reduction of 1,2-dicarbonyl substrates [6]. In this study we demonstrate that certain OYEs can catalyse the non-conventional diastereoselective mono-reduction of 1,2-dicarbonyls to the corresponding hydroxy carbonyl enantiomer (Figure 1). Purification of these enzymes, use of stoichiometric amounts of native cofactor, synthetic cofactor, and mechanistic studies confirmed formation of the α-hydroxy ketone product with full conversion, in high enantiomeric and diastereomeric excess. We also highlight the serendipitous discovery of promiscuous GDH activity on these vicinal dicarbonyls. |
| References: | [1] C. Palomo, M. Oiarbide, J.M. Garcia, Chem. Soc. Rev. 2012, 41, 4150-4164. [2] P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcantara, P. Dominguez de Maria, Acc. Chem. Res. 2010, 43, 288-299. [3] M. Rabuffetti, P. Cannazza, M. L. Contente, A. Pinto, D. Romano, P. Hoyos, A. R. Alcantara, I. Eberini, T. Laurenzi, L. Gourlay, F. Di Pisa, F. Molinari, Bioorg. Chem. 2021, 108, 104644. [4] S. Velikogne, W.B. Breukelaar, F. Hamm, R.A. Glabonjat, W. Kroutil, ACS Catal. 2020, 10, 13377-13382. [5] K. Heckenbichler, A. Schweiger, L. A. Brandner, A. Binter, M. Toplak, P. Macheroux, K. Gruber, R. Breinbauer, Angew. Chem. Int. Ed. 2018, 57, 7240-7244. [6] P. Schweiger, H. Gross, S. Wesener, U. Deppenmeier, Appl. Microbiol. Biotechnol. 2008, 80, 995-1006. |
| Figures: | 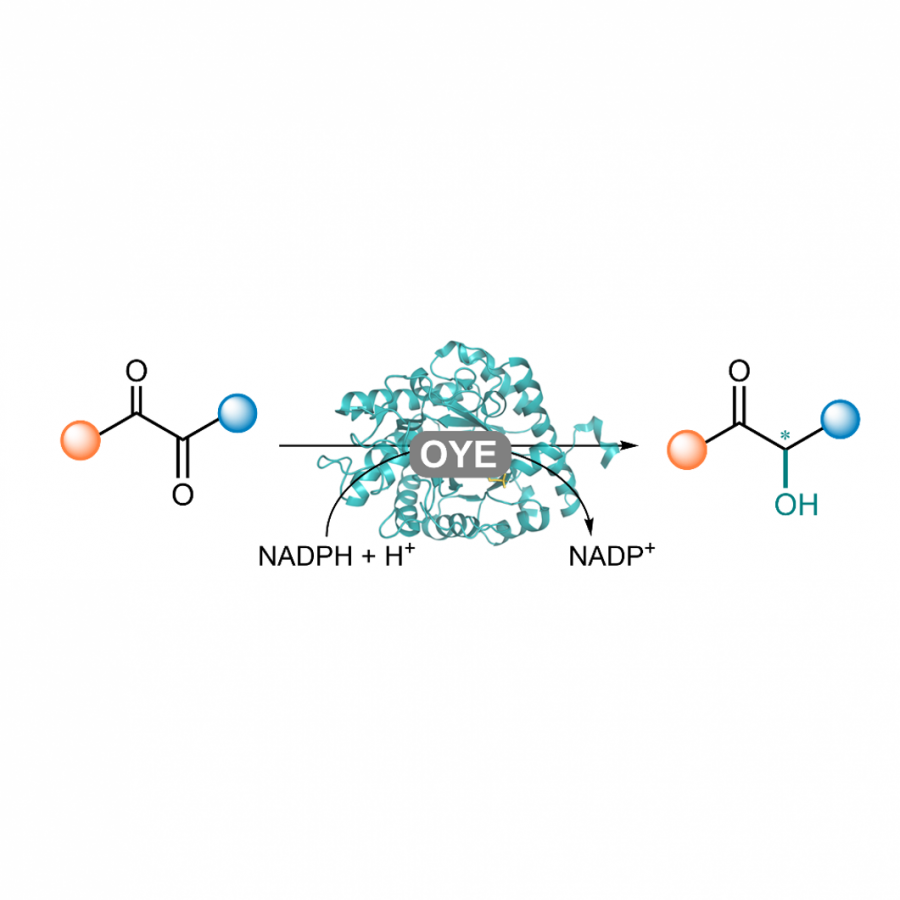 Figure 1 Stereoselective mono-reduction of vicinal dicarbonyls
catalysed by an OYE to the corresponding
alpha-hydroxyl carbonyl. |
#1762 | Novel Metallopterin-Dependent Sulfurase from the Abyss |
|
| Presenting author: | Mariia BELIAEVA of EUROPEAN MOLECULAR BIOLOGY LABORATORY |
| Other authors: | Florian SEEBECK of UNIVERSITY OF BASEL |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | sulfurase / ergothioneine / DMSO reductase / metallopterin-dependent enzyme |
| Purpose: | Metallopretin-dependent proteins contain transition metals molybdenum or tungsten in the active site and catalyze essential processes in living organisms. The most diverse class of these enzymes, the dimethyl sulfoxide (DMSO) reductase superfamily, facilitates a wide range of chemical transformations that mainly implicate oxygen atom installation, removal, and transfer [1]. Here, we describe a novel enzyme from the DMSO reductase superfamily involved in the anaerobic biosynthesis of the natural antioxidant ergothioneine [2]. Metallopterin-dependent ergothioneine synthase (MES) contains two domains: an N-terminal metallopterin-binding sulfurase, and a C-terminal domain, which is a functional cysteine desulfurase. The two modules interact to transfer sulfur from cysteine onto the imidazole ring of trimethylhistidine (TMH) via an intramolecular persulfide relay (Figure 1). The C−S bond-forming activity of MES is unprecedented among metallopretin-containing enzymes and adds to the potential utility of these enzymes in biocatalysis. In addition, this discovery documents the second case of the independent emergence of anaerobic ergothioneine biosynthesis along with two alternative oxygen-dependent pathways [3], providing additional evidence for the importance of this widely-distributed metabolite in early anaerobic cell life. |
| References: | [1] C. Le, M. Bae, S. Kiamehr, E. P. Balskus, Annu. Rev. Biochem., 2022, 91, 475-504. [2] M. A. Beliaeva, F. P. Seebeck, JACS Au, 2022, 2, 2098-2107. [3] A. R. Stampfli, W. Blankenfeldt, F. P. Seebeck, COSB, 2020, 65, 1-8. |
| Figures: | 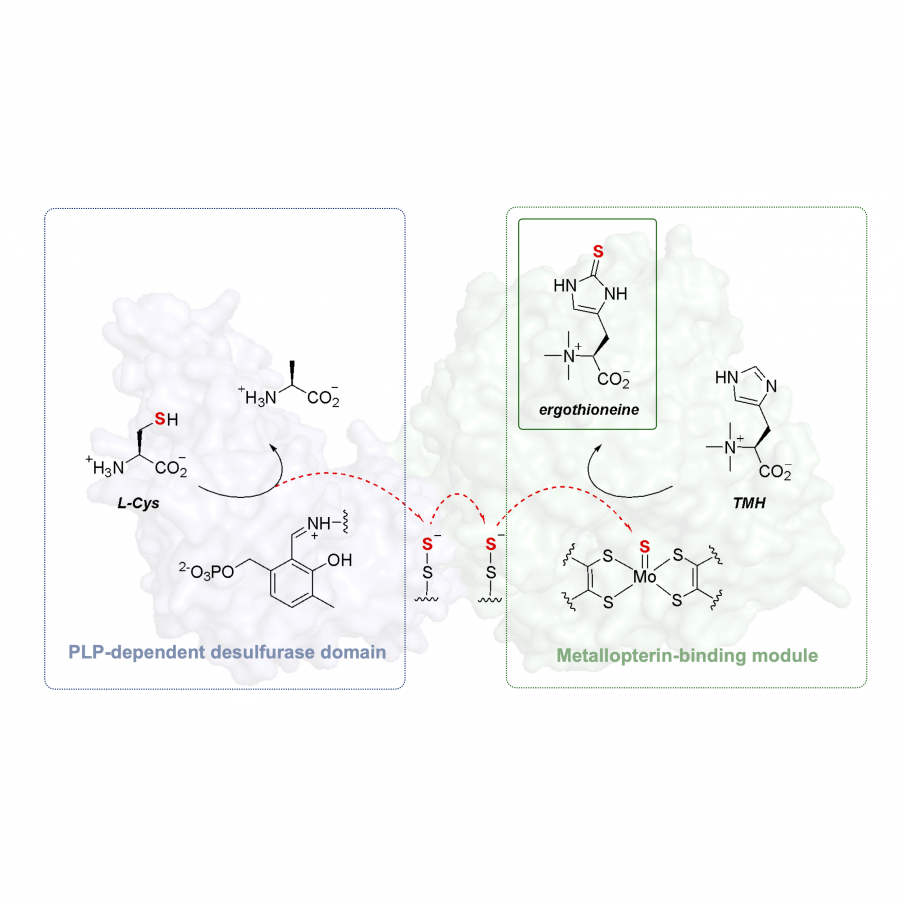 Scheme of ergothioneine biosynthesis by MES. PLP-dependent desulfurase domain extracts sulfur from L-cysteine, which is then transported via the transient formation of persulfides on two cysteine residues to the N-terminal metallopterin-binding module that mediates oxidative sulfurization of TMH. |
#1763 | Diversification of carotenoid cleaving enzymes serving as ‘ozonylases’ for generation of aldehydes from a lignin-based feedstock |
|
| Presenting author: | Lukas SCHOBER of TU GRAZ |
| Other authors: | Astrid SCHIEFER of TU WIEN Florian RUDROFF of TU WIEN Margit WINKLER of TU GRAZ |
| Topic: | Industrial biocatalysis |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Oxidative alkene cleavage / Ozonolysis / AromaticDiOxygenase / |
| Purpose: | Carotenoid cleaving enzymes (CCOs or CCDs) are non-heme iron-dependent oxygenases that can be used as an alternative to ozonolysis. Unlike ozonolysis, they use molecular oxygen instead of ozone to cleave alkenes to form two equivalents of carbonyl compound (aldehyde or ketone, depending on the substitution pattern of the alkene). The catalytic center is an Fe2+ that is bond by 4 histidine residues. Based on the known enzymes Aromatic DiOxygenase (ADO) from Thermothelomyces thermophila[1] we conducted a sequence similarity search and found several promising enzymes. A selected set of enzymes was tested in a mono-phasic whole cell reaction to convert several substrates, including 4-vinyl-guaiacol and isoeugenol, to vanillin. Among the tested enzymes, AspADO (Altererythrobacter sp.), TsADO (Talaromyces stipitatus), PaADO (Podospora anserina), VsADO (Valsa sordida), and CpADO from (Coniochaeta pulveracea) showed significant yields of product. However, MapADO from the marine fungus Moesziomyces aphidis was found to be the most effective in converting isoeugenol to vanillin. Using whole cells containing MapADO, we achieved quantitative conversion of 60 mM isoeugenol to the corresponding aldehyde overnight. Furthermore, 1 mg/mL of purified MapADO converted 20 mM of the same substrate completely in less than 30 minutes. In conclusion, the novel ‘ozonylases’ produced up to more than 75 times more product compared to the already described ADO. |
| References: | [1] J. Ni, Y. T. Wu, F. Tao, Y. Peng, P. Xu, J. Am. Chem. Soc. 2018, 140, 16001-16005. |
| Figures: | 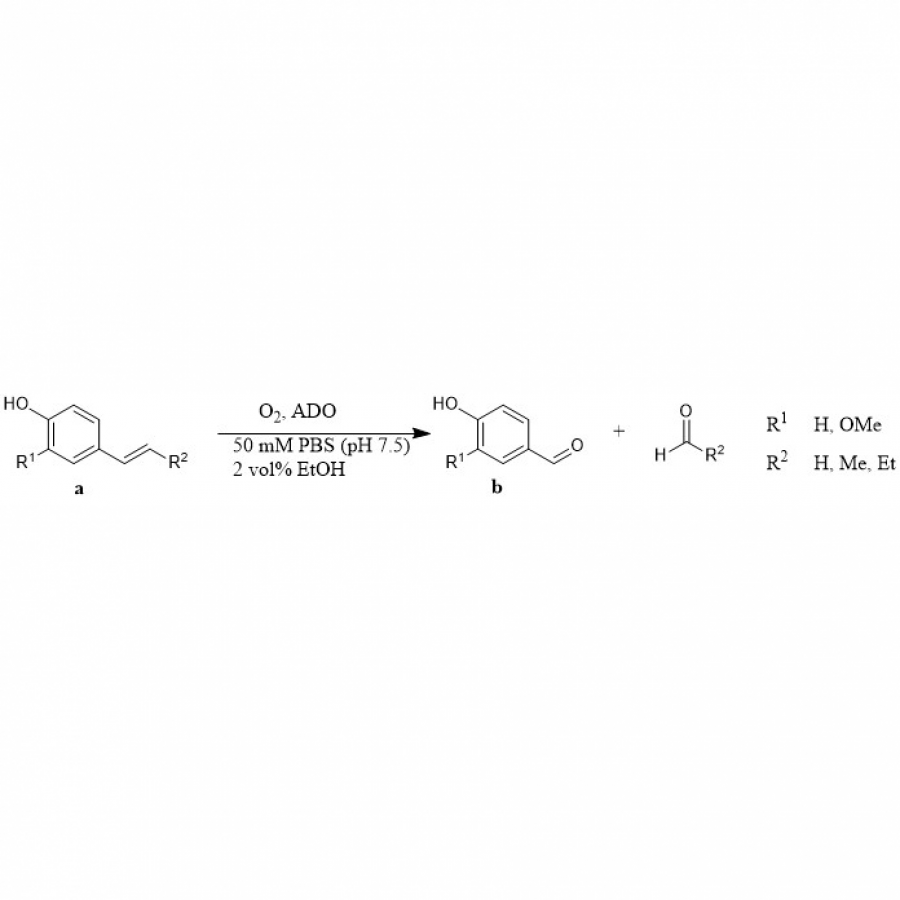 Reaction scheme: Enzymatic oxidative alkene cleavage |
#1770 | anti-selective Epoxidation of α,β-unsaturated Aldehyde Enabled by Engineered Aldolase DERA |
|
| Presenting author: | Hangyu ZHOU of UNIVERSITY OF GRONINGEN |
| Corresponding author: | Gerrit POELARENDS of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | anti-selectivity / enzyme engineering / catalytic promiscuity / peroxidation |
| Purpose: | Chiral epoxides are versatile building blocks which are essential for the synthesis of biologically active compounds and chiral pharmaceuticals. The development of efficient catalysts for the synthesis of chiral epoxides has received considerable attention in recent times. We herein describe the anti-selective epoxidation of α,β-unsaturated aldehydes promoted by an engineered 2-deoxy-D-ribose-5-phosphate aldolase (DERA). After 5 rounds of directed evolution, the archetypical class I aldolase DERA was engineered into an efficient non-natural cofactor-independent peroxygenase. The resulting mutant DERA-EP has 277-fold enhanced peroxygenase activity over the DERA wild-type. In addition, DERA-EP accepts a broad array of α,β-unsaturated aldehydes and enables the 100 milligram-scale synthesis of the epoxy aldehyde in good to excellent crude yield (75-98%) with excellent conversion (>96%), enantiopurity (>95% e.r.) and high anti-diastereoselectivities for most of the cinnamaldehyde derivatives analyzed. This enzymatic strategy is complementary to numerous methodologies previously developed, such as syn-selective metal and amine catalysis, thus opening up a manifold of novel prospects for producing previously inaccessible compounds. From a broader perspective, these studies represent attractive starting points for further expanding the toolbox of enzymes for the practical synthesis of important chiral synthons. |
| References: | [1] G. Xu, M. Crotti, T. Saravanan, K. M. Kataja, G. J. Poelarends, Angew. Chem. Int. Ed. 2020, 59, 10374-10378; Angew. Chem. 2020, 132, 10460-10464. [2] M. Sigmund, G. Xu, E. Grandi, G. J. Poelarends, Chem. - A Eur. J. 2022, 28, e202201651. [3] G. Xu, A. Kunzendorf, M. Crotti, H. J. Rozeboom, A. W. H. Thunnissen, G. J. Poelarends, Angew. Chem. Int. Ed. 2022, 61, e202113970; Angew. Chem. 2022, 134, e202113970. [4] A. Kunzendorf, G. Xu, J. J. H. van der Velde, H. J. Rozeboom, A. W. H. Thunnissen, G. J. Poelarends, ACS Catal. 2021, 11, 13236-13243. [5] L. Biewenga, M. Crotti, M. Saifuddin, G. J. Poelarends, ACS Omega 2020, 5, 2397-2405. [6] Ł. Albrecht, L. K. Ransborg, B. Gschwend, K. A. Jørgensen, J. Am. Chem. Soc. 2010, 132, 17886-17893. |
| Figures: | 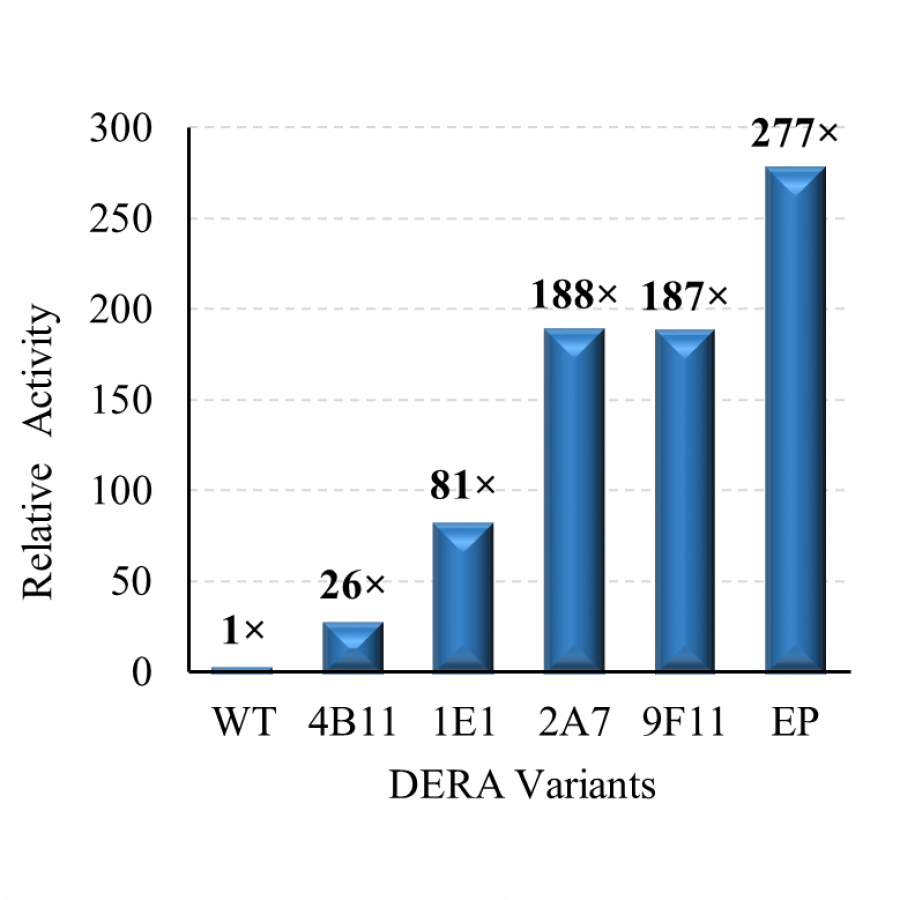 Comparison of the peroxygenase activity of wild-type DERA (WT) and engineered DERA variants. no |
#1784 | Novel fluorescent compounds for screening and characterizing PET-degrading enzymes |
|
| Presenting author: | Georgios TAXEIDIS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Other authors: | EFSTRATIOS NIKOLAIVITS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS VESELIN MASLAK of UNIVERSITY OF BELGRADE JASMINA NIKODINOVIC-RUNIC of INSTITUTE OF MOLECULAR GENETICS AND GENETIC ENGINEERING EVANGELOS TOPAKAS of NATIONAL TECHNICAL UNIVERSITY OF ATHENS |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | enzyme screening / polyethylene terephthalate / fluorescent substrates / plastic degradation |
| Purpose: | Poly(ethylene terephthalate) (PET) is one of the most commonly used polymer resins, especially in the food and beverage industry, as it has great barrier properties, excellent chemical resistance, and stability over a broad temperature range. Even though most people are aware of recycling benefits and PET recycling rates are quite high for some European countries, in other cases, PET bottle waste ending up in landfills remains above 50%. Traditional recycling after mechanical and chemical treatment can completely convert PET waste into new products, although the resulting recycled polymer is of lower quality due to mechanical stress and oxidation during the process [1]–[3]. Regarding chemical treatment, this alternative requires high temperatures in conjunction with toxic chemicals, making the process cost-effective and energy-intensive [4]. On the other hand, enzymatic degradation of PET is highly dependent on material properties such as crystallinity [5], but still, it is the most eco-friendly alternative for handling PET debris, as it requires mild reaction conditions, reducing the energy and reagent consumption [6]. Although the development of new polyester hydrolases that act on highly crystalline PET materials remains a challenge, the enzymatic hydrolysis of PET is a process that is ever-improving [7]. The main enzymes involved in PET degradation are carboxylesterases, cutinases, and lipases [8]. Discovering novel PET-degrading enzymes can be a time-consuming and resource-intensive process. This involves screening metagenomic libraries using appropriate substrates or following conventional methods of microorganism isolation and cultivation under optimal conditions [9]. Directed enzyme evolution and semi-rational strategies comprise other sources for discovering novel PETases, although the major impediment in this approach is the absence of efficient high-throughput screening [9], [10]. A usual approach followed by several reports often includes short (C2-C6), medium (C8-C10), or long-chain (>C10) p-nitrophenyl acyl esters (p-NP esters), which aim to predict and/or compare putative PET hydrolyzing enzymes [11]–[13]. Nonetheless, in that case, no correlation between esterase and depolymerizing activity should be taken into consideration [14], as PET polymer structure substantially differs from these substrates. Alternative approaches perform colorimetric, spectrophotometric, and fluorometric high-throughput assays using substrates, such as synthetic PET oligomers, or PET powder in suspension [17]–[21], although these techniques are often subjected to experimental restrictions such as the use of highly corrosive solvents or the need for secondary analyses for the quantification of released products. In this work, novel fluorescent compounds were synthesized in order to screen and characterize PET-degrading enzymes. In more detail, the structural design of these compounds mimics the structure of PET oligomers and contains a fluorescent moiety (Figure 1), whose release allows real-time monitoring of enzyme activity. The performance of these substrates was evaluated after conducting kinetic studies using various polyesterases including LCC-ICCG variant, IsPETase from Ideonella sakaiensis, and MoPE from Moraxella sp., as well as three enzymes that are not considered PET hydrolyses and cannot degrade PET. The kinetic parameters were further correlated with the ability of the enzymes to degrade virgin PET, proving that some of these PET model substrates can be utilized not only for a rapid, and sensitive high-throughput screening of novel PET-degrading enzymes but also in evaluating the performance of the enzymes that have been mutated using protein engineering tools. |
| References: | [1] P. Falkenstein et al., “UV Pretreatment Impairs the Enzymatic Degradation of Polyethylene Terephthalate,” Front Microbiol, vol. 11, p. 689, Apr. 2020, doi: 10.3389/FMICB.2020.00689/BIBTEX. [2] A. Maurya, A. Bhattacharya, and S. K. Khare, “Enzymatic Remediation of Polyethylene Terephthalate (PET)-Based Polymers for Effective Management of Plastic Wastes: An Overview,” Front Bioeng Biotechnol, vol. 8, p. 1332, Nov. 2020, doi: 10.3389/FBIOE.2020.602325/BIBTEX. [3] E. Pinter et al., “Circularity Study on PET Bottle-To-Bottle Recycling,” 2021, doi: 10.3390/su13137370. [4] “World Economic Forum ,” The new plastics economy: Rethinking the future of plastics., 2016. https://www.greenpeace.org/static/planet4-philippines-stateless-release/2019/05/b1e5a437-b1e5a437-wef_the_new_plastics_economy.pdf (accessed Mar. 27, 2023). [5] T. B. Thomsen, C. J. Hunt, and A. S. Meyer, “Influence of substrate crystallinity and glass transition temperature on enzymatic degradation of polyethylene terephthalate (PET),” N Biotechnol, vol. 69, pp. 28-35, Jul. 2022, doi: 10.1016/J.NBT.2022.02.006. [6] B. Zhu, D. Wang, and N. Wei, “Enzyme discovery and engineering for sustainable plastic recycling,” Trends Biotechnol, vol. 40, no. 1, pp. 22-37, Jan. 2022, doi: 10.1016/J.TIBTECH.2021.02.008. [7] F. Kawai, T. Kawabata, and M. Oda, “Current State and Perspectives Related to the Polyethylene Terephthalate Hydrolases Available for Biorecycling,” ACS Sustain Chem Eng, vol. 8, no. 24, pp. 8894-8908, Jun. 2020, doi: 10.1021/ACSSUSCHEMENG.0C01638/ASSET/IMAGES/MEDIUM/SC0C01638_0003.GIF. [8] V. Tournier et al., “Enzymes’ Power for Plastics Degradation,” Chem Rev, Mar. 2023, doi: 10.1021/ACS.CHEMREV.2C00644/ASSET/IMAGES/LARGE/CR2C00644_0009.JPEG. [9] B. Zhu, D. Wang, and N. Wei, “Enzyme discovery and engineering for sustainable plastic recycling,” Trends Biotechnol, vol. 40, no. 1, pp. 22-37, Jan. 2022, doi: 10.1016/J.TIBTECH.2021.02.008. [10] H. S. Zurier and J. M. Goddard, “A high-throughput expression and screening platform for applications-driven PETase engineering,” Biotechnol Bioeng, vol. 120, no. 4, pp. 1000-1014, Apr. 2023, doi: 10.1002/BIT.28319. [11] D. Danso et al., “New insights into the function and global distribution of polyethylene terephthalate (PET)-degrading bacteria and enzymes in marine and terrestrial metagenomes,” Appl Environ Microbiol, vol. 84, no. 8, Apr. 2018, doi: 10.1128/AEM.02773-17. [12] S. Chen, X. Tong, R. W. Woodard, G. Du, J. Wu, and J. Chen, “Identification and characterization of bacterial cutinase,” Journal of Biological Chemistry, vol. 283, no. 38, pp. 25854-25862, Sep. 2008, doi: 10.1074/JBC.M800848200. [13] F. Kawai et al., “Comparison of polyester-degrading cutinases from genus thermobifida,” ACS Symposium Series, vol. 1144, pp. 111-120, 2013, doi: 10.1021/BK-2013-1144.CH009. [14] C. M. Carr, D. J. Clarke, and A. D. W. Dobson, “Microbial Polyethylene Terephthalate Hydrolases: Current and Future Perspectives,” Front Microbiol, vol. 11, p. 2825, Nov. 2020, doi: 10.3389/FMICB.2020.571265/BIBTEX. [15] P. Pérez-García, D. Danso, H. Zhang, J. Chow, and W. R. Streit, “Exploring the global metagenome for plastic-degrading enzymes,” Methods Enzymol, vol. 648, pp. 137-157, Jan. 2021, doi: 10.1016/BS.MIE.2020.12.022. [16] M. Hajighasemi et al., “Screening and Characterization of Novel Polyesterases from Environmental Metagenomes with High Hydrolytic Activity against Synthetic Polyesters,” Environ Sci Technol, vol. 52, no. 21, pp. 12388-12401, Nov. 2018, doi: 10.1021/acs.est.8b04252 [17] J. Arnling Bååth, K. Borch, and P. Westh, “A suspension-based assay and comparative detection methods for characterization of polyethylene terephthalate hydrolases,” Anal Biochem, vol. 607, p. 113873, Oct. 2020, doi: 10.1016/J.AB.2020.113873. [18] A. M. de Castro, A. Carniel, J. Nicomedes Junior, A. da Conceição Gomes, and É. Valoni, “Screening of commercial enzymes for poly(ethylene terephthalate) (PET) hydrolysis and synergy studies on different substrate sources,” J Ind Microbiol Biotechnol, vol. 44, no. 6, pp. 835-844, Jun. 2017, doi: 10.1007/S10295-017-1942-Z. [19] J. Lusty Beech et al., “A flexible kinetic assay efficiently sorts prospective biocatalysts for PET plastic subunit hydrolysis,” RSC Adv, vol. 12, no. 13, pp. 8119-8130, Mar. 2022, doi: 10.1039/D2RA00612J. [20] S. Heumann et al., “New model substrates for enzymes hydrolysing polyethyleneterephthalate and polyamide fibres,” J Biochem Biophys Methods, vol. 69, no. 1-2, pp. 89-99, Nov. 2006, doi: 10.1016/J.JBBM.2006.02.005. [21] L. Pfaff, D. Breite, C. P. S. Badenhorst, U. T. Bornscheuer, and R. Wei, “Fluorimetric high-throughput screening method for polyester hydrolase activity using polyethylene terephthalate nanoparticles,” Methods Enzymol, vol. 648, pp. 253-270, Jan. 2021, doi: 10.1016/BS.MIE.2020.11.003. |
| Figures: | 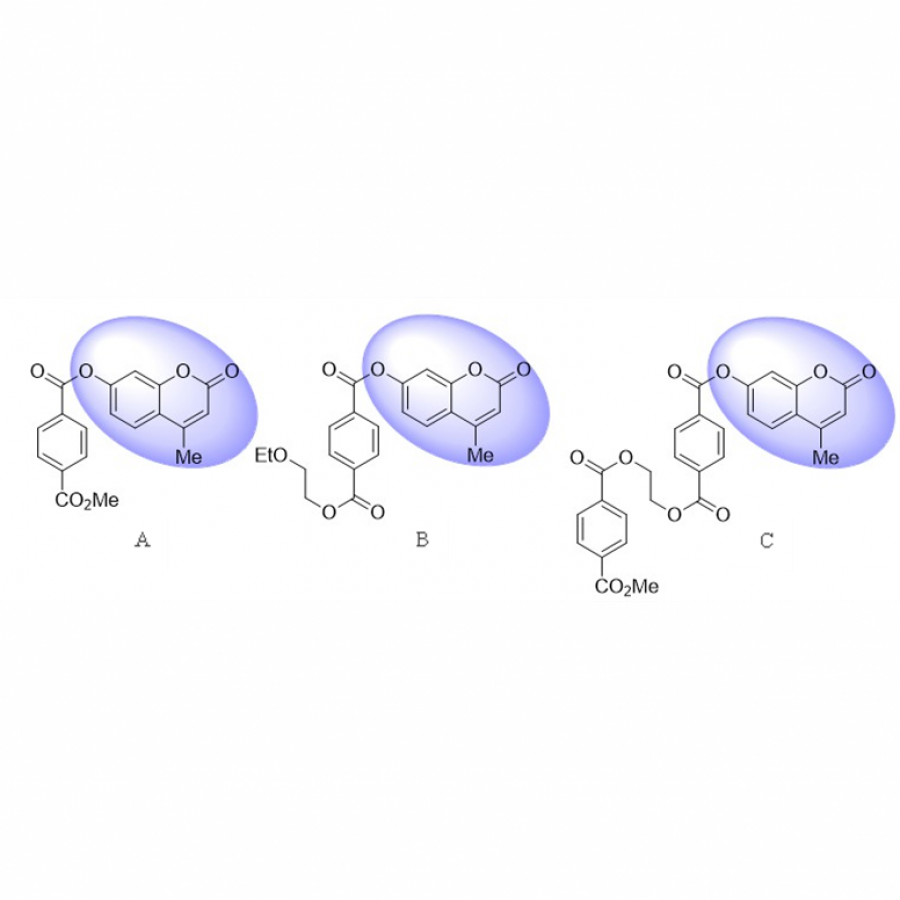 Figure 1 Structure of model substrate (A) mUPET1, (B) mUPET2 and (C) mUPET3. The fluorogenic moieties, which are released due to the enzymatic hydrolysis, are depicted in blue circles. |
#1792 | Dimethylsulfoniopropionate lyase from Pelagibacter ubique HTCC1062 as catalysts for aza-Michael reactions of amines to acrylic and methacrylic acids |
|
| Presenting author: | Diletta ARCERI of IQAC-CSIC |
| Topic: | Enzyme engineering & Discovery |
| Date: | 04:30 pm - 07:00 pm Poster session |
| Keywords: | Aza-Michael reactions / alpha,beta-Unsaturated carboxylic acids / Dimethylsulfoniopropionate lyase / beta-Amino acids |
| Purpose: | The construction of C-N bonds is an important chemical transformation for the preparation of amines, amino alcohols and amino acids. These compounds have been widely employed as chiral building blocks in the pharmaceutical and agrochemical industries. Furthermore the presence of a chiral amine in active pharmaceutical ingredients is estimated to be around 40% and this percentage is larger when considering only the amino groups (chiral and achiral).(1) Several strategies have been employed for the biocatalytic synthesis of C-N bonds, e.g., transaminases, iminoreductases and aza-Michael additions. Particularly, the biocatalytic aza-Michael addition reactions have been performed using promiscuous hydrolases (e.g., proteases, lipases, amylases) using alkyl acrylates, acrylonitrile and enones, and with specific ammonia lyases.(2) However, the use of acrylic and methacrylic acids as Michael acceptors in biocatalysis is not documented. alpha,beta-Unsaturated carboxylic acids are difficult substrates that must be efficiently activated to increase the electrophilicity. In this communication, we report the catalytic properties of dimethylsulfoniopropionate (DMSP) lyase from Pelagibacter ubique HTCC1062 (DddK) as catalyst for Michael type reactions. In nature, DddK catalyzed the cleavage of DMSP to dimethyl sulphide and acrylate. We envisaged that the enzyme can operated in the synthetic direction and catalyze both hetero C-X and C-C Michael additions using acrylic, and methacrylic and other alpha,beta-unsaturated carboxylic acids as acceptors. In this work, we assayed DddK as catalyst for aza-Michael addition of primary and secondary amines to acrylic and methacrylic acids (Scheme 1). Dddk activity, reaction conversion of the beta-carboxylic acid derivatives, and the stereochemical outcome of the products from methacrylic acid will be presented and discussed. Acknowledgements: This project has received funding from the European Union's Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 956631 |
| References: | [1] Breuer, M.; Ditrich, K.; Habicher, T., et al., Angew. Chem. Int. Ed. 2004, 43, 788-824. [2] (a) Steunenberg, P.; Sijm, M.; Zuilhof, H., et al., J. Org. Chem. 2013, 78, 3802-3813; (b) Torre, O.; Alfonso, I.; Gotor, V., Chem. Commun. 2004, 1724-1725; (c) Dutt, S.; Goel, V.; Garg, N., et al., Adv. Synth. Catal. 2020, 362, 858-866; (d) Zhang, J.; Abidin, M. Z.; Saravanan, T., et al., ChemBioChem 2020, 21, 2733-2742. |
| Figures: | 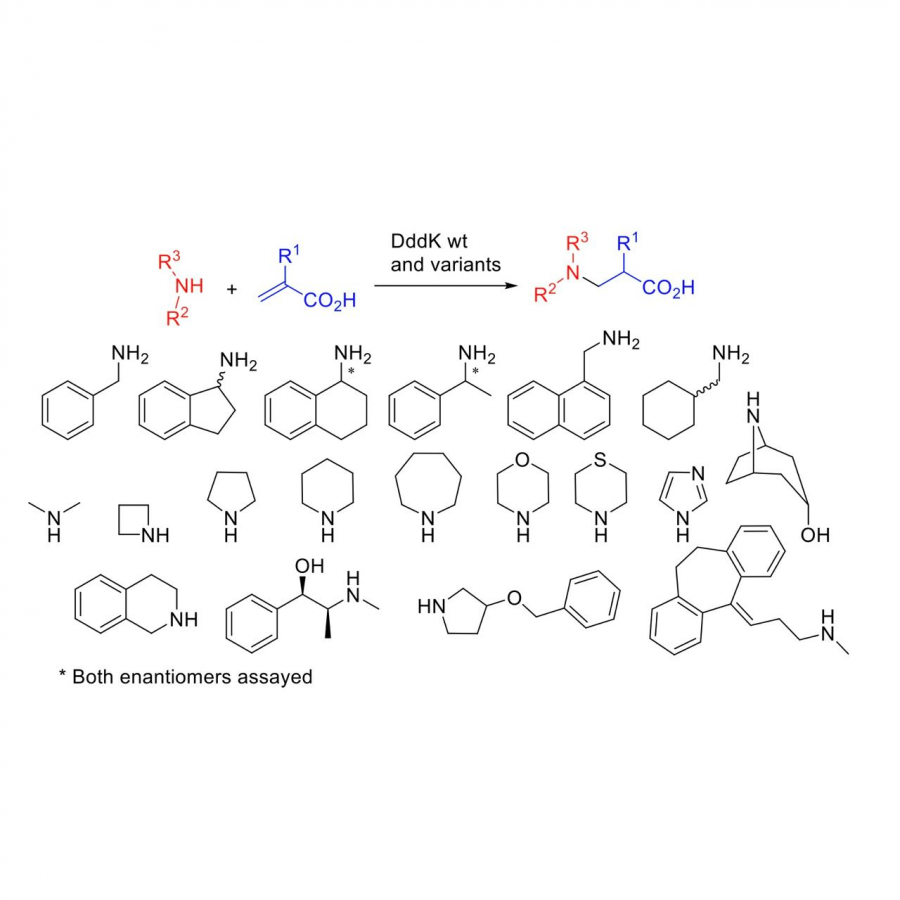 Figure 1 DddK catalyzed aza-Michael additions of primary and secondary amines to acrylic (R1 = H) and methacrylic acids (R2 = CH3).
|