
#38 | Enzyme-mediated C-N Bond Formation via Ene Reactions and Diels-Alder Cycloadditions |
|
| Presenting author: | Jan DESKA of UNIVERSITY OF HELSINKI |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 11:45 am - 12:00 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | promiscuity / ene reaction / amination / heterocycles |
| Purpose: | The incorporation of nitrogen into organic building blocks through C-N bond forming reactions represents one of the most crucial methodology goals in modern organic chemistry. Especially nitrogen-containing heterocycles, an absolute go-to motif in today's pharmaceuticals, pose a critically important synthetic target. Biocatalytic methodologies to facilitate C-N bond formations are therefore highly desireable. However, Nature's repertoire to introduce nitrogen moieties into molecular frameworks is generally limited to direct functional group interconversions. Here, the toolbox of synthetically valuable biocatalytic methods has been expanded steadily in the past years through a number of nitrogen-fixation strategies based on reductive aminations and additions catalyzed by transaminases, imine reductases, and amine dehydrogenases, to name a few.[1] In a complementary approach mimicking chemistry by biological means, most recently the biocatalytic imitation of non-natural but synthetically relevant reactions was moved into the spotlight,[2] and nitrene insertion-based transformations utilizing engineered heme proteins introduced whole new pathways for the biocatalytic C–N bond formation.[3] As a yet untapped template from the world of traditional synthesis, another attractive alternative approach for selective C-N bond formations engages reactive nitroso. First explored in the 1960´s for the synthesis of allylamines using nitrosobenzene, nitroso ene reactions, and the related nitroso-Diels-Alder cycloadditions, are nowadays found in a wide variety of synthetic strategies towards natural products and pharmaceuticals.[4] In this study, we highlight the potential of established oxidoreductase systems as biological mediators for the generation of reactive nitroso species. The biocatalytic oxidation of acylated hydroxylamines enables the direct and selective introduction of nitrogen functionalities via activation of allylic C-H bonds or [4+2]-cycloaddition with dienes.[5] Utilizing either laccases or an oxidase/peroxidase couple for the formal dehydrogenation of N-hydroxycarbamates and hydroxamic acids with air as terminal oxidant, acylnitroso species are generated under particularly mild aqueous conditions. The reactive intermediates undergo C-N bond formation through an ene-type mechanism and provide high yields both in intramolecular and intermolecular enzymatic aminations. Alternatively, hetero-Diels-Alder cycloadditions offer access to six-membered N,O-heterocycles. In addition to extensive studies on the product scope and stereoselectivity aspects, investigations on different pathways of the two biocatalytic systems and labelling studies provide more insights into this unprecedented promiscuity of classical oxidoreductases as catalysts for nitroso-based transformations. |
| References: | [1] a) J. H. Schrittwieser, S. Velikogne, W. Kroutil, Adv. Synth. Catal. 2015, 357, 1655; b) F. Guo, P. Berglund, Green Chem. 2017, 19, 333; c) I. Slabu, J. L. Galman, R. C. Lloyd, N. J. Turner, ACS Catal. 2017, 7, 8263. [2] a) J. B. Siegel, A. Zanghellini, H. M. Lowick, G. Kiss, A. R. Lambert, J. L. St. Clair, J. L. Gallaher, D. Hilvert, M. H. Gelb, B. L. Stoddard, K. N. Houk, F. E. Michael, D. Baker, Science 2010, 329, 309; b) P. S. Coelho, E. M. Brustad, A. Kannan, F. H. Arnold, Science 2013, 339, 307; c) D. Thiel, D. Doknić, J. Deska, Nat. Commun. 2014, 5, 5278. [3] N. W. Goldberg, A. M. Knight, R. K. Zhang, F. H. Arnold, J. Am. Chem. Soc. 2019, 141, 19585. [4] W. Adam, O. Krebs, Chem. Rev. 2003, 103, 4131 [5] C. Jäger, M. Haase, K. Koschorreck, V. B. Urlacher, J. Deska, Angew. Chem. Int. Ed. 2023, 62, e202213671. |
| Figures: | 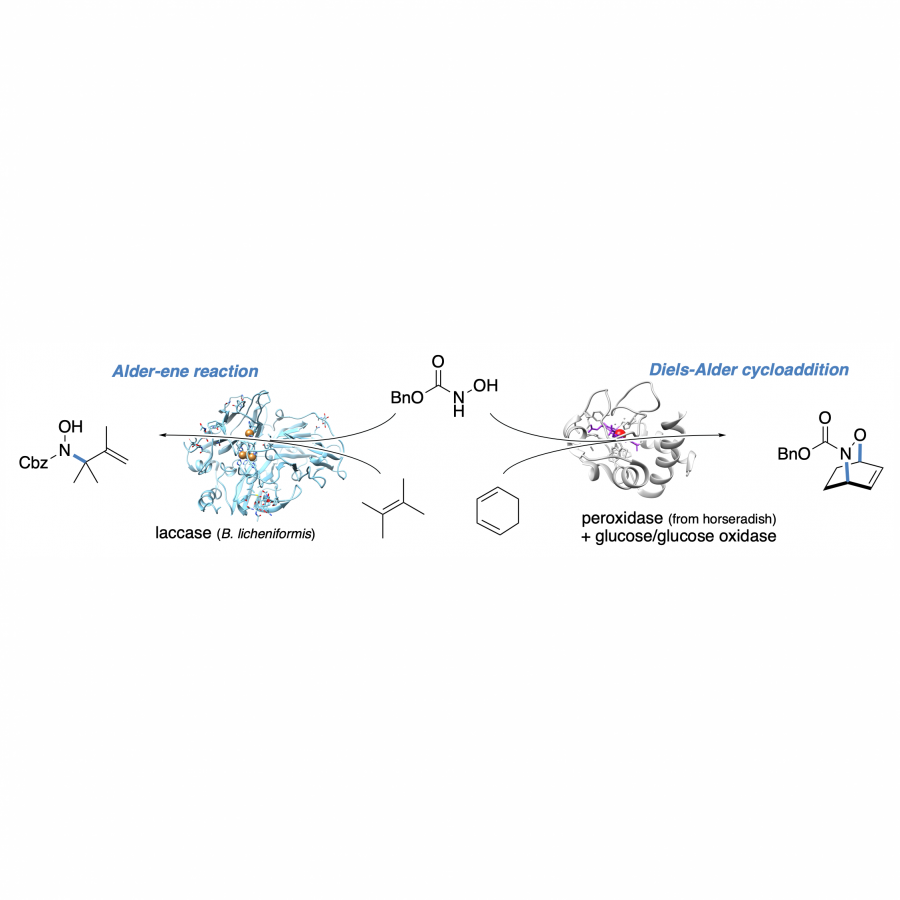 Figure 1 Ene reactions and Diels-Alder cycloadditions catalyzed by oxidoreductases |
#43 | Generating evolvable, artificial metalloenzymes containing organometallic cofactors via ligand exchange |
|
| Presenting author: | Oskar James KLEIN of UNIVERSITY OF CAMBRIDGE |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 04:15 pm - 04:18 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | Artificial metalloenzymes / Transition metal catalysis / / |
| Purpose: | In this work, ruthenium cofactors were introduced into protein scaffolds by ligand exchange between ruthenium compounds and peptidic, Lewis basic entities (Figure 1). This method ensures an intimate link between the first and second coordination-sphere, with the latter being dictated by the protein fold. By systematically adapting the electronic and steric properties of the ligands on the original ruthenium complex, the exchange process was optimised to give good yields of the desired mononuclear species rapidly using only small excess of ruthenium. The resulting modified protein was shown to be catalytically active, effectively reducing a pre-fluorescent quinolonium substrate in aqueous solution via transfer-hydrogenation from formate. [1] With a new ArM at hand, further work was started to evolve it using a high-throughput microfluidic approach. Multiple pre-fluorescent substrates were developed or adapted for use in droplets, allowing for screening of different new-to nature reactions. We further present various transition metal complexes that undergo ligand exchange in the presence of protein to yield ArMs bearing different organometallic cofactors, including iridium and ruthenium-NHC based moieties. Initial findings have shown distinct catalytic activities depending on the cofactor prior to any directed evolution, thus highlighting the potential of a combinatorial approach where informed choice of transition metal complex can install a biorthogonal reactivity into a protein that subsequently could be evolved to yield highly functional catalysts. |
| References: | [1] G. S. Biggs*, O. J. Klein*, S. L. Maslen, J. M. Skehel, T. J. Rutherford, S. M. V. Freund, F. Hollfelder, S. R. Boss, P. D. Barker, Angew. Chem. Int. Ed. 2021, 60, 10919. |
| Figures: | 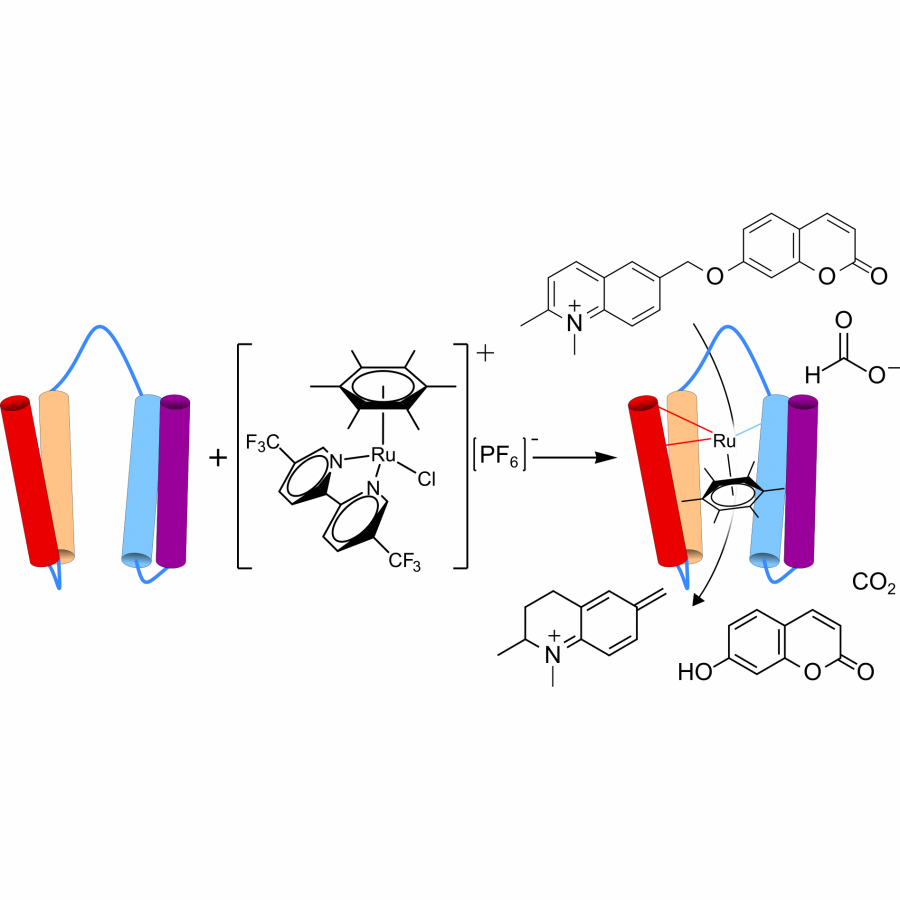 Overview Overview of ArM formation and activity, taken from [1]. |
#77 | Directed Evolution of De Novo Designed Artificial Metatheases |
|
| Presenting author: | Zhi (Robin) ZOU of UNIVERSITY OF BASEL |
| Corresponding author: | Thomas R WARD of UNIVERSITY OF BASEL |
| Other authors: | Indrek KALVET of INSTITUTE FOR PROTEIN DESIGN, UNIVERSITY OF WASHINGTON Boris LOZHKIN of UNIVERSITY OF BASEL David BAKER of INSTITUTE FOR PROTEIN DESIGN, UNIVERSITY OF WASHINGTON |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 11:30 am - 11:45 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | Artificial Metalloenzyme / De Novo Protein Design / Directed Evolution / Metathesis |
| Purpose: | Artificial metalloenzymes are metalloproteins made in the laboratory which aim to catalyze highly selective and efficient abiotic chemical reactions. These biohybrid catalysts are often designed by tethering a catalytic metal cofactor to a preexisting (or repurposed) protein scaffold. Owning to the advances in computational biology, de novo protein design is emerging as a powerful methodology to generate proteins with user-defined functions. Developing artificial metalloenzymes using de novo designed protein scaffolds is of high interest. Here we report the designing and evolution of novel artificial metalloenzymes for highly efficient and selective olefin metathesis, one of the most widely used reactions in chemical synthesis, using modular and robust de novo designed proteins as scaffolds. Taking advantage of protein engineering methodologies, especially directed evolution campaigns, we optimized the supramolecular interactions toward the metal cofactor and improved the activity of the de novo designed artificial metathases (DeNovoArMs). We envision that this strategy will also evolve the regio-/enantio-selectivity of the DeNovoArMs in a wide range of olefin substrates. |
| References: | [1]: Doyle, Lindsey, et al. Nature. 528.7583 (2015): 585-588. [2]: Schwizer, Fabian, et al. Chem. Rev. 118.1 (2018): 142-231. [3]: Jeschek, Markus, et al. Nature. 537.7622 (2016): 661-665. [4]: Sabatino, Valerio, et al. J. Am. Chem. Soc. 141.43 (2019): 17048-17052. |
| Figures: | 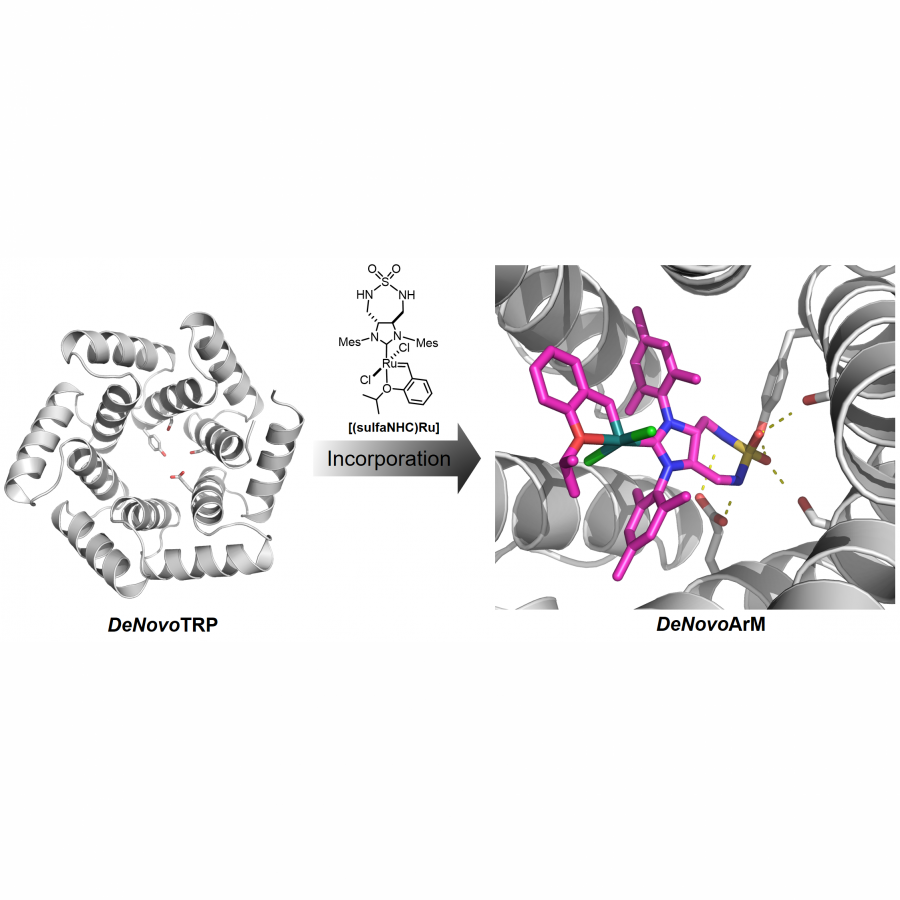 Figure 1 Assembling of the de novo designed artificial metathase (DeNovoArM) via supramolecular interactions of DeNovoTRP host (left, marked in grey) and [(sulfaNHC)Ru] cofactor (right, marked in magenta). 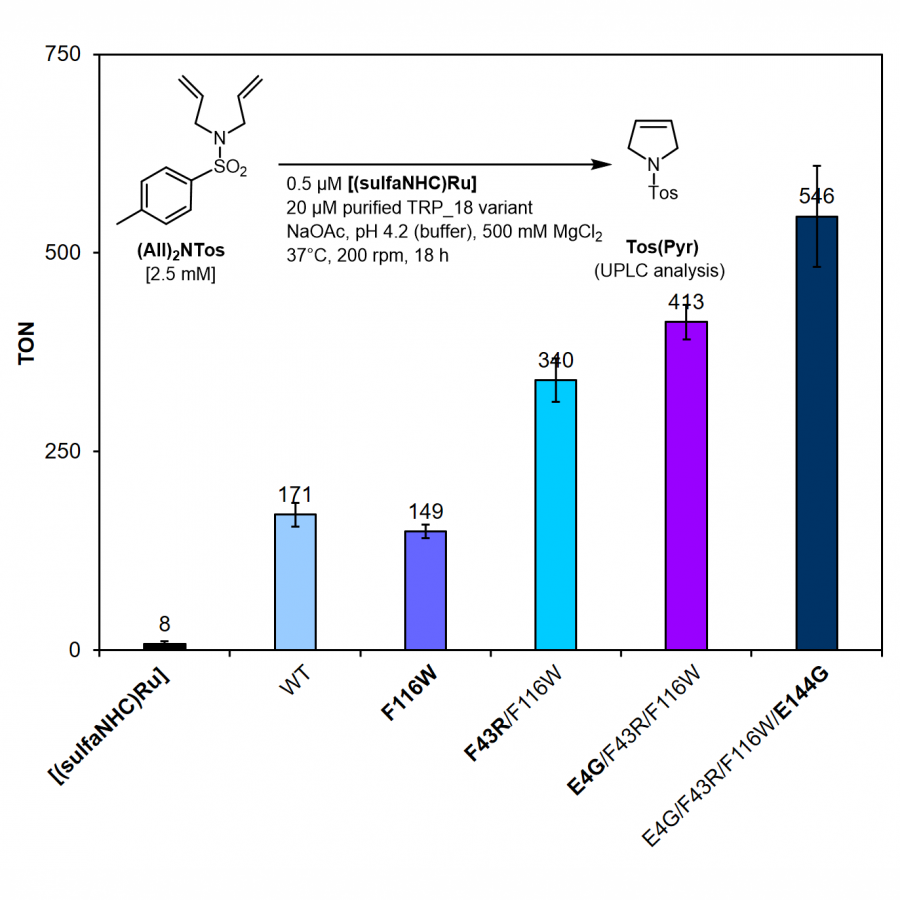 Figure 2 Directed evolution of the DeNovoArM using an (AII)2NTos as the substrate. Screening of three iterative rounds of site-saturation mutagenesis libraries has generated a tetra-variant (E4G/F43R/F116W/E144G) which gains 3.7-fold TON. |
#101 | Enzyme Discovery & Engineering to Create Biocatalysts Suitable for Efficient Applications |
|
| Presenting author: | Uwe BORNSCHEUER of UNIVERSITY OF GREIFSWALD |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:30 pm - 05:30 pm Open Lecture - Chair : S. ROY, Sanofi |
| Keywords: | BIOCATALYSIS / CHIRAL COMPOUNDS / PROTEIN ENGINEERING / HIGH-THROUGHPUT SCREENING |
| Purpose: | This lecture will cover recent achievements in the discovery, protein engineering and application of enzymes in biocatalysis [1]. For the asymmetric synthesis of chiral amines, we created (S)-selective amine transaminases for the acceptance of bulky ketones [2]. Most recently, we have developed a sophisticated growth selection method and could create highly active and selective enzymes from three classes to make important chiral precursors for pharmaceutical building blocks [3]. For the regioselective methylation/alkylation, we have explored SAM-dependent O-methyltransferases to make flavonoids and related compounds [4] and developed engineered halide methyltransferases to transfer alkyl residues such as ethyl-, propyl- or allyl-, substantially expanding the repertoire of target compounds [5]. In addition, we have engineered a P450 enzyme for the highly selective formation of ursodeoxycholic acid (UDCA) from lithocholic acid [6]. For the recycling of plastics, we have investigated PET [7], for which we determined the first structure of an MHETase in complex with a substrate analogue [8] and also provided important adjustments of a published PETase structure [9]. We also used various methods of protein engineering to improve several PET-hydrolase for higher activity and thermostability [9]. Most recently, we have identified the first urethanases in a metagenomic library able to degrade polyurethanes [10] and designed an enzyme cascade to degrade poly(vinylalcohols) [11]. |
| References: | [1] Yi., D. et al., Chem. Soc. Rev., 50, 8003-8049 (2021); Wu, S. et al. Angew. Chem. Int. Ed., 60, 88-119 (2021); Rudroff, F. et al., Nat. Catal. 1, 12-22 (2018); Badenhorst C.P.S., Bornscheuer, U.T., Trends Biochem. Sci., (2018), 43, 180-198; Bornscheuer, U.T. et al., Nature, 485, 185-194 (2012) [2] Pavlidis, I. et al., Nature Chem., 8, 1076-1082 (2016) [3] Wu, S. et al. Nature Commun., 13, 7458 (2022) [4] Tang, Q. et al., ChemBioChem, 22, 2584-2590 (2021); Tang, Q. et al., ChemCatChem, 12, 3721-3727 (2020); Tang, Q. et al., ChemCatChem, 11, 3227-3233 (2019) [5] Tang, Q. et al., Angew. Chem. Int. Ed., 60, 1524-1527 (2021) [6] Grobe, S. et al., Angew. Chem. Int. Ed., 60, 753-757 (2021) [7] Wei, R. et al. (2022), ACS Catal., 12, 3382-3396 (2022); Wei, R. et al., Nature Catal., 3, 867-871 (2020); Bornscheuer, U.T., Science, 351, 1155-1156 (2016) [8] Palm, G.J. et al., Nature Commun., 10, 1717 (2019) [9] Wei, R. et al., Nature Commun., 10, 558 (2019); Pfaff, L. et al., ACS Catal., 12, 9790-9800 (2022); von Haugwitz et al., ACS Catal., 12, 15259-15270 (2022) [10] Branson, Y. et al., Angew. Chem. Int. Ed., 62, e202216220 (2023) [11] von Haugwitz, G. et al., Angew. Chem. Int. Ed., 62, e202216962 (2023) |
#107 | An arylamine-diazotizing enzyme catalyses N-N bond formation in the biosynthesis of tasikamides |
|
| Presenting author: | Zhao-Xun LIANG of NANYANG TECHNOLOGICAL UNIVERSITY |
| Topic: | Enzyme discovery and engineering |
| Date: | 03:45 pm - 04:00 pm Session Enzyme engineering & Discovery #2 - Chair : T. WARD, University of Basel |
| Keywords: | Diazonium / cyclic peptide / biosynthesis / pathway coupling |
| Purpose: | An arylamine-diazotizing enzyme catalyses N-N bond formation in the biosynthesis of tasikamides Guang-Lei Ma, Hartono Candra, Li Mei Pang, Juan Xiong, Yichen Ding, Hoa Thi Tran, Zhen Jie Low, Hong Ye, Min Liu, Jie Zheng, Mingliang Fang, Bin Cao, and Zhao-Xun Liang* School of Biological Sciences & School of Civil and Environmental Engineering, Nanyang Technological University Abstract We discovered a family of novel cyclic peptides (tasikamides) that share a unique cyclic pentapeptide scaffold. Tasikamides A–C (1–3) contain a rare hydrazone group (C═N─N) that connects the cyclic peptide scaffold to an alkyl 5-hydroxylanthranilate (AHA) moiety. We found that the biosynthesis of 1–3 requires two biosynthetic gene clusters with one encoding a nonribosomal peptide synthetase (NRPS) pathway for assembling the cyclic peptide scaffold and another encoding the AHA-synthesizing pathway. The AHA gene cluster encodes three ancillary enzymes (Aha1, 2 and 11) that catalyse the diazotization of AHA to yield an aryl diazonium species (diazo-AHA), which undergoes Japp–Klingemann coupling with a cyclic peptide precursor to furnish the hydrazone group and yield 1–3. In vitro enzymatic assays suggest the N─N bond in the hydrazone group of 1–3 is forged via a HNO2-mediated mechanism, with Aha1 and Aha2 producing NO2–/HNO2 and Aha11 catalysing the diazotization of AHA to generate the reactive diazo-AHA. The findings raise the prospect of exploiting the arylamine-diazotizing enzymes for the in vivo synthesis of aryl compounds and modification of biological macromolecules. References: 1. Biosynthesis of Tasikamides via Pathway Coupling and Diazonium-Mediated Hydrazone Formation, Ma, GL., Candra, H., Pang, LM, Xiong, J., Ding, Y., Tran, HT., Low, ZJ., Ye, H., Liu, M., Zheng, J., Fang, M., Cao, B., Liang, Z.-X. Journal of the American Chemical Society, 2022, 144, 4, 1622–1633. 2. Enaminone formation drives the coupling of biosynthetic pathways to generate cyclic lipopeptides, Candra, H., Ma, GL., En, SLQ, Liang, Z.-X., ChemBioChem 2022, 23 (22), e202200457. |
| References: | Ma et al, Biosynthesis of Tasikamides via Pathway Coupling and Diazonium-Mediated Hydrazone Formation, Journal of the American Chemical Society, 2022, 144, 4, 1622-1633. Candra et al, Enaminone formation drives the coupling of biosynthetic pathways to generate cyclic lipopeptides, ChemBioChem 2022, 23 (22), e202200457. |
| Figures: | 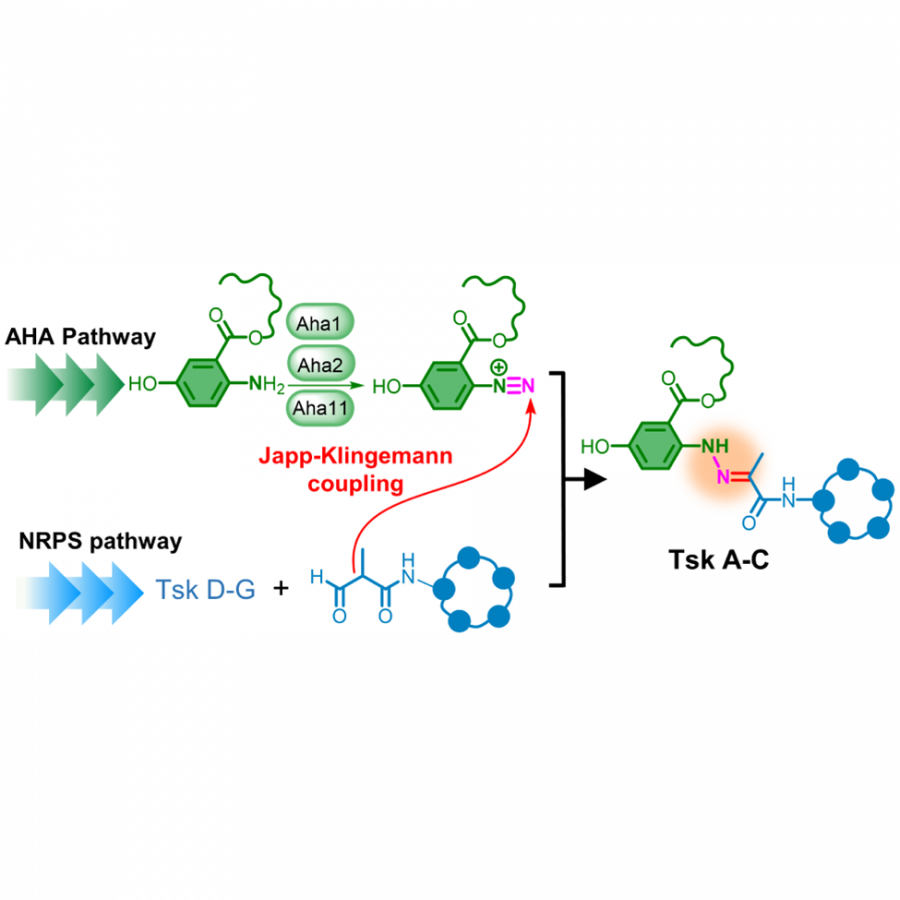 Diazonium-mediated hydrazone formation in biosynthesis The AHA gene cluster encodes three enzymes (Aha1, 2 and 11) that catalyse the diazotization of AHA to yield an aryl diazonium species (diazo-AHA), which undergoes Japp–Klingemann coupling with a cyclic peptide precursor to furnish the hydrazone group and |
#109 | Fueling the Digital Revolution in Biocatalysis with Language Models |
|
| Presenting author: | Teodoro LAINO of IBM RESEARCH EUROPE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 05:30 pm - 06:00 pm Invited Lecture - Chair : S. ROY, Sanofi |
| Keywords: | / / / |
| Purpose: | Artificial Intelligence (AI) and Data are driving one of the most notable revolutions in organic chemistry. The use of experimental data, extracted from public documents or collected using ELNs, emerged as one of the most effective, scalable approaches for capturing human knowledge, modelling and improving chemical processes. Machine learning tasks demonstrated high quality and ease of use in problems such as predicting chemical reactions [1-2], retrosynthetic routes [3], digitizing chemical literature [4], predicting detailed experimental procedures [5], designing new fingerprints [6] and yield predictions [7]. In this talk, I'll briefly introduce the use of AI as the cornerstone of the accelerated discovery method, in which intelligent workflows are used to collect and synthesize known information, augment the known data with rich simulation, predict potential solutions with the desired attributes based on data-driven models. The rest of the presentation will focus on recent applications of language models to the field of enzymatic catalysis [8], including the development of retrosynthetic strategies as a mean to promote sustainability and green chemistry at scale [9]. [1] IBM Research Europe, Chem. Sci., 2018, 9, 6091-6098 [2] IBM Research Europe, ACS Cent. Sci. 2019, 5, 9, 1572-1583 [3] IBM Research Europe, Chem. Sci., 2020, 11, 3316-3325 [4] IBM Research Europe, Nat. Comm., 2020, 11, 3601 [5] IBM Research Europe, Nat. Comm., 2021, 12, 2573 [6] IBM Research Europe, Nat. Mach. Intel., 2021, 3, 144–152 [7] IBM Research Europe, Mach. Learn.: Sci. Technol., 2021, 2, 015016 [8] IBM Research Europe, Nat Comm., 2022, 13, 964 [9] https://rxn.res.ibm.com |
#112 | Accelerating Discovery of Substrate Promiscuity in Cytochrome P450 BM3 |
|
| Presenting author: | Joelle PELLETIER of UNIVERSITÉ DE MONTRÉAL |
| Topic: | Enzyme discovery and engineering |
| Date: | 09:00 am - 09:45 am Session Enzyme engineering & Discovery #1 - Chair : M. REMAUD-SIMEON, Toulouse Biotechnology Institute |
| Keywords: | / / / |
| Purpose: | Enzymes collectively display a great breadth of catalytic properties yet are individually confined to one or a few specific catalytic tasks. Despite key advances in enzyme engineering, our capacity to predict the effects of mutations on function remains nebulous. Here we present advances in engineering non-native substrate recognition for biocatalyzed transformation into useful products. We examine cytochrome P450 oxidase from Bacillus megaterium (P450 BM3) in its capacity to functionalize C-H bonds. Cost-effective, high-throughput colorimetric screening at the whole-cell level had previously suggested a correlation between the production of indigo and increased substrate promiscuity, in a small number of P450 BM3 variants. We greatly expand the diversity of indigo-producing P450 BM3 variants and demonstrate a correlation with promiscuous aromatic hydroxylation reactions. We look ahead to the potential for large experimental datasets to train smarter design algorithms for enzyme engineering. |
| Figures: |  High-throughput colorimetric screening P450 BM3 in cell lysate was screened for hydroxylation of non-native substrates |
#138 | Dimethylsulfoniopropionate lyase from Pelagibacter ubique HTCC1062 as catalysts for aza-Michael reactions of amines to acrylic and methacrylic acids |
|
| Presenting author: | Diletta ARCERI of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA (IQAC-CSIC) |
| Other authors: | Karel HERNANDEZ of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC, Angela MOURELLE of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC, Jesus JOGLAR of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC, Jordi BUJONS of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC, Pere CLAPES of INSTITUTE FOR ADVANCED CHEMISTRY OF CATALONIA, DEPT. OF BIOLOGICAL CHEMISTRY, IQAC-CSIC, |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:12 pm - 04:15 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | Aza-Michael reactions / alpha,beta-Unsaturated carboxylic acids / Dimethylsulfoniopropionate lyase / beta-Amino acids |
| Purpose: | The construction of C-N bonds is an important chemical transformation for the preparation of amines, amino alcohols and amino acids. These compounds have been widely employed as chiral building blocks in the pharmaceutical and agrochemical industries. Furthermore the presence of a chiral amine in active pharmaceutical ingredients is estimated to be around 40% and this percentage is larger when considering only the amino groups (chiral and achiral).(1) Several strategies have been employed for the biocatalytic synthesis of C-N bonds, e.g., transaminases, iminoreductases and aza-Michael additions. Particularly, the biocatalytic aza-Michael addition reactions have been performed using promiscuous hydrolases (e.g., proteases, lipases, amylases) using alkyl acrylates, acrylonitrile and enones, and with specific ammonia lyases.(2) However, the use of acrylic and methacrylic acids as Michael acceptors in biocatalysis is not documented. alpha,beta-Unsaturated carboxylic acids are difficult substrates that must be efficiently activated to increase the electrophilicity. In this communication, we report the catalytic properties of dimethylsulfoniopropionate (DMSP) lyase from Pelagibacter ubique HTCC1062 (DddK) as catalyst for Michael type reactions. In nature, DddK catalyzed the cleavage of DMSP to dimethyl sulphide and acrylate. We envisaged that the enzyme can operated in the synthetic direction and catalyze both hetero C-X and C-C Michael additions using acrylic, and methacrylic and other alpha,beta-unsaturated carboxylic acids as acceptors. In this work, we assayed DddK as catalyst for aza-Michael addition of primary and secondary amines to acrylic and methacrylic acids (Scheme 1). Dddk activity, reaction conversion of the beta-carboxylic acid derivatives, and the stereochemical outcome of the products from methacrylic acid will be presented and discussed. Acknowledgements: This project has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement No 956631 |
| References: | [1] Breuer, M.; Ditrich, K.; Habicher, T., et al., Angew. Chem. Int. Ed. 2004, 43, 788-824. [2] (a) Steunenberg, P.; Sijm, M.; Zuilhof, H., et al., J. Org. Chem. 2013, 78, 3802-3813; (b) Torre, O.; Alfonso, I.; Gotor, V., Chem. Commun. 2004, 1724-1725; (c) Dutt, S.; Goel, V.; Garg, N., et al., Adv. Synth. Catal. 2020, 362, 858-866; (d) Zhang, J.; Abidin, M. Z.; Saravanan, T., et al., ChemBioChem 2020, 21, 2733-2742. |
| Figures: | 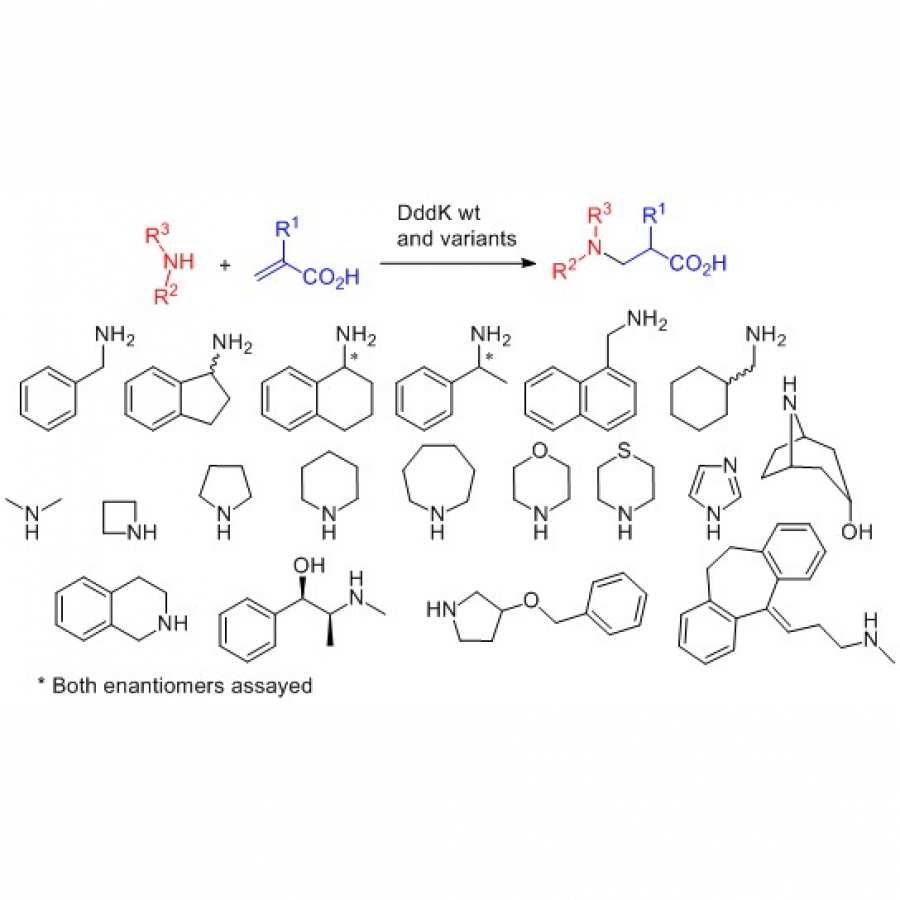 Scheme 1 DddK catalyzed aza-Michael additions of primary and secondary amines to acrylic (R1 = H) and methacrylic acids (R2 = CH3). |
#149 | Enantioselective Biocascade Catalysis with a Single Multifunctional Enzyme |
|
| Presenting author: | VASILEIOS TSELIOU of ICIQ |
| Corresponding author: | Paolo MELCHIORRE of DIPARTIMENTO DI CHIMICA INDUSTRIALE "TOSO MONTANARI'' |
| Topic: | Biocatalytic cascade reactions |
| Date: | 12:00 am - 12:15 pm Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | biocatalysis / enantioselectivity / cascade reactions / organocatalysis |
| Purpose: | Asymmetric catalytic cascade processes are powerful synthetic tools for rapidly generating structural and stereochemical complexity from simple substrates and in a single step [1]. In biocatalysis, cascades are generally designed by combining multiple enzymes, each catalyzing individual steps of a sequence [2]. Herein, we report a different strategy for biocascades based on a single multifunctional enzyme that can promote multiple stereoselective steps of a domino process by mastering distinct catalytic mechanisms of substrate activation in a sequential way. Specifically, we report the use and further optimization, via fusion and internal His-Tag insertion, of an engineered 4-oxalocrotonate tautomerase (4-OT) with the ability to form both enamines and iminium ions [3] and combine their mechanisms of catalysis in a complex sequence. This approach allowed us to activate aldehydes and enals toward the synthesis of enantiopure cyclohexene carbaldehydes. The multifunctional 4-OT enzymes could promote both a two-component reaction and a triple cascade characterized by different mechanisms and activation sequences demonstrating that biocatalysis can match and even surpass in efficiency the potential of organocascade catalysis [4]. |
| References: | [1] a) L. F. Tietze, G. Brasche, K. M. Gericke (Eds.), Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006; b) H. Pellissier, Chem. Rev. 2013, 113, 442-524; c) W. Kroutil, M. Rueping, ACS Catal. 2014, 4, 2086-2087 [2] a) A. I. Benítez-Mateos, D. Roura Padrosa, F. Paradisi, Nat. Chem. 2022, 14, 489-499; b) S. P. France, L. J. Hepworth, N. J. Turner, ACS Catal. 2017, 7, 710-724. [3] a) J.-Y. van der Meer, et al. Nat. Comm. 2016, 7, 10911. b) C. Guo, M. Saifuddin, T. Saravanan, M. Sharifi, G. J. Poelarends, ACS Catal. 2019, 9, 4369-4373. [4] a) D. Enders, M. R. M. Hüttl, C. Grondal, G. Raabe, Nature 2006, 441, 861-863. b) D. Enders, M. Jeanty, J. Bats, W. Synlett 2009, 19, 3175-3178. |
| Figures: | 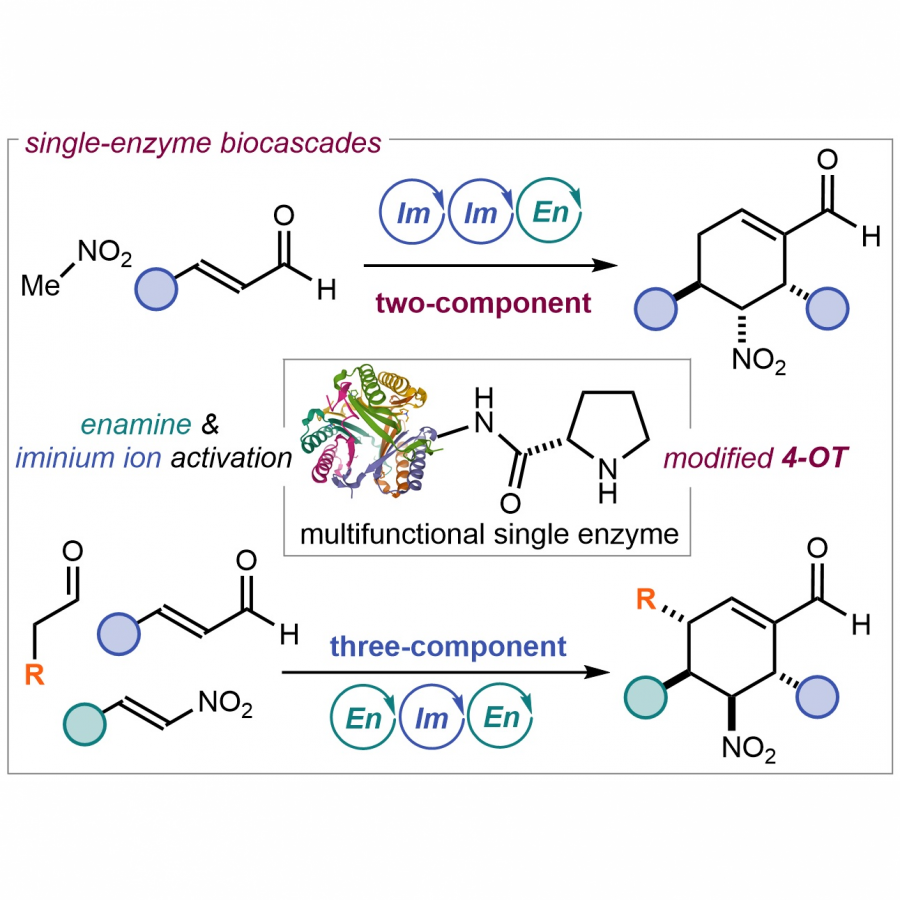 A single multifunctional enzyme can promote biocatalytic cascades based on multiple stereoselective steps a 4-oxalocrotonate tautomerase (4-OT) enzyme can form enamine and iminium ion intermediates from aldehydes and enals to promote both a two-component reaction and a triple cascade characterized by different mechanisms and activation sequences. 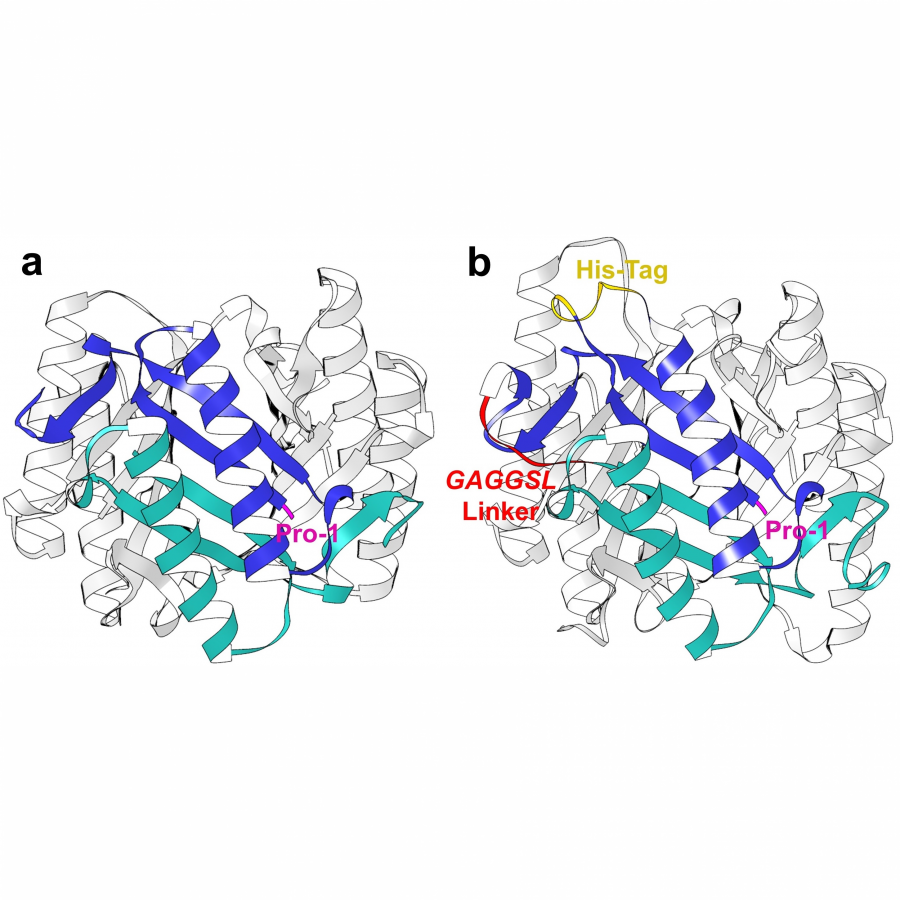 A genetically fused 4OT with internal His-Tag a) The homohexameric wild type Pp-4OT in which two monomers (blue and light green) form a dimer. b) Homology model of Pp-4OT-F3 in which the His-Tag (yellow) and the linker (red) have been genetically inserted. |
#156 | Terpene factory |
|
| Presenting author: | Létitia LEYDET of AIX-MARSEILLE UNIVERSITE |
| Other authors: | Gilles IACAZIO of AIX-MARSEILLE UNIVERSITE Katia DUQUESNE of AIX-MARSEILLE UNIVERSITE Agnès AMOURIC of AIX-MARSEILLE UNIVERSITE |
| Topic: | Biocatalytic cascade reactions |
| Date: | 04:18 pm - 04:21 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | Biocatalysis / Terpene / Synthetic biology / In vivo |
| Purpose: | Terpenoid are one of the most represented families of natural molecules on Earth. To date, more than 100,000 compounds [1] whose structural, biological (antibiotic, anticancer, anti-inflammatory, etc.) and physicochemical (cleaner, flavor, dye, etc.) properties hold the attention of the scientific community [2-3]. However, their access is limited because of the low available quantity by extraction from natural sources ; an often expensive and laborious chemical synthesis ; and long biosynthetic pathways. We have developed a new in vitro production pathway[4] : two enzymes making it possible to obtain diphosphates (DMAPP and IPP the universal precursors of terpenes), a prenyl transferase and finally the enzyme allowing cyclization. This synthesis is carried out from two alcohols with five carbon atoms in order to give different terpene compounds. In our case, one terpene from each family is selected: santalene, squalene, taxadiene and lycopene (respectively for sesquiterpene, triterpene, diterpene and teraterpene synthases). By combining bioinformatics, biochemical and molecular biology approaches, we have developed this approach in vivo, by joining together two plasmids comprising the two genes necessary for this mini-pathway. Playing on the gene position, but also on the different regulatory elements(operonic organization, RBS shuffling,...), the quantity of terpenes obtained can vary : it is a question of optimising the experimental conditions, from genetic construction to bioconversion. In the future the objective is to apply theses approaches to all terpenoids and that it may represent a new biosynthetic tool of interest to optimize and facilitate access to current terpenes, but also to explore biodiversity and characterize new terpene synthases. |
| References: | [1] Xiaoqiang Ma, Hong Liang, Qiuchi Pan, Kristala L. J. Prather, Anthony J. Sinskey, Gregory Stephanopoulos, and Kang Zhou, Optimization of the Isopentenol Utilization Pathway for Isoprenoid Synthesis in Escherichia coli, J. Agric. Food Chem. 2022, 70, 3512-3520 [2] Bohlmann J, Keeling CI. Terpenoid biomaterials. Plant J. (2008) 54(4):656-69. [3] Paramasivan K, Mutturi S. Progress in Terpene Synthesis Strategies through Engineering of Saccharomyces cerevisiae. Crit. Rev. Biotechnol. (2017) 37(8):974-989. [4] Couillaud J, Rico J, Rubini A, Hamrouni T, Courvoisier-Dezord E, Petit JL, Mariage A, Darii E, Duquesne K, de Berardinis V, Iacazio G. Simplified in Vitro and in Vivo Bioaccess to Prenylated Compounds. ACS Omega. (2019) 4(4), 7838-7849. |
| Figures: | 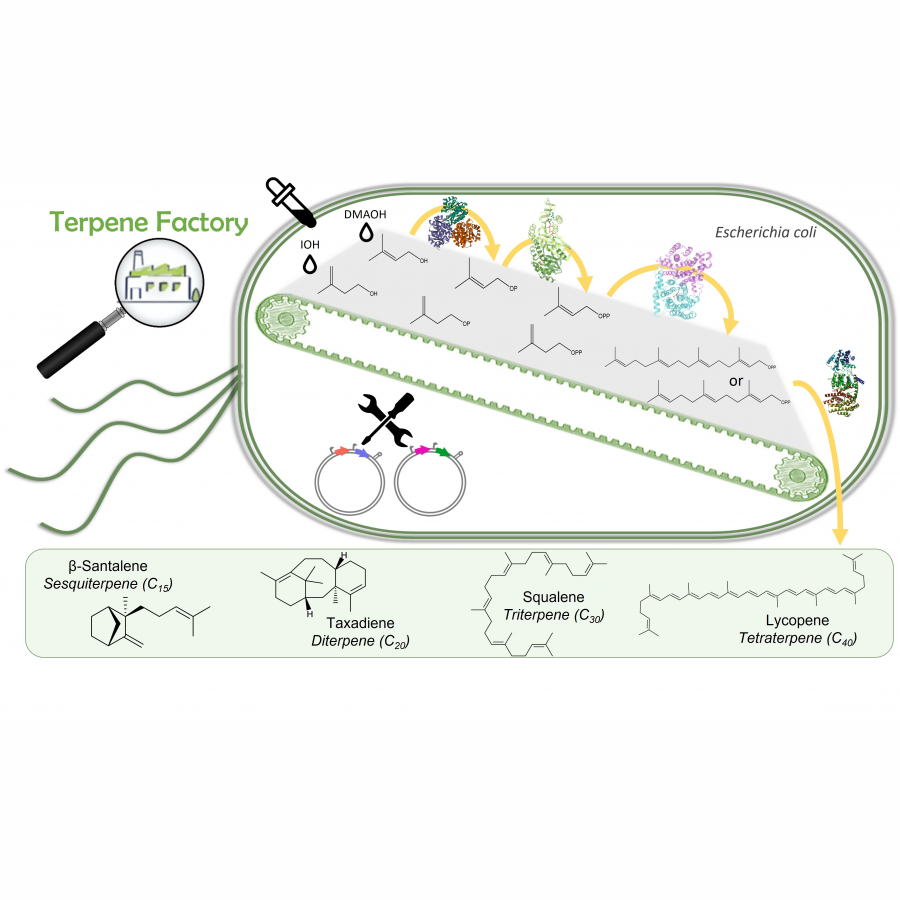 Terpene factory Genetically modified bacteria capable of producing terpenes through associated enzymes. |
#175 | Concurrent Hybrid Catalysis at acidic pH for Rare Monosaccharide Production by Combining Aldolase and N-Heterocyclic Carbene Gold Catalysts |
|
| Presenting author: | Cédric GASTALDI of UCL |
| Other authors: | Christine GUERARD-HELAINE of UCA Virgil HELAINE of UCA Claude FORANO of UCA Arnaud GAUTIER of UCA Muriel JOLY of UCA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 12:12 am - 12:15 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | Hybrid Catalysis / Layered Double Hydroxide / Organometallic Catalyst / Biocatalysis |
| Purpose: | Hybrid catalysis has been increasingly developed in recent years. Indeed, it takes advantage of the robustness and the broad range of applications of the chemical catalysts combined with the high regio, chemo and stereoselectivity of the enzymes. Among chemical catalysts, transition-metal complexes are considered as one of the most powerful . [1] Furthermore, metal catalysts working in water are recently emerging, giving opportunities to couple them with enzymes whose natural medium is aqueous. Thus, various examples of combination of biocatalysts and metal-catalysts for linear cascades have been reported, but often in a sequential mode, because of various incompatibilities that could not be solved (for instance: pH, temperature, substrate concentrations, inhibition problems, etc). [2] Indeed, few concurrent processes, i.e., involving the addition of all the reagents and catalysts from the beginning without any changes in the operating conditions till the end, are described. The reason is often an incompatibility of the catalysts with each other, or with the operating conditions. Several techniques have overcome these difficulties by compartmentalization using either cells, biphasic systems, artificial metallo-enzymes or supramolecular hosts. [3] Combined with techniques of enzymes evolution, in order to make biocatalysts more robust, in particular with respect to the temperature of the medium or the concentrations of substrates, these methods have made it possible to solve almost all the reasons of incompatibilities of the catalysts between them, except, to our knowledge, when the incompatibility comes from the pH. Here, a chemical catalyst, a N-heterocyclic carbene gold complex, only capable of operating at pH 3, was successfully coupled to an aldolase, Fructose-6-phosphate Aldolase (FSA), [4] previously confined in cells to protect it from this extreme pH. The catalyst working better at 60°C, cells were in turn immobilized on Layered Double Hydroxides (LDH), specially designed to resist to pH 3. Finally, to avoid any mutual deleterious interactions between chemical catalyst and the immobilized cells harboring the aldolase, a compartmentalization has been set up thanks to a semi-permeable membrane. Thus, as proof of principle, propargyl alcohol, a cheap and achiral compound, could be hydrated, using a fair range of substrates concentrations (200-500mM) for easier scale-up, and subsequently converted to the corresponding aldol, a high added-value compound with one asymmetric center with fixed configuration, at pH 3 and 60°C, in 68% isolated yield (Figure 1). [5] Interestingly, this process could be exemplified to another concurrent reaction involving an acidic resin catalyzed acetal hydrolysis to the corresponding aldehyde, as aldolase electrophilic substrate, particularly useful in the case of unstable aldehydes. This latter system, even simpler than the previous one, gave the corresponding aldol in 98% yield, with two asymmetric centers with fixed configurations. |
| References: | [1] Huang, X.; Cao, M.; Zhao, H. Curr. Opin. Chem. Biol. 2020, 55, 161-170. [2] a) Simons, C.; Hanefeld, U.; Arends, I. W. C. E.; Maschmeyer, T.; Sheldon, R. A. Top. Catal. 2006, 40 (1-4), 35-44. b) Prastaro, A.; Ceci, P.; Chiancone, E.; Boffi, A.; Cirilli, R.; Colone, M.; Fabrizi, G.; Stringaro, A.; Cacchi, S. Green Chem. 2009, 11 (12), 1929-1932. c) Ríos-Lombardía, N.; Vidal, C.; Cocina, M.; Morís, F.; García-Álvarez, J.; González-Sabín, J. Chem. Commun. 2015, 51 (54), 10937-10940. d) Burda, E.; Hummel, W.; Gröger, H. Angew. Chem. Int. Ed. 2008, 47 (49), 9551-9554. [3] a) Foulkes, J. M.; Malone, K. J.; Coker, V. S.; Turner, N. J.; Lloyd, J. R. E. ACS Catal. 2011, 1 (11), 1589-1594. b) Denard, C. A.; Huang, H.; Bartlett, M. J.; Lu, L.; Tan, Y.; Zhao, H.; Hartwig, J. F. Angew. Chem. Int. Ed. 2014, 53 (2), 465-469. c) Köhler, V.; Wilson, Y. M.; Dürrenberger, M.; Ghislieri, D.; Churakova, E.; Quinto, T.; Knörr, L.; Häussinger, D.; Hollmann, F.; Turner, N. J.; Ward, T. R. Nat. Chem. 2013, 5 (2), 93-99. d) Wang, Z. J.; Clary, K. N.; Bergman, R. G.; Raymond, K. N.; Toste, F. D. A. Nat. Chem. 2013, 5 (2), 100-103. [4] Gastaldi, C.; Mekhloufi, G.; Forano, C.; Gautier, A.; Guérard-Hélaine, C. Green Chem. 2022, 24 (9), 3634-3639. [5] Gastaldi, C.; Hélaine, V.; Joly, M.; Gautier, A.; Forano, C.; Guérard-Hélaine, C. Catal. Sci. Technol., 2023, Accepted Manuscript |
| Figures: | 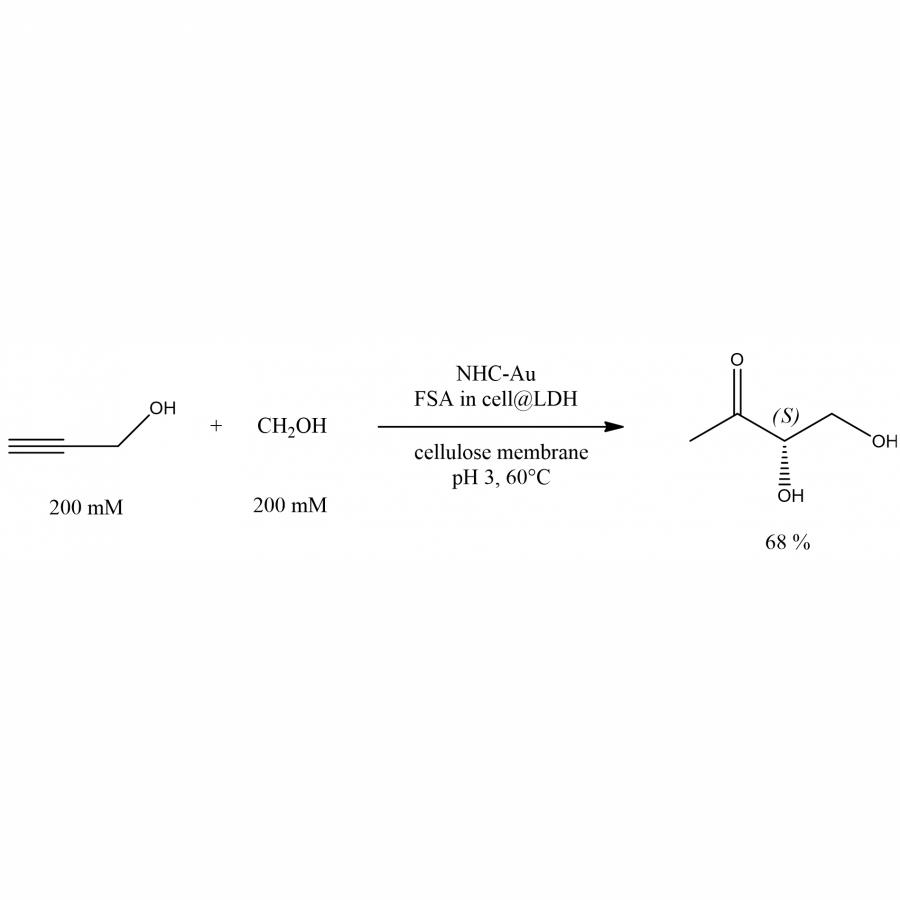 Hybrid system optimized conditions for the one-pot concurrent hydration-aldolisation reaction |
#235 | How Do We Find New Functional Proteins in Sequence Space? |
|
| Presenting author: | Florian HOLLFELDER of UNIVERSITY OF CAMBRIDGE |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 02:00 pm - 02:45 pm Session Enzyme engineering & Discovery #2 - Chair : P. CLAPES, Institute for Advanced Chemistry of Catalonia |
| Keywords: | directed evolution / functional metagenomics / enzyme mechanism / droplet microfluidics |
| Purpose: | Functional proteins for a variety of useful applications, as binders and catalysts, are required, but currently not known. Functional metagenomics and directed evolution promise access to such new proteins, but the chances of finding them are low. Therefore high-throughput technologies are crucial to beat the odds: screening in picoliter water-in–oil emulsion droplets produced in microfluidic devices allow screening of >10e7 clones and permit successful selections While potentially faster, the vastness of sequence space (and the scarcity of ‘solutions’ in it) require strategies for the identification and interconversion of enzymes. In this context the role of ‘promiscuous’ enzymes, sequencing of full length of genes at high throughput (UMIC-seq) and insertion/deletion mutagenesis (using the transposon-based method TRIAD) will be discussed. Together with a molecular and mechanistic understanding new routes to functional can be charted. • Schnettler, J. D.; et al & Hollfelder, F. Ultrahigh-Throughput Directed Evolution of a Metal-Free alpha/beta-Hydrolase with a Cys-His-Asp Triad into an Efficient Phosphotriesterase. J Am Chem Soc 2023, 145, 1083-1096. • Knyphausen, P.; et al Hollfelder, F. Evolution of protease activation and specificity via alpha-2-macroglobulin-mediated covalent capture. Nat Commun 2023, 14, 768 • Neun, S.; Brear, P.; Campbell, E.; Tryfona, T.; El Omari, K.; Wagner, A.; Dupree, P.; Hyvonen, M.; Hollfelder, F. Functional metagenomic screening identifies an unexpected beta-glucuronidase. Nat Chem Biol 2022, doi: 10.1038/s41589-022-01071-x • Scheele, R. A.; Lindenburg, L. H.; Petek, M.; Schober, M.; Dalby, K. N.; Hollfelder, F. Droplet-based screening of phosphate transfer catalysis reveals how epistasis shapes MAP kinase interactions with substrates. Nat Commun 2022, 13, 844. • Zurek, P. J.; Knyphausen, P.; Neufeld, K.; Pushpanath, A.; Hollfelder, F., UMI-linked consensus sequencing enables phylogenetic analysis of directed evolution. Nat Commun 2020, 11 (1), 6023. • Emond, S.; Petek, M.; Kay, E. J.; Heames, B.; Devenish, S. R. A.; Tokuriki, N.; Hollfelder, F., Accessing unexplored regions of sequence space in directed enzyme evolution via insertion/deletion mutagenesis. Nat Commun 2020, 11 (1), 3469. • Skamaki, K.; Emond, S.; Chodorge, M.; Andrews, J.; Rees, D. G.; Cannon, D.; Popovic, B.; Buchanan, A.; Minter, R. R.; Hollfelder, F., In vitro evolution of antibody affinity via insertional scanning mutagenesis of an entire antibody variable region. Proc Natl Acad Sci U S A 2020, 117 (44), 27307-27318. • van Loo, B.; Bayer, C. D.; Fischer, G.; Jonas, S.; Valkov, E.; Mohamed, M. F.; Vorobieva, A.; Dutruel, C.; Hyvonen, M.; Hollfelder, F., Balancing Specificity and Promiscuity in Enzyme Evolution: Multidimensional Activity Transitions in the Alkaline Phosphatase Superfamily. J Am Chem Soc 2019, 141 (1), 370-387. • Miton, C. M.; Jonas, S.; Fischer, G.; Duarte, F.; Mohamed, M. F.; van Loo, B.; Kintses, B.; Kamerlin, S. C. L.; Tokuriki, N.; Hyvonen, M.; Hollfelder, F., Evolutionary repurposing of a sulfatase: A new Michaelis complex leads to efficient transition state charge offset. Proc Natl Acad Sci U S A 2018, 115 (31), E7293-E7302. • Ultrahigh-throughput-directed enzyme evolution by absorbance-activated droplet sorting (AADS). Gielen F, Hours R, Emond S, Fischlechner M, Schell U, Hollfelder F. Proc Natl Acad Sci U S A. 2016;113(47):E7383-E7389. • Colin, P.-Y.; Kintses, B.; Gielen, F.; Miton, C. M.; Mohamed, M. F.; Fischer, G.; Hyvonen, M.; Morgavi, D. P.; Janssen, D. B.; Hollfelder, F., Ultrahigh-throughput Discovery of Promiscuous Enzymes by Picodroplet Functional Metagenomics. Nature Communications 2015, 6:10008. doi: 10.1038/ncomms10008. • Fischlechner, M.; Schaerli, Y.; Mohamed, M. F.; Patil, S.; Abell, C.; Hollfelder, F., Evolution of enzyme catalysts caged in biomimetic gel-shell beads. Nat Chem 2014, 6 (9), 791-6 |
| References: | Florian Hollfelder is Professor for Chemical and Synthetic Biology at the Biochemistry Department of the University of Cambridge/UK. Born in Berlin he was trained at TU Berlin, was a visiting fellow at Stanford (with D. Herschlag), obtained his MPhil and PhD Degrees at Cambridge (with AJ Kirby) and worked at as a postdoc at Harvard Medical School (with C. T. Walsh). His group was started in 2001 at Cambridge and employs a broad multi-disciplinary approach that combines methods and ideas ranging from physical-organic chemistry to biophysics, molecular biology and directed evolution. High- and low-throughput approaches are combined with classical kinetic and thermodynamic analysis. For directed evolution, the group has developed microfluidic devices to carry our screening of up to 108 clones via assays in emulsion droplets at a picolitre scale. Such high throughput experiments are used to gain insight into the process of protein evolution for binders and catalysts, into strategies to identify new enzymes from metagenomic sources and, on a fundamental level, to investigate the origins of enzymatic rate accelerations. The mechanistic principles that emerge from this work will form a basis for development of transferable, general rules to guide future enzyme evolution and protein engineering. Website: http://www2.bio.cam.ac.uk/~fhlab// |
#261 | The Fatty Acid Photodecarboxylase from Chlorella Variabilis - A Challenging Enzyme with Surprising Reactivity |
|
| Presenting author: | Christoph K. WINKLER of UNIVERSITY OF GRAZ |
| Other authors: | Stefan SIMIC of UNIVERSITY OF GRAZ Florian WEISSENSTEINER of UNIVERSITY OF GRAZ Valentina JURKAS of UNIVERSITY OF GRAZ Maria Emilia IGLESIAS MONCAYO of UNIVERSITY OF GRAZ Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Topic: | Enzyme discovery and engineering |
| Date: | 03:15 pm - 03:30 pm Session Enzyme engineering & Discovery #2 - Chair : T. WARD, University of Basel |
| Keywords: | photobiocatalysis / continnous flow / enzyme engineering / decarboxylase |
| Purpose: | The fatty acid photodecarboxylase from Chlorella variabilis (CvFAP) is capable to decarboxylate carboxylic acids in a light-dependent radical process (Figure 1, A) [1]. Since its first description, its potential as biocatalyst has been broadly explored, unraveling an extensive substrate scope of the wild type enzyme and its variants.[2] Despite these research efforts, two limiting factors remain: the enzymes poor photostability and the challenges associated with the scalability of light-dependent reactions [3, 4]. In this talk, we report our work on CvFAP, tackling the before-mentioned challenges, which led to the discovery of an unprecedented reactivity. In detail, we increased the stability and reusability of CvFAP using reaction engineering and immobilization. Applying the enzyme in continuous flow allowed to overcome the inefficient illumination of larger volumes and led to an increased productivity of the photodecarboxylation reaction (Figure 1, B) [4]. In addition to this, we developed a fascinating novel photo-biocatalytic coupling reaction (Figure 1, C). We demonstrate that terminal radical, that is generated when carboxylic acids are decarboxylated via the enzyme’s native mechanism, is able to form a new C-C bond with a range of different radical acceptors. The overall reaction represents a novel method for biocatalytic C-C bond formations. |
| References: | [1] D. Sorigue, B. Legeret, S. Cuine, S. Blangy, S. Moulin, E. Billon, P. Richaud, S. Brugiere, Y. Coute, D. Nurizzo, P. Muller, K. Brettel, D. Pignol, P. Arnoux, Y. Li-Beisson, G. Peltier, F. Beisson, Science 2017, 357, 903-907. [2] J. Xu, Y. Hu, J. Fan, M. Arkin, D. Li, Y. Peng, W. Xu, X. Lin, Q. Wu, Angew. Chem. Int. Ed. 2019, 58, 8474-8478; J. Xu, J. Fan, Y. Lou, W. Xu, Z. Wang, D. Li, H. Zhou, X. Lin, Q. Wu, Nat. Commun. 2021, 12, 3983. [3] B. Lakavath, T. M. Hedison, D. J. Heyes, M. Shanmugam, M. Sakuma, R. Hoeven, V. Tilakaratna, N. S. Scrutton, Anal. Biochem. 2020, 600, 113749; Y. Wu, C. E. Paul, F. Hollmann, ChemBioChem 2021, 22, 2420-2423. [4] S. Simic, M. Jakstaite, W. T. S. Huck, C. K. Winkler, W. Kroutil, ACS Catal. 2022, 12, 14040-14049. |
| Figures: | 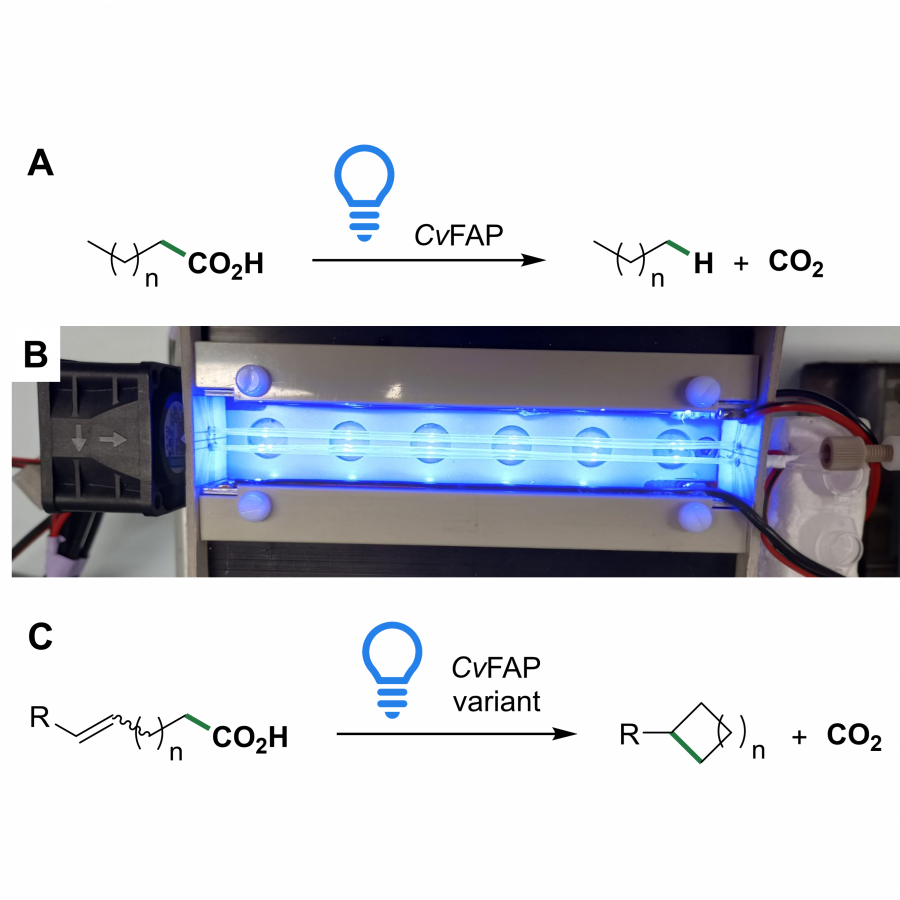 Reactivity and Reaction Engineering of the Photodecarboxylase CvFAP (A) Light-dependent decarboxylation of fatty acids. (B) Photodecarboxylation in continuous flow. (C) Unprecedented promiscuous C-C bond formation reaction. |
#279 | Stronger, tighter and faster: designing improved protein functions |
|
| Presenting author: | Olga KHERSONSKY of WEIZMANN INSTITUTE OF SCIENCE |
| Topic: | Artifical intelligence / computational methods |
| Date: | 11:00 am - 11:15 am Session AI & Computational methods - Chair : R. KOURIST, University of Technology of GRAZ |
| Keywords: | protein stability / atomistic calculations / function design / phylogenetic information |
| Purpose: | The evolution of altered or improved function is constrained by stability-function tradeoffs, whereby mutations accumulated along an evolutionary trajectory increasingly reduce stability and functional expression. A further constraint is the epistatic relationships among the spatially close positions that make up an active site. To address these problems, we have developed a methodology that combines phylogenetic information and Rosetta atomistic simulations to stabilize proteins and to improve their function. PROSS method stabilizes enzymes and other proteins without altering their functions. It was applied to several “tough-nut” cases, resulting in improvement in bacterial expression and substantial increases in thermal resistance. FuncLib method introduces combinations of mutations in the active site without impacting the core catalytic residues. Testing as few as several dozens of mutants can yield variants with various activity profiles, simultaneously increasing activity on several substrates. FuncLib designs epistatic active-site mutations that are likely to be inaccessible by natural and laboratory evolution, and opens the way to design highly efficient and diverse catalytic repertoires. In addition, our new method, HT-FuncLib (high throughput FuncLib) relies on machine learning and allows to sample large sequence spaces in an efficient way, Our design algorithms are currently available as webservers and are being extensively used. The methods are fully automated, requiring only a structure or a model and a sequence alignment of homologues, and they can be applied to proteins, whether or not they are amenable to high-throughput screening. I will describe several recent cases of protein stabilization and improvement of function, which were achieved using PROSS and FuncLib methods. |
| References: | [1] Weinstein JJ et al, Bioinformatics, 2020, 37(1): 123-125 [2] Khersonsky O et al, Mol Cell, 2018, 72(1): 178-186 [3] Weinstein JJ et al, Nat Comm, 2023, accepted |
| Figures: | 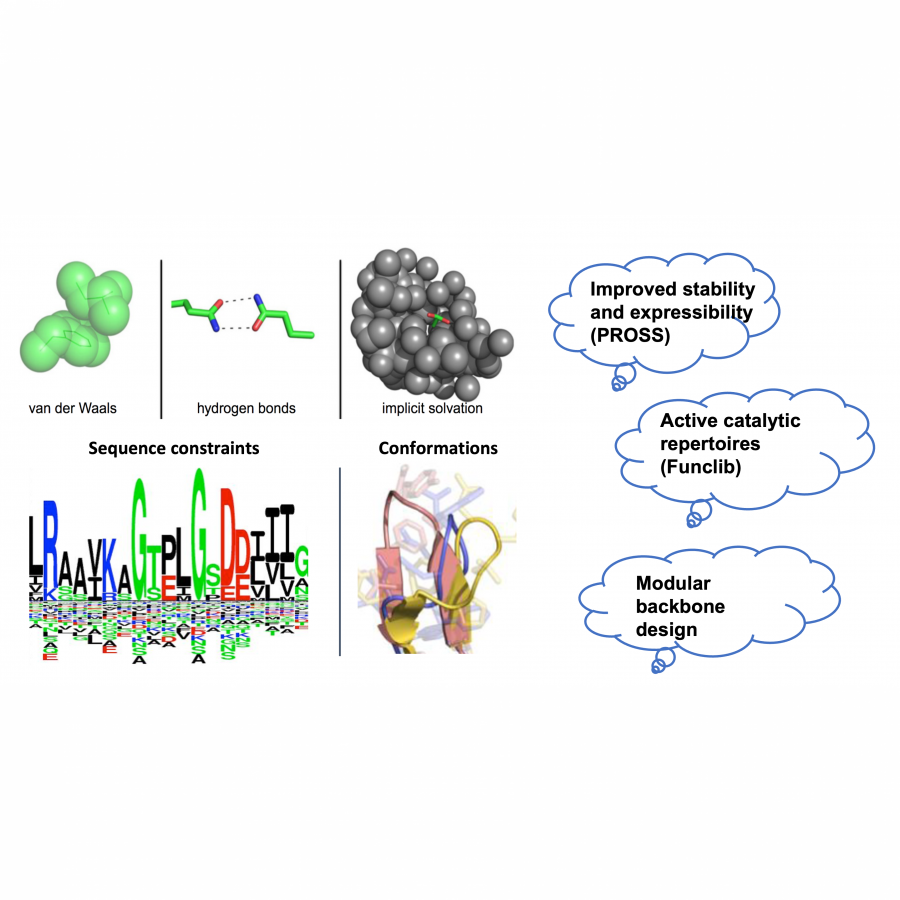 Computational design strategy Our computational methods rely on the combination of phylogenetic data and native protein conformations with explicit energy calculations by Rosetta |
#282 | Metallopterin-Dependent Sulfurase From The Abyss |
|
| Presenting author: | Mariia BELIAEVA of EUROPEAN MOLECULAR BIOLOGY LABORATORY |
| Other authors: | Florian SEEBECK of UNIVERSITY OF BASEL |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:00 pm - 04:03 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | sulfurase / ergothioneine / sulfur metabolism / metallopterin-dependent enzyme |
| Purpose: | Metallopretin-dependent proteins contain transition metals molybdenum or tungsten in the active site and catalyze essential processes in living organisms. The most diverse class of these enzymes, the dimethyl sulfoxide (DMSO) reductase superfamily, facilitates a wide range of chemical transformations that mainly implicate oxygen atom installation, removal, and transfer [1]. Here, we describe a novel enzyme from the DMSO reductase superfamily involved in the anaerobic biosynthesis of the natural antioxidant ergothioneine [2]. Metallopterin-dependent ergothioneine synthase (MES) contains two domains: an N-terminal metallopterin-binding sulfurase, and a C-terminal domain, which is a functional cysteine desulfurase. The two modules interact to transfer sulfur from cysteine onto the imidazole ring of trimethylhistidine (TMH) via an intramolecular persulfide relay (Figure 1). The C−S bond-forming activity of MES is unprecedented among metallopretin-containing enzymes and adds to the potential utility of these enzymes in biocatalysis. In addition, this discovery documents the second case of the independent emergence of anaerobic ergothioneine biosynthesis along with two alternative oxygen-dependent pathways [3], providing additional evidence for the importance of this widely-distributed metabolite in early anaerobic cell life. |
| References: | [1] C. Le, M. Bae, S. Kiamehr, E. P. Balskus, Annu. Rev. Biochem., 2022, 91, 475-504. [2] M. A. Beliaeva, F. P. Seebeck, JACS Au, 2022, 2, 2098-2107. [3] A. R. Stampfli, W. Blankenfeldt, F. P. Seebeck, COSB, 2020, 65, 1-8. |
| Figures: | 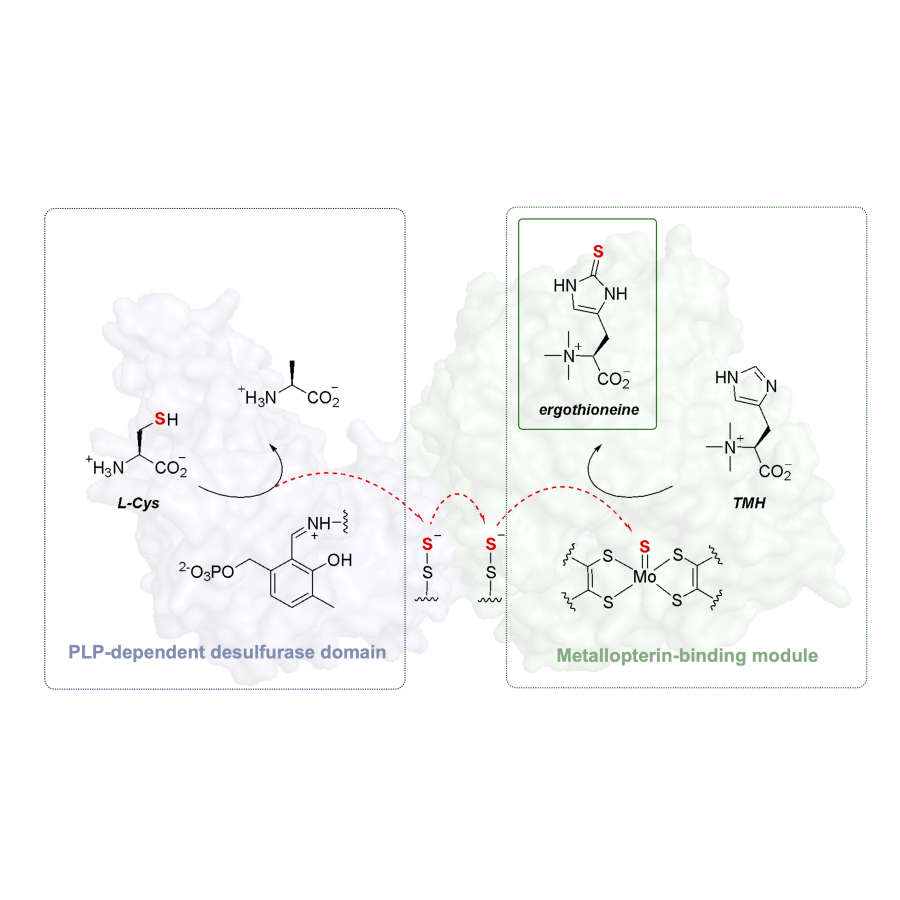 Scheme of ergothioneine biosynthesis by MES. PLP-dependent desulfurase domain extracts sulfur from L-cysteine, which is then transported via the transient formation of persulfides on two cysteine residues to the N-terminal metallopterin-binding module that mediates oxidative sulfurization of TMH. |
#409 | Digitalization of biocatalysis: the data exchange format EnzymeML |
|
| Presenting author: | Jürgen PLEISS of UNIVERSITY OF STUTTGART |
| Other authors: | Jan RANGE of UNIVERSITY OF STUTTGART Max HÄUSSLER of UNIVERSITY OF STUTTGART Torsten GIESS of UNIVERSITY OF STUTTGART |
| Topic: | Artifical intelligence / computational methods |
| Date: | 12:00 am - 12:03 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | research data management / FAIR data principles / enzyme kinetics / reproducibility |
| Purpose: | The design of biocatalytic reaction systems is highly complex due to the dependency of the estimated kinetic parameters on the enzyme, the reaction conditions, and the modelling method. Consequently, reproducing enzymatic experiments and reusing enzymatic data are challenging. To enable storage, retrieval, and exchange of enzymatic data such as the reaction conditions, the measured time courses of substrate and product, the selected kinetic model, and the estimated kinetic parameters, the XML–based markup language EnzymeML has been developed [1]. EnzymeML is based on SBML and the community Standards for Reporting Enzyme Data (STRENDA). The EnzymeML toolbox supports biocatalysis research by making enzymatic data findable, accessible, interoperable, and reusable [2]. An EnzymeML document contains information about reaction conditions and the measured time course of substrate or product concentrations (Fig. 1). It is generated from an EnzymeML spreadsheet or by the webtool BioCatHub. Kinetic modelling is performed by uploading EnzymeML documents to the modelling platforms COPASI or PySCeS. The estimated kinetic parameters are then added to the EnzymeML document. The EnzymeML document containing the experimental and modelling results is then uploaded to a Dataverse installation or to the reaction kinetics database SABIO-RK. The workflow of a project is encoded as Jupyter Notebook, which can be re-used, modified, or extended. EnzymeML serves as a seamless communication channel between experimental platforms, electronic lab notebooks, tools for modelling of enzyme kinetics, publication platforms, and enzymatic reaction databases (Fig. 2). The feasibility and usefulness of the EnzymeML toolbox was demonstrated in six scenarios, where data and metadata of different enzymatic reactions are collected, analysed, and uploaded to public data repositories for future re-use [3]. Because EnzymeML documents are structured and standardized, the experimental results encoded in an EnzymeML document are interoperable and reusable by other groups. Because an EnzymeML document is machine-readable, it can be used in an automated workflow for storage, visualization, data analysis, and re-analysis of previously published data, without limitations of the size of each dataset or the number of experiments. Thus, EnzymeML contributes to the digitalization of (bio)catalytic sciences. EnzymeML is open, transparent, and invites the community to contribute. Tools, documentation, and examples are available at https://enzymeml.org/. |
| References: | 1. Pleiss J, 2021. Standardized data, scalable documentation, sustainable storage - EnzymeML as a basis for FAIR data management in biocatalysis. ChemCatChem 13: 3909-3913; doi: 10.1002/cctc.202100822 2. Range J, Halupczok C, Lohmann J, Swainston N, Kettner C, Bergmann FT, Weidemann A, Wittig U, Schnell S, Pleiss J, 2022. EnzymeML - a data exchange format for biocatalysis and enzymology. FEBS J 289: 5864-5874; doi: 10.1111/febs.16318 3. Lauterbach S, Dienhart H, Range J, Malzacher S, Spöring JD, Rother D, Pinto MF, Martins P, Lagerman CE, Bommarius AS, Vang Høst A, Woodley JM, Ngubane S, Kudanga T, Bergmann FT, Rohwer JM, Iglezakis D, Weidemann A, Wittig U, Kettner C, Swainston N, Schnell S, Pleiss J, 2023. EnzymeML: seamless data flow and modeling of enzymatic data. Nat Methods; doi: 10.1038/s41592-022-01763-1 |
| Figures: | 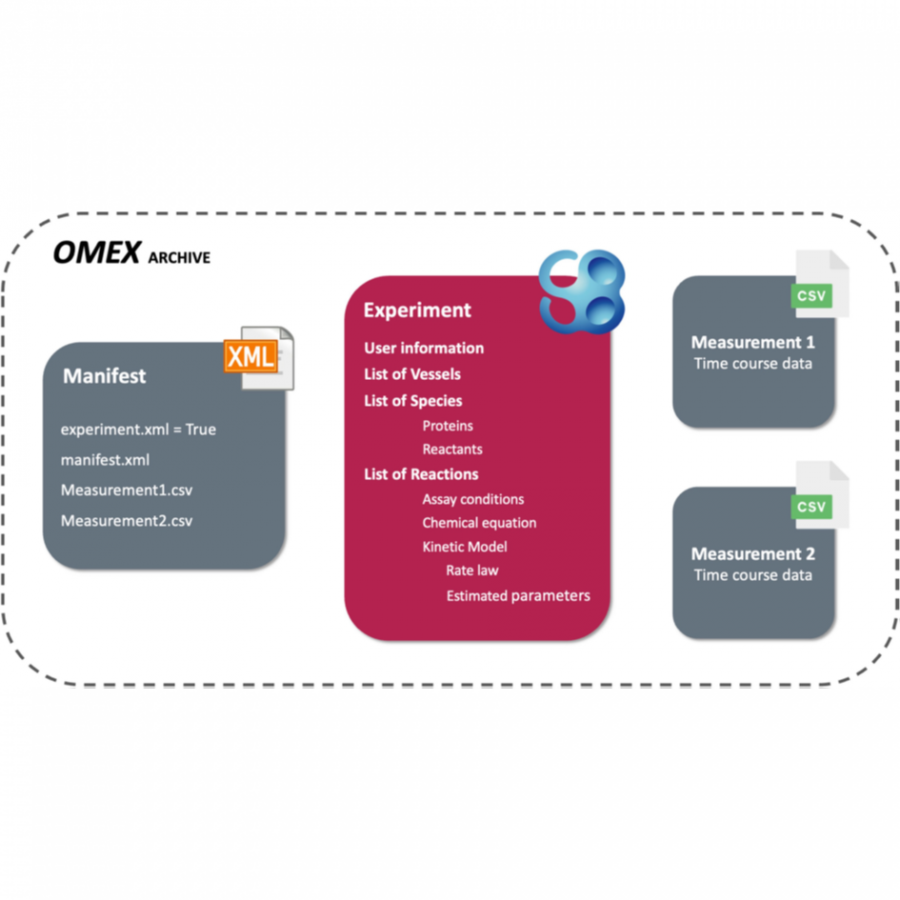 Structure of an EnzymeML document An EnzymeML document is a ZIP container in OMEX format and contains the experiment file (SBML) and the measurement files (CSV). 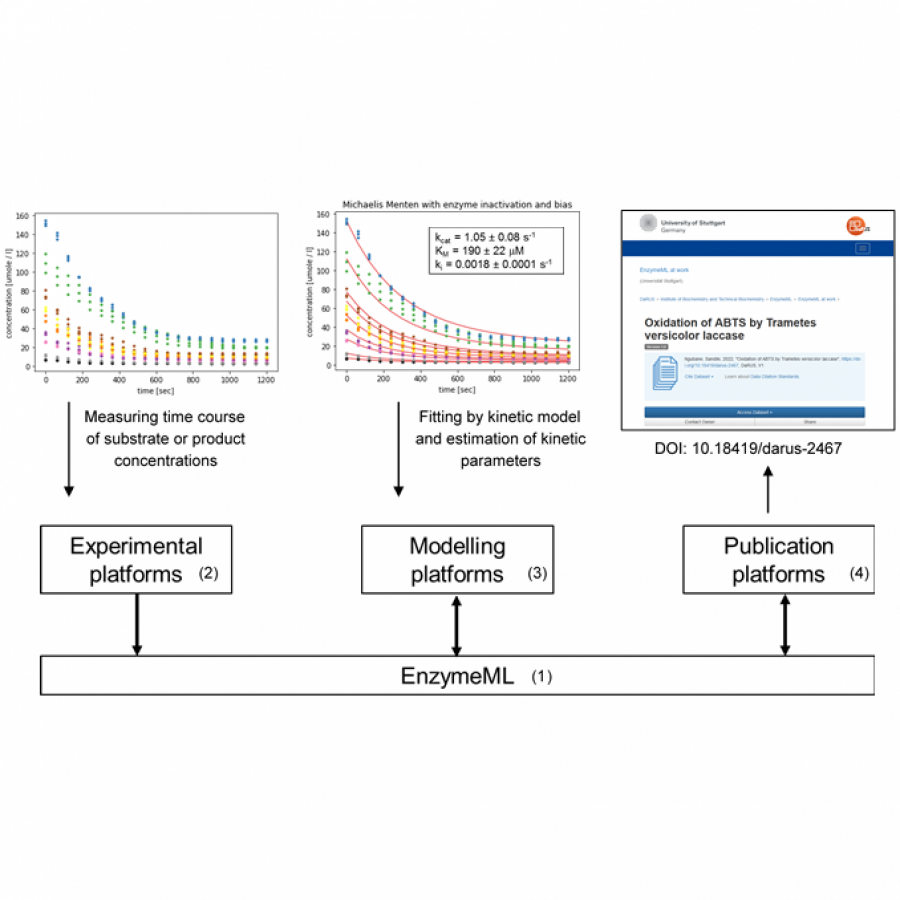 Seamless data transfer An EnzymeML document integrates (meta-)data from experiment and modelling), and serves as a communication channel between experimental, modelling, and publication platforms. |
#439 | Selective hydrogenation of nitro groups to amines using a heterogeneous biocatalyst |
|
| Presenting author: | Daria SOKOLOVA of UNIVERSITY OF OXFORD |
| Corresponding author: | Kylie VINCENT of UNIVERSITY OF OXFORD |
| Other authors: | Tim SUDMEIER of UNIVERSITY OF OXFORD Tara LURSHAY of UNIVERSITY OF OXFORD Jack ROWBOTHAM of UNIVERSITY OF MANCHESTER Sarah CLEARY of HYDREGEN LIMITED Holly REEVE of HYDREGEN LIMITED Georgia STONADGE of UNIVERSITY OF OXFORD |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 12:00 am - 12:15 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | nitro reduction / biocatalysis / aryl amines / |
| Purpose: | This study describes the development of a heterogeneous biocatalyst for the reduction of aryl nitro compounds to corresponding amines. Aryl amines are important molecules often present in pharmaceuticals[1] and used as synthons in azo dyes, which have applications such as pigments and food additives.[2] To prepare these compounds, the amine functional group is often installed into an aromatic molecule ‘masked’ as a nitro group, which is reduced at a later stage to provide the desired amine.[3,4] These organic nitro chemicals are prepared through well-established and efficient, albeit often hazardous methods,[5,6] and recent efforts have lowered the associated risks through process engineering (e.g., in flow).[7] There are many methods to reduce the nitro product to provide the corresponding amine. Precious metal (Pd, Pt, Au, Ag)[8-11] and base metal (Ni, Mn, Cu)[12-14] catalysts have been used with hydrogen gas, however, they are not general in terms of chemoselectivity and functional group tolerance. Moreover, the amine product can sometimes poison the catalyst, adding further complexity to metal-catalysed strategies. Conversely, biocatalytic nitro reductions are gentle alternatives to metal-based ones. For instance, nitroreductases, which have been engineered to facilitate process intensification and widen substrate scope with demonstrated chemoselectivity, operate under mild reaction conditions.[15-16] However, when used for amine synthesis, nitroreductases rely on 3 or more equivalents of a reducing agent, typically in the form of cofactors NAD(P)H, which in turn are commonly re-generated from their oxidised form using 3 or more equivalents of glucose. To overcome the limitations of the existing approaches, we have developed a heterogeneous hydrogenation biocatalyst that enables the reduction of nitroaromatic compounds to the corresponding amines (Fig. 1). The biocatalyst is easily accessible, stable and selective, and can be handled similarly to typical hydrogenation catalysts (e.g., Pd/C) but avoids using toxic metals that are often not functional group tolerant. Moreover, it opens up an opportunity to translate the nitro reduction process into a continuous flow reactor. |
| References: | [1] Heravi, M. M. & Zadsirjan, V., RSC Adv., 2020, 10, 44247-44311. [2] Mezgebe, K. & Mulugeta, E., RSC Adv., 2022, 12, 25932-25946. [3] Orlandi, M., Brenna, D., Harms, R., Jost, S. & Benaglia, M., Org. Process Res. Dev., 2016, 22, 430-445. [4] Sukhorukov, A. Y., Front. Chem., 2020, 8, 215. [5] Yan, G. & Yang, M., Org. Biomol. Chem., 2013, 11, 2554-2566. [6] Qian, Y. E., Zheng, L., Xiang, H. Y. & Yang, H., Org. Biomol. Chem., 2021, 19, 4835-4851. [7] Brocklehurst, C. E., Lehmann, H. & La Vecchia, L., Org. Process Res. Dev., 2011, 15, 1447-1453. [8] Comeau, A. B., Critton, D. A., Page, R. & Seto, C. T., J. Med. Chem., 2010, 53, 6768-6772. [9] Mori, A. et al., Adv. Synth. Catal., 2008, 350, 406-410. [10] Sledeski, A. W. et al., J. Org. Chem., 2000, 65, 8114-8118. [11] Crook, R., Deering, J., Fussell, S. J., Happe, A. M. & Mulvihill, S., Tetrahedron Lett., 2010, 51, 5181-5184. [12] Formenti, D., Ferretti, F., Scharnagl, F. K. & Beller, M., Chem. Rev., 2019, 119, 2611-2680. [13] Zubar, V., Dewanji, A. & Rueping, M., Org. Lett., 2021, 23, 2742-2747. [14] Xu, F. et al., RSC Adv., 2020, 10, 28585-28594. [15] Bornadel, A. et al., Org. Process Res. Dev., 2021, 25, 648-653. [16] Bisagni, S. et al., Curr. Res. Chem. Biol., 2022, 2, 100026. |
| Figures: | 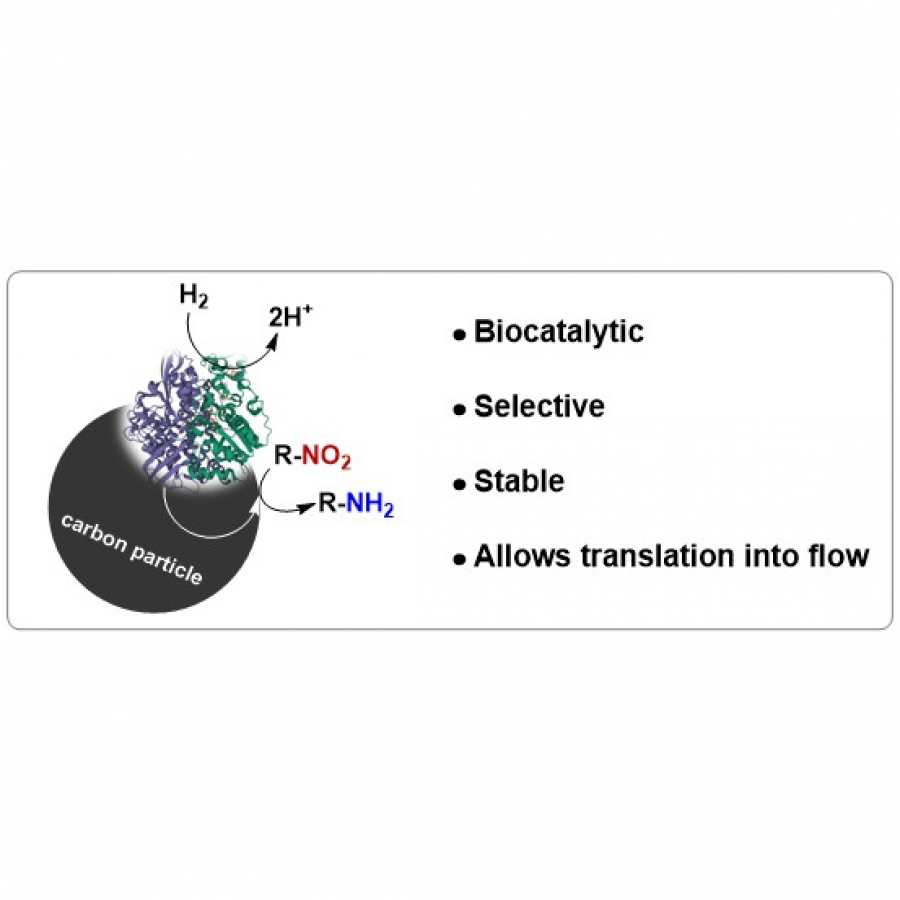 Figure 1. Novel heterogeneous hydrogenation biocatalyst for selective reduction of aromatic nitro compounds to corresponding amines. |
#440 | Engineering Enzymes for an End-to-End Nucleic Acid Manufacturing Platform |
|
| Presenting author: | Stefan LUTZ of CODEXIS |
| Topic: | Biocatalytic cascade reactions |
| Date: | 12:00 am - 12:15 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | Oligonucleotide / Nucleic acid / TdT polymerase engineering / enzyme cascade |
| Purpose: | Codexis’ CodeEvolver® directed evolution technology has been applied to improve enzymes for specialized functions for well over a decade. The fully integrated workflow leverages computational (design) tools, advances in high-throughput gene & protein synthesis (build), and biochemical screening (test) in combination with cutting-edge data analytics (learn) to optimize enzymes and drive increasing complexity in developing novel biocatalysts for pharma manufacturing, the life sciences, and biotherapeutics. In the life sciences, new solutions are needed to address growing needs for scalable, sustainable, and economical manufacturing of high-quality therapeutic oligonucleotides. We are deploying CodeEvolver® to develop a (novel) enzyme-catalyzed oligonucleotide manufacturing process for de novo synthesis of nucleic acid sequences. The synthesis platform leverages a proprietary Terminal Deoxynucleotidyl Transferase (TdT) engineered to efficiently incorporate 2’-and backbone modified nucleotides in an iterative process. The TdT action is complemented with engineered enzyme solutions for remaining steps in oligo synthesis and reagent production, together offering a complete, highly integrated solution to meet the demands of future nucleic acid synthesis. |
#445 | Enzyme engineering for the recycling of C1 gas into plastic monomers |
|
| Presenting author: | Jin-Byung PARK of EWHA WOMANS UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 12:15 am - 12:30 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | ethylene glycol / glycolic acid / carboligase / enzyme catalysis |
| Purpose: | C1 gas (e.g., CH4) can be converted into multi-carbon products such as ethylene glycol, glycolic acid, and ethanol amine via methanol, formaldehyde, and glycolaldehyde (Fig. 1). Here, we will present engineering of carboligases for the efficient production of the C2 products from formaldehyde. The thermostable glyoxylate carboligase from Escherichia coli K-12 (EcGCL), which is able to condense two molecules of formaldehyde into one molecule of glycolaldehyde [1], was selected as a C1 carboligase. The EcGCL was engineered to improve catalytic efficiency based on the crystal structure of EcGCL-glycolaldehyde complex [2]. The structure-guided rationale generated several mutants, one of whose catalytic activity reached 15.6 M-1·s-1, almost 10 folds greater than the wild-type enzyme. The combination of EcGCL variants with a lactaldehyde reductase of E. coli K-12, an aldehyde dehydrogenase of E. coli K-12, and omega-transaminase of Silicibacter pomeroyi led to the production of ethylene glycol, glycolic acid, and ethanol amine, respectively, (Fig. 1) to a high conversion from formaldehyde [1,3]. Besides, combination of the quintuple mutant EcGCL_R484M/N283Q/L478M/M488L/R284K with a decarboxylating E1 subunit of the alpha-ketoglutarate dehydrogenase complex of Vibrio vulnificus [4] allowed to produce C4-erythrulose from formaldehyde via glycolaldehyde without byproduct formation (Fig. 2). This study will contribute to valorization of C1 gases into industrially relevant multi-carbon products in an environment-friendly way. |
| References: | [1] Jo, H. J., Kim, J. H., Kim, Y. N., Seo, P. W., Kim, C. Y., Kim, J. W., Yu, H. N., Cheon, H. J., Lee, E. Y., Kim, J. S., & Park, J. B., Green Chem. 2021, 24, 218-226. [2] Cheon, H., Kim, J.-H., Jo, H.-J., Kim, G.Y., Kim, J.-S. & Park, J. B. Submitted in Green Chem. [3] Jo, H. J., Yu, H. N., Cheon, H. J., Kim, J. H., Kim, G. Y., Kim, B. S., Kim, J. S., & Park, J. B., ACS Sustainable Chem. Eng. 2023, 11, 1078-1086. [4] Seo, P. W., Jo. H. J., Hwang, I. Y., Jeong, H. Y., Kim, J. H., Kim, J. W., Lee, E. Y., Park., J. B., & Kim, J. S., Catal. Sci. Technol. 2020, 10, 79-85. |
| Figures: | 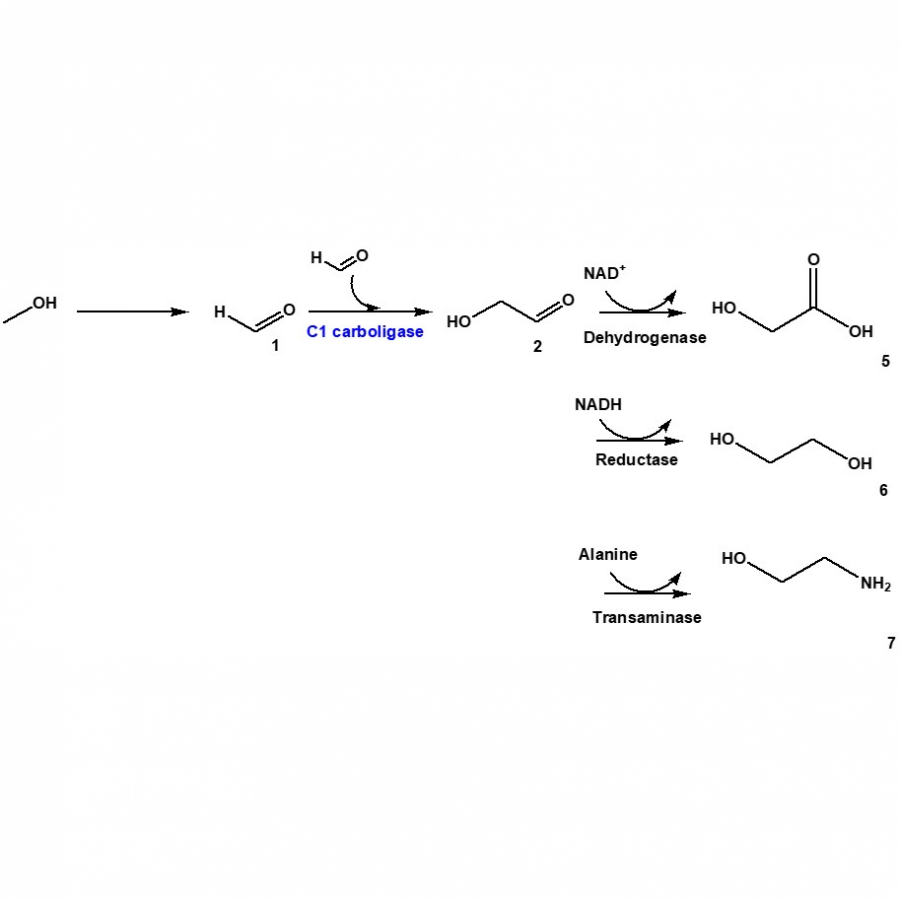 Biotransformation of formaldehyde 1 Biotransformation of formaldehyde (1) into glycolaldehyde (2) by C1 carboligases, which can be further converted into glycolic acid, ethylene glycol, and ethanolamine.
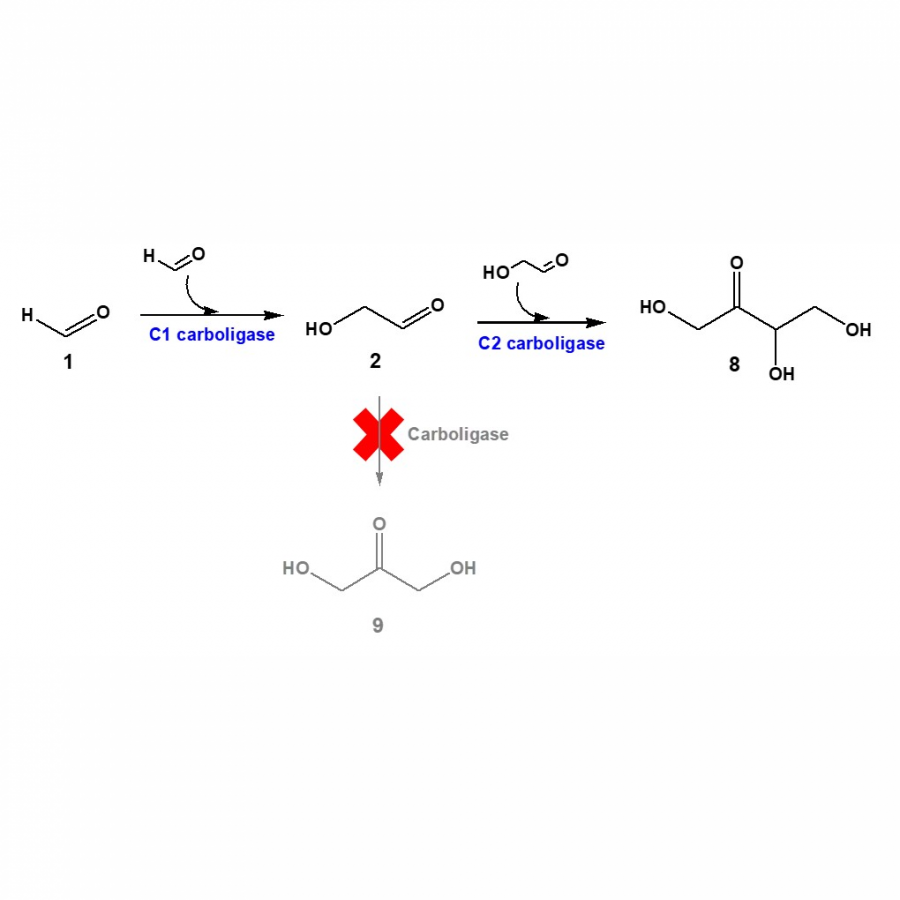 Biotransformation of formaldehyde 2 Biotransformation of formaldehyde (1) into erythrulose (8) via glycolaldehyde (2).
The C2 compound (2) can be converted into C3-dihydroxyacetone (9) by side reactions.
|
#498 | Biocatalytic hydrogenation: robust biotechnologies for sustainable chemical manufacturing |
|
| Presenting author: | CLEARY Sarah of HYDREGEN LIMITED |
| Corresponding author: | Holly REEVE of HYDREGEN |
| Other authors: | Sarah CLEARY of HYDREGEN |
| Topic: | Industrial biocatalysis |
| Date: | 02:30 pm - 03:00 pm Session Bio-photo - Electro-catalysis - Chair : B. DOUMECHE, University of Lyon |
| Keywords: | biocatalytic hydrogenation / immobilised enzyme / continuous processes / |
| Purpose: | The use of enzymes as tools in chemical synthesis is growing thanks to technological breakthroughs, such as enzyme engineering and developments toward scalability. These advances make biocatalysis useful for a range of applications, from early stage drug discovery to chemical manufacturing. HydRegen’s “black powder” biocatalytic hydrogenation systems, which are composed of enzymes immobilized on a carbon material, make biocatalysis simple to implement in chemistry settings due to similar catalyst handling to that of commonplace hydrogenation catalysts (e.g. Pd/C). The biocatalyst systems are powered by atom economical H2 gas, thus swapping out waste-intensive reductants that the enzymes typically rely on (e.g. glucose) while still taking advantage of the enzyme properties (mild operating conditions, excellent selectivity and functional group tolerance). The HydRegen biocatalytic hydrogenation tools are useful across a range of applications: from route scouting and building chemical libraries, through to bespoke chemical syntheses demonstrated in scalable batch and continuous reactors. Case examples involve the use of different NADH-dependent enzymes to achieve a variety of asymmetric hydrogenation of double bonds (carbonyl, alkene) and reductive aminations. More recently we developed a broad scope tool for mild nitro reductions to the corresponding amines. An array of specialty chemicals have been prepared, including at multi-gram scale (100%, >99.9% ee) with simple catalyst removal and chromatography-free purifications. Fed-batch and continuous flow reactions have shown working week catalyst stability (5 days, > 1 million catalyst turnovers). Overall, we find our biocatalytic hydrogenation systems provide several advantages: avoidance of precious metals, decarbonization of traditional biocatalysis, excellent selectivity, functional group tolerance, no catalyst deactivation by typical poisons (e.g. sulfur), and slotting-in to existing hydrogenation infrastructure. |
| References: | Reeve, H.A., Lauterbach, L., Lenz, O., Vincent, K.A. 'Enzyme-Modified Particles for Selective Bio-Catalytic Hydrogenation via H2-driven NADH Recycling' ChemCatChem, 2015, 7, 21, 3480-3487 DOI:10.1002/cctc.201500766 Cleary SE, Kazantzi S, Trenchard JA, Monedero M, Allman JW, Lurshay TC, Zhao X, Kenny MBC and Reeve HA (2023), Preparation of (3R)-quinuclidinol using heterogeneous biocatalytic hydrogenation in a dynamically-mixed continuous flow reactor. Front. Catal. 3:1114536. doi:10.3389/fctls.2023.1114536 |
| Figures: | 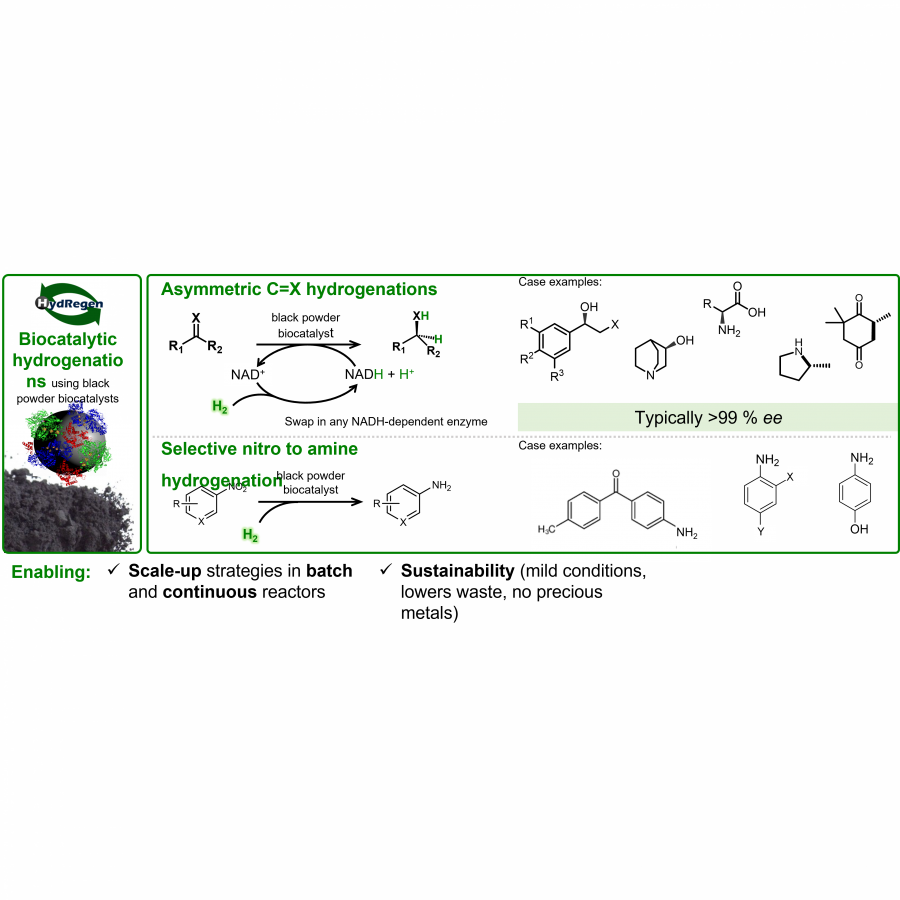 Heterogeneous biocatalysts enable mild and highly selective hydrogenations, from screening tools to scale-up strategies for sustainable chemical synthesis Heterogenous biocatalysts for H2-powered NADH recycling coupled to asymmetric reductions, or nitro to amine conversions. Reactions proceed with excellent selectivity and allow operation of biocatalysis using standard chemical protocols. |
#499 | Reaction engineering of an unspecific peroxygenase towards kg production of KA-oil |
|
| Presenting author: | Thomas HILBERATH of DELFT UNIVERSITY OF TECHNOLOGY |
| Other authors: | Juliet VICTORIA of TECHNICAL UNIVERSITY OF DENMARK Remco VAN OOSTEN of DELFT UNIVERSITY OF TECHNOLOGY Miguel ALCALDE of INSTITUTE OF CATALYSIS, CSIC John M. WOODLEY of TECHNICAL UNIVERSITY OF DENMARK Frank HOLLMANN of DELFT UNIVERSITY OF TECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 04:00 pm - 04:15 pm Session Industrial biocatalysis - Chair : J. Martin, SEQENS |
| Keywords: | peroxygenase / cyclohexane / reaction engineering / upscaling |
| Purpose: | Unspecific peroxygenases (UPOs, EC 1.11.2.1) are H2O2-dependent, heme-thiolate enzymes catalysing oxyfunctionalisation reactions of typically hydrophobic substrates.[1] Their robustness, high activities and simple use make UPOs attractive biocatalysts for chemical production at a reasonable preparative scale.[2] However, to date, peroxygenases, among many other biocatalysts, have been mainly applied for syntheses of high value-added chiral products, whereas biocatalytic syntheses of bulk chemicals are scarce.[3] “KA-oil” consisting of cyclohexanol/cyclohexanone is an example of a highly demanded bulk chemical used for polymer production such as nylon on a billion kg-scale.[4] With this contribution, we report the evaluation of reaction conditions that govern the use the recombinant unspecific peroxygenase from Agrocybe aegerita (rAaeUPO, PaDa-I variant) for the synthesis of KA-oil on a kg-scale (Figure 1). Suitable conditions for a reaction system enabling high substrate loadings are identified and were used to screen process-relevant parameters including H2O2-feeding rate and enzyme concentration on a 100 mL scale. Transferring the optimal conditions to a 10 L scale enabled synthesis of KA-oil with promising productivities of 16.5 g *L-1* h-1 and 480 g of product. To the best of our knowledge, this is the first time UPOs have been used at this scale to produce oxyfunctionalized products. |
| References: | [1] Hobisch M., Holtmann D., Gomez de Santos P., Alcalde M., Hollmann F., Kara S., Biotechnol. Adv. 2020, 107615. [2] Xu X., Hilberath T., Hollmann F., Curr. Opin. Green Sustain. Chem. 2023, 39, 100745. [3] Hanefeld,U., Hollmann F., Paul CE., Chem. Soc. Rev. 2022, 51.2, 594-627. [4] Musser MT., Ullmann's Encyclopedia of Industrial Chemistry,2011. |
| Figures: | 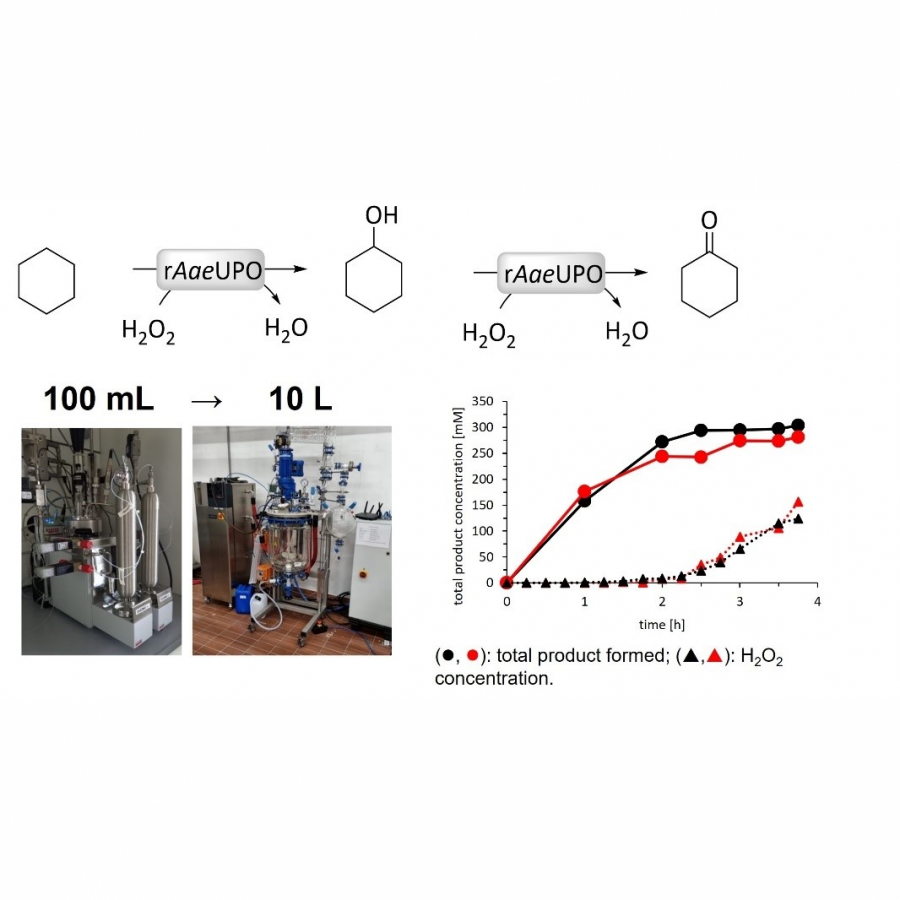 Figure 1 rAaeUPO-mediated oxidation of cyclohexane on a 10L-scale. |
#500 | SCALE-UP OF AN ENZYMATIC PRODUCTION OF CYRENE FROM LEVOGLUCOSENONE (LGO) |
|
| Presenting author: | Kévin AMRANI of URD ABI |
| Other authors: | Florent ALLAIS of URD ABI Louis MOUTERDE of URD ABI |
| Topic: | Industrial biocatalysis |
| Date: | 04:15 pm - 04:30 pm Session Industrial biocatalysis - Chair : J. Martin, SEQENS |
| Keywords: | CyreneTM / Levoglucosenone (LGO) / Biocatalysis / Scale-up |
| Purpose: | SCALE-UP OF AN ENZYMATIC PRODUCTION OF CYRENETM FROM LEVOGLUCOSENONE (LGO) This work is part of the European BBI JU Flagship ReSolute project. The overall project objective is to build the first-of-its-kind industrial plant and downstream value chain for the adoption of 99% pure Dihydrolevoglucosenone (CyreneTM). CyreneTM is a clear, colorless-to-light-yellow liquid with a slight ketone odor and has proven to be a multi-purpose aprotic solvent that can be a high-performance alternative to NMP, DMF, DMSO, DMAc etc. The Resolute plant feedstock is locally sourced renewable cellulosic biomass waste meaning manufacture is close to net-zero, sustainable and circular. The ReSolute plant will produce its CyreneTM offtake via the reduction of the platform molecule Levoglucosenone (LGO) which itself is a product of the flash pyrolysis of wood cellulose (FuracellTM process). The CyreneTM reduction is carried out using a heterogeneous catalyst and, although it is efficient in terms of yield and cost, there can be limitations. For example, whilst tests have proven essentially zero-to-minimal presence of metal ion impurities, even the idea of a low 100 parts per billion (ppb) metal ion presence can be unpalatable for certain high-value industry applications, such as electronics or pharmaceuticals. Therefore, to give downstream end-users from all industry sectors – including those sensitive to metals – full commercial access to CyreneTM as a bio-based, environmentally-benign solvent, a biocatalytic process has been developed. It involves an alkene reductase from Pichia stipitis – the OYE 2.6 (Old Yellow Enzyme family) - and has produced a guaranteed metal-free CyreneTM material with the potential for large scale manufacture. The aim of this work is to adapt and scale-up this enzymatic reaction to prove its feasibility at a pilot scale. Several difficulties must be addressed, such as LGO solubility in the aqueous medium, its inhibition on the OYE 2.6 at certain concentrations and the denaturation of the cofactor regeneration enzyme, the Glucose Dehydrogenase (GDH), at a certain concentration of CyreneTM. In the course of this work, we went from milliliter scale in a microplate to liter scale in a bioreactor by multiplying the final concentration of CyreneTM by 30 and from an overnight reaction to a few hours. Improvement in productivity allowed us to reach a 1483-fold scale-up. |
| References: | G. R. Court, C. H. Lawrence, W. D. Raverty and A. J. Ducan, US 20120111714, 2011 Mouterde L. M. M.; Allais F.; Stewart, J.D. Green Chem. 2018, 20 (24), 5528-5532. Patterson-Orazem A, Sullivan B, Stewart JD. Bioorganic & Medicinal Chemistry. 2014 Oct;22(20):5628-5632. |
| Figures: | 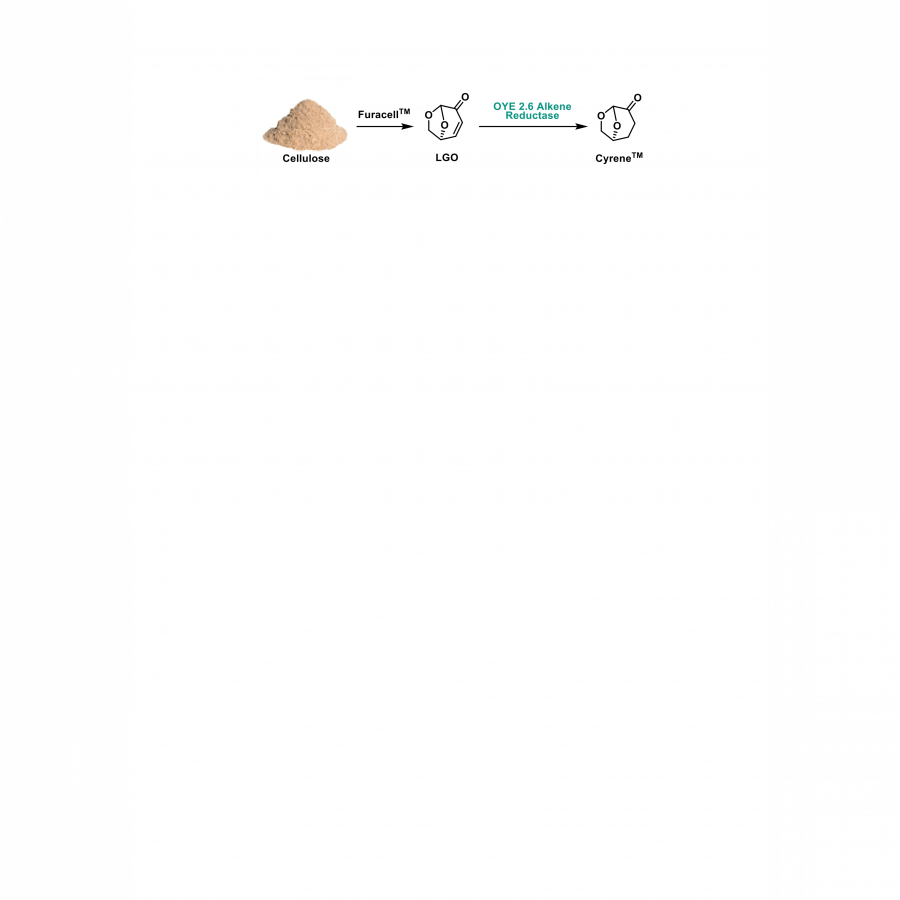 Enzymatic production route of CyreneTM from LGO This figure describes the enzymatic production route of CyreneTM from wood cellulose. |
#531 | Glucosylated mycosporines for innovative UV-filters: a strategy inspired by marine organisms. |
|
| Presenting author: | Elodie BASCANS of TBI - IPREM |
| Corresponding author: | Claire MOULIS of TBI |
| Other authors: | David GUIEYSSE of TBI Etienne SEVERAC of TBI Magali REMAUD-SIMEON of TBI Susana FERNANDES of IPREM |
| Topic: | Biocatalytic cascade reactions |
| Date: | 12:06 am - 12:09 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | Mycosporine-like Amino Acids / Transglucosylases / UV-Filter / Biomaterials |
| Purpose: | Preventing people from UV damages is a major concern and challenge of the 21st century. As living beings have evolved their own defense strategies, Nature could be a great source of inspiration for developing new UV-filters(1). Some organisms accumulate small molecules with unique UV-absorption and antioxidant properties, the mycosporines and mycosporine-like amino acids (MAAs). Interestingly, metazoans such as fish accumulate MAAs especially in sensitive tissues or organs like the eyes, either in free or associated form(2). Having MAAs associated to a carbohydrate moiety com-pound is of relevant interest for photostability(3) and formulation purposes. Glycosylated MAAs exist in terrestrial cyanobacteria, but their synthesis pathways remain unknown4. In our work, we focused on the enzymatic glucosylation of the mycosporine-serinol MSer(OH) naturally found in a lichen from the medio-littoral zones of Western European coasts, and, to date, never described as glycosyl-ated. This reaction would allow the development of innovative anti-UV products such as biosourced and biodegradable sunscreens or ophthalmic products. As such, we relied on enzymes called GH70 a-transglucosylases that use sucrose as cheap glucosyl donor to transfer it onto a large variety of hydroxylated acceptor moieties(5). After a first screen of our lab collection, a sole enzyme (GS-D) efficiently recognized MSer(OH) by adding one or two gluco-syl units (95% conversion). In order to elongate the glucidic head, an enzymatic cascade involving a second GH70 enzyme was then developed. Now, a vast array of glucidic chains were added, varying in term of linkage specificity and size (from 3 to 24,000 glucosyl units) depending on the second enzyme used. Once glucosylated, these compounds keep their strong absorbance and photostability as well as their interesting antioxidant properties. Moreover, the enzymatic cascade works equally whether it is starting from purified MSer(OH) or from a raw extract, paving the way for the devel-opment of biomedical applications with a refined compound having texturing, moisturizing, anti-UV and antioxidant properties(6). |
| References: | [1] Fernandes S.C.M., Alonso-Varona A., Palomares T., Zubillaga, V., Labidi, J., Bulone, V.; ACS Applied Materials & Interfaces 2015 30, 16558-16564 [2] Carreto J.; O Carignan M.; Marine Drugs, 2011, 3, 387-446 [3] Thomas M.G.; Samalens F.; Blanc S.; Pigot T.; Fernandes S.C.M.; (submitted) [4] Ishihara K.; Wanatabe R.; Uchida H.; Journal of Photochem. & Photobio., 2017, 172, 102-108 [5] Moulis C.; Guieysse D.; Morel S. Severac E. Remaud-Simeon M. Current Opinion in Chemical Biology, 2021, 61, 90-106 [6] Fernandes S., Moulis C., Bascans E., Claverie M., Severac E., Remaud-Simeon M. Patent application n°2208098 (pending), 2022 |
| Figures: | 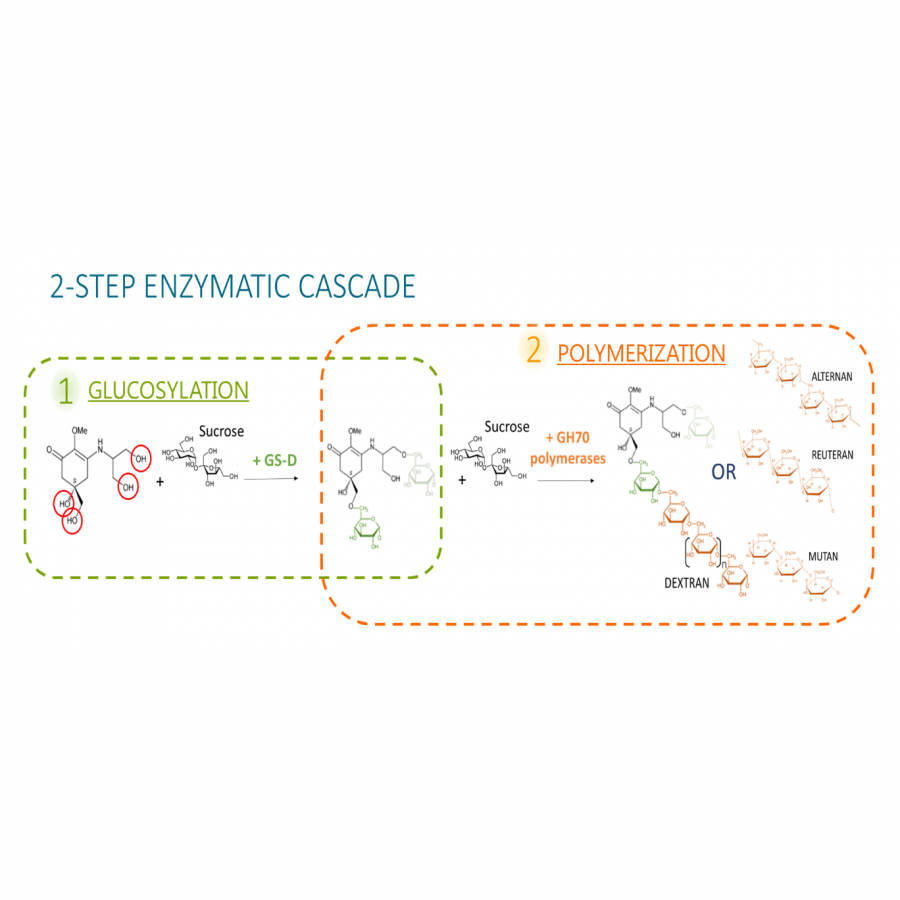 Figure 1 : Representation of the enzymatic cascade set up it this work. |
#549 | Unlocking a modular platform for bioacylation reactions enabling amide bond synthesis |
|
| Presenting author: | Christian SCHNEPEL of KTH ROYAL INSTITUTE OF TECHNOLOGY |
| Other authors: | Laura RODRIGUEZ-PEREZ of THE UNIVERSITY OF MANCHESTER Yuqi YU of THE UNIVERSITY OF MANCHESTER Antonio ANGELASTRO of THE UNIVERSITY OF MANCHESTER Max LUBBERINK of THE UNIVERSITY OF MANCHESTER Francesco FALCIONI of ASTRAZENECA Keith MULHOLLAND of ASTRAZENECA Martin HAYES of ASTRAZENECA Nicholas TURNER of THE UNIVERSITY OF MANCHESTER Sabine FLITSCH of THE UNIVERSITY OF MANCHESTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 11:30 am - 11:45 am Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | Cascade / Cofactor regeneration / Carboxylic acid reductase / Amide synthesis |
| Purpose: | Acylation reactions, in particular amide bond formations, account for a vast number of transformations in organic synthesis. Undoubtedly, they are of utmost importance in medicinal chemistry and fine chemical synthesis to assemble complex molecules. Hence, there is a huge interest in the development of green methodologies that allow for the efficient formation of amides, whereas conventional approaches typically suffer from hazardous conditions, low atom economy as well as require toxic reagents. Enzyme-catalysed approaches that enable direct activation and functionalisation of carboxylic acids to amides are most sought after. Thioesters play a central and unique role as acyl carriers in enzyme catalysis due to their chemical properties as sufficiently stable, but highly reactive acyl building blocks. They are particularly relevant for the biochemistry of coenzyme A (CoA-SH) that functions as an essential cofactor for biocatalytic N-, O, and C-acylations. It is worth mentioning that a vast number of acyltransferases is available from natural sources, yet their applications are prohibited by restricted access to thioester substrates. Cognate coenzyme A (CoA-SH) ligases that are able to provide acyl-S-CoA substrates are typically rather specialised for their native carboxylic acid substrate and of limited use for biocatalysis . This dilemma demands for a more generic thioester generation and recycling system that can be applied in amide synthesis, for example. We found that the adenylation domain of a carboxylic acid reductase is able to function as a generic thioester synthetase, which can be utilised for the synthesis of acyl-S-CoA and other thioesters. This provides a widely applicable system for the in-situ-recycling of thioesters (Figure 1). Coupling the CoA-SH recycling system with different N-acyltransferases facilitates scalable and selective synthesis of a range of challenging amides in water at mild conditions. Exploiting this viable platform opens up manifold applications towards thioester-dependent, enzyme-catalysed C-N couplings and beyond. Recent developments of modular amide synthesis applying enzyme mining, engineering and cascade development in the context of pharmaceutical synthesis are being demonstrated. |
| References: | [1] S. R. Derrington, N. J. Turner, S. P. France, J. Biotechnol. 2019, 304, 78-88. [2] M. Lubberink, C. Schnepel, J. Citoler, S. R. Derrington, W. Finnigan, M. A. Hayes, N. J. Turner, S. L. Flitsch, ACS Catal. 2020, 10, 10005-10009. [3] M. Lubberink, W. Finnigan, C. Schnepel, C. Baldwin, N. J. Turner, S. L. Flitsch, Angew. Chem. Int. Ed. 2022, 61, e202205054 [4] C. Schnepel, L. R. Pérez, Y. Yu, A. Angelastro, R. S. Heath, M. Lubberink, F. Falcioni, K. Mulholland, M. A. Hayes, N. J. Turner, S. L. Flitsch, Nat. Catal. 2023, 6, 89-99. |
| Figures: | 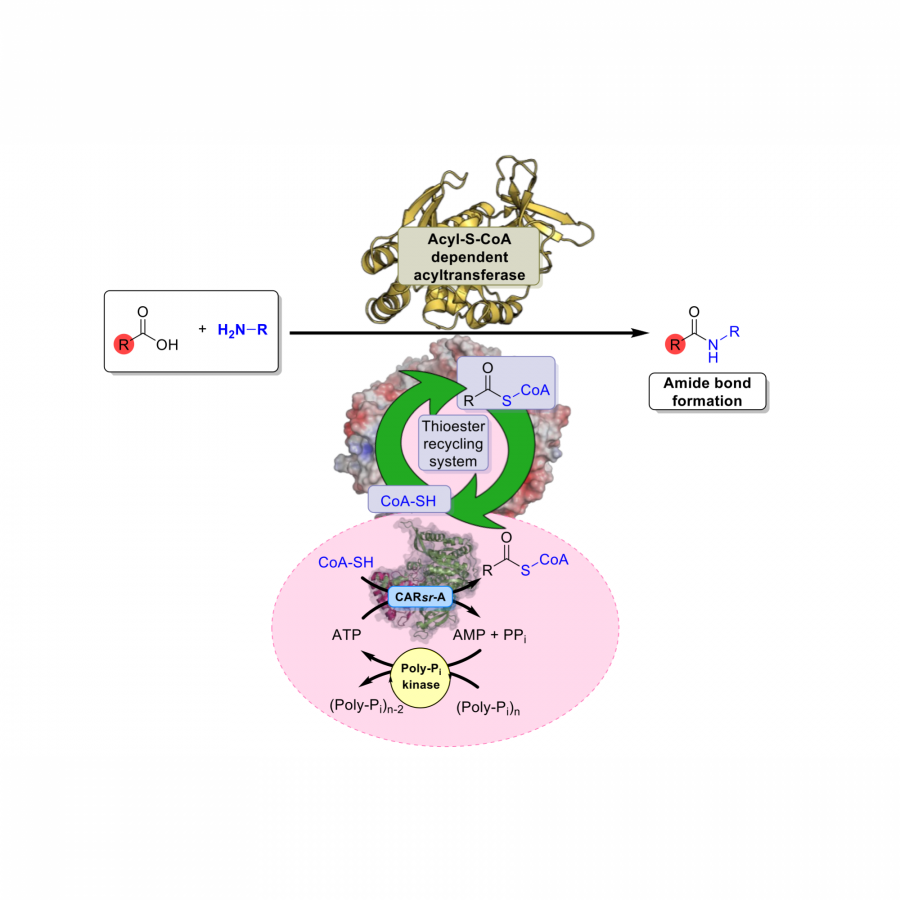 Figure 1 Adenylation domain of a carboxylic acid reductase functions as a versatile thioester synthetase providing a promiscuous in situ recycling system that can be exploited for amide synthesis. |
#581 | Accelerating the implementation of Biocatalysis in API synthesis: Contribution to Sanofi eco-design commitment |
|
| Presenting author: | Alain RABION of SANOFI |
| Topic: | Industrial biocatalysis |
| Date: | 03:15 pm - 03:30 pm Session Industrial biocatalysis - Chair : J. Martin, SEQENS |
| Keywords: | Biocatalytic platform / eco-design / API synthesis / commercial routes |
| Purpose: | Sanofi has committed to improving the impact of its processes on the global environment. Sanofi Process Chemistry organization utilizes rigorous metrics to prioritize, guide the development of processes toward meeting these important goals. Biocatalysis is widely regarded as a sustainable technology to innovate API synthesis. Sanofi is accelerating the development of strong mindset and efficient capabilities in biocatalysis to foster eco-design and promote greener solution for its synthetic processes. In this presentation, we will describe how Sanofi is building its internal end to end Biocatalytic platform from gene identification to biotransformation reaction. We will highlight the successful implementation of biocatalytic approaches for synthesizing chiral Sanofi pharmaceutical intermediates in early development phase and late stage for potential commercial routes. Positive contribution of biocatalysis towards green metrics will be presented for Sanofi API synthesis. |
#588 | Understanding alcohol dehydrogenase catalysis and enzyme-substrate-binding in non-conventional reaction media |
|
| Presenting author: | ZHANG Ningning of LEIBNIZ UNIVERSITY HANNOVER |
| Corresponding author: | Jan Philipp BITTNER of HAMBURG UNIVERSITY OF TECHNOLOGY / INSTITUTE OF THERMAL SEPARATION PROCESSES |
| Other authors: | Ningning ZHANG of LEIPNIZ UNIVERSITY HANNOVER / INSTITUTE OF TECHNICAL CHEMISTRY Pablo DOMÍNGUEZ DE MARÍA of SUSTAINABLE MOMENTUM S.L. Irina SMIRNOVA of HAMBURG UNIVERSITY OF TECHNOLOGY / INSTITUTE OF THERMAL SEPARATION PROCESSES Selin KARA of LEIPNIZ UNIVERSITY HANNOVER / INSTITUTE OF TECHNICAL CHEMISTRY Sven JAKOBTORWEIHEN of HAMBURG UNIVERSITY OF TECHNOLOGY / DEPARTMENT FOR CHEMICAL REACTION ENGINEERING |
| Topic: | Artifical intelligence / computational methods |
| Date: | 12:03 am - 12:06 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | molecular dynamics simulations / alcohol dehydrogenase / deep eutectic solvents / experimental analysis |
| Purpose: | Understanding the enzymatic behavior of alcohol dehydrogenase (ADH) in non-conventional reaction media is vital to establish its catalysis in these systems and overcome the limitations of aqueous solutions. Those include a limited solubility of hydrophobic reactants and water-induced side effects. Solvent selections for ADH catalysis are pivotal to optimize the biotransformation in non-conventional reaction media. In this context, the hydration of ADHs is crucial to maintain its activity in non-aqueous media and is difficult to access experimentally. However, molecular dynamics (MD) simulations can shed light on the hydration of ADHs in different solvents and their influence on the enzyme behavior. Moreover, in conjunction with experimental analysis, MD simulations offer a much deeper understanding of the underlying phenomena on the atomistic level. In this work, we present the investigation of horse liver alcohol dehydrogenase (HLADH) as a model enzyme in different organic solvents [1] and deep eutectic solvents (DESs) [2,3] – a novel solvent class, which recently got attention in biocatalysis. With the help of MD simulations, the solvation effects of DESs, individual DES components and organic solvents and their impact on the specific activity and stability of HLADH in these systems could be identified. For example, a high affinity of the HLADH surface towards glycerol could be linked to a drastically improved enzymatic stability in ChCl-Gly (1:2)-water mixtures. This could even be improved by shifting the molar ratio of glycerol in ChCl-Gly from 1:2 to 1:9 (Fig. 1) [3]. Moreover, the spatial distribution of the organic solvent and DES molecules around HLADH could identify the regions on the enzyme surface that are strongly affected by a specific solvent [1,3]. This helps to unravel the solvent impact on the molecular flexibility and regional/global structural changes of HLADH [2,3]. In addition, for an organic-aqueous phase boundary, the MD simulations could identify the position and orientation of HLADH at this interface and highlighted the usage of methyl tert-butyl ether (MTBE) and cyclopentyl methyl ether (CPME) as suitable organic solvents for ADH catalysis [1]. A particular focus is laid on the active center of HLADH and its structural changes in different reaction environments. The MD simulations can hereby help to unravel the enzyme substrate interactions in different solvent environments and can even quantify them by calculating free energy profiles of a substrate molecule along the substrate-binding tunnel from the bulk phase to the active center. Understanding the enzyme behavior in non-conventional reaction media and its underlying phenomena on the atomistic level is crucial for guiding solvent selection for biocatalysis. Thereby, MD simulations provide deep insights into the enzyme-solvent interactions and enzyme-substrate binding in non-conventional reaction media, which can ultimately help to guide solvent engineering and the necessary addition of water to maintain enzymatic activity. |
| References: | [1] Zhang, N., Bittner, J. P., Fiedler, M., Domínguez de María, P., Beretta, T., Jakobtorweihen, S., Kara, S., ACS Catal. 2022, 12, 15, 9171-9180. [2] Huang, L., Bittner, J. P., Domínguez de María, P., Jakobtorweihen, S., Kara, S., ChemBioChem 2020, 21, 6, 811-817. [3] Bittner, J. P., Zhang, N., Huang, L., P., Domínguez de María, P., Jakobtorweihen, S., Kara, S., Green Chem. 2022, 24, 1120-1131. |
| Figures: |  Figure 1. Spatial distribution function of glycerol molecules (green) and choline ions (blue) in the proximity of horse liver alcohol dehydrogenase (gray) in the MD simulations of (a) ChCl-Gly (1:2, mol:mol) and (b) ChCl-Gly (1:9, mol:mol) in mixtures with 20 vol.% |
#604 | Iterative metagenome mining leads to expanded substrate scope of the LEH for biocatalytic epoxide opening |
|
| Presenting author: | Bethany HOGG of UNIVERSITY OF MANCHESTER |
| Corresponding author: | Nicholas TURNER of UNIVERSITY OF MANCHESTER |
| Other authors: | Emily KEMPA of ASTRAZENECA Charlotte MORRILL of UNIVERSITY OF MANCHESTER Christian SCHNEPEL of KTH ROYAL INSTITUTE OF TECHNOLOGY James FINNIGAN of PROZOMIX LTD Martin HAYES of ASTRAZENECA |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:03 pm - 04:06 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | metagenome mining / LEH / epoxide opening / |
| Purpose: | Recently, the use of metagenome mining to discover novel enzymes has become of increasing interest in biocatalysis.[1] The process has been integral in accelerating the field through identification of enzymes for specific molecule synthesis, particularly drug target synthesis, and exploring sequence space.[2,3] Interrogating genomic data from environmental samples and comparing these against known, previously characterised sequences through a bioinformatics platform has previously allowed for screening and characterisation of a 384-panel of imine reductases, thus offering a broad screening tool for the synthesis of chiral amines. More recently, this method is being applied to find novelty in well-studied enzyme classes, in the early stage functionalisation of high value intermediates.[4] In this work, we describe the discovery of more than 50 novel limonene epoxide hydrolase (LEH) enzymes from interrogating metagenomic data, adding to this small subset of the epoxide hydrolase (EH) family. Furthermore, we discovered that some candidates demonstrated high activity towards a vastly different substrate scope and displayed good tolerance for more sterically hindered epoxides. These ‘hit’ enzymes introduce novelty to a well-studied but poorly classified enzyme class. Computational studies rationalised the high activity of two both LEHs in particular, which prompted structural analysis by X-ray crystallography revealing substrate and product binding modes which are currently harnessed for further enzyme engineering. A second search of the metagenomic data using the most active LEHs from the first panel as reference points generated a second panel of homologs, which possessed more active and enantioselective candidates for the aforementioned substrate scope. This shows that iterative metagenome mining can be used to walk along protein sequence space and be a powerful, complementary tool in directed evolution and biocatalysis. |
| References: | [1] M. Ferrer, M. Martínez-Martínez, R. Bargiela, W. R. Streit, O. V. Golyshina, P. N. Golyshin, Microb. Biotechnol. 2016, 9, 22-34. [2] D. Baud, J. W. E. Jeffries, T. S. Moody, J. M. Ward, H. C. Hailes, Green Chem. 2017, 19, 1134-1143. [3] L. Leipold, D. Dobrijevic, J. W. E. Jeffries, M. Bawn, T. S. Moody, J. M. Ward, H. C. Hailes, Green Chem. 2019, 21, 75-86. [4] J. R. Marshall, P. Yao, S. L. Montgomery, J. D. Finnigan, T. W. Thorpe, R. B. Palmer, J. Mangas-Sanchez, R. A. M. Duncan, R. S. Heath, K. M. Graham, et al., Nat. Chem. 2021, 13, 140-148. |
| Figures: |  Figure 1 Iterative metagenome mining leads to expanded enzymes families and novel biocatalysts.  Figure 2 Flowchart showing metagenomic panel creation from the initial round of mining metagenomic data. |
#624 | Bicatalytic mediated synthesis of siRNA |
|
| Presenting author: | Jill CASWELL of ALMAC SCIENCES |
| Other authors: | Stephanie PAUL of ALMAC SCIENCES Darren GRAY of ALMAC SCIENCES |
| Topic: | Industrial biocatalysis |
| Date: | 03:45 pm - 04:00 pm Session Industrial biocatalysis - Chair : J. Martin, SEQENS |
| Keywords: | RNA ligase / oligonucleotide synthesis / Industrial biocatalysis / |
| Purpose: | Biocatalysis has transformed chemical synthesis of small molecule APIs in recent years by shortening routes, lowering waste and by reducing costs. Enzymes are now seeing applications beyond small molecules, such as exploited in oligonucleotide synthesis. Novel technologies are required to overcome the challenges that are faced with manufacturing commercial quantities of oligonucleotide drug substances which are required for treating large patient populations[1]. The current preferred method for oligonucleotide production utilizes the well-established solid-phase synthesis platform to produce milligram and kilogram quantities[2]. However this platform, being a linear process can have limitations around scalability and purity of the product . This presentation will highlight a hybrid strategy that utilizes RNA ligase enzymes in combination with the classical solid phase synthesis as a path forward to overcome some of these limitations and can be used for large scale manufacture. This hybrid approach complements existing oligo synthesis technologies and utilizes engineered RNA ligases and is supported by polynucleotide kinase enzymes and nucleases. |
| References: | [1] Sasso, J.M., et al, J. Med. Chem. 2022, 65 (10), 6975-7015 [2] Andrews B.I., et al, J. Org.Chem. 2021 86 (1), 49-61 |
#638 | Increasing the Utility of Biocatalytic Methylation Reactions through a Two-Enzyme Cascade |
|
| Presenting author: | John REED of THE UNIVERSITY OF BASEL |
| Topic: | Biocatalytic cascade reactions |
| Date: | 11:45 am - 12:00 am Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | methyltransferase / biocatalysis / enzymatic cascade / S-adenosyl methionine |
| Purpose: | S-adenosylmethionene (SAM)-dependent methyltransferases catalyse the methylation of a broad array of primary and secondary metabolites, often with exquisite chemo-, regio-, and stereoselectivity.1 However, the reliance of these enzymes on SAM as a stoichiometric methyl donor restricts their utility for large-scale applications, due to this reagents high cost and instability. Our laboratory has reported a dual-enzyme cascade involving a halide methyltransferase (HMT) and a C-, N-, or O-methyltransferase that enables biocatalytic methylation reactions to proceed with only a catalytic quantity of SAM and a stoichiometric amount of a structurally simpler, cheaper methyl donor (Figure 1).2 Remarkably, a number of abiotic methyl donor reagents, including sulfates and sulfonates, are accepted by the halide methyltransferase for the in situ regeneration of SAM.3 Our current research efforts are exploring this unexpected result to further optimize the biocatalytic reaction according to parameters such as reactivity, cost, atom-efficiency, and safety. |
| References: | 1] Bennett, M. R., Shepherd, S. A., Cronin, V. A., Micklefeld, J. Curr. Opin. Chem. Biol. 2017, 37, 97-106. 2] Liao, C., Seebeck, F. P. Nature Catalysis, 2019, 2, 696-701. 3] Wen, X., Leisinger, F., Leopold, V. Seebeck, F. P. Angew. Chem. Int. Ed. 2022, 61, e202208746. |
| Figures: |  Figure 1 A two-enzyme cascade enables in situ regeneration of S-adenosyl methionine from S-adenosyl homocysteine and a methyl donor, while concurrently catalyzing the methylation of a target molecule. |
#639 | Challenging enzymes with non-natural substrates and reaction conditions to reveal unexplored activities |
|
| Presenting author: | Wolfgang KROUTIL of UNIVERSITY OF GRAZ |
| Other authors: | Erna ZUKIC of UNIVERSITY OF GRAZ Willelm BREUKELAAR of UNIVERSITY OF GRAZ Nakia POLIDORI of UNIVERSITY OF GRAZ Amit SINGH of UNIVERSITY OF GRAZ Mathias PICKL-FARNBERGER of UNIVERSITY OF GRAZ Francesco MASCIA of UNIVERSITY OF GRAZ Valerio FERRARIO of BASF Christian WILLRODT of BASF Anton GLIEDER of TU GRAZ Silvia GLUECK of UNIVERSITY OF GRAZ Karl GRUBER of UNIVERSITY OF GRAZ Michael BREUER of BASF |
| Topic: | Enzyme discovery and engineering |
| Date: | 09:00 am - 09:45 am Session Reaction design - Chair : A. BOMMARIUS, Georgia Tech |
| Keywords: | promiscuity / amines / oximes / amide |
| Purpose: | Enzymes have been recognized as rather flexible not only in their structure but also concerning their substrate tolerance. Even the functional groups which are transformed may be varied. We will show in the talk, that (1) by challenging enzymes with functional groups not known as part of metabolic pathways, novel activities can be identified. Furthermore, (2) by providing non-natural reagents and conditions, novel activities can be launched and (3) by exploiting kinetic effects, reactions can be performed not expected in water. The reduction of oximes has until recently been elusive using a defined enzyme. Interestingly, we found that several ene-reductases reduce the oxime functionality, thus a C=N bond, of β-keto-α-oximo esters to the corresponding amino group [1]. Due to the functionalization of the amine formed, subsequent non-enzymatic cyclization and oxidation occurred yielding tetrasubstituted pyrazines. Analyzing the exact pathway of the two-step enzyme-catalyzed reduction by mechanistic studies including crystallography and MD simulation revealed, that the imine must be the intermediate [2]. Subsequent studies showed even more functional groups which can be transformed [3]. A demethylation of methyl phenyl ethers was enabled by a cobalamin-dependent enzyme under anaerobic conditions to diversify natural substrates including wood derived building blocks [4,5]. While demethylation is described under oxidative conditions, anaerobic conditions would be preferred to avoid possible side reactions of the functionalized phenol products. Amide formation is one of the most important reactions in industrial pharmaceutical synthesis [6]. In general amide formation using hydrolases from acids requires organic solvents [7]. Alternatively, one may start from the ester. By choosing appropriate conditions and substrates anilides were formed in buffer [8]. |
| References: | [1] S. Velikogne; W. B. Breukelaar; F. Hamm; R. A. Glabonjat; W. Kroutil, ACS Catal. 2020, 10, 13377-13382. [2] W. B. Breukelaar, N. Polidori, A. Singh, B. Daniel, S. M. Glueck, K. Gruber, W. Kroutil, ACS Catal. 2023, 13, 2610-2618. [3] unpublished results. [4] C. Grimm, S. Pompei, K. Egger, M. Fuchs, W. Kroutil, RSC Adv. 2023, 13, 5770-5777. [5] S. Pompei, C. Grimm, C. Schiller, L. Schober, W. Kroutil, Angew. Chem. Int. Ed. 2021, 60, 16906-16910. [6] D. J. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks, T. Y. Zhang, Green Chem. 2007, 9, 411-420. [7] J. Pitzer, K. Steiner, C. Schmid, V. K. Schein, C. Prause, C. Kniely, M Reif, M. Geier, E. Pietrich, T. Reiter, P. Selig, C. Stückler, P. Pöchlauer, G. Steinkellner, K. Gruber, H. Schwab, A. Glieder, W. Kroutil, Green Chem. 2022, 24, 5171-5180. [8] A. Żądło-Dobrowolska, N. G. Schmidt, W. Kroutil, Chem. Commun. 2018, 54, 3387-3390. |
#641 | PLURIZYMES AS ALTERNATIVE FOR CASCADE REACTIONS: A TRANSAMINASE-ESTERASE PLURIZYME AS STUDY CASE |
|
| Presenting author: | Laura FERNANDEZ LOPEZ of CSIC - ICP |
| Other authors: | Sergi RODA of BARCELONA SUPERCOMPUTING CENTER Marius BENEDENS of CENTER FOR STRUCTURAL STUDIES, HEINRICH-HEINE-UNIVERSITY Alexander BOLLINGER of INSTITUTE OF MOLECULAR ENZYME TECHNOLOGY, HEINRICH-HEINE-UNIVERSITY Stephan THIES of INSTITUTE OF MOLECULAR ENZYME TECHNOLOGY, HEINRICH-HEINE-UNIVERSITY Julia SCHUMACHER of CENTER FOR STRUCTURAL STUDIES, HEINRICH-HEINE-UNIVERSITY Cristina COSCOLIN of CSIC - ICP Cristoph G.W. GERTZEN of CENTER FOR STRUCTURAL STUDIES, HEINRICH-HEINE-UNIVERSITY Jose L. GONZALEZ-ALFONSO of CSIC - ICP Francisco J. PLOU of CSIC -ICP Karl-Erich JAEGER of INSTITUTE OF MOLECULAR ENZYME TECHNOLOGY, HEINRICH-HEINE-UNIVERSITY Sander H.J. SMITS of CENTER FOR STRUCTURAL STUDIES, HEINRICH-HEINE-UNIVERSITY Victor GUALLAR of BARCELONA SUPERCOMPUTING CENTER Manuel FERRER of CSIC - ICP |
| Topic: | Enzyme discovery and engineering |
| Date: | 12:15 am - 12:18 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | PluryZymes / Enzyme design / Biocatalysis / Transaminase-esterase |
| Purpose: | Nowadays, efficiency and environmental sustainability are major issues of the circular economy. One-pot cascade reactions are the preferable choice for the synthesis and assembly of numerous molecules, as they can minimize chemical waste, save time and money and simplify technical aspects. However, they require the engineering of the biocatalysts of each internal chemical reaction that conform the cascade reaction. Enzymes are the key to this handicap, as they are selective, specific and a greener option than chemical catalysts, and through protein engineering and machine-learning methods these biocatalysts properties can be adjusted. In this regard, a novel alternative to the use and engineering of several enzymes for one-pot reactions would be PluriZymes, enzymes that maintain their native active site whilst having others artificially introduced ones supporting similar [1] or complementary [2] chemistry. In this study we designed TR2E2, a PluriZyme with amine transferase and esterolytic activity [3]. In brief, a Ser-Glu-His catalytic triad (which provides esterase activity) was introduced, unprecedentedly, into a class III ω-transaminase, to produce the first transaminase-esterase PluriZyme that could perform one-pot bioinspired cascade reactions that allow the one-pot synthesis of chiral amino acids from keto-esters. This is achieved through the conversion of a β-keto ester into a β-keto acid at the artificially introduced hydrolytic site and its subsequent conversion into a β-amino acid (e.e. >99 %) at the native transaminase site. Some limitations and challenges related to cascade reactions and lessons learnt from the recent design of PluriZymes for non-cascade and cascade reactions are discussed. We also provided biochemical and structural information demonstrating that new active sites supporting esterolytic activity can be effectively introduced into transaminase scaffolds to design PluriZymes for bioinspired cascade reactions of interest. Acknowledgements. This research was funded by the FuturEnzyme Project, funded by the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 101000327 and 101000607. We also acknowledge the financial support under Grants PCIN-2017-078, PID2020-112758RB-I00, PID2019-106370RB-I00, PDC2021-121534-I00, PID2019-105838RB-C31 and TED2021-130544B-I00, from the Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación (AEI) (Digital Object Identifier 10.13039/501100011033), and the European Union (“NextGenerationEU/PRTR”). |
| References: | [1] Alonso S., Santiago G. et al. Genetically engineered proteins with two active sites for enhanced biocatalysis and synergistic chemo- and biocatalysis (2020). Nature Catalysis, 3:319-328. DOI: 10.1038/s41929-019-0394-4. [2] Fernandez-Lopez L., Roda S. et al. Design and characterization of in-one protease-esterase PluriZyme (2022). International Journal of Molecular Sciences, 23:13337. DOI: 10.3390/ijms232113337. [3] Roda S., Fernandez-Lopez L. et al. A Plurizyme with transaminase and hydrolase activity catalyzes cascade reactions (2022). Angewandte Chemie International Edition, 61(37):e202207344. DOI: 10.1002/anie.202207344. |
#655 | Controlled Biocatalytic Synthesis of Metal Nanoparticle-Enzyme Hybrids: Demonstration for Catalytic Hydrogen-Driven NADH or Flavin Recycling |
|
| Presenting author: | Lucy BROWNE of UNIVERSITY OF OXFORD |
| Other authors: | Kylie VINCENT of UNIVERSITY OF OXFORD |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 04:06 pm - 04:09 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | biocatalysis / biohybrid metal nanoparticles / chemoenzymatic / cofactor regeneration |
| Purpose: | Having control over the shape and size of metal nanoparticles (NPs) is key for making them suitable for specific applications: ranging from drug delivery, biosensing to catalysis. This has given rise to substantial effort into exploring different synthesis routes for improving the control over the NPs polydispersity as well as the sustainability of the process.[1] Here, we demonstrate the controlled synthesis of metal NPs under mild conditions. An isolated enzyme, an NAD+ reductase, oxidizes a nicotinamide cofactor (either the native NADH or the cheaper, synthetic cofactors: BNAH or AmNAH)[2] which provides electrons for metal reduction (Fig. 1A). These metal NPs can subsequently be used as H2 oxidation catalysts, supplying electrons back to the enzyme, which can then selectively reduce NAD+ to the bioactive cofactor 1,4-NADH (Fig. 1B). This biohybrid was then utilized as a H2-driven NADH recycling catalyst for an alcohol dehydrogenase to carry out an enantioselective ketone reduction. We have also explored the synthesis of another metal NP-enzyme hybrid, which uses H2 as the reductant and therefore leads to no carbon-containing by-products. Here, the hydrogenase enzyme reduces metal salts while oxidising H2 (Fig. 2A). Initial studies have shown these metal NP-enzyme hybrids can give increased activities in comparison to the enzyme alone, likely due to the metal NP increasing the electronic surface area between the hybrid catalyst and the substrate (Fig. 2B). In both systems we have observed the enzymes’ compatibility with the metal NPs; we hope this is an area which can help bring about new reactivities for more sustainable processes by using both the enzyme and metal NP properties either in a cascade reaction or synergistically. |
| References: | [1]J. E. Ortiz-Castillo, R. C. Gallo-Villanueva, M. J. Madou, V. H. Perez-Gonzalez, Coord. Chem. Rev. 2020, 425, 213489. [2]H. A. Reeve, J. Nicholson, F. Altaf, T. H. Lonsdale, J. Preissler, L. Lauterbach, O. Lenz, S. Leimkuhler, F. Hollmann, C. E. Paul, K. A. Vincent, Chem. Commun. 2022, 58, 10540-10543. |
| Figures: |  NAD+ reductase used to make metal NPs, subsequently creating a metal biohybrid catalyst. Using NAD+ reductase enzyme to reduce metal salts, leading to the controlled synthesis of metal NPs and utilising the resulting metal NP-enzyme hybrids as catalysts for selective NAD+ reduction.  Using hydrogenase for H2-driven metal NP synthesis and flavin recycling. Hydrogenase oxidises H2, releasing electrons for metal reduction. The resulting metal NP-enzyme hybrid can then be used for catalytic applications such as flavin recycling. |
#658 | Highly active and enantioselective HheG variants obtained by flexible loop engineering |
|
| Presenting author: | Anett SCHALLMEY of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG |
| Other authors: | Marcel STAAR of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG Lina AHLBORN of TECHNISCHE UNIVERSITÄT BRAUNSCHWEIG |
| Topic: | Enzyme discovery and engineering |
| Date: | 11:15 am - 11:30 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | protein engineering / enantioselectivity / halohydrin dehalogenase / epoxide ring opening |
| Purpose: | G-type halohydrin dehalogenases, such as HheG from Ilumatobacter coccineus, are privileged enzymes among halohydrin dehalogenases (HHDHs) due to their ability to convert also sterically more demanding internal epoxide substrates (cyclic as well as acyclic ones) with a variety of anionic C-, N-, O-, S- and halide nucleophiles [1-3]. Their rather low stability and often only moderate enantioselectivity, however, constitute major limitations for practical application. While we recently solved the stability issue by generation of highly stable and active cross-linked enzyme crystals of HheG [4,5], we now attempted to create also highly enantioselective variants by protein engineering. Thus, a highly flexible active-site loop was found to influence HheG’s enantioselectivity tremendously. A site-saturation library containing all possible amino acid exchanges at three selected residues was generated and screened in ring opening reactions of different epoxide substrates in combination with two different nucleophiles. This way, several variants with largely increased or even inverted enantioselectivity towards the tested substrates could be obtained. The most impressive one did not only yield (1S,2S)-2-azidocyclohexan-1-ol with ≥96% enantiomeric excess, but displayed also a 10-fold higher specific activity compared to wild-type HheG (Figure 1). Kinetic data indicate that this improvement in enzymatic activity is caused by a largely increased kcat as well as enhanced binding of the nucleophile. Overall, we are convinced that this loop represents a major target for enantioselectivity engineering not only in HheG but also other related G-type halohydrin dehalogenases. Currently, we are trying to rationalize on a structural level the impact of that loop on enzyme activity and enantioselectivity by performing molecular dynamics (MD) simulations in collaboration with the group of Silvia Osuna. |
| References: | [1] J. Koopmeiners et al., ACS Catal., 2017, 7, 6877-6886 [2] E. Calderini et al., ChemCatChem, 2019, 11, 2099-2106 [3] J. Solarczek et al., Chem. Eur. J., 2022, 28, e202202343 [4] M. Staar et al., ChemCatChem, 2022, 14, e20220014 [5] M. Staar et al., Catalysts, 2022, 12, 1553 |
| Figures: | 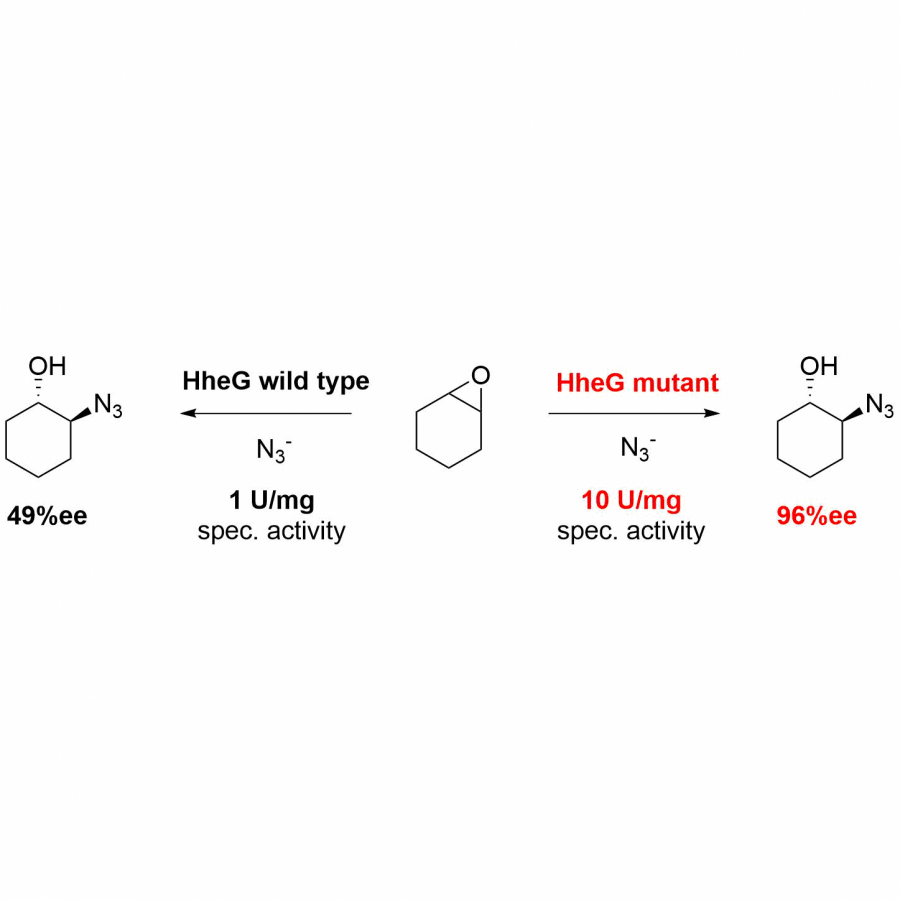 Figure 1. Azidolysis of cyclohexene oxide catalyzed by HheG wild type and a mutant with enhanced enantioselectivity. |
#711 | Biosynthesis of 3-Hydroxy Acids Through an ATP-Independent and Cell-Free Enzymatic Cascade Using Vinyl Esters as Smart Substrates |
|
| Presenting author: | ALEJANDRO H. ORREGO of CIC BIOMAGUNE |
| Corresponding author: | FERNANDO LÓPEZ-GALLEGO of CIC BIOMAGUNE |
| Other authors: | MARIA GRAZIA RUBANU of CIC BIOMAGUNE IDANIA L. LÓPEZ of CIC BIOMAGUNE DANIEL ANDRÉS-SANZ of CIC BIOMAGUNE GUILLERMO GARCÍA-MARQUINA of CIC BIOMAGUNE GERMAN E. PIESLINGER of DONOSTIA INTERNATIONAL PHYSICS CENTER LUCA SALASSA of DONOSTIA INTERNATIONAL PHYSICS CENTER |
| Topic: | Biocatalytic cascade reactions |
| Date: | 11:00 am - 11:15 am Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | Multi-enzyme systems / Cell-free biocatalysis / Coenzyme A / Thiolases |
| Purpose: | The production of compounds through in vitro biosynthetic cascades demands redox power and energy, usually in the form of NAD(P)H and ATP, respectively. The necessity of regenerate those cofactors, through chemicals, electricity or light, strongly increases the cost of the whole process, decreasing the atom economy or increasing the energy consumption. Nice examples of biosystems in which both cofactors are regenerated in situ could be found on the production of β-hydroxy acids or its polymers. These systems usually contain CoA-dependent Claisen condensation and a NAD(P)H-dependent asymmetric reduction steps. In vivo results, which this system was widely explored, reached up to 3 g/L[1,2]. On the contrary, in vitro systems barely reached 40 mM of poly(3-hydroxy butyrate)[3], alternatively artificial cell-free circular metabolisms, which includes those reactions, are emerging to fix CO2 into acyl-CoA, yielding C2-C3 compounds in the µM range[4,5]. In this work[6], we propose a new approach to produce β-hydroxy acids through an ATP-independent cascade by exploiting vinyl esters as dual acyl and electron donor. In that way, the substrate embedded the chemical energy to activate the acyl group and the redox power. The cell-free enzymatic cascade is wisely designed in 4 steps, an abiotic thiolysis, a non-decarboxylative Claisen condensation, an asymmetric reduction and a hydrolysis, including a NADH recycling step. The whole cascade is enzymatically catalyzed by 4 enzymes, achieving a titer of 3-hydroxy butyrate of 24 mM without ATP requirements, showing the potential of in vitro biocatalysis to transform simple molecules into multi-functional ones. |
| References: | [1] H.-C. Tseng, C. H. Martin, D. R. Nielsen, K. L. J. Prather, Applied and environmental microbiology 2009, 75, 3137-3145. [2] S. Cheong, J. M. Clomburg, R. Gonzalez, Nature biotechnology 2016, 34, 556-561. [3] F. Li, X. Wei, L. Zhang, C. Liu, C. You, Z. Zhu, Angewandte Chemie 2022, 134, e202111054 [4] S. Sundaram, C. Diehl, N. S. Cortina, J. Bamberger, N. Paczia, T. J. Erb, Angewandte Chemie International Edition 2021, 60, 16420-16425. [5] T. Schwander, L. Schada von Borzyskowski, S. Burgener, N. S. Cortina, T. J. Erb, Science 2016, 354, 900-904. [6] A. H. Orrego, M. G. Rubanu, I. L. Lopez, D. Andres-Sanz, G. Garcia-Marquina, G. E. Pieslinger, L. Salassa, F. Lopez-Gallego, Angewandte Chemie International Edition, n/a, e202218312. |
| Figures: | 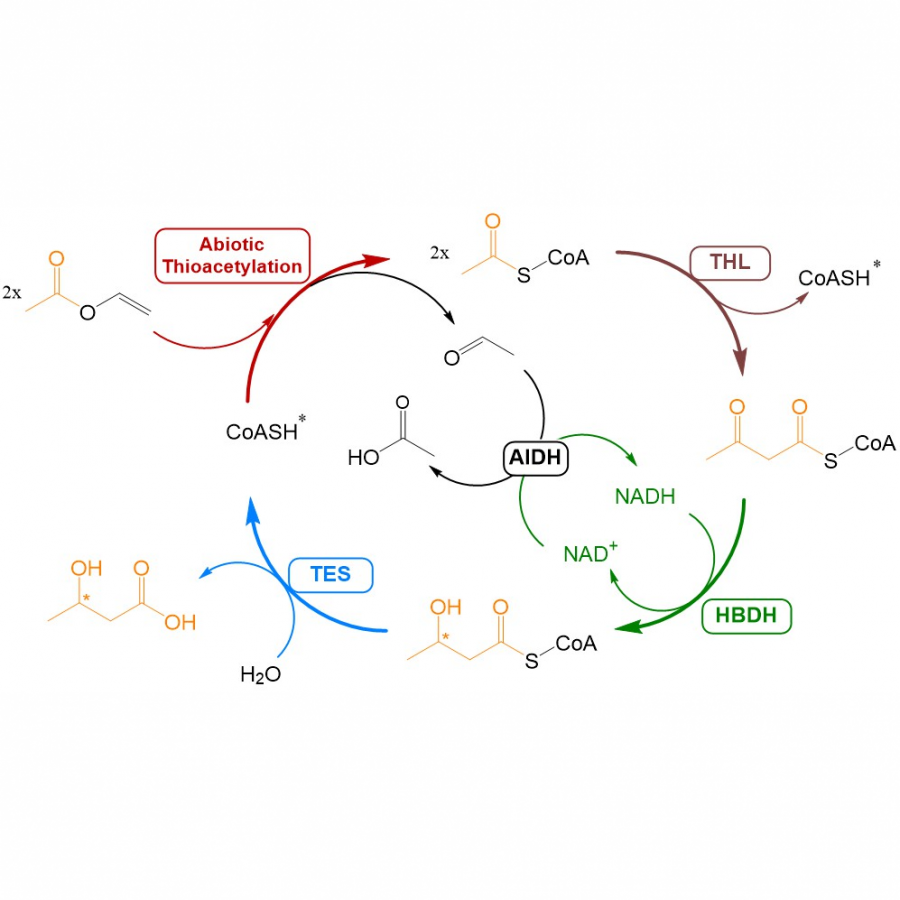 Reaction scheme of the Cell-Free Cascade for 3-Hydroxy Acids Biosynthesis 4-enzyme artificial biosynthetic pathway to synthesize 3-hydroxy butyrate through sequential abiotic thiolysis, Claisen condensation, reduction and hydrolysis. 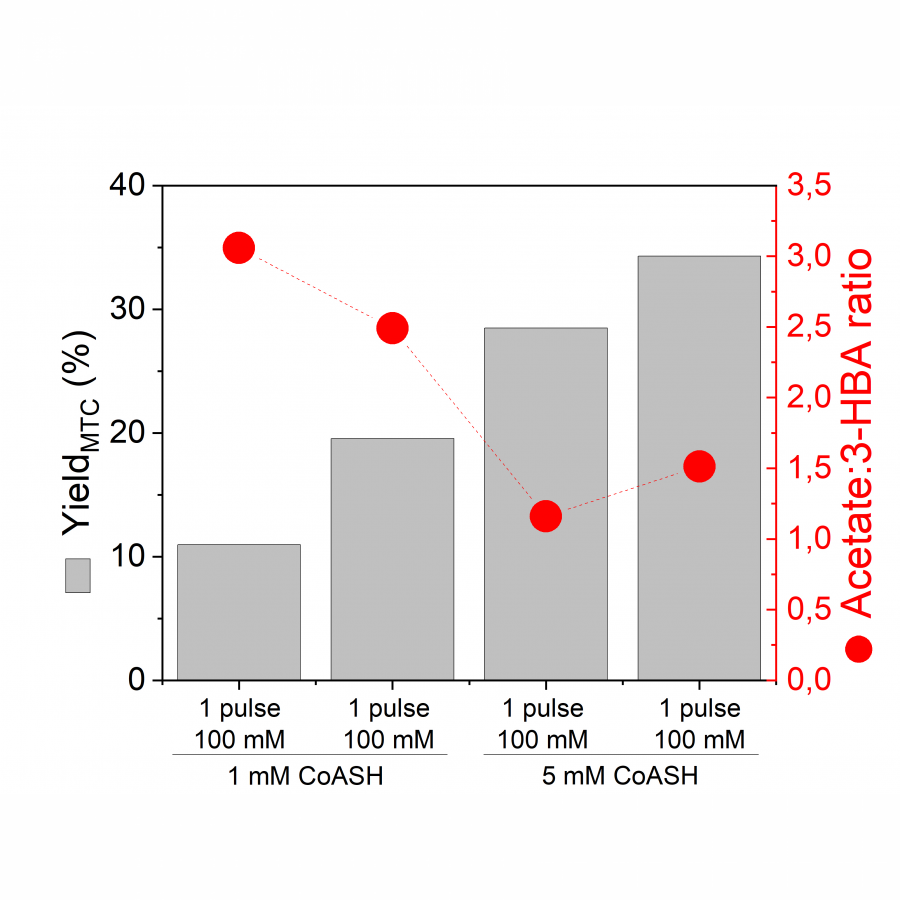 Theoretical maximum conversion and acetate:3-HB molar ratio Theoretical maximum conversion (YieldMTC) (grey bars) and acetate:3-HB molar ratio (red circles) using the cell-free enzyme systems starting at different CoASH concentration (1-5 mM) and adding the VA in different pulses. |
#720 | Methyltransferases – Towards a universal catalyst for tailoring of drug molecules |
|
| Presenting author: | Pascal SCHNEIDER of DISCOVERY SCIENCES, BIOPHARMACEUTICALS R&D, ASTRAZENECA |
| Other authors: | Jordi CHI of DISCOVERY SCIENCES, BIOPHARMACEUTICALS R&D, ASTRAZENECA David ÖLING of DISCOVERY SCIENCES, BIOPHARMACEUTICALS R&D, ASTRAZENECA Francesco FALCIONI of PHARMACEUTICAL SCIENCES, BIOPHARMACEUTICALS R&D, ASTRAZENECA Bernhard HAUER of INSTITUTE OF BIOCHEMISTRY AND TECHNICAL BIOCHEMISTRY, UNIVERSITY OF STUTTGART Martin HAYES of DISCOVERY SCIENCES, BIOPHARMACEUTICALS R&D, ASTRAZENECA |
| Topic: | Industrial biocatalysis |
| Date: | 03:30 pm - 03:45 pm Session Industrial biocatalysis - Chair : J. Martin, SEQENS |
| Keywords: | Biocatalysis / enzyme immobilization / protein engineering / late-stage functionalization |
| Purpose: | A wide variety of drug products and chemical building blocks (BBs) contain 1°, 2° or 3° amines. Often regio- and chemoselective N-methylation of these compounds is required to improve desired physchem properties in drug design.[1] Classical organic synthesis often lacks selectivity resulting in low yields due to side product formation. To overcome this limitation, we are implementing N-methyltransferases as a bio-inspired alternative to classic organic alkylations.[2, 3] N-MTs are widely known for their role in biology, but their potential for chemical synthesis remains underexplored.[4] To unlock the full potential of N-MTs for tailoring of drug molecules the issue was tackled from multiple angles to find the most suitable catalyst and experimental set-up for screening and subsequent upscaling. The nicotinamide N-methyltransferase (NNMT) was chosen as a starting point. The NNMT was reported to be a versatile enzyme accepting a broad range of small molecules while being able to be evolved for regio-selective N-methylation of various N-heterocycles.[2, 5] To implement the NNMT in our high-throughput screening workflow the enzyme was first immobilized on Co2+-resin, which did lead to a drastic increase in enzyme stability over cell free extract obtained from cell lysis. On top of that, the immobilized NNMT was able to be used for multiple screenings decreasing hands on time by removal of multiple operational steps. Screening of a diverse small molecule library highlighted NNMT promiscuity and we were able to demonstrate that a piperazine containing BB was accepted as a substrate with > 20 % conversion. By screening NNMT against a small panel of desmethyl-precurser of drug molecules containing a terminal piperazine moiety we successfully demonstrated translatability of the small molecule screening onto drug molecules. Here, 3 out 4 of examples were methylated regioselectivly towards the final drug molecule with up to 33 % conversion. Detailed analysis of the NNMT crustal structure (3ROD) suggested loop engineering of the NNMT wild type as a potential handle to optimize enzyme activity for drug molecule methylation. This was probed by site saturation mutagenesis of 11 target residues along the loop region leading to a 2-5-fold increase in target substrate conversion. Future work will feature combinatorial mutagenesis and rational protein design to generate highly active and selective NNMT variants for application in late-stage functionalization of drug molecules. In summary, we were able to demonstrate direct and regioselective biocatalytic methylation of drug molecules by using a single enzyme. Process optimization was achieved by enzyme immobilization leading to a more robust screening approach. Further, enzyme activity was improved by site saturation mutagenesis of a crucial loop region. Future work will focus on the exploration on natures reservoir of N-MTs as a source for novel catalyst to access a broader substrate scope and thereby implement biocatalytic methylation as a useful tool for tailoring of drug molecules in the future. |
| References: | [1] E. Vitaku et al. J. Med. Chem. 2014, 57, 10257-10274. [2] L. L. Bengel et al. Angew. Chem., Int. Ed. 2021, 60, 5554-5560. [3] F. Ospina et al. ChemPlusChem 2022, 87, e202100454. [4] A. W. Struck et al. ChemBioChem 2012, 13, 2642-2655. [5] F. Ospina et al. Angew. Chem., Int. Ed. 2022, 61, e202213056. |
| Figures: | 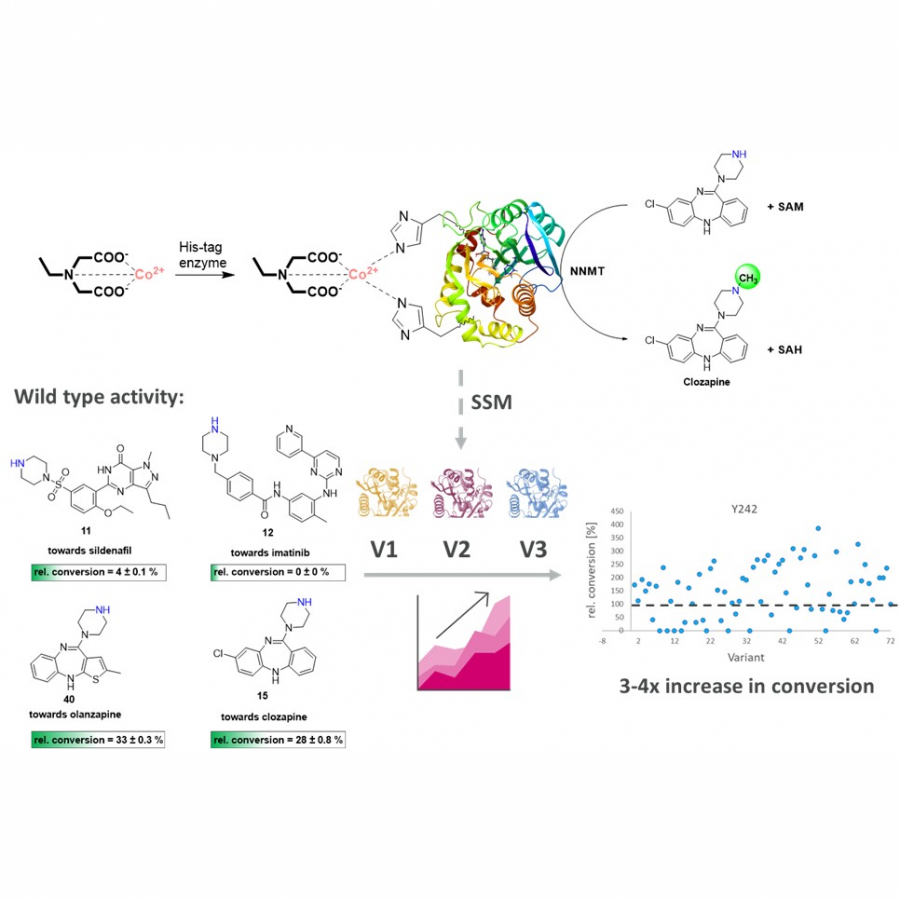 Project overview Workflow for high-throughput screening of immobilized Nicotinamide N-Methyltransferase (NNMT) for late-stage drug molecule methylation and semi-rational protein engineering by Site Saturation Mutagenesis (SSM) to improve enzyme activity. |
#731 | In vivo redox and multistep biocatalysis coupled to hetero- and/or phototrophic metabolism |
|
| Presenting author: | Bruno BÜHLER of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ |
| Other authors: | Lisa BRETSCHNEIDER of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Carolin BERTELMANN of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Adrian TÜLLINGHOFF of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Anna HOSCHEK of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Sara LUPACCHINI of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ Jörg TOEPEL of HELMHOLTZ CENTRE FOR ENVIRONMENTAL RESEARCH - UFZ |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 10:45 am - 11:00 am Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | in vivo redox biocatalysis / oxygenases / cofactor recycling / light-driven biocatalysis |
| Purpose: | In the past three decades, efficient oxygenase biocatalysis concepts have been developed relying on the metabolism of living cells of heterotrophic organisms (e.g. E. coli or Pseudomonas) [1, 2]. These organisms utilize organic compounds such as glucose as source of carbon, energy, and reduction equivalents which implies that carbon and energy metabolism are coupled. As examples, in vivo cascades for monomer production and efficient steroid hydroxylation in engineered Pseudomonads and E. coli, respectively, will be presented [3-7]. Phototrophic organisms on the other hand acquire high-energy reduction equivalents from water as highly attractive electron donner, i.e., via photosynthetic water oxidation, and thus gained interest as host systems for redox biocatalysis [8, 9]. The photosynthetic light reaction provides both co-substrates of oxygenases, i.e., electrons and O2, via a path orthogonal to the carbon metabolism. The fast photosynthetic water oxidation thus augurs well for resource-efficient oxygenation and redox biocatalysis in general (Figure 1). We present results on photosynthesis-driven redox reactions catalyzed by recombinant Synechocystis sp. PCC 6803, on their dependency on the phototrophic metabolism in terms of electron and O2 supply, as well as on reaction engineering options. Light-driven biocatalysis based on a CYP450 monooxygenase [10, 11], a BVMO [12], and an oxygen-tolerant hydrogenase will be highlighted. The latter recently has successfully been introduced into Synechocystis with the perspective to enable photosynthetic H2 production [13]. |
| References: | [1] e. theodosiou et al., front. bioeng. biotechnol. 2022, 10, 855715. [2] m. schrewe et al., chem. soc. rev. 2013, 42, 6346-6377. [3] c. bertelmann et al., front. catal. 2022, 2, 887458. [4] l. bretschneider et al., metab. eng. 2022, 79, 206-217. [5] l. bretschneider et al., front. catal. 2021, 1, 683248. [6] l. bretschneider et al., microb. biotechnol 2021, 14, 1011-1025. [7] l. schaefer et al., biotechnol. j. 2020, 15, 2000091. [8] j. toepel et al., curr. opin. biotechnol. 2023, 80, 102892. [9] a. hoschek et al., chemcatchem 2018, 10, 5366-5371. [10] a. hoschek et al., biotechnol. j. 2019, 14, 1800724. [11] a. hoschek et al., bioresour. technol. 2019, 282, 171-178. [12] a. tuellinghoff et al., front. catal. 1, 2022, 780474. [13] s. lupacchini et al., metab. eng. 2021, 68, 199 - 209. |
| Figures: | 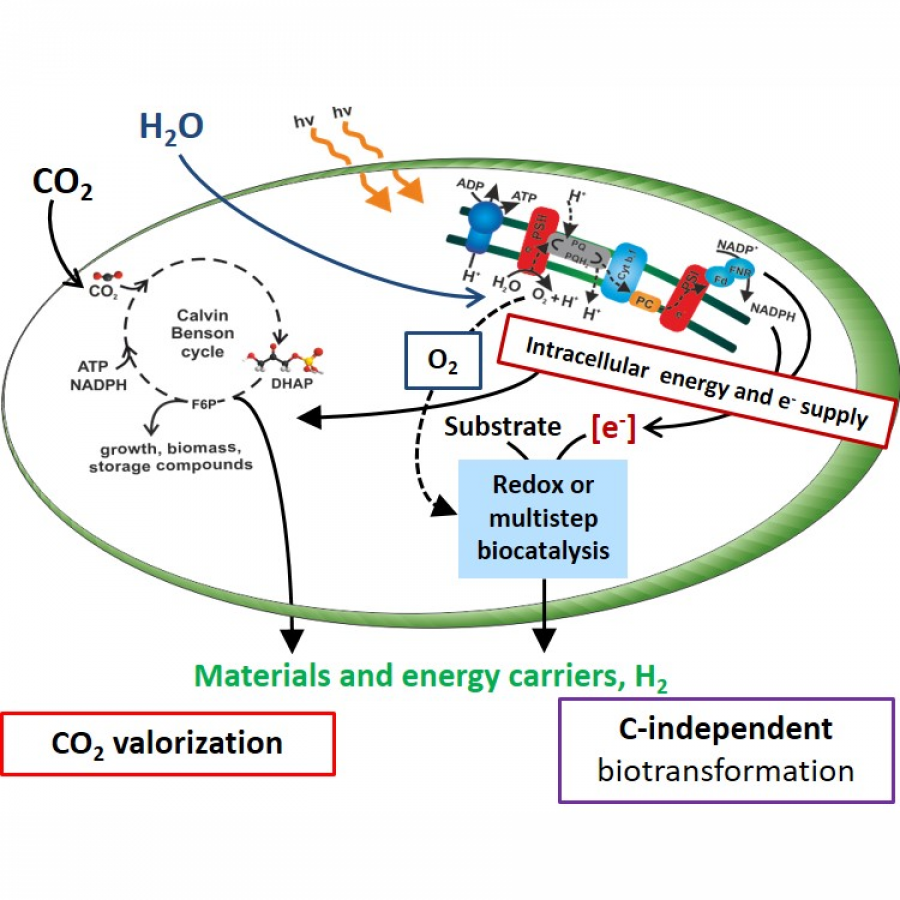 Figure 1 Light-driven biocatalysis based on cyanobacteria. |
#735 | Asymmetric mono-reduction of 1,2-dicarbonyls by Old Yellow Enzyme and glucose dehydrogenase |
|
| Presenting author: | Allison WOLDER of TU DELFT |
| Other authors: | Caroline PAUL of TU DELFT |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 12:09 am - 12:12 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | asymmetric reduction / ene reductases / hydroxy ketones / dicarbonyls |
| Purpose: | Chiral α-hydroxy ketones are valuable molecules that can be used as fine chemicals or as building blocks for asymmetric synthesis to obtain bioactive compounds [1]. These enantiopure molecules can be obtained via asymmetric reduction of one carbonyl from a vicinal diketone or other (enzymatic) approaches [2]. Alcohol dehydrogenases typically catalyse the double reduction of diketones to the corresponding diols, with some exception for the mono-reduction of certain aliphatic and aromatic 1,2-diketones [3]. Recent studies on flavin-dependent ene reductases from the Old Yellow Enzyme family (OYE) have revealed compelling non-conventional activity without illumination, in particular oxime reduction [4] and C-C bond formation [5], that are distinct from the typical asymmetric reduction of α,β-unsaturated compounds. Evidence alludes to yet another variant reaction, where OYEs might catalyse the reduction of 1,2-dicarbonyl substrates [6]. In this study we demonstrate that certain OYEs can catalyse the non-conventional diastereoselective mono-reduction of 1,2-dicarbonyls to the corresponding hydroxy carbonyl enantiomer (Figure 1). Purification of these enzymes, use of stoichiometric amounts of native cofactor, synthetic cofactor, and mechanistic studies confirmed formation of the α-hydroxy ketone product with full conversion, in high enantiomeric and diastereomeric excess. We also highlight the serendipitous discovery of promiscuous GDH activity on these vicinal dicarbonyls. |
| References: | [1] C. Palomo, M. Oiarbide, J.M. Garcia, Chem. Soc. Rev. 2012, 41, 4150-4164. [2] P. Hoyos, J. V. Sinisterra, F. Molinari, A. R. Alcantara, P. Dominguez de Maria, Acc. Chem. Res. 2010, 43, 288-299. [3] M. Rabuffetti, P. Cannazza, M. L. Contente, A. Pinto, D. Romano, P. Hoyos, A. R. Alcantara, I. Eberini, T. Laurenzi, L. Gourlay, F. Di Pisa, F. Molinari, Bioorg. Chem. 2021, 108, 104644. [4] S. Velikogne, W.B. Breukelaar, F. Hamm, R.A. Glabonjat, W. Kroutil, ACS Catal. 2020, 10, 13377-13382. [5] K. Heckenbichler, A. Schweiger, L. A. Brandner, A. Binter, M. Toplak, P. Macheroux, K. Gruber, R. Breinbauer, Angew. Chem. Int. Ed. 2018, 57, 7240-7244. [6] P. Schweiger, H. Gross, S. Wesener, U. Deppenmeier, Appl. Microbiol. Biotechnol. 2008, 80, 995-1006. |
| Figures: | 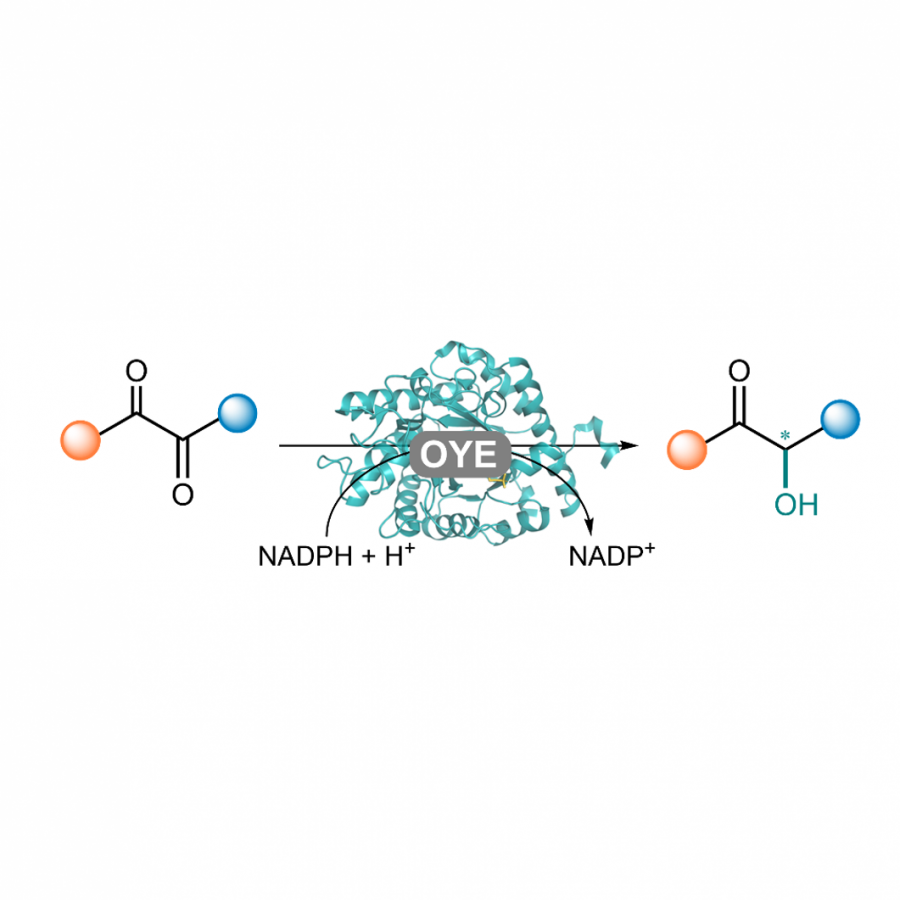 Figure 1 Stereoselective mono-reduction of vicinal dicarbonyls catalysed by an OYE to the corresponding alpha-hydroxyl carbonyl. |
#736 | Enzyme adaptation and habitat thermal legacy: from microbiome adaptation to climate change to smart selection of enzymes with thermal characteristics |
|
| Presenting author: | Cristina COSCOLÍN of CSIC-ICP |
| Other authors: | Ramona MARASCO of KING ABDULLAH UNIVERSITY OF SCIENCE AND TECHNOLOGY (KAUST) MARCO FUSI of KING ABDULLAH UNIVERSITY OF SCIENCE AND TECHNOLOGY (KAUST) ALAN BAROZZI of KING ABDULLAH UNIVERSITY OF SCIENCE AND TECHNOLOGY (KAUST) DAVID ALMENDRAL of CSIC-ICP RAFAEL BARGIELA of BANGOR UNIVERSITY Christina GOHLKE NEÉ NUTSCHEL of INSTITUTE OF BIO- AND GEOSCIENCES FORSCHUNGSZENTRUM JÜLICH CHRISTOPHER PFLEGER of MATHEMATISCH-NATURWISSENSCHAFTLICHE FAKULTÄT, INSTITUT FÜR PHARMAZEUTISCHE UND MEDIZINISCHE CHEMIE, HEINRICH-HEINE-UNIVERSITÄT DÜSSELDORF Jonas DITTRICH of HEINRICH-HEINE-UNIVERSITÄT DÜSSELDORF Holger GOHLKE of JOHN VON NEUMANN INSTITUTE FOR COMPUTING (NIC) AND JÜLICH SUPERCOMPUTING CENTRE (JSC) RUTH MATESANZ of SPECTROSCOPY LABORATORY, CENTRO DE INVESTIGACIONES BIOLOGICAS MARGARITA SALAS (CIB), CSIC Sergio SANCHEZ-CARRILLO of CENTRO DE BIOLOGIA MOLECULAR SEVERO OCHOA (CBM), CSIC-UAM Francesca MAPELLI of DEPARTMENT OF FOOD ENVIRONMENTAL AND NUTRITIONAL SCIENCES, UNIVERSITY OF MILAN TATYANA N CHERNIKOVA of CENTRE FOR ENVIRONMENTAL BIOTECHNOLOGY, SCHOOL OF NATURAL SCIENCES, BANGOR UNIVERSITY Peter N GOLYSHIN of CENTRE FOR ENVIRONMENTAL BIOTECHNOLOGY, SCHOOL OF NATURAL SCIENCES, BANGOR UNIVERSITY MANUEL FERRER of CSIC-ICP Daniele DAFFONCHIO of KING ABDULLAH UNIVERSITY OF SCIENCE AND TECHNOLOGY (KAUST) |
| Topic: | Artifical intelligence / computational methods |
| Date: | 11:15 am - 11:30 am Session AI & Computational methods - Chair : R. KOURIST, University of Technology of GRAZ |
| Keywords: | enzyme discovery / computational methods / thermal legazy / marine microbiome |
| Purpose: | Global warming is one of the biggest concerns of modern society as it is one of the main hazards for healthcare. But, climate change is also altering the ecological patterns and the way temperature would select the skills of cellular building blocks, such as enzymes. We hypothesize that environmental mean annual temperature (MAT) contributes to drive the adaptability of marine microbial communities by tuning the thermal plasticity of their enzymes, and that such plasticity is fine-tuned further by environmental temperature variability [1]. To test this hypothesis, we studied the enzymatic thermal response of marine microbial communities at a global geographical scale across different locations within a broad latitudinal gradient and different MAT conditions (-1.4 to 28.5 °C). Here, the following meta-data are presented for active proteomes and 233 individual enzymes: 1) the optimal temperature (Topt) for activity of seven functionally-independent enzyme classes; 2) the thermo-stability, by meaning of the denaturation temperature (Td) patterns; and 3) the phase transition temperature (Tp), a computational measure of structural flexibility. The generated meta-data were analyzed as a function of the environmental parameters at the site from which the enzymes originated, namely, the variability in MAT, salinity, pH, hydrostatic pressure or light received. To also consider the thermal variability (TV), the response of enzyme activity (Tp and Td) to TV was evaluated and compared with the microbial growth characteristics in three adjacent marine sites with same MAT but with different TV throughout all the seasons. Our results showed that microorganisms better adapt to different thermal conditions by selecting thermally-adapted enzymes (through genetic adaptation, e.g., new genes) rather than selecting enzymes with broad thermal tolerances. At the same time, microbial communities living in environments with a wide temperature variability have higher thermal plasticity, in consonance with the thermal legacy of their enzymes. The fine-tuned response of microbial communities to temperature change is thus controlled, at least in part, by the selection of enzyme variants that are capable of being active under more variable temperatures. From a climate change adaptation point of view, this implies that while the adaptation of microbial communities to temperature change appears to be mediated by the selection of thermally adapted enzymes, the extent of this adaptation also depends on temperature variability. This should be considered when modelling the response of marine and coastal microbiota to climate change, as projections suggest that more than 85% of ecosystems and 16% of their microbes will be in influenced either directly or indirectly by climate change [2]. From a more technical point of view, the fact that a significant correlation (p < 0.05) between the computed Topt, Td, Tp, and the MAT and TV from the site where each enzyme was retrieved, implies that these parameters, particularly the Tp, which can be calculated from sequence information through constraint network analysis, and the environmental MAT/TV information, can be used as motifs/parameters for implementing sequence-based smart search of enzymes fitting thermal needs with which to implement the industrial reconversion needed to complete the green transition and to achieve climate neutrality. Acknowledgements. The presenters acknowledge the financial support by the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 101000327, and the grants PID2020-112758RB-I00, PDC2021-121534-I00 (M.Fe.) and TED2021-130544B-I00 (M.Fe.) from the Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación (AEI) (Digital Object Identifier 10.13039/501100011033), and the European Union (“NextGenerationEU/PRTR). |
| References: | 1. Marasco, R., Fusi, M., Coscolín, C. et al. Enzyme adaptation to habitat thermal legacy shapes the thermal plasticity of marine microbiomes. Nat Commun 14, 1045 (2023). 2. Guerra, C.A., Delgado-Baquerizo, M., Duarte, E. et al. Global projections of the soil microbiome in the Anthropocene. Glob Ecol Biogeogr 30, 987-99 (2021). |
#738 | Enzyme engineering approaches on a stereospecific indole C-methyltransferase – challenges, new functions and solutions for practical applications |
|
| Presenting author: | Diana-Alexandra AMARIEI of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF IN FORSCHUNGSZENTRUM JÜLICH |
| Other authors: | Julia TENHAEF of IBG-1: BIOTECHNOLOGY, FORSCHUNGSZENTRUM JÜLICH Tobias ROSCH of IBG-1: BIOTECHNOLOGY, FORSCHUNGSZENTRUM JÜLICH Mona HAASE of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF IN FORSCHUNGSZENTRUM JÜLICH Pascal SCHNEIDER of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF IN FORSCHUNGSZENTRUM JÜLICH Stephan NOACK of IBG-1: BIOTECHNOLOGY, FORSCHUNGSZENTRUM JÜLICH Jörg PIETRUSZKA of INSTITUTE FOR BIOORGANIC CHEMISTRY, HEINRICH HEINE UNIVERSITY DÜSSELDORF IN FORSCHUNGSZENTRUM JÜLICH; IBG-1: BIOTECHNOLOGY, FORSCHUNGSZENTRUM JÜLICH |
| Topic: | Enzyme discovery and engineering |
| Date: | 04:09 pm - 04:12 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | methyltransferase / enzyme engineering / indole methylation / library screening |
| Purpose: | The indole and pyrroloindoline structural motifs are common among bioactive natural product alkaloids with various biological activities.[1] AChE inhibitor physostigmine was the inspiration for the development of a stereoselective enzymatic methylation platform for these scaffolds. A key step in the biosynthesis of physostigmine is the enzymatic C-methylation of an indole derivative, which drives the subsequent intramolecular cyclization, forming a stereogenic centre.[2] By using a chemo-enzymatic approach, we were able to produce physostigmine analogs in an enantiopure fashion. However, the limits of the enzyme are preventing application on a broader scope.[3] Site-specific and saturation mutagenesis approaches allowed the modification of the catalytic site, with implications on the product size and chemistry. Our mechanistic study, as well as an analysis of known methyltransferases containing a Rossmann-fold motif are a source of inspiration for targeted modifications, while directed evolution techniques were able to fill in the gaps towards a substrate promiscuous – but stereoselective engineered catalyst. Further enzymatic alkylations of the indole scaffold were also enabled by this approach, using SAM analogs. In order to support the screening effort, we developed an automated process for the cultivation, expression and activity testing of the resulting mutant libraries. Our results lead us to an enhanced biocatalytic approach for the production of physostigmine analogs. |
| References: | [1] de Sa Alves, F. R.; Barreiro, E. J.; Manssour Fraga, C. A. Mini Rev. Med. Chem. 2009, 9, 782-793. [2] Liu, J.; Tailun, Ng.; Rui, Z.; Ad, O.; Zhang, W. Angew. Chem. Int. Ed. 2014, 53, 136-139. [3] Amariei, D. A.; Pozhydaieva, N.; David, B.; Schneider, P.; Classen, T.; Gohlke, H.; Weiergräber, O. H; Pietruszka, J. ACS Catal. 2022, 12(22), 14130-14139. |
| Figures: | 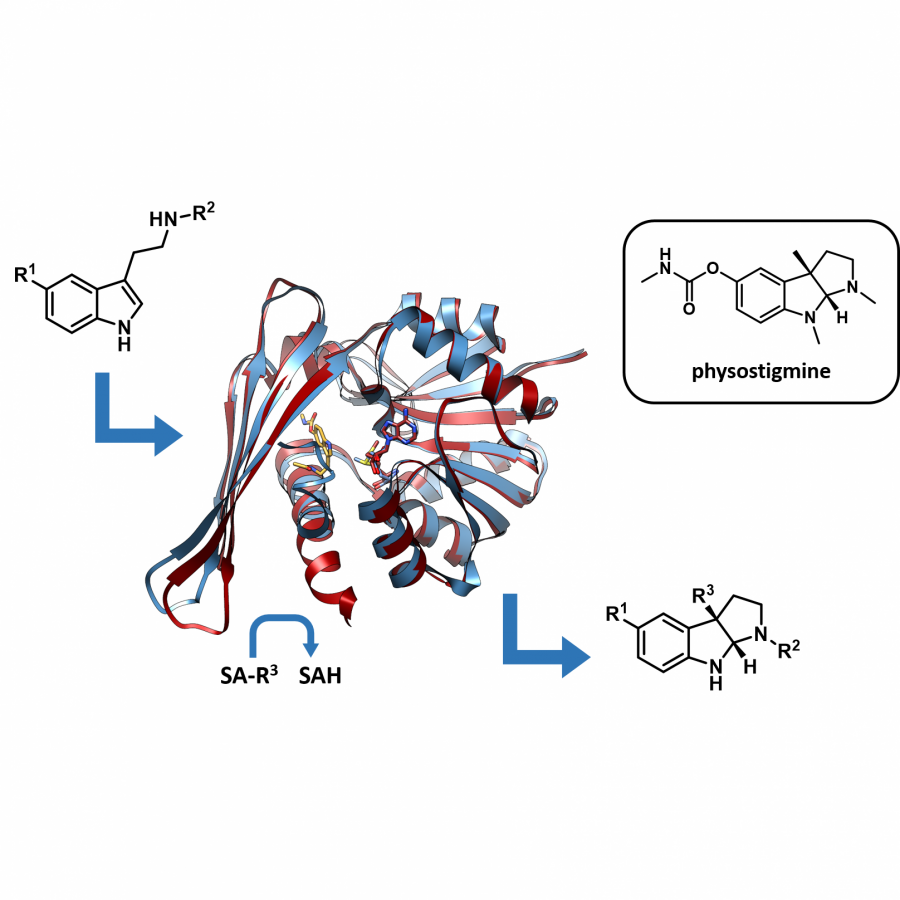 General process catalyzed by the engineered methyltransferase 3 points of interest were selected for the structural diversification of the products. SAM analogs were used for alkylation in the 3 possition (ex. R3=Me for methylation) |
#754 | Unspecific Peroxygenases UPOs: Modular Yeast-based Expression and Directed Evolution Towards Chemoselective Transformations |
|
| Presenting author: | Martin WEISSENBORN of MARTIN-LUTHER UNIVERSITY HALLE-WITTENBERG |
| Topic: | Enzyme discovery and engineering |
| Date: | 11:00 am - 11:15 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | Unspecific Peroxygenase / protein engineering / directed evolution / yeast |
| Purpose: | Fungal unspecific peroxygenases (UPOs) can transfer peroxide-borne oxygen to a vast diversity of substrates, including aromatic and sp3-carbons. They are independent of external electron sources and additional reductase units and have demonstrated outstanding activities.[1] These highly glycosylated proteins currently have two significant challenges for their broader applicability: 1) The heterologous expression in a fast-growing host permitting directed evolution endeavors and 2) the lack of chemo- and regioselectivity for specific substrates. We aimed to address these challenges by developing several techniques. To enable the heterologous expression of UPOs in S. cerevisiae and P. pastoris, a modular system consisting of i) the signal peptide, ii) the UPO gene, and iii) a C-terminal tag for split-GFP detection was established. With a Golden Gate protocol, these three biobricks can be rapidly shuffled and enable the expression of eleven UPOs.[2] The S. cerevisiae system was utilized for the directed evolution of MthUPO from a thermophilic fungus.[3] This protein engineering led to increased oxyfunctionalization activity, with improved kcat/Km values of up to 16.5-fold for the model substrate 5-nitro-1,3-benzodioxole (NBD). Variants were identified with high chemo-, regio- and enantioselectivities in the oxyfunctionalization of aromatic and benzylic carbons, respectively (Fig. 1). My talk will be about the whole process, from developing the UPO expression system to protein engineering culminating in preparative scale conversions. |
| References: | [1] J. Münch, P. Püllmann, W. Zhang, M. J. Weissenborn, ACS Catal. 2021, 11, 9168-9203. [2] P. Püllmann, A. Knorrscheidt, J. Münch, P. R. Palme, W. Hoehenwarter, S. Marillonnet, M. Alcalde, B. Westermann, M. J. Weissenborn, Commun. Biol. 2021, 4, 562. [3] A. Knorrscheidt, J. Soler, N. Hünecke, P. Püllmann, M. Garcia-Borràs, M. J. Weissenborn, ACS Catal. 2021, 11, 7327-7338. |
| Figures: | 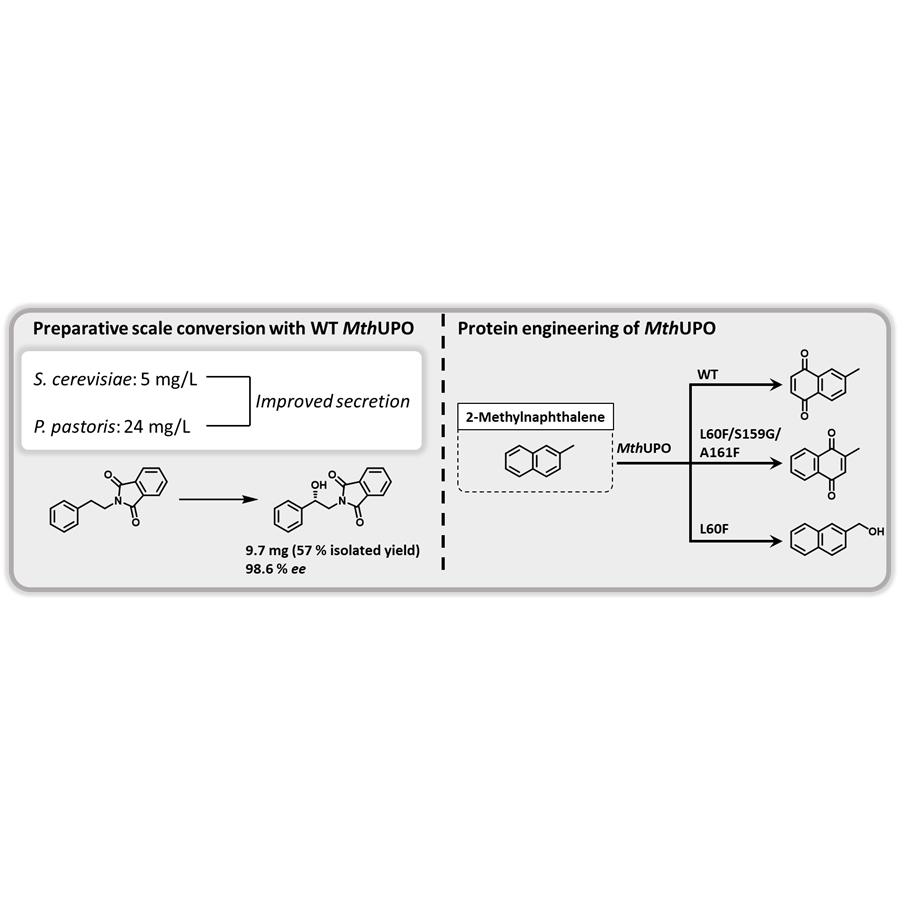 Engineered UPO: By improving the UPO secretion in P. pastoris, preparative scale conversions were possible. The S. cerevisiae system allowed the screening of more than 5,000 transformants leading to MthUPO variants with new chemoselectivities. |
#762 | Next-Gen Enzyme Engineering – A wet lab data-driven approach to identify and recombine key point mutations with EnzyMAP AI and EnzyREC AI for superior enzyme performance |
|
| Presenting author: | David SCHÖNAUER of AMINOVERSE |
| Other authors: | Eduardo OLIVEIRA of AMINOVERSE Kerstin CARDINIER of AMINOVERSE |
| Topic: | Enzyme discovery and engineering |
| Date: | 11:30 am - 11:45 am Session AI & Computational methods - Chair : R. KOURIST, University of Technology of GRAZ |
| Keywords: | #artificialintelligence / #enzymeevolution / #aminoverse / #recombination |
| Purpose: | Directed evolution of enzymes has evolved towards increasing reliance on in silico designs and lessened dependence on expensive experimental screens. In the last few years we are witnessing the emergence of in silico Machine learning (ML) strategies with the promise to propel the field of enzyme evolution further, eventually reaching the goal of function-guided de novo enzyme design . Aminoverse seeks to fulfill that promise with its proprietary EnzyMAP AI and EnzyREC AI. EnzyMAP AI is a modern deep learning architecture that uses a small wet-lab data input of 3 to 5 representative amino acids, combined with evolutionary and structural information, to predict the fitness of all 20 amino acid mutations in all positions of any given enzyme. For training we collected and generated datasets with >150,000 mutants spanning diverse proteins. With a wet lab data input of only 20%, EnzyMAP AI predicts the best performing point mutations (hotspots) across the entire enzyme sequence with >90% accuracy visualized by a fitness heatmap (Fig.1). This approach is especially useful to gather holistic knowledge about the effect of mutations towards a variety of enzyme properties while reducing screening efforts and costs by up to 85%. Guided by the knowledge base generated via EnzyMAP AI, EnzyREC AI employs the latest advancements in large language models and protein generative models conditioned on backbone coordinates , to cherry-pick and efficiently recombine key point mutations in final variants exhibiting superior enzyme performance. We exemplify the synergies of EnzyMAP AI and EnzyREC AI with an internal case study revolving around an Alcohol Dehydrogenase engineered towards enhanced thermostability and activity. The performance of Aminoverse’s AI platform is benchmarked against traditional approaches such as random mutagenesis and in silico design and more recent publicly reported machine learning algorithms . |
| References: | [1] Wittmann et al. 2021, https://doi.org/10.1016/j.sbi.2021.01.008. [2] ESM - Rives et al. 2021, https://doi.org/10.1073/pnas.2016239118 [3] ProteinMPNN - Dauparas et al. 2022, https://doi.org/10.1126/science.add2187 [4] MSATransformer - Rao et al. 2020, https://doi.org/10.1101/2020.12.15.422761. [5] FRESCO - Wijma et al. 2014, https://doi.org/10.1093/protein/gzt061 [6] DDGun - Montanucci et al. 2019, https://doi.org/10.1186/s12859-019-2923-1 |
| Figures: | 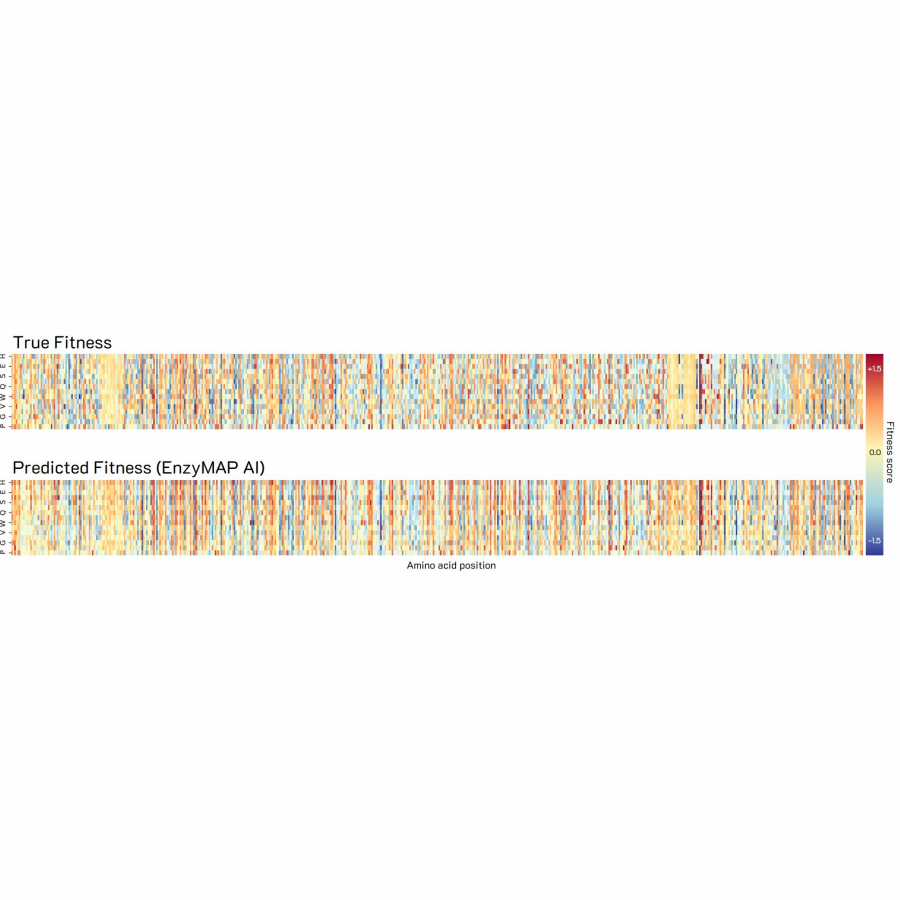 EnzyMAP AI Fitness Landscape Prediction Fitness landscape prediction of EnzyMAP AI (bottom) on unseen experimental data set (top). Red color marks fitness improving mutations (hotspots), blue color marks fitness decreasing mutations (coldspots), while yellow color indicates mutations not affect 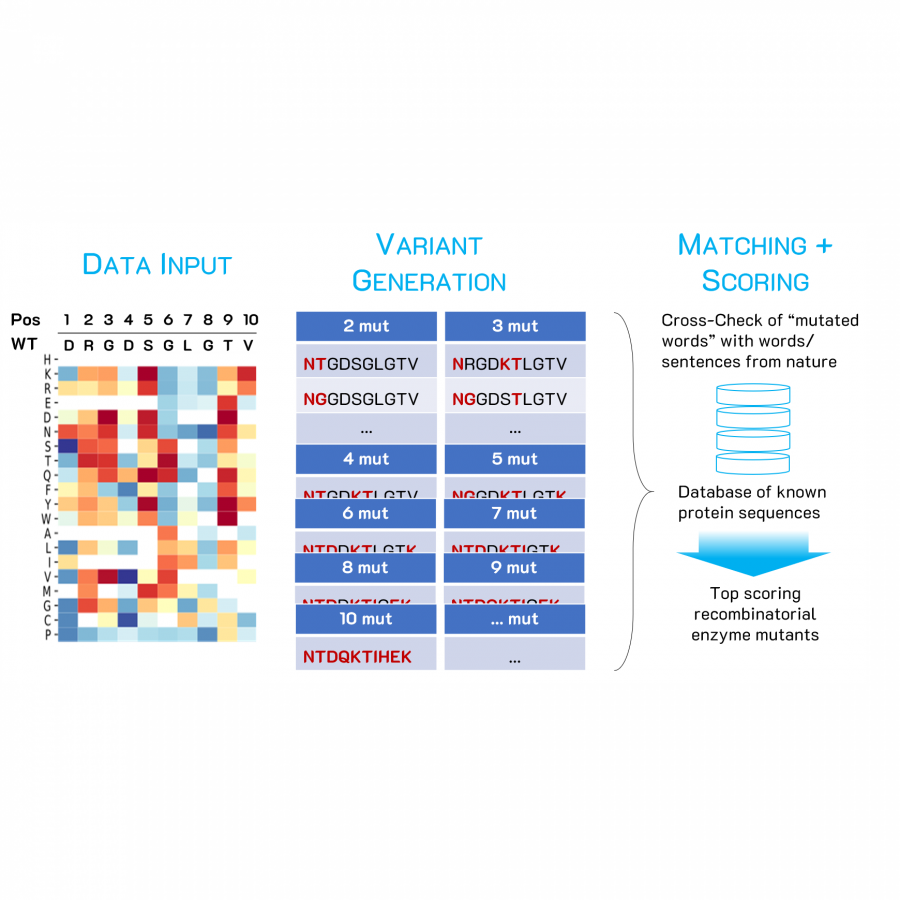 EnzyREC AI workflow in LLM framework Starting from the data input by EnzyMAP AI, EnzyREC AI first generates all beneficial recombinatorial variants and subsequently scores their folding probability against all protein sequences known in nature. |
#765 | One-step production and immobilization approach for unspecific peroxygenases |
|
| Presenting author: | Niklas TEETZ of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN |
| Other authors: | Selina LANG of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN Dirk HOLTMANN of UNIVERSITY OF APPLIED SCIENCES MITTELHESSEN |
| Topic: | Enzyme production, immobilization |
| Date: | 04:21 pm - 04:24 pm Session Pitch Talks - Chair : J.C. LEC, ARKEMA |
| Keywords: | unspecific peroxygenases / immobilization / cell surface display / |
| Purpose: | One-step production and immobilization approach for unspecific peroxygenases Niklas Teetz, Department of Bioprocess Intensification, THM Gießen, Department of Technical Biology, Karlsruhe Institute of Technology, Karlsruhe; Selina Lang, Department of Bioprocess Intensification, THM Gießen; Dirk Holtmann, Department of Bioprocess Intensification, THM Gießen, Department of Technical Biology, Karlsruhe Institute of Technology, Karlsruhe Unspecific peroxygenases (UPOs) are regarded as a “dream catalyst” [1] for a variety of selective oxyfunctionalization reactions like hydroxylations, epoxidations and oxygenations [2]. The catalyzed reactions are similar to those of the well-known P450 monooxygenases [3] but UPOs offer independence from reduced nicotinamide cofactors and electron transport chains by using the easily produced hydrogen peroxide as only cofactor [1]. Here, we present the display of the model UPO rAaeUPO (PaDaI) on the cell surface of the commonly used heterologous production host Pichia pastoris (Komagataella phaffii) as a 1-step production and immobilization approach. Multiple genes were cloned, combining the coding sequence for PaDaI with genes or part of genes coding for cell wall proteins from Saccharomyces cerevisiae. The genes were transformed into P. pastoris via an integration vector for production of the fusion proteins and subsequent comparison of the different systems among each other and with secreted, free PaDaI. All used systems yielded active UPOs, immobilized on the cell surface. Only minimal to no enzyme activity was detected in the supernatant, indicating a near complete surface display of the produced enzymes. One system in particular, a C-terminal fusion of PaDaI and SAG1, which is a glycoprotein involved in cell-cell contact during yeast mating, yielded identical ABTS activity per volume culture broth to the secreted PaDaI with ~90 % of the activity being in the cell pellet. The presented cell surface system of UPOs offers even easier downstream processing than systems with secreted UPOs do and already includes immobilization on a cheap, easily retainable and replacable matrix, that is the cells of the production host themselves. |
| References: | [1] Y. Wang et al, Current Opinion in Chemical Biology 2017, 37:1-9 [2] Hofrichter et al, Antioxidants 2022, 11, 163 [3] S. Bormann et al, Molecular Catalysis 2020, 492, 110999 |
| Figures: | 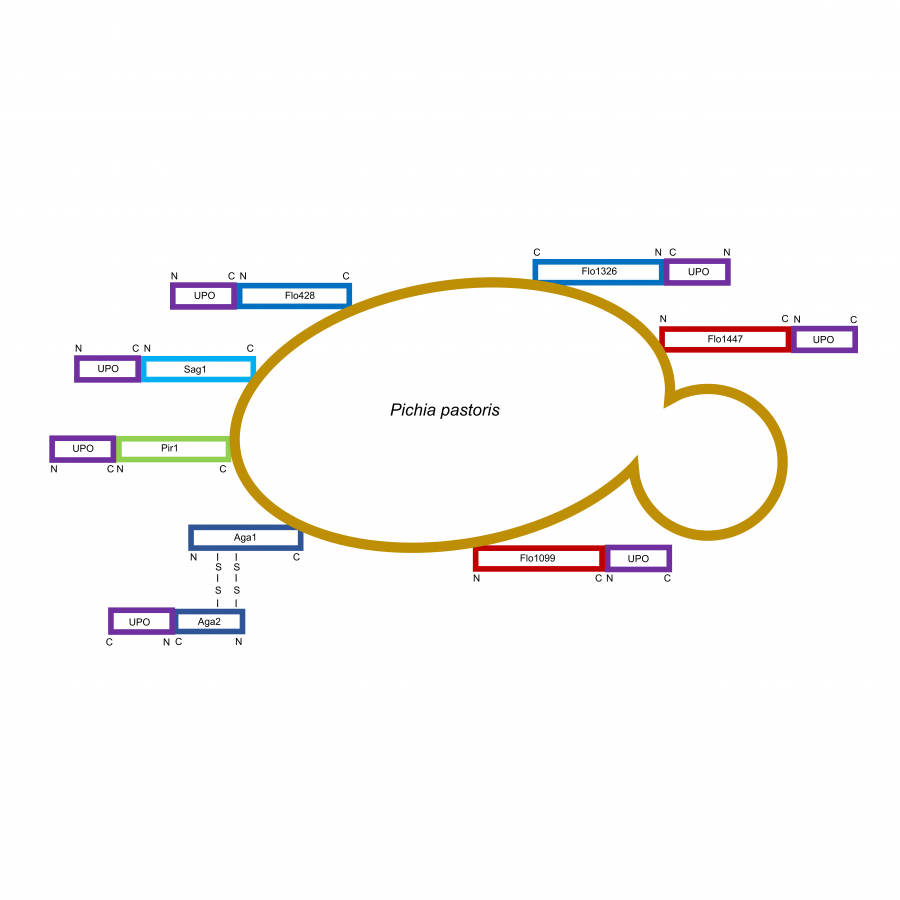 Scheme 1: Overview over the used cell surface display systems N: N-terminus
C: C-terminus
S--S: disulfide bond
SAG1: alpha-agglutinin
AGA1: anchoring subunit of a-agglutinin
AGA2: receptor binding subunit of a-agglutinin
PIR1: protein containing internal repeats
FLO: protein involved in flocculation 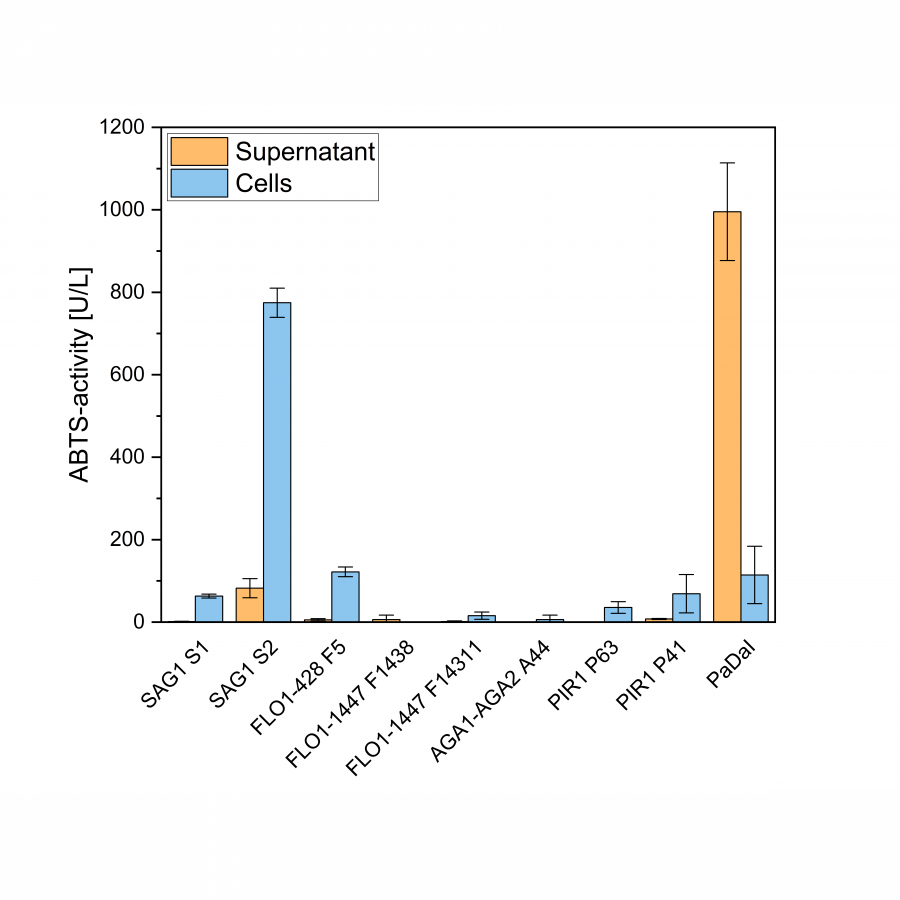 Comparison of ABTS-activity of different cell surface display systems and secreted PaDaI orange bars: activity in supernatant
blue bars: activity in cell pellet, resuspended in 100 mM KPi and normalized to initial culture volume |
#771 | Biocatalytic Utilization of the Gaseous Substrate Butane in a Bubble Column Reactor |
|
| Presenting author: | Paul BUBENHEIM of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF TECHNICAL BIOCATALYSIS |
| Corresponding author: | Andreas LIESE of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF TECHNICAL BIOCATALYSIS |
| Other authors: | Florian KELSCH of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF TECHNICAL BIOCATALYSIS Frederic PERZ of HAMBURG UNIVERSITY OF TECHNOLOGY, INSTITUTE OF TECHNICAL BIOCATALYSIS Hans-Georg HENNEMANN of EVONIK CREAVIS GMBH Felix NISSEN of EVONIK CREAVIS GMBH Dirk HOLTMANN of TECHNISCHE HOCHSCHULE MITTELHESSEN, INSTITUT FÜR BIOVERFAHRENSTECHNIK UND PHARMAZEUTISCHE TECHNOLOGIE Sebastian BORMANN of DECHEMA-FORSCHUNGSINSTITUT Frank HOLLMANN of TU DELFT, DEPARTMENT OF BIOTECHNOLOGY |
| Topic: | Industrial biocatalysis |
| Date: | 12:21 am - 12:24 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | Enzyme catalysis / Whole cell catalysis / / |
| Purpose: | Utilization of short chain alkanes is restrained by their low chemical reactivity, due to their structure of C-C and C-H bonds. However, this resource is abundant, easily accessible and can serve as a starting material for biocatalytic synthesis. For this reason two different promising approaches of biocatalytic reaction systems were investigated and characterized in a bubble column reactor for a highly selective activation of butane by monooxygenases: 1) Application of unspecific peroxygenases, AaeUPO[1], as a free enzyme catalysing the sub-terminal hydroxylation of butane to 2-butanol. As stoichiometric co-substrate only hydrogen peroxide is applied [2]. 2) A whole cell approach with the membrane bound alkBGT enzyme system expressed in E. coli[3]. The hydroxylation of butane to 1-butanol is catalysed with subsequent oxidation to the final product butyric acid. The selection of suitable process parameters with a design of experiment approach (e.g. butane and oxygen supply, glucose feed and biomass) can yield high productivities. However, detailed knowledge on the effect of individual process variables (e.g. gassing rate, butane content, pressure and temperature) is mandatory. |
| References: | [1] P. Molina-Espeja, E. Garcia-Ruiz, D. Gonzalez-Perez, R. Ullrich, M. Hofrichter, M. Alcalde, Applied and environmental microbiology 2014, 80, 3496, DOI: 10.1128/AEM.00490-14 [2] Perz F., Bormann S., Ulber R., Alcalde M., Bubenheim P., Hollmann F., Holtmann D., Liese A., 2020, ChemCatChem 2020, 12, 3666-3669, DOI: 10.1002/cctc.202000431 [3] G. Sluyter, J. Kleber, F. Perz, B. Grund, S. Leuchs, S. Sieberz, P. Bubenheim, O. Thum, A. Liese, Biochemical Engineering Journal 2020, 155, 107486, DOI: 10.1016/j.bej.2020.107486 |
| Figures: | 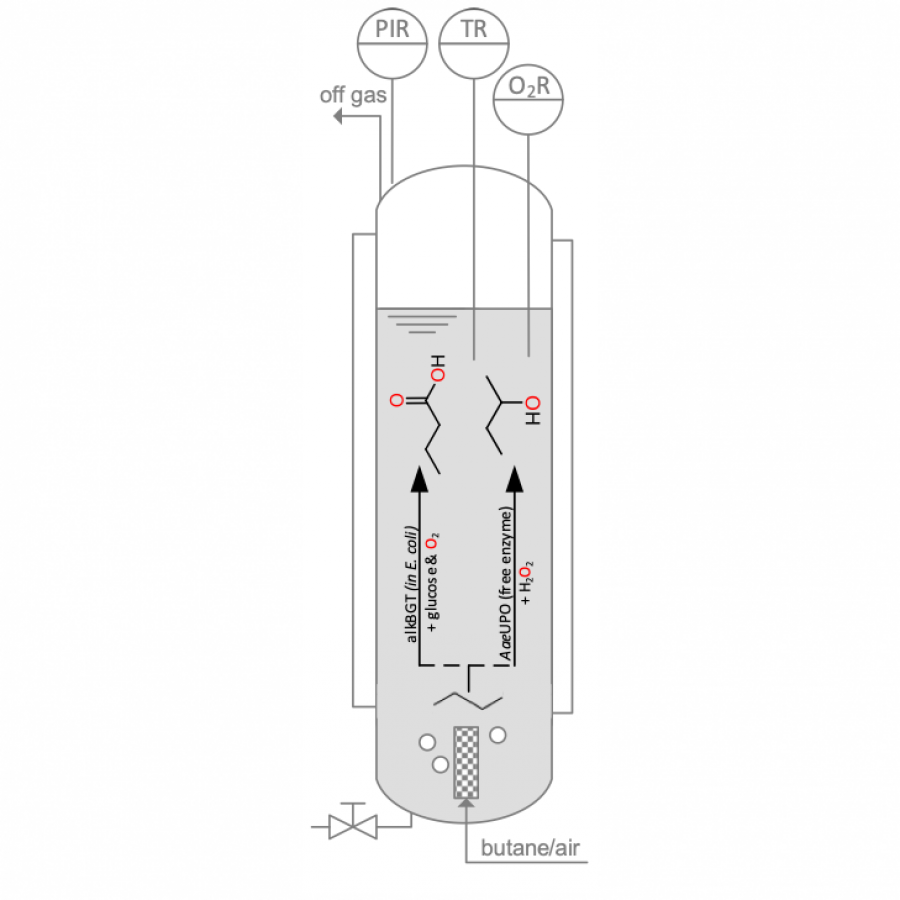 Figure 1 Scheme of the two applied reaction systems |
#844 | Data-driven protein engineering through machine learning models |
|
| Presenting author: | Mehdi D. DAVARI of LEIBNIZ INSTITUTE OF PLANT BIOCHEMISTRY HALLE |
| Other authors: | Alexander-Maurice ILLIG of RWTH AACHEN UNIVERSITY Niklas E. SIEDHOFF of RWTH AACHEN UNIVERSITY Ulrich SCHWANEBERG of RWTH AACHEN UNIVERSITY |
| Topic: | Artifical intelligence / computational methods |
| Date: | 10:45 am - 11:00 am Session AI & Computational methods - Chair : R. KOURIST, University of Technology of GRAZ |
| Keywords: | Protein engineering / machine learning / Data-driven / enzyme engineering |
| Purpose: | Protein engineering is nowadays used routinely in developing biocatalysts for biotechnological, biomedicine, and life sciences applications. Recently, data-driven strategies attracted attention in protein design and engineering because of advances in large experimental databanks of proteins, next-generation sequencing (NGS), high-throughput screening (HTS) methods, and the development of artificial intelligence algorithms [1-3]. However, the reliable prediction of beneficial amino acid substitutions, their combination, and effect on functional properties still pose significant challenges in machine learning assisted protein engineering [4]. In this presentation, we describe a general-purpose framework (PyPEF: Pythonic Protein Engineering Framework[5-6]) for data-driven protein engineering by combination of machine learning methods with signal processing and statistical physics techniques. PyPEF assist in the identification and selection of beneficial proteins of a given sequence space by either systematically or randomly exploring the fitness of variants and by sampling random evolution pathways. The predictive accuracy and throughput performance of PyPEF was evaluated based on four public protein and enzyme datasets using common regression models. It turns out that the PyPEF could efficiently predict the fitness of protein sequences for different target properties (predictive models with coefficient of determination values ranging from 0.58 to 0.92). By combining machine learning and protein evolution, PyPEF enabled the screening of proteins with various functions reaching a screening capacity of more than 500,000 protein sequence variants in the timeframe of only a few minutes on a standard PC. PyPEF exhibited significant accuracies on four public datasets (different proteins and properties) and underlined the potential of integrating data-driven technologies for covering different philosophies by either predicting the fitness of the variants to the highest accuracy or capturing the general trend of introduced mutations on the fitness in directed protein evolution campaigns. In essence, PyPEF provide a powerful solution to current sequence exploration and combinatorial problems present in protein engineering through exhaustive in silico screening of the protein sequence space. |
| References: | [1] N. E. Siedhoff, U. Schwaneberg and M. D. Davari, Methods in Enzymology 2020, 643, 281-315. [2] B. J. Wittmann, K. E. Johnston, Z. Wu and F. H. Arnold, Current Opinion in Structural Biology 2021, 69, 11-18. [3] K. K. Yang, Z. Wu and F. H. Arnold, Nature methods 2019, 16, 687-694. [4] S. Mazurenko, Z. Prokop and J. Damborsky, ACS Catalysis 2019, 10, 1210-1223. [5] N. E. Siedhoff, A.-M. Illig, U. Schwaneberg and M. D. Davari, Journal of Chemical Information and Modeling 2021. [6] A.-M. Illig, N. E. Siedhoff, U. Schwaneberg and M. D. Davari, bioRxiv 2022, 2022.2006. 2007.495081. |
#850 | A microfluidics-enabled workflow for rapid large-scale fitness data generation informs imine reductase engineering |
|
| Presenting author: | Maximilian GANTZ of UNIVERSITY OF CAMBRIDGE |
| Other authors: | Friederike NINTZEL of UNIVERSITY OF CAMBRIDGE Florian HOLLFELDER of UNIVERSITY OF CAMBRIDGE |
| Topic: | Enzyme discovery and engineering |
| Date: | 12:18 am - 12:21 am Session Pitch Talks - Chair : A. ZAPARUCHA, University of Evry |
| Keywords: | / / / |
| Purpose: | Directed evolution is the method of choice for optimizing biocatalysts but is often time consuming and its success is unpredictable. To meet the fast pace of product development in industry the turnaround time of engineering enzymes needs to become shorter. Directed evolution can be especially slow and challenging when fitness peaks are rare or poorly accessible in sequence space. Here we show that the availability of large-scale sequence–function data in enzymes relevant to biocatalysis can guide and thereby accelerate enzyme engineering. As a model system we chose an imine reductase (IRED) from S. roseum (SrIRED) catalysing reductive amination. To rapidly assess the fitness of >10000 IRED variants, we developed a cheap and versatile absorbance-activated droplet sorting-based ultra-high throughput screening setup. Coupled to a novel deep sequencing protocol combining next generation- and long-read-sequencing, this workflow allows the generation of large-scale fitness data using unbiased mutagenesis by error-prone PCR and the resolution of epistasis in less than 2 weeks. Using this novel workflow hot spots for engineering substrate specificity in the active site and at the periphery of the enzyme were identified. We used higher-order mutation data to score the combinability of individual improving mutations and rationally engineered highly active variants (10-fold improved kcat/KM) displaying positive epistasis showing that our workflow can give rapid access to highly improving variants. Due to its versatility and broad applicability, we envision that this workflow will become a valuable part of the protein engineer’s toolkit for rapid enzyme engineering. |
#854 | CaADH, An Enzyme with Broad Substrate Promiscuity, Yet Stereochemical Fidelity - An Efficient DYRKR Entry Into Myriad Taxoid Side Chains |
|
| Presenting author: | David BERKOWITZ of UNIVERSITY OF NEBRASKA |
| Other authors: | Gaurav KUDALKAR of UNIVERSITY OF NEBRASKA Nivesh KUMAR of UNIVERSITY OF NEBRASKA Jared HASS of UNIVERSITY OF NEBRASKA Peter MADZELAN of UNIVERSITY OF NEBRASKA Wei NIU of UNIVERSITY OF NEBRASKA Mark WILSON of UNIVERSITY OF NEBRASKA |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 12:15 am - 12:30 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | dynamic reductive kinetic resolution / hybrid biocatalysis/cross-coupling / Taxotere analogues / CaADH |
| Purpose: | Our laboratory has heterologously expressed a Clostridium acetobutylicum alcohol dehydrogenase (CaADH) enzyme that, in our hands, displays remarkable substrate promiscuity and yet also quite impressive stereochemical fidelity. This presentation will focus on the deployment of this enzyme toward the construction of twenty potential "aryl isoserine" side chains for the Taxotere family of tubulin-binding chemotherapeutics. The approach involves dynamic reductive kinetic resolution (DYRKR) upon the corresponding alpha-chloro-beta-keto esters. The enzyme displays both a distinct preference for the (S)-stereochemistry at the alpha-C-Cl bond, essentially deracemizing the substrate, while also exhibiting very high facial selectivity for carbonyl reduction, providing the D-stereochemistry almost exclusively at the new beta-C-OH center. The active site of the enzyme has been probed with both heteroaryl and aryl substrates bearing a panoply of both electron-donating and electron-withdrawing substituents. In addition, the ability of the enzyme to take fused bicyclic arene systems has been examined with both sp2- and sp3-type fusions being investigated. A half-dozen of the enzymatic products are taken to the final aryl isoserine side chains in three steps. The addition of a range of cross-coupling chemistries performed upon the p-bromophenyl isoserine side chain greatly enhances the structural diversity and functional group breadth that can be introduced into these taxoid side chains, and highlights the value of such hybrid biocatalysis/cross-coupling approaches in medicinal chemistry/chemical biology. This presentation will also describe the first crystal structure of the CaADH enzyme. The structure reveals two particularly compelling features: (i) the nicotinamide cofactor is bound in the anti-conformation with the amide carbonyl of the cofactor occupying the ketone binding pocket and thus giving important insight on substrate binding and (ii) the structure reveals a flexible loop near the active site which likely underlies the remarkable substrate promiscuity exhibited by CaADH. We will present our view of the dynamic nature of the enzyme active site through molecular dynamics simulation and discuss potential structural origins of the remarkable (S)-enantioselectivity and D-facial selectivity displayed by this exceptionally useful short chain dehydrogenase enzyme. |
| Figures: |  DYRKR/Cross-Coupling Entry into Taxoid Side Chains Depiction of the hybrid biocatalysis/cross-coupling entry into novel taxoid side chains with control of both absolute and relative stereochemistry |
#892 | Regioselective Ring-Opening Reactions with Non-Natural Substrates using Engineered Halohydrin Dehalogenase |
|
| Presenting author: | Xinhang YANG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO |
| Corresponding author: | Haibin CHEN of ENZYMASTER (NINGBO) BIO-ENGINEERING CO |
| Other authors: | Xiao LUO of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Junxia ZHAO of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Kuifang HE of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Qinli PENG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Ruimei HONG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Marco BOCOLA of ENZYMASTER DEUTSCHLAND GMBH Neha VERMA of ENZYMASTER DEUTSCHLAND GMBH Hao YANG of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Fenshai SUN of ENZYMASTER (NINGBO) BIO-ENGINEERING CO Osama MAHMOUD of ENZYMASTER DEUTSCHLAND GMBH Thomas DAUSSMANN of ENZYMASTER DEUTSCHLAND GMBH |
| Topic: | Enzyme discovery and engineering |
| Date: | 11:45 am - 12:00 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | Epoxides / Halohydrin dehalogenase / (S)-4-phenyl-2-oxazolidinone / Ring-opening |
| Purpose: | Epoxides are important building blocks for chemical and pharmaceutical synthesis [1], and many of their ring-opening derivatives have been used as chiral building blocks or auxiliary agents such as chiral diols [2], halohydrins, glycinols [3], and Evans-type auxiliaries [4]. The conventional methods for epoxide ring-opening suffers from poor regioselectivity, which poses a challenge for the direct utilization of eopxide derivatives. The oxazolidines are well known as Evans-type auxiliaries in the organic synthesis since 1981. A lot of medicine were developed by using oxazolidines as starting materials [4]. In industry, expensive amino acids are traditionally used as starting points for the above-mentioned chiral building blocks whose production involves NaBH4 reductions, phosgene/ethyl chloroformate cyclizations, and high pollutant emissions like hydrogen chloride, hydrogen sulfide, and methylbenzene [5,6]. These traditional approaches result in higher costs and pollution compared to epoxide dervatives [7–9]. Halohydrin dehalogenases (HHDH) are industrially relevant enzymes that catalyze the reversible dehalogenation of vicinal haloalcohols, yielding the corresponding epoxides [10]. In the reverse reaction, non-native nucleophiles like NaOCN [11] and NaSCN [12] may lead to the enzymatic SN2 ring-opening and spontaneous ring re-closing processes for desired products such as oxazolidines. As a result, such non-native reactions would give 100% conversion. In recent research, many HHDHs were discovered to accept various non-native nucleophiles for ring-opening (Figure 1) [13]. Most of the ring opening reactions were triggered by nucleophiles at the beta carbon position (beta type), providing secondary alcohols as final products [3]. In nature, however, several HHDHs exhibit special regioselective alpha ring-opening reactions (alpha type), producing a new series of heterocyclic products [11]. Unfortunately, like other traditional catalysts for chemical ring opening [14], most of the wild-type (WT) enzymes had insufficient regio- and stereo-selectivity towards the desired alpha ring-opening products, leaving a large number of beta ring-opening by-products. At the same time, the process efficiency of HHDH-catalyzed ring-opening reactions is quite poor due to HHDHs’ poor substrate tolerance and low catalyitc acitivity. Here, a novel HHDH has been engineered by computer-aided direct enzyme evolution in our enzyme engineering lab which enables highly regio- and stereo-selective synthesis of Evans-type auxiliary reagents and other chiral glycinols at high substrate loading. For our engineered HHDH variants, their substrate tolerance has been improved by 100fold [144 g/L] for full conversion, while the alpha ring-opening products have excellent chirality (ee >99%) with minimum by-products (<1%). Until now, our best HHDH variants were well suited for the SN2 ring-opening reactions with (R)-styrene oxide, giving 4-phenyl-2-oxazolidinone and (S)-styrene oxide, both in high enantiopurity. From our research, a panel of HHDH variants was developed to enable chemoenzymatic synthesis of a series of high-value products, including chiral glycinols, halohydrins, epichlorohydrins and mandelic acid. These enzymatic synthesis routes will provide more economic and environmental-friendly technologies to the chemical industry. |
| References: | (1) Moschona, F.; Savvopoulou, I.; Tsitopoulou, M.; Tataraki, D.; Rassias, G. Epoxide Syntheses and Ring-Opening Reactions in Drug Development. Catalysts 2020, 10 (10), 1117. https://doi.org/10.3390/catal10101117. (2) Kamble, M. P.; Yadav, G. D. Biocatalytic Resolution of (R,S)-Styrene Oxide Using a Novel Epoxide Hydrolase from Red Mung Beans. Catalysis Today 2018, 309, 236-241. https://doi.org/10.1016/j.cattod.2017.06.013. (3) Wang, H.-H.; Wan, N.-W.; Miao, R.-P.; He, C.-L.; Chen, Y.-Z.; Liu, Z.-Q.; Zheng, Y.-G. Identification and Structure Analysis of an Unusual Halohydrin Dehalogenase for Highly Chemo-, Regio- and Enantioselective Bio-Nitration of Epoxides. Angewandte Chemie International Edition 2022, 61 (37), e202205790. https://doi.org/10.1002/anie.202205790. (4) Heravi, M. M.; Zadsirjan, V.; Farajpour, B. Applications of Oxazolidinones as Chiral Auxiliaries in the Asymmetric Alkylation Reaction Applied to Total Synthesis. RSC Adv. 2016, 6 (36), 30498-30551. https://doi.org/10.1039/C6RA00653A. (5) Jiqiu Ren. An method of synthesis of 4-phenyl-2-oxazolidinone in microchannel reactor. CN111995593A, November 27, 2020. https://patents.google.com/patent/CN111995593A/en?oq=CN+111995593+A (accessed 2023-03-09). (6) Feihong Ma; Zhengxin Cha; Jinfeng Du; Zaipei Tan. Preparation Method of (S) -4-Phenyl-2-Oxazolidinone. CN112500361A, March 16, 2021. https://patents.google.com/patent/CN112500361A/en (accessed 2023-03-09). (7) Diekema, D. I.; Jones, R. N. Oxazolidinones: A Review. Drugs 2000, 59 (1), 7-16. https://doi.org/10.2165/00003495-200059010-00002. (8) Shaw, K. J.; Barbachyn, M. R. The Oxazolidinones: Past, Present, and Future. Ann N Y Acad Sci 2011, 1241, 48-70. https://doi.org/10.1111/j.1749-6632.2011.06330.x. (9) Pulla, S.; Felton, C. M.; Ramidi, P.; Gartia, Y.; Ali, N.; Nasini, U. B.; Ghosh, A. Advancements in Oxazolidinone Synthesis Utilizing Carbon Dioxide as a C1 Source. Journal of CO2 Utilization 2013, 2, 49-57. https://doi.org/10.1016/j.jcou.2013.07.005. (10) Fauzi, A. M.; Hardman, D. J.; Bull, A. T. Biodehalogenation of Low Concentrations of 1,3-Dichloropropanol by Mono- and Mixed Cultures of Bacteria. Appl Microbiol Biotechnol 1996, 46 (5), 660-666. https://doi.org/10.1007/s002530050877. (11) Wan, N.; Tian, J.; Zhou, X.; Wang, H.; Cui, B.; Han, W.; Chen, Y. Regioselective Ring-Opening of Styrene Oxide Derivatives Using Halohydrin Dehalogenase for Synthesis of 4-Aryloxazolidinones. Advanced Synthesis & Catalysis 2019, 361 (20), 4651-4655. https://doi.org/10.1002/adsc.201900786. (12) Ma, R.; Hua, X.; He, C.-L.; Wang, H.-H.; Wang, Z.-X.; Cui, B.-D.; Han, W.-Y.; Chen, Y.-Z.; Wan, N.-W. Biocatalytic Thionation of Epoxides for Enantioselective Synthesis of Thiiranes. Angewandte Chemie International Edition 2022, 61 (52), e202212589. https://doi.org/10.1002/anie.202212589. (13) Schallmey, A.; Schallmey, M. Recent Advances on Halohydrin Dehalogenases—from Enzyme Identification to Novel Biocatalytic Applications. Appl Microbiol Biotechnol 2016, 100, 7827-7839. https://doi.org/10.1007/s00253-016-7750-y. (14) Mohseni, S.; Bakavoli, M.; Morsali, A. Theoretical and Experimental Studies on the Regioselectivity of Epoxide Ring Opening by Nucleophiles in Nitromethane without Any Catalyst: Nucleophilic-Chain Attack Mechanism. Progress in Reaction Kinetics and Mechanism 2014, 39 (1), 89-102. https://doi.org/10.3184/97809059274714X13874723178403. |
| Figures: |  Figure 1 Reaction scopes of epoxides driven by HHDH and epoxide hydrolase  Figure 2 Scheme of enzymatic synthesis of 4-phenyl-2-oxazolidinone from styrene and NaOCN |
#929 | Oriented immobilization of ferredoxin NADP+ reductase by site-directed exchanged cysteine residues. |
|
| Presenting author: | Juan Cruz ALMADA of UNIVERSIDAD DE ZARAGOZA |
| Corresponding author: | Susana VELASCO LOSANO of UNIVERSIDAD DE ZARAGOZA |
| Other authors: | Yasmine KADRI of UNIVERSIDAD DE ZARAGOZA Cristina ALIERTA of UNIVERSIDAD DE ZARAGOZA |
| Topic: | Enzyme production, immobilization |
| Date: | 11:30 am - 11:45 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | ferredoxin NADP+ reductase / immobilization / cofactor recycling / direct electron transfer |
| Purpose: | The cost-efficient production of sustainably derived biochemicals and biofuels by redox-biosynthetic pathways, is a critical goal of modern biotechnology for establishing a bio-based economy. Moreover, oxidoreductases, which are the second largest class of enzymes [1], are especially valuable in the production of pharmaceutical compounds due to their unique enantioselectivity [2]. However, these enzymes require stoichiometric amounts of expensive nicotinamide cofactors hampering its exploitation at large scale applications. To solve this limitation, many efforts have been described aiming at the sustainable regeneration and recycling of redox cofactors; where bioelectrocatalytic approaches have harnessed the direct electron transfer (DET) capability of electroactive enzymes to achieve this goal [3]. During photosynthesis, ferredoxin NADP+ reductase (FNR) is the main flavoenzyme responsible for the regeneration of NADPH. The catalytic ability of this enzyme has been exported outside the photosynthetic cell where it has been attached to an electrode surface thus managing the reversible interconversion of NADP+ to NADPH by directly receiving the injected electrons [4]. When confined on the electrode, the orientation of FNR plays a crucial role on the effective DET process [5], where the cofactor must be located at a compatible distance from the electro-active surface to accomplish an effective electron transfer. To this aim, FNR has been immobilized on functionalized gold electrodes through an oriented immobilization strategy by introducing genetically engineered metal binding sites (histidine clusters) on selected regions of the protein surface, yielding a reversible anchoring of the enzyme through metal chelation. However, such enzyme-electrode binding chemistry requires previous functionalization of the electrode gold surface with a self-assembled monolayer of thiols displaying nitrilotriacetic acid groups complexed with metal transition ions [6]. Therefore, in this work, we propose a direct and straightforward strategy allowing the site-directed immobilization of FNR on bare gold electrodes by introducing mono and multi cysteine residues on strategic positions along FNR’s surface, thus attaining an irreversible enzyme-electrode attachment. To this aim we introduced three single mutations and several combinations of these mutations allowing the location of the catalytic site of the FNR at less than 14 Å apart from the active electrode surface. We have performed the genome mining, cloning, heterologous expression and purification of a functional FNR from the cyanobacteria Anabaena sp. PCC7119 as a model flavoenzyme. Finally, we evaluated the Michaelis-Menten kinetics of the cys-variants in solution and when working confined on a gold electrode surface, where only two of the designed mutants achieve a direct electron transfer with the gold electrode. This work provides one step forward in the incorporation of oxidoreductases in the pool of industrially available biocatalytic tools. |
| References: | [1] Zhou, S., & Wang, L. Unraveling the structural and chemical features of biological short hydrogen bonds. Chemical Science, 2019, 10(33), 7734-7745. [2] Martínez, A. T., Ruiz-Dueñas, F. J., Camarero, S., Serrano, A., Linde, D., Lund, H., … & Alcalde, M. Oxidoreductases on their way to industrial biotransformations. Biotechnology advances, 2017, 35(6), 815-831. [3] Armstrong, F. A.; Cheng, B.; Herold, R. A.; Megarity, C. F.; Siritanaratkul, B., From Protein Film Electrochemistry to Nanoconfined Enzyme Cascades and the Electrochemical Leaf. Chem. Rev. 2022. [4] Megarity, C. F.; Siritanaratkul, B.; Heath, R. S.; Wan, L.; Morello, G.; FitzPatrick, S. R.; Booth, R. L.; Sills, A. J.; Robertson, A. W.; Warner, J. H.; Turner, N. J.; Armstrong, F. A. Electrocatalytic Volleyball: Rapid Nanoconfined Nicotinamide Cycling for Organic Synthesis in Electrode Pores. Angewandte Chemie International Edition, 2019, 58 (15), 4948-4952. [5] Reginald, S. S., Lee, H., Fazil, N., Sharif, B., Lee, M., Kim, M. J., ... & Chang, I. S. Control of carbon monoxide dehydrogenase orientation by site-specific immobilization enables direct electrical contact between enzyme cofactor and solid surface. Communications Biology, 2022, 5(1), 390. [6] Madoz-Gurpide, J., Abad, J. M., Fernandez-Recio, J., Velez, M., Vazquez, L., Gomez-Moreno, C., & Fernandez, V. M. Modulation of electroenzymatic NADPH oxidation through oriented immobilization of ferredoxin: NADP+ reductase onto modified gold electrodes. Journal of the American Chemical Society, 2000, 122(40), 9808-9817. |
#935 | Debottlenecking of multi-enzyme cascades with data-driven optimization tools |
|
| Presenting author: | Katrin ROSENTHAL of CONSTRUCTOR UNIVERSITY |
| Other authors: | Regine SIEDENTOP of TU DORTMUND UNIVERSITY Martin BECKER of TU DORTMUND UNIVERSITY Maximilian SISKA of FORSCHUNGSZENTRUM JÜLICH Eric VON LIERES of FORSCHUNGSZENTRUM JÜLICH Stephan LÜTZ of TU DORTMUND UNIVERSITY of |
| Topic: | Biocatalytic cascade reactions |
| Date: | 11:45 am - 12:00 am Session AI & Computational methods - Chair : R. KOURIST, University of Technology of GRAZ |
| Keywords: | enzyme cascade / kinetic models / Bayesian optimization / one-pot synthesis |
| Purpose: | The combination of several enzymes in multi-step reaction cascades enables the synthesis of complex molecules with often shorter reaction pathways compared to chemical routes, thus supporting the development of ecologically sustainable processes [1]. Multi-enzyme reactions have even more advantages such as the shift of the equilibrium towards the product side, no intermediate isolation, and the synthesis of complex molecules in one reaction pot. Recently, several cascades have been developed that led to efficient synthetic pathways that could be of great relevance for the synthesis of anti-viral and anti-cancer compounds [2]. Among these compounds are cyclic dinucleotides, for which we have developed an enzyme cascade that catalyzes the synthesis of cyclic GMP-AMP (cGAMP) [3]. The cascade consists of four enzymes, namely an adenosine kinase and two polyphosphate kinases for ATP synthesis from adenosine, coupled with cyclic GMP-AMP synthase (cGAS), which catalyzes the formation of cGAMP. The cascade was rationally optimized in an iterative manner by adjusting the enzyme concentrations. It was found that minor changes can lead to improvement but also failure of the cascade, illustrating the difficulty of optimizing enzyme cascades. The optimization of an enzyme cascade means that cooperative effects between several design parameters must be taken into account, which involves a high experimental effort. One way to overcome the challenge of optimizing such multi-parameter systems is to combine in silico models with experimental design, model calibration and validation (Figure 1). We have therefore investigated computational methods to increase the efficiency of the optimization process. For example, to identify bottlenecks of a complex cascade involving ten enzymes for the synthesis of farnesyl pyrophosphate from acetate, a combination of in silico modeling and wet-lab experiments was applied [4]. The goal was to identify the limiting steps and subsequent adaptation of the enzyme cascade composition. Using a kinetic model, bottlenecks and performance-influencing parameters were identified. Subsequent wet-lab experiments validated their relevance for the cascade's performance. However, the more complex the cascade becomes, the more interactions take place between the reaction partners, which are increasingly difficult to describe by mechanistic models. We therefore also applied empirical surrogate models for Bayesian optimization [5]. As a proof-of-concept system, we have used a comparatively small cascade of three enzymes that catalyze the phosphorylation of mevalonate to mevalonate phosphate, including the synthesis and regeneration of ATP. Bayesian optimization was used to adapt the enzyme and co-substrate concentrations to improve the synthesis rate of mevalonate phosphate. With this approach, we were able to iteratively optimize the initial composition of the enzyme cascade components to increase the product synthesis rate to 10.2 µmol/min/mg that even exceeded the results of the reference reaction with stoichiometrically added ATP by 16%. Simultaneously, the product concentrations were improved so that high yields were achieved. In summary, the application of these data-driven optimization methods in combination with experiments helps to identify bottlenecks and limiting parameters of multi-enzyme cascades, taking cooperative effects into account. |
| References: | [1] Siedentop R., Rosenthal K., Int. J. Mol. Sci. 2022, 23(7), 3605. [2] Rosenthal K., Bornscheuer U., Lütz S., Angew. Chem.Int. Ed. 2022, 61, e2022083. [3] Becker M., Nikel P., Andexer J., Lütz S., Rosenthal K., Biomolecules 2021, 11(4), 590. [4] Siedentop R., Dziennus M., Lütz S., Rosenthal K., Chem. Ing. Tech. 2023, 95(4), 1-7. [5] Siedentop R., Siska M., Möller N., Lanzrath H., von Lieres E., Lütz S., Rosenthal K., Catalysts 2023, 13(3), 468. |
| Figures: | 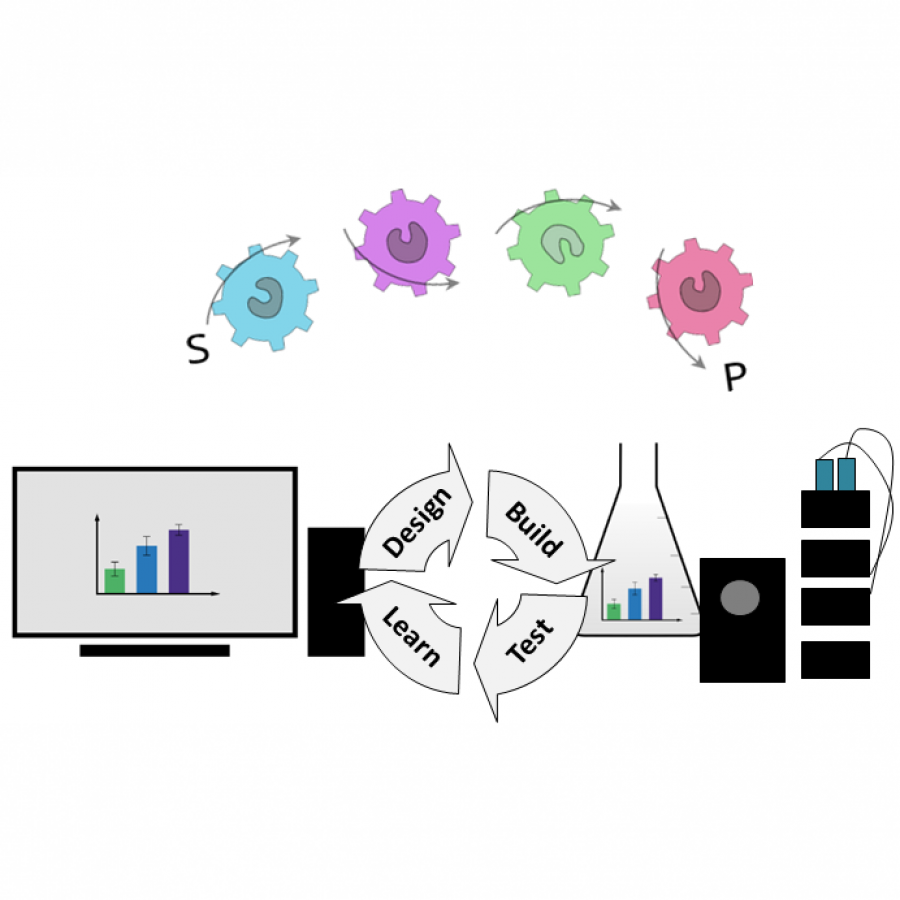 Figure 1 Figure 1: Design-Build-Test-Learn cycle to optimize multi-enzyme cascades with a combination of models and experiments. |
#946 | Selecting better biocatalysts by complementing recoded bacteria |
|
| Presenting author: | Clemens MAYER of UNIVERSITY OF GRONINGEN |
| Topic: | Enzyme discovery and engineering |
| Date: | 10:45 am - 11:00 am Session Enzyme engineering & Discovery #1 Chair : D. ROTHER, University of Jülich |
| Keywords: | directed evolution / in vivo selection / genetic-code expansion / enzyme engineering |
| Purpose: | Assaying enzymatic activities often proves a critical bottleneck for the directed evolution of useful biocatalysts. Commonly-employed, multi-well screens that evaluate enzyme variants one-by-one are laborious and slow when compared to nature’s approach to select improved variants from large populations through adaptation to a selection pressure. To apply analogous selections for the directed evolution of industrially-useful biocatalysts, I will present an in vivo directed evolution platform that leverages recoded organisms addicted to non-canonical amino acids (ncAAs) to evolve biocatalysts that can provide these building blocks from synthetic precursors.[1] Repurposing recoded organisms to link enzymatic activities to bacterial proliferation requires the introduction of three readily-available genetic components into Escherichia coli (see Figure 1A): (1) an enzyme able to convert an appropriate precursor to a ncAA (=input); (2) an orthogonal translation system that enables the site-selective incorporation of this ncAA (=sensor); and (3) a β-lactamase featuring an in-frame stop-codon, whose activity to degrade the hydrolytically-stable penicillin derivative, carbenicillin, is strictly dependent on the incorporation of the same ncAA. Critically, in our selection platform, the growth rates of bacteria in presence of carbenicillin and the synthetic precursor correlate with the activities of the enzyme they produce. As such, improved biocatalysts are readily identified by subjecting bacteria harboring vast enzyme libraries to continuous growth-dilution cycles in presence of increasing carbenicillin concentrations (=selection pressure, Figure 1B). In my talk, I will showcase how our selection platform can be employe for the directed evolution of hydrolases as well as biocatalysts that catalyze C-H activation or C-C-bond-forming reactions. Lastly, I will discuss how our platform that requires minimal human intervention and no specialized equipment will enable the autonomous exploration of many evolutionary trajectories in a continuous fashion. |
| References: | [1] Rubini, R., Jansen, S. C., Beekhuis, H., Rozeboom, H. J., Mayer, C., Angew. Chem. Int. Ed. 2023, 62, e202213942; Angew. Chem. 2023, 135, e202213942. |
| Figures: |  Principles of selecting better biocatalysts by complementing recoded bacteria A: Blueprint of our directed evolution platform that functions by complementation of recoded bacteria. B: Selection of improved biocatalysts from vast libraries by serial passaging. |
#960 | Self-Assembling Enzyme Materials and Additive Manufacturing for Flow Biocatalysis |
|
| Presenting author: | Kersten RABE of KARLSRUHE INSTITUTE OF TECHNOLOGY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 12:15 am - 12:30 pm Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | flow biocatalysis / enzyme immobilization / thermostabilization / additive manufacturing |
| Purpose: | Applications employing catalysts in fluidic, cascaded setup are becoming increasingly relevant.[1] In order to also incorporate biocatalysts in such setups, efficient immobilization strategies are in demand. Employing the genetically encoded, covalent SpyTag/SpyCatcher (ST/SC) system, we have recently developed an all-enzyme hydrogel (Figure 1), which can be prepared from many enzymes, enabling the continuous conversion of a wide variety of substrates.[2-5] In such carrier-free formulations the enzymes themselves form a material, which for example stabilizes the enzymes and also offer the retention of expensive cofactors such as NAD(P)H. Depending on the application, the same immobilization strategy using the ST/SC system can also be used to immobilize enzymes onto epoxy-modified beads[6,7] or fully biogenic SC-modified magnetosomes.[7] In all cases the biocatalytic conversions in the resulting flow reactors converted the corresponding substrates continuously for several days with high efficiency. As an alternative approach, biocatalysts can also be implemented in flow biocatalysis employing additive manufacturing. Such bioprinting applications call for thermotolerant organisms and proteins. We have recently showed reported how to employ guided protein evolution for engineering biocatalysts for such applications.[8-10] In this context new methods using correlation analysis and machine learning have been developed which greatly aid the identification of thermostable enzymes.[11, 12] With this approach, we have successfully evolved enzyme variants that reveal significantly increased stability and activity at elevated temperatures. These improved variants along with naturally thermostable proteins were then used for direct 3D printing of bioinks to manufacture reaction modules (Figure 2).[8,9] These modules can be produced on demand from bioinks, which are stable over weeks inside the printing cartridge. Reactor modules containing different enzymes can be arranged into cascades and show a tunable behavior. Different enzyme classes have been successfully utilized including alcohol dehydrogenases, esterases, ketoisovalerate decarboxylases, phenolic acid decarboxylases and benzaldehyde lyases. |
| References: | [1] Rabe, K. S. et al. Angew Chem 2017, 56 (44), 13574-13589. [2] Peschke, T. et al Angew Chem 2018, 30 (52), 17274-17278 [3] Bitterwolf, P. et al Chemical Science 2018, 10 (42), 9752-9757 [4] Peschke, T. et al Catalysts 2019, 9 (2), 164 {5] Mittmann E. et al Micromachines 2019, 10 (12), 795 [6] Peng, M. et al Chembiochem 2021, 23 (2), e202100472 [7] Mittmann, E. et al ACS Applied Materials & Interfaces 2022, 14 (19), 22138-22150 [8] Maier, M. et al Angew Chem 2018, 57 (19), 5638-5642. [9] Peng, M. et al Chemistry-A European Journal 2019, 25, 15998-16001 [10] Buss, O. Chembiochem 2018, 19 (4), 379-387. [11] Peng, M. et al. Biological Chemistry 2019, 400 (11), 1519-1527 [12] Gang, L.; Rabe, K. S.; Nielsen, J.; Engqvist, M. K. M. ACS Synthetic biology 2019, 8, 6, 1411-1420 |
| Figures: |  All-enzyme hydrogel formation Genetically encoded ST/SC system enables the formation of defined all-enzyme hydrogels.  3D printing of biocatalytic reaction modules Thermostabile enzyme allow the direct printing of biocatalytic reaction modules. |
#966 | On demand activity regulation of enzymes in one-pot multi-step catalysed processes using online analytics, automated feeding and light induced enzyme inactivation |
|
| Presenting author: | Dörte ROTHER of RESEARCH CENTER JÜLICH |
| Other authors: | Stephan MALZACHER of RESEARCH CENTER JÜLICH Tim GERLACH of RESEARCH CENTER JÜLICH Douglas WEBER of RESEARCH CENTER JÜLICH Christiane CLAASSEN of RESEARCH CENTER JÜLICH Morten M. C. H. VAN SCHIE of RESEARCH CENTER JÜLICH Thomas DREPPER of HEINRICH-HEINE UNIVERSITY DÜSSELDORF |
| Topic: | Biocatalytic cascade reactions |
| Date: | 11:15 am - 11:30 am Session Cascade reaction - Chair : C. PAUL, Technology University of Delft |
| Keywords: | activity regulation / multi-enzyme catalysed processes / automation / cross-reactivity prevention |
| Purpose: | There is an urgent need for the development of greener syntheses procedures if we want to maintain an environment worth living in and keep a high standard in material comfort (or reach a higher one in developing countries). The establishment of more biocatalytic steps in chemical syntheses is one possible solution, as enzymes and whole cells offer sustainable advantages, such as biodegradability, intoxicity, high selectivity, and many more. As a myriad of enzymatic reactions exist for almost any product, their potential is immense. Great scientific achievements and new techniques have enabled the design of economically and ecologically feasible one-step and multi-step enzyme catalysed reactions. However, with these new opportunities, also new challenges arise. The more enzyme steps are combined, the higher the risk of undesired cross-reactivity. Spatial or temporal separation can solve this problem. In the 'LightCas' project, we are investigating the possibilities of avoiding cross-reactivity in one-pot systems by separating the reaction steps over time. Using sequential enzyme addition and on-demand light-induced enzyme inactivation [1] each step can be turned on and off individually. To achieve selective inactivation of the enzymes, a genetically encoded photosensitizer is coupled to the enzymes. Exposure to (blue) light produces reactive oxygen species. The most suitable photosensitizers produce predominantly singlet oxygen. Due to the short lifetime of these reactive oxygen species, only the bound enzyme is inactivated, but not other enzymes in the same reaction vessel. In combination with suitable online analytics (benchtop NMR) [2], we have exemplarily operated a three-stage light-controlled single-pot enzyme reactor for the production of tetrahydroisoquinolines [3] from cheap (renewable) substrates. The spatially resolved activity control makes it possible to obtain the desired product in high purity even in a one-pot system. Furthermore, we have recently achieved our ultimate goal of a technically self-controlled process in which an automated and FAIR data principles based data collection process is implemented. Time-resolved online concentration determination of substrates, intermediates and products allowed each enzymatic step to be automatically started and stopped upon completion without the need for manual intervention. As long as NMR-signal separation is achieved, this method can in principle be used for the optimisation and control of other one-pot reaction systems. |
| References: | [1] Gerlach T, Schain J, Söltl S, van Schie M M C H, Hilgers F, Bitzenhofer N L, Drepper T, Rother D. 2022. Frontiers Catal. 2:835919; Gerlach T, Nugroho D L, Rother D. 2021. ChemCatChem 13: 2398-2406 [2] Claaßen C, Mack K, Rother D. 2020.. ChemCatChem. 12 (4): 1190-119 [3] Erdmann V, Lichman B R, Zhao J, Simon R C, Kroutil W, Ward J M, Hailes H C, Rother D. 2017. Angew. Chem. Int. Ed. 56 (41): 12503-07 |
| Figures: | 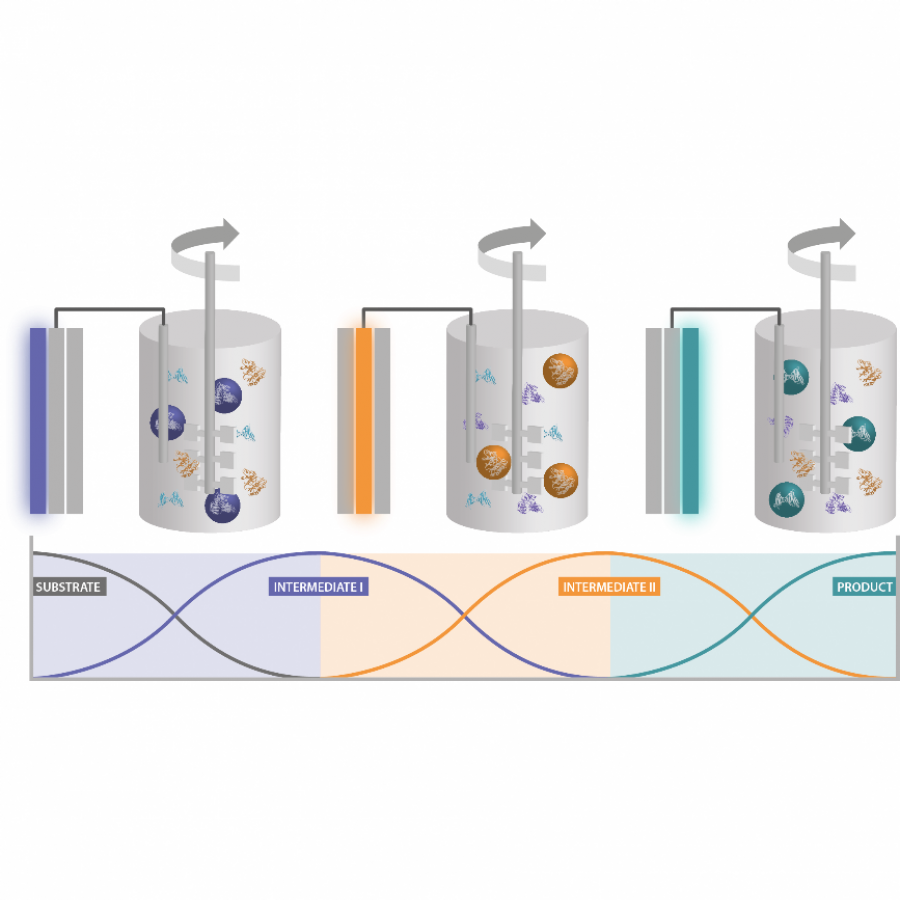 Figure 1. Activity controlled one-pot multi-step reactor. With benchtop NMR online analytics, the reactant concentrations can be determined. This makes it possible to start subsequent steps, e.g. by enzyme supply or substrate feeding, and to inactivate the activity of a specific enzyme after completion of a step |
#979 | Stabilization and application of a Baeyer-Villiger monooxygenase as protein scaffold in photochemical reactions |
|
| Presenting author: | Ioannis PAVLIDIS of UNIVERSITY OF CRETE |
| Other authors: | Christina MICHALOPOULOU of UNIVERSITY OF CRETE Tzesi MERTIKA of UNIVERSITY OF CRETE Charitomeni ANGELI of UNIVERSITY OF CRETE |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 11:15 am - 11:30 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | BVMO / photocatalysis / stabilization / singlet oxygen |
| Purpose: | Baeyer-Villiger-Monooxygenases (BVMOs), belong to the large family of Flavoprotein-Monooxygenases (FPMOs) and can catalyze the enzymatic Baeyer-Villiger oxidation reaction under mild conditions, leading to a sustainable chemical process, often accompanied with high enantio- and regioselectivity. However, there are many limitations to the application of BVMOs on an industrial scale, due to their low thermo-, pH-, solvent- and oxidative stability. Herein, we propose that flavin-dependent Type I BVMOs can act as protein-based photosensitizers for singlet oxygen generation, which can be utilized in synthetically useful photochemical reactions. The biochemical characterization of several wild-type enzymes available in house, highlighted the instability of these enzymes under conditions employed in photochemical reactions. Thus, Rational Design Mutagenesis was performed on the residues susceptible to oxidation, in order to enhance the stability of the proteins. We managed to construct a thermostable variant of the 2-oxo-Δ3-4,5,5-trimethylcyclopentenylacetyl-CoA monooxygenase from Pseudomonas putida (OTEMO), with the single mutation C444S. Applying this variant in photochemical reactions, we were able to provide the proof-of-principle of our hypothesis, which was the generation of singlet oxygen in the active site of the BVMO. The photochemical reactions with furan substrates consisted the benchmark reactions, that helped us to understand the principles governing these reactions and expand the substrate portfolio on nonfuran substrates. |
#1033 | Expanding the portfolio of native Amine Dehydrogenases by extensive biodiversity screening |
|
| Presenting author: | Carine VERGNE-VAXELAIRE of GENOSCOPE (CEA CNRS UPSACLAY) |
| Topic: | Enzyme discovery and engineering |
| Date: | 03:30 pm - 03:45 pm Session Enzyme engineering & Discovery #2 - Chair : T. WARD, University of Basel |
| Keywords: | amine dehydrogenases / biodiversity screening / metagenomic data / in silico analysis |
| Purpose: | In the course of the discovery of biocatalysts for amine production, interest in oxidoreductases has grown, particularly for enzymes catalyzing the NAD(P)H-reductive amination of ketones with ammonia to primary chiral amines. Previously restricted to engineered α-aminoacid dehydrogenases, this enzymatic toolbox has been recently extended by the discovery of genes coding for native AmDHs (nat-AmDHs) by our group[1] [2], in addition to reductive aminases and engineered ɛ-deaminating L-lysine dehydrogenase. Following the preliminary success of identification of other nat-AmDHs among metagenomic databases[3], a deep exploration of available sequenced biodiversity has been conducted. This gave rise to an extended nat-AmDH family of 17k sequences for which representative enzyme products were experimentally tested for reductive amination activity. The exploration of their active site diversity based on an Active Site Modelling and Clustering analysis[4], supported by crystallographic structures[2] [5] and 3D-models, led to the discovery of homologs with key structural variations that will be highlighted. Biocatalytic transformations with novel ketone and amine substrates will also be detailed (Figure 1). Furthermore, we will briefly present the in silico strategy carried out to isolate new distant homologs among a generated pool of ~ 20M NAD(P)-binding protein sequences, using HMM-HMM profile comparison. This work is supported by the Agence Nationale de la Recherche through the MODAMDH (ANR-19-CE07-0007) and ALADIN (ANR-21-ESRE-0021) projects. |
| References: | [1] L., Ducrot et al.; Advanced Synthesis and Catalysis, 2020, 363, 328-351. [2] O., Mayol et al.; Nature Catalysis, 2019, 2, 324-333. [3] A., Caparco et al.; Advanced Synthesis and Catalysis, 2020, 362, 2427-2436. [4] R. C. de Melo-Minardi et al., Bioinformatics, 2010, 26, 3075-3082 [5] M. Bennett et al., ChemBioChem, 2022, 23 (10), e202200136 |
| Figures: |  Identification of key enzymes among the extended nat-AmDH-family no legend |
#1186 | Novel approaches towards industrial bulk and specialty chemicals through chemoenzymatic cascades |
|
| Presenting author: | Harald GRÖGER of BIELEFELD UNIVERSITY |
| Topic: | Industrial biocatalysis |
| Date: | 02:00 pm - 02:45 pm Session Industrial biocatalysis - Chair : R. WOHLGEMUTH, University of Technology of Lodz |
| Keywords: | Industrial applications / Aldoxime dehydratases / Chemoenzymatic synthesis / Nitriles |
| Purpose: | Enzyme catalysis has emerged towards a key technology in the chemical industry, in particular for the production of chiral fine chemicals and pharmaceuticals. In contrast, applications of enzyme catalysis in the field of bulk chemicals and specialty chemicals are still rare. In general, among current challenges in enzyme catalysis related to production of products in these high-volume low-cost product segments are the realization of production processes running at high substrate loading (being in a multi-100 g/L range) together with economic work-up steps as well as the efficient combination of biotransformations with chemocatalytic reactions towards multi-step synthetic sequences. The development of chemoenzymatic one-pot cascades, being a young but emerging field of research,[1] avoids the need for tedious work-up steps such as isolation and purification of intermediates, thus offering a perspective for decreasing the overall number of unit operation steps, needed amount of solvent (used for extraction) and consequently lowering the amount of formed waste. In this presentation, examples from our recent research collaborations with academic and industrial project partners to develop technically feasible biocatalytic and chemoenzymatic processes for applications in the field of specialty and bulk chemicals starting from biorenewable feedstocks will be given. One case-study is related to the development of a new technology platform for chiral and achiral nitriles.[2-6] By means of aldoxime dehydratases as a biocatalyst, a cyanide-free approach to nitriles starting from readily available aldehydes was established and a broad substrate scope was demonstrated. This process technology can be used for synthesizing the polymer building block adiponitrile, benzonitriles as specialty chemicals as well as fatty nitriles as precursors for detergents and lubricants,[4-6] The enzymatic process for fatty nitriles, which was developed jointly with Klüber Lubrication, runs at a high substrate loading of up to 1.4 Kg per L of aqueous reaction medium, thus representing one of the highest substrate loadings in enzyme catalysis with hydrophobic substrates in aqueous media.[6] Furthermore, a chemoenzymatic process consisting of an initial metal-catalyzed hydroformylation of an alkene as high pressure reaction under formation of the corresponding aldehyde, which was then converted into the desired nitrile by means of enzyme catalysis, has been developed.[7] A second case-study is the development of a chemoenzymatic process to produce the polymer-building block caprolacton starting from biorenewable raw materials. Polycaprolacton is of interest as a polymer also due to its favorable biodegradability properties, but is today produced from fossil feedstocks. A chemoenzymatic process has been developed based on metal-catalyzed hydrogenation of phenol (which is available in bio-based form on large scale today), followed by a biocatalytic "double oxidation" of cyclohexanol with molecular oxygen as only reagent.[8] Currently, the further development of this process concept is done within a joint research project with BYK-Chemie/ALTANA. In a third case study, preliminary results on the combination of metal-catalyzed syngas high pressure chemistry, hydrogenation and enzymatic esterification within a chemoenzymatic process for producing tailor-made lubricants is presented. Within a joint project with OQ Chemicals (previously OXEA) and Klüber Lubrication, oligomeric esters are accessible based on enzymatic esterification within a solvent-free process under neat conditions, which enables a fine-tuning of the performance properties of the lubricant molecules and resulted in the formation of a new generation of bio-based lubricants with proven biodegradability.[9] |
| References: | [1] H. Gröger, F. Gallou, B. H. Lipshutz, Chem. Rev. 2023, in print, DOI: 10.1021/acs.chemrev.2c00416. [2] T. Betke, P. Rommelmann, K. Oike, Y. Asano, H. Gröger, Angew. Chem. 2017, 129, 12533-12538; Angew. Chem. Int. Ed. 2017, 56, 12361-12366. [3] H. Yavuzer, Y. Asano, H. Gröger, Angew. Chem. 2021, 133, 19311-19317; Angew. Chem. Int. Ed. 2021, 60, 19162-19168. [4] T. Betke, M. Maier, H. Gruber-Wölfler, H. Gröger, Nature Commun. 2018, 9, 5112. [5] M. Hinzmann, H. Yavuzer, M. Bittmann, H. Gröger, Chem Catal. 2023, 3, 100572. [6] A. Hinzmann, S. Glinski, M. Worm, H. Gröger, J. Org. Chem. 2019, 84, 4867-4872. [7] C. Plass, A. Hinzmann, M. Terhorst, W. Brauer, K. Oike, H. Yavuzer, Y. Asano, A. J. Vorholt, T. Betke, H. Gröger, ACS Catal. 2019, 9, 5198-5203. [8] S. Wedde, P. Rommelmann, C. Scherkus, S. Schmidt, U. T. Bornscheuer, A. Liese, H. Gröger, Green Chem. 2017, 19, 1286-1290. [9] L. Koch, A. Guntermann, C. Plass, T. Betke, L. Ma, K. Hirschbichler, T. Kilthau H. Gröger, manuscript submitted for publication. |
#1239 | ILLUMINATING BIOCATALYSIS: COMBINING CHEMICAL AND ENZYMATIC TRANSFORMATIONS FUELED BY LIGHT |
|
| Presenting author: | Sandy SCHMIDT of UNIVERSITY OF GRONINGEN |
| Topic: | (Chemo)enzymatic strategies |
| Date: | 02:00 pm - 02:30 pm Session Bio-photo - Electro-catalysis - Chair : B. DOUMECHE, University of Lyon |
| Keywords: | Biocatalysis / Photocatalysis / Rieske oxygenase / Cascade |
| Purpose: | Enzyme catalysis and photocatalysis are two research areas that have become of great interest in organic synthesis. This is mainly because both represent attractive strategies for making chemical synthesis more efficient and sustainable. It is therefore not surprising that bio- and photocatalytic approaches are now often combined to exploit the exquisite selectivity of enzymes and the unique chemical transformations accessible to photocatalysis.[1–3] In recent work, we have investigated the application of photobiocatalytic strategies using different photosensitizers and sacrificial electron donors to drive Rieske oxygenase (RO)-catalyzed hydroxylations in vivo,[4] demonstrating that light-induced electron transfer leads to similar catalytic activities as in whole-cell reactions supplemented with glucose. We are currently exploring the possibility of using this light-induced electron transfer via photosensitizers as a simple way of overcoming the need for the natural redox partners of ROs. This is important because the identification of the physiological redox partner(s) of a given RO is often hampered by the fact that host genomes typically contain numerous candidate genes encoding redox partner proteins, most of which are not located near the RO gene(s). Thus, a light-driven approach to screen for RO activity without the physiological redox partner(s) would provide an effective surrogate electron supply system for functional characterization and/or biocatalytic application of ROs. In another example, we have shown that the combination of bio- and photocatalysis provides a highly valuable approach to building molecular complexity from simple, cheap and widely available starting materials.[5] By combining photocatalytic C-C bond formation with enzymatic asymmetric reduction, we have demonstrated the direct conversion of simple aldehydes and acrylates or unsaturated carboxylic acids to chiral gamma-lactones. The photochemoenzymatic synthesis of aliphatic and aromatic gamma-lactones was thereby achieved with up to >99% ee and 99% yield. |
| References: | [1] L. Schmermund, V. Jurkaš, F. F. Özgen, G. D. Barone, H. C. Büchsenschütz, C. K. Winkler, S. Schmidt, R. Kourist, W. Kroutil, ACS Catal. 2019, 9, 4115-4144. [2] W. Harrison, X. Huang, H. Zhao, Acc. Chem. Res. 2022, 55, 1087-1096. [3] F. F. Özgen, M. E. Runda, S. Schmidt, ChemBioChem 2021, 22, 790-806. [4] F. Feyza Özgen, M. E. Runda, B. O. Burek, P. Wied, J. Z. Bloh, R. Kourist, S. Schmidt, Angew. Chem. Int. Ed. 2020, 59, 3982-3987. [5] F. F. Özgen, A. Jorea, L. Capaldo, R. Kourist, D. Ravelli, S. Schmidt, ChemCatChem 2022, 14, e202200855. |
#1264 | Production of non-natural products by microbial cell factories using artificial metabolic pathways |
|
| Presenting author: | Akihiko KONDO of KOBE UNIVERSITY |
| Topic: | Biocatalytic cascade reactions |
| Date: | 09:45 am - 10:15 am Session Cascade reaction - Chair : M. HALL, University of Graz |
| Keywords: | Biofoundry / Engineering Biology / Artificial Enzyme / Metabolic Pathway |
| Purpose: | We have developed rapid cell factory construction technology (biofoundry platform), which is an integrated system (DBTL) of advanced technologies such as metabolic design system (Design), rapid breeding technology using advanced genome engineering technologies (Build), rapid and accurate metabolic evaluation technology (Test), and machine learning or mathematical modelling for further improvement and a new metabolic pathway design (Learn). The biofoundry platform, a fusion of biotechnology and digital technology, is expected to be a major force not only in the construction of cell factories but also in the development of various biotechnologies. In the biofoundry platform, to design and construct microorganisms that produce useful compounds, it is essential to have the technology to optimally design "metabolism," which includes not only the flow of carbon within the cell, but also the production and consumption of energy and the balance of redox. The limitation of the previous design tools is that it cannot design artificial metabolic pathways for the production of non-natural compounds that are not produced by living organisms, because they use only natural metabolic pathways existing in KEGG. Therefore, the BioProV method was newly devised to describe only chemical reaction patterns from the databases of metabolic and enzymatic reactions, such as KEGG and BRENADA. In this methods, similar chemical reaction patterns were reclassified as a single chemical reaction and trained by a computer. Then, the precursors were reverse synthesized from the target compounds in a random and exhaustive manner. The simulation is successful when the reverse-synthesized precursor contains a compound that is known to exist in vivo. With this BioProV, it is now possible to design artificial metabolic pathways to synthesize non-natural compounds. However, since we focused only on chemical reaction patterns, we need a technology to rapidly create artificial enzymes with specificity that does not exist in nature. Here, the important thing in the construction of artificial metabolic pathways became the problem of enzyme engineering: how to quickly create highly active artificial enzymes with the desired reaction specificity. As an example of microbial production of non-natural compounds by designing artificial metabolic pathways, I describe the case of butadiene. Using BioProV, an artificial metabolic pathway design tool, we found a pathway to produce 1,3-butadiene via a two-step decarboxylation reaction using cis,cis-muconic acid (ccMA), as a starting compound. The next challenge is to create an artificial enzyme that performs a two-step decarboxylation reaction of cis,cis-muconic acid (ccMA), which does not exist in nature. We modified the substrate specificity of ferulic acid decarboxylase (FDC), which has very wide substrate specificity, to create an artificial enzyme that converts ccMA to 1,3-butadiene and incorporated to create an artificial metabolic pathway for 1,3-butadiene production in Escherichia coli. Using this E, coli which has artificial metabolic pathway with artificial enzyme, we have succeeded to produce 1,3-butadiene from glucose. As shown in this example, it is very important to rapidly create artificial metabolic pathways with artificial enzymes to construct microbial cell factories for the production of non-natural products by using biofoundry platform. To solve this problem, we are now constructing the new enzyme and pathway database for biofoundry platform. I will describe the current progress of our approach. |
| References: | [1] Mori, Y., Noda, S., Shirai, T., Kondo, A., Nature communications, 12: 2195 (2021) [2] Shirai, T., Kondo, A., biomolecules, 12(5), 620 (2022) |
#1657 | Search & Optimization of Novel Proteins by 3D Point-Cloud Catalophores |
|
| Presenting author: | Bettina NESTL of INNOPHORE GMBH |
| Topic: | Artifical intelligence / computational methods |
| Date: | 09:45 am - 10:15 am Session AI & Computational methods - Chair : M. FRAAIJE, University of Gröningen |
| Keywords: | / / / |
| Purpose: | Biocatalysis is increasingly becoming a key technology for developing a more environmentally friendly and efficient chemical industry. Some natural enzymes do not have the required activity, stability, selectivity, or specificity for many desired chemical transformations. Recent advances in computational technology offer a potential roadmap for the search, identification, and development of new biocatalysts. Innophore, a TechBio company, is using AI-powered point cloud technology to identify and optimize drugs and proteins. Point clouds provide a structurally independent approach to representing the properties of voids or surfaces. Each point in the cloud corresponds to a property at a specific location, making it a valuable tool for separating structural information from function. In addition, these point clouds serve as search templates for identifying proteins with similar binding features. We focus on volumetric voids and surface volumes to predict function, properties, and reactivity. Using our Catalophores™ point cloud, we are accelerating the discovery of new and better enzymes by translating biomolecules into machine-readable datasets. This talk will review the strategies and opportunities for applying point clouds to enzyme and drug discovery. |
#1659 | Biocatalytic Aerobic Oxidations for the Synthesis of Pharmaceuticals |
|
| Presenting author: | Juan E VELASQUEZ of PROCESS RESEARCH AND DEVELOPMENT |
| Topic: | Industrial biocatalysis |
| Date: | 02:45 pm - 03:15 pm Session Industrial biocatalysis - Chair : R. WOHLGEMUTH, University of Technology of Lodz |
| Keywords: | Biocatalysis / Hydroxylation / Oxygenases / Oxidases |
| Purpose: | Aerobic oxidations using biocatalysts offer a promising solution for addressing key challenges in the chemical industry, such as selective C-H functionalization and C-C or C=O bond formation, while improving atom economy, process safety, and reducing environmental impact. Despite their potential, the large-scale implementation of oxygen-dependent biotransformations is limited due to the need for extensive protein engineering and efficient oxygen delivery methods to achieve optimal reaction performance. In this talk, we will summarize recent developments in the discovery and engineering of oxygenases and oxidases, as well as their application in biocatalytic processes for manufacturing Belzutifan, Islatravir, and Molnupiravir. We will highlight the challenges associated with enzyme-catalyzed oxidations and the strategies employed to overcome them. Our work expands the scope of biocatalytic aerobic oxidations and highlights their potential for sustainable and efficient chemical synthesis. |
#1661 | New Insights into Anthocyanin Biosynthesis |
|
| Presenting author: | Rebecca BULLER of ZURICH UNIVERSITY OF APPLIED SCIENCES |
| Topic: | Enzyme engineering & Discovery |
| Date: | 02:45 pm - 03:15 pm Session Enzyme engineering & Discovery #2 - Chair : P. CLAPES, Institute for Advanced Chemistry of Catalonia |
| Keywords: | anthocyanin biosynthesis / enzyme mechanism / yeast engineering / protein structure |
| Purpose: | Anthocyanins are ubiquitous plant pigments, which participate in the attraction of pollinators and seed dispersers as well as in stress response. The food, nutraceutical, and cosmetic industries increasingly adopt the use of these bioactive molecules due to their diverse color palette and their many proposed health benefits. To produce the pigments on a large scale, plant raw materials with high levels of stable anthocyanins are typically extracted, despite the associated issues of sustainability and supply. With over a century of research focused on the biochemistry and regulation of the natural colorants’ biosynthesis, the route toward anthocyanins is often considered the best understood plant secondary metabolic pathway. Nevertheless, construction of microbial cell factories to produce anthocyanins with commercially viable product titers has not yet been achieved due to issues arising from the late steps of the biosynthetic pathway. In our study, we focus on the penultimate biosynthetic step to the colored anthocyanidins which is thought to be catalyzed by leucoanthocyanidin dioxygenase. However, this reaction has never been fully reconstituted in vitro or in heterologous microorganisms. Based on biochemical, structural, and computational evidence, we present new insights into key enzymatic transformations. Based on this knowledge, we succeeded to incorporate a heterologous pathway into baker's years leading to greatly increased anthocyanin production. Our elucidation of the long-elusive late biosynthesis of anthocyanins paves the way for the construction of microbial production platforms for these relevant natural colorants and will impact the breeding of industrial and ornamental plants. |
#1662 | Fatty Acid Photodecarboxylases: mechanism, substrate specificity and use in biocatalysis and biotransformation |
|
| Presenting author: | Frédéric BEISSON of INSTITUTE OF BIOSCIENCES AND BIOTECHNOLOGIES OF AIX-MARSEILLE |
| Topic: | Reaction design |
| Date: | 09:45 am - 10:15 am Session Reaction design - Chair : A. BOMMARIUS, Georgia Tech |
| Keywords: | Hydrocarbons / Fatty acids / Photoenzymes / Decarboxylation |
| Purpose: | Fatty acid photodecarboxylase (FAP, EC 4.1.1.106) is a natural photoenzyme converting fatty acids into hydrocarbons that we originally isolated from the green microalgae Chlorella variabilis (1). FAP activity has been shown to be conserved in a variety of algae and is thought to play a role in photosynthetic membranes (2). Being members of the glucose-methanol-choline (GMC) oxidoreductase family, FAPs harbor a flavin adenine dinucleotide (FAD) as their chromophore. The structure and photocycle of Chlorella variabilis FAP (CvFAP) have been characterized in detail (3). CvFAP has also been shown to be an interfacial enzyme that prefers fatty acids present at organized lipid-water interfaces such as liposomes and microemulsions (4). We also recently showed that CvFAP is highly active on shorter fatty acids than previously thought and provided spectroscopic evidence for an autocatalytic effect on octanoic acid (5). CvFAP appears to represent an attractive light-focused and redox-neutral medium for the production of not only n-alkanes and n-alkenes, but also other specialty chemicals such as enantiomerically pure alpha-amino and alpha-hydroxy carboxylic acids, secondary fatty alcohols and aliphatic amines and esters. In this talk, I will present our structural, mechanistic and enzymology studies on CvFAP. Some of the potential applications of CvFAP in biocatalytic or biotransformation processes that have been explored by several groups since its discovery will also be reviewed. |
| References: | (1) D. Sorigué, B. Légeret, S. Cuiné, S. Blangy, S. Moulin, ..., F. Beisson (2017) Science 357, 903-907. (2) S. L. Y. Moulin, A. Beyly-Adriano, S. Cuiné, S. Blangy, B. Légeret,..., F. Beisson (2021) Plant Physiology 186, 1455-1472. (3) D. Sorigué, K. Hadjidemetriou, S. Blangy, G. Gotthard, A. Bonvalet, ….,, F. Beisson (2021) Science 372, eabd5687. (4) C. Aselmeyer, B. Légeret, A. Bénarouche, D. Sorigué, G. Parsiegla, F. Beisson, F. Carrière (2021) Biochemistry 60, 3200-3212. (5) P.P. Samire, B. Zhuang, B. Légeret, Á. Baca-Porcel, G. Peltier, D. Sorigué, A. Aleksandrov, F. Beisson, P. Müller (2023) Science Advances 9, eadg3881. |
| Figures: | 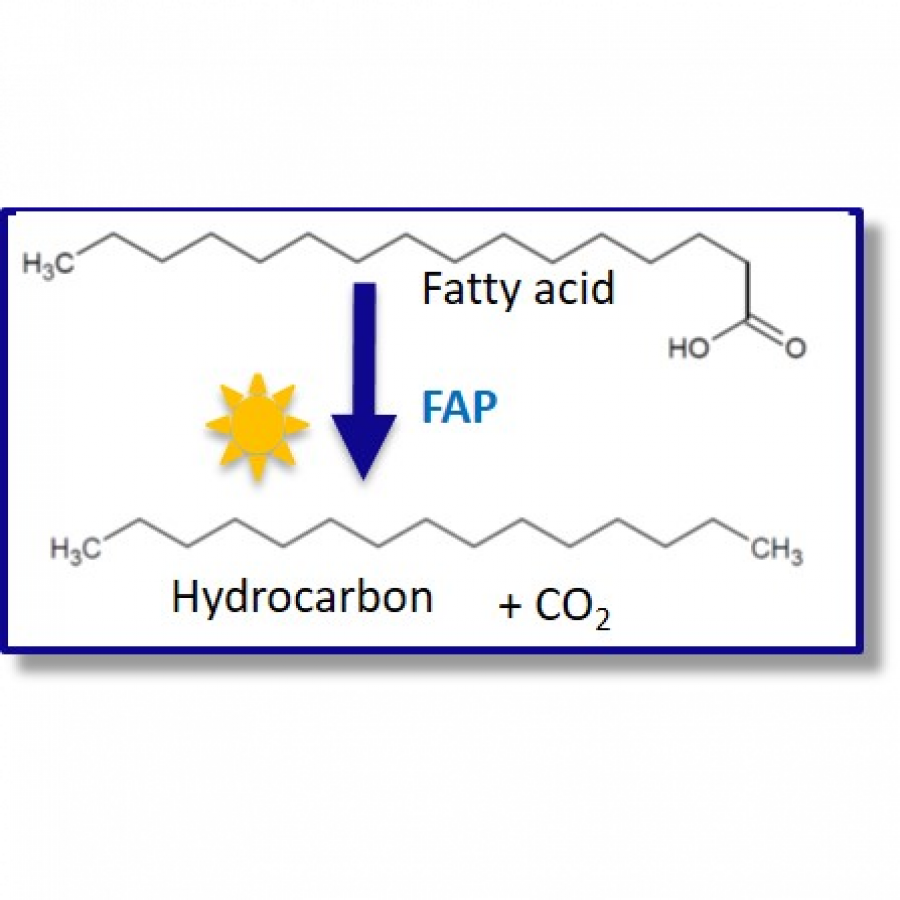 Figure 1 Reaction catalyzed by Fatty Acid Photodecarbxylase |
#1663 | PET RECYCLING: FROM ENZYME AND PROCESS OPTIMIZATION TO AN INDUSTRIAL PLANT |
|
| Presenting author: | Alain MARTY of CARBIOS |
| Topic: | Enzyme engineering & Discovery |
| Date: | 10:45 am - 11:15 am Session Reaction design - Chair : H. HAILES, University College London |
| Keywords: | PET recycling / enzyme engineering / PETase / industrial plant. |
| Purpose: | Plastics are found everywhere in our daily life due to exceptional properties. The worldwide market reaches 400 million tons. However, they represent a major environmental issue with 125 million tons of generated plastic waste annually. Only 10% of collected plastics are recycled, and, at best, plastic wastes are incinerated but an unacceptable quantity are lost in nature, with 9 million tons ending each year in the oceans. Carbios (http://www.carbios.com), a young innovative green chemistry company, in collaboration with the laboratory TBI (Toulouse Biotechnology institute; INSA/CNRS/INRAE; http://www.toulouse-biotechnology-institute.fr), developed an enzymatic process to recycle one of the main plastics, PET (~100 million tons per year). A first breakthrough was reached with the optimization of an extraordinary PETase used to break down PET returning to monomers (Nature; Vol. 580 Issue 7802, 9 April 2020). Since then, we continue to optimize this enzyme, to improve kinetics and yields and the performances of our best enzymes will be presented. The scale-up of the process in an industrial demonstrator will be presented with a 20m3 reactor and all the downstream processing to purify both terephthalic acid and ethylene glycol. Carbios is building a first industrial unit in France, operational in 2025, which will recycle 50,000 tonnes of PET waste per year. |
#1664 | Coupling retrosynthesis with automated in vitro and in vivo metabolic pathway engineering |
|
| Presenting author: | Jean-Loup FAULON of INRAE |
| Topic: | Synthetic biology, metabolic engineering |
| Date: | 09:00 am - 09:45 am Session Cascade reaction - Chair : M. HALL, University of Graz |
| Keywords: | Biochemical reaction networks / Computational platforms and environments / Metabolic engineering / Synthetic biology |
| Purpose: | This presentation will introduce Galaxy-SynBioCAD [1], a Galaxy toolshed for synthetic biology, metabolic engineering, and industrial biotechnology. The tools and workflows currently shared on the toolshed enable one to build libraries of strains producing desired chemical targets covering an end-to-end metabolic pathway design and engineering process from the selection of strains and targets, the generation of pathways producing the targets, the design of DNA parts to be assembled, to the generation of scripts driving liquid handlers for plasmid assembly and strain transformations (cf. Fig. 1). Pathways are designed using retrosynthesis methods [2], and several template-based and template-free alternatives to perform that task will be compared. Data standards to enforce compatibility and to chain up tools into workflows will also be presented. The link between pathway design and engineering will be illustrated with the build-up of a library of E. coli lycopene-producing strains in a study carried out at four different sites. The workflows were also benchmarked on literature and expert-validated pathways. Overall, 83% success rate was found in retrieving the validated pathways among the top 10 pathways generated by the workflows (cf. Fig 2). Ongoing developments within the Galaxy-SynBioCAD toolshed will also be presented, these include integration of DoE and active learning modules for bioproduction in vivo and in cell-free [3,4]. |
| References: | 1. Hérisson J, et al.. The automated Galaxy-SynBioCAD pipeline for synthetic biology design and engineering. Nat Commun., 2022, 13(1):5082. | DOI: 10.1038/s41467-022-32661-x 2. Koch M, et al. Reinforcement Learning for Bioretrosynthesis. ACS Synthetic Biology, 2020, 9(1): 15. | DOI: 10.1021/acssynbio.9b00447 3. Pandi A, et al.. A versatile active learning workflow for optimization of genetic and metabolic networks. Nat Commun., 2022, 13(1):3876. | DOI: 10.1038/s41467-022-31245-z 4. Borkowski O, et al.. Large scale active-learning-guided exploration for in vitro protein production optimization. Nature Communications, 2020, 11(1): 1872. | DOI: 10.1038/s41467-020-15798-5 |
| Figures: |  Galaxy-SynBioCAD nodes and worflows Main nodes in the Galaxy toolshed 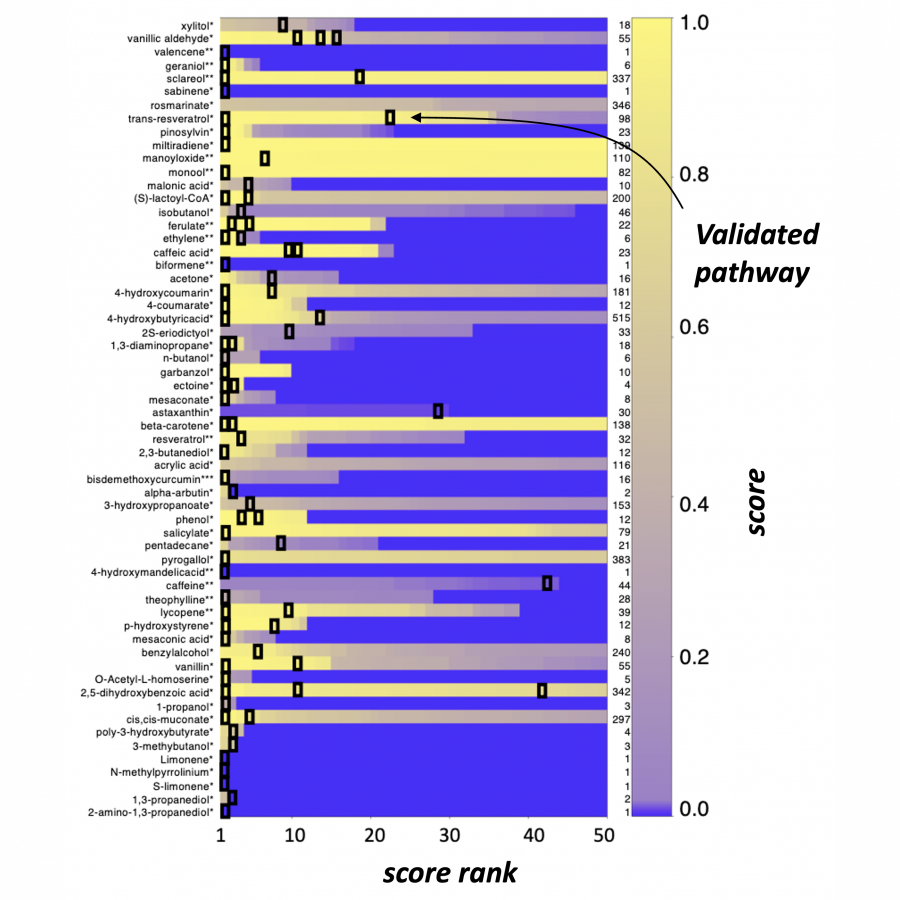 Ranking predicted pathways with machine learning global score. Color code on the right side shows the machine learning global score (from 1 top to 0). Black boxes show the location of the literature or expert-selected pathways for a set 60 literature target engineered in E. coli (*), yeast (**) or P. putida (***) |
#1665 | Biocatalysts for New-to-Nature Reactions: Engineered Metalloenzymes for Asymmetric C-C and C–N Bond Formation via Carbene Transfer |
|
| Presenting author: | Rudi FASAN of UNIVERSITY OF ROCHESTER |
| Topic: | Enzyme engineering & Discovery |
| Date: | 03:00 pm - 03:30 pm Award Junior - Chair : S. RIVA, University of Milan |
| Keywords: | / / / |
| Purpose: | Expanding the reaction scope of biological catalysts beyond the realm of enzymatic transformations occurring in nature can offer new opportunities for the exploitation of biocatalysis for organic chemistry and asymmetric synthesis. Our group has been engaged in the design, investigation, and application of engineered hemoproteins for enabling the construction of new carbon-carbon and carbon-nitrogen bonds via ‘abiological’ enzyme-catalyzed carbene and nitrene transfer reactions. These efforts have led to the development of efficient and highly stereoselective biocatalysts for the synthesis of optically active cyclopropanes and amines as well as the asymmetric C(sp3)–H functionalization of heterocycles and other valuable scaffolds for medicinal chemistry. As illustrated by representative examples discussed during this talk, these biocatalytic strategies are amenable to gram-scale synthesis and can be applied to enable the chemoenzymatic synthesis and late-stage C–H functionalization of pharmaceuticals and other bioactive molecules. |
#1666 | To be determined |
|
| Presenting author: | Donald HILVERT of ETH |
| Topic: | Enzyme engineering & Discovery |
| Date: | 03:30 pm - 04:00 pm Award Senior - Chair : W.D. FESSNER, Technical University of Darmstadt |
| Keywords: | / / / |
| Purpose: | To be determined |
#1668 | opening |
|
| Presenting author: | Fred LE GALL of LORD |
| Topic: | Reaction design |
| Date: | Welcome introduction : L. HECQUET - Chair BIOTRANS,UCA - 04:00 pm - 04:15 pm |
| Keywords: | / / / |
| Purpose: | e |
#1746 | e |
|
| Presenting author: | Fred LE GALL of LORD |
| Topic: | Biocatalytic cascade reactions |
| Date: | Opening adress : S. SAGAN - Deputy CSO, INC-CNRS - 04:15 pm - 04:30 pm |
| Keywords: | / / / |
| Purpose: | e |
#1772 | The impact of AlphaFold on structural biology |
|
| Presenting author: | Alexander PRITZEL of DEEPMIND |
| Topic: | Artifical intelligence / computational methods |
| Date: | 09:00 am - 09:45 am Session AI & Computational methods - Chair : M. FRAAIJE, University of Gröningen |
| Keywords: | / / / |
| Purpose: | Machine learning systems have grown to be an invaluable tool in computational biology. In particular structural biology has been transformed in recent years since the release of AlphaFold, with protein structure prediction tools becoming a core part of the workflow in structural biology. In this talk I will discuss recent advances in the application of machine learning methods to computational biology, with a specific focus on structural biology. |
#1773 | Building Enzymes with New Function |
|
| Presenting author: | Anthony GREEN of MANCHESTER |
| Topic: | Artificial enzymes and de-novo enzyme design |
| Date: | 09:45 am - 10:15 am Session Enzyme engineering & Discovery #1 - Chair : M. REMAUD-SIMEON, Toulouse Biotechnology Institute |
| Keywords: | biocatalysis / enzyme design and engineering / non-canonical amino acids / enzymes with new functions |
| Purpose: | Enzyme design and evolution strategies typically rely exclusively on Nature’s standard amino acid alphabet of twenty canonical residues which contain limited functionality. Here we demonstrate that incorporation of non-canonical amino acids into enzyme active sites provides a fruitful avenue to probe complex biological mechanisms and can lead to the creation of designed enzymes with wholly new catalytic functions. Significantly, optimization of enzyme activity can be achieved using directed evolution workflows adapted to an expanded genetic code. We are optimistic that this integration of enzyme design, genetic code expansion and laboratory evolution can provide a versatile strategy for creating enzymes with catalytic functions not accessible to Nature. |
| References: | [1] J. S. Trimble, R. Crawshaw, F. J. Hardy, C. W. Levy, M. J. B. Brown, D. E. Fuerst, D. Heyes, R. Obexer, A. P. Green, A Designed Photoenzyme for Enantioselective [2+2]-Cycloadditions. Nature 2022, 611, 709. [2] E. Bell, R. Smithson, S. Kilbride, J. Foster, F. J. Hardy, S. Ramachandran, A. Tedstone, S. Haigh, A. Garforth, P. Day, C. Levy, M. Shaver, and A. P. Green, Directed Evolution of an Efficient and Thermostable PET Depolymerase. Nature Catalysis, 2022, 5, 673. [3] S. L. Lovelock, R. Crawshaw, S. Basler, C. Levy, D. Baker, D. Hilvert, A. P. Green, The Road to Fully Programmable Protein Catalysis. Nature, 2022, 606, 49. [4] R. Crawshaw, A. E. Crossley, L. Johannissen, A. J. Burke, S. Hay, C. Levy, D. Baker, S. L. Lovelock, A. P. Green, Engineering an Efficient and Enantioselective Enzyme for the Morita-Baylis-Hillman Reaction. Nature Chemistry, 2022, 14, 313-320. [5] A. J. Burke, W. R. Birmingham, Y. Zhuo, T. W. Thorpe, B. Zucoloto da Costa, R. Crawshaw, I. Rowles, J. D. Finnigan, C. Young, G. M. Holgate, M. P. Muldowney, S. J. Charnock, S. L. Lovelock, N. J. Turner, A. P. Green, An Engineered Cytidine Deaminase for Biocatalytic Production of a Key Intermediate of the Covid-19 Antiviral Molnupiravir. J. Am. Chem. Soc., 2022, 144, 3761-3765. |